

Abstract
Objective:
Annular pancreas is an uncommon cause of duodenal obstruction in children. In this study, the clinical, radiological, and prognostic findings related to this disorder over a 12-year review period were analyzed.
Materials and Methods:
A retrospective review of 22 patients with annular pancreas who were treated with surgical repair between April 1998 and February 2010 was performed at two different pediatric surgical units. Presenting symptoms, associated anomalies, radiological findings, the type of surgery performed, postoperative outcomes, and complications were analyzed.
Results:
Twenty-two patients were identified. Thirteen of the 22 patients (59.1%) were born prematurely, 11 patients (50%) had low birth weight, 2 patients (9.1%) had very low birth weight and 1 patient (4.5%) had extremely low birth weight. The mean birth weight was 2285.23±675.12 g. (970-3300). All patients presented with vomiting, which was bilious in nine (40.9%). Nine patients (40.9%) had chromosomal anomalies. Corrective surgery consisted of duodenoduodenostomy in 9 patients (40.9 %), duodenojejunostomy in 9 patients (40.9%), and gastrojejunostomy in 4 patients (18.1%). Fourteen of the 22 patients have survived (63.6%). The causes of death were combinations of sepsis, pneumonia, brain hemorrhage, and cardiac anomaly.
Conclusion:
Infants with annular pancreas associated with duodenal obstruction were often born prematurely and/or had low birth weights; many had cardiovascular anomalies. Annular pancreas associated with duodenal obstruction correlated strongly with the trisomy 21 karyotype among the chromosomal anomalies, as did duodenal atresia. The oral feeding tolerance time was nearly the same for all patients regardless of the surgical procedure used.
Keywords: Child, Duodenal obstruction, Pancreas
Özet
Amaç:
Annüler pankreas çocuklarda duodenum tıkanıklığının ender bir nedenidir. Bu çalışmada, 12 yıllık zaman periyodunda hastalıkla ilgili klinik, prognostik ve radyolojik bulgular analiz edildi.
Gereç ve Yöntem:
İki farklı cerrahi klinik de Nisan 1998 ve Temmuz 2010 tarihleri arasında annüler pankreas nedeni ile cerrahiye maruz kalan 22 hastanın kayıtları geriye dönük olarak incelendi. Başvuru semptomları, hastalığa eşlik eden anomaliler, radyolojik bulgular, uygulanan ameliyat tipleri, ameliyat sonrası sonuçlar ve komplikasyonlar analiz edildi.
Bulgular:
Toplam 22 hasta çalışmaya dahil edildi. 22 hastanın 13’ü (%59.1) prematüre, 11’i (%50) düşük doğum ağırlıklı, 2’si (%9.1) çok düşük doğum ağırlıklı ve 1’i (%4.5) çok çok düşük doğum ağırlıklı idi. Ortalama doğum ağırlığı 2285.2±675.1 gramdı (970-3300). Hastaların tamamında başvuru esnasında kusma mevcuttu. Kusma içerik olarak 9 (%40.9) hastada safralıydı. 9 (%40.9) hastada kromozom anomalisi tespit edildi. 9 (40.9 %) hastaya düzeltici ameliyat olarak duodenoduodenostomi, 9 hastaya duodeno-jejunostomi (%40.9) ve 4 (%18.1) hastaya da gastro-jejunostomi yapıldı. Toplamda 22 hastanın 14’ü sağlıklı şekilde taburcu edildi (%63.6). Ölüm nedenleri, sepsis, pnömoni, beyin kanaması ve kardiyak anomalilerin bir kombinasyonu olarak gözlendi.
Sonuç:
Duodenum tıkanıklığı ile kendini gösteren, annüler pankreasa sahip olan yeni doğanlar genelde prematür ve düşük doğum ağırlıklıydı. Aynı zamanda bu hastaların büyük bir çoğunluğunda kardiyo vasküler anomaliler mevcuttu. Hastalarımızdan kromozom anomalisine sahip olanların nerdeyse tamamı Trizomi 21 karyotipine sahipti. Farklı ameliyat teknikleri kullanılan hastaların beslenmeye geçiş zamanları benzerlik gösterdi.
Introduction
Annular pancreas is an uncommon congenital anomaly and is usually asymptomatic. However, symptomatic patients most commonly present in infancy or early childhood [1, 2]. Past and recent reports have also described various presentations of annular pancreas in adults [3, 4]. To date, a few articles about isolated annular pancreas have been published in the English literature [1, 3, 5–7]. For this reason, we planned this study to document the characteristic features of 22 cases with symptomatic annular pancreas diagnosed during exploration. In addition, to our knowledge, this is the largest reported body of experience with this anomaly in children presented in the English literature. Other large studies of pediatric duodenal obstruction included patients with annular pancreas but did not analyze them separately [8, 9]. Our review aimed specifically to define the clinical, radiographic, therapeutic, and outcome characteristics of patients with duodenal obstruction.
Materials and Methods
We retrospectively reviewed the medical records of all patients with annular pancreas treated between 1998 and 2010. Sex, gender, gestational age, age at diagnosis, birth weight, presence and type of vomiting (bilious vs. non bilious), presence and type of intestinal obstruction (partial vs. complete), use of a prenatal ultrasound scan, type of radiographic tests done to confirm the diagnosis and additional congenital anomalies were recorded. Additionally, the type of surgical intervention, the number of postoperative ventilator days, the total number of days on parenteral nutrition, the number of days until oral feedings were initiated (and until the goal was reached), and the length of hospitalization were tabulated. Finally, immediate and short-term outcomes were identified.
Statistical analysis was performed using commercially available software (Statistical Package for the Social Sciences, version 11.0, SPSS Inc., Chicago, IL, USA).
Descriptive statistical analysis included the calculation of means, medians, and standard deviations of the obtained data. The chi-squared test was used for the statistical analysis of differences between groups. Student’s t test and one-way ANOVA were used for continuous variables. When small numbers invalidated the chi-squared approximation, the likelihood quotient was used. P values below.05 were considered statistically significant in all analyses, and the confidence interval (CI) was accepted as 95%.
Results
A total of 22 pediatric patients with annular pancreas were treated over a 12-year period. Ten were boys, and 12 were girls. Of the patients, 20 (90.9%) presented symptoms during the first week of life, 1 (4.5%) during the second week of life, 1 (4.5%) during the first month of life. The mean gestational age for all patients was 35.1±3.8 weeks (range: 28–40 weeks). Thirteen of the 22 patients (59.1%) were premature, 11 patients (50%) had low birth weight, 2 patients (9.1%) had very low birth weight and 1 patient (4.5%) had extremely low birth weight. The mean birth weight was 2285.2±675.1 g (range: 970-3300 g). Chromosomal anomalies existed in 9 patients (40.9%). Eight (36.3 %) patients had trisomy 21, and one (4.5%) had trisomy 18. Identified congenital gastrointestinal anomalies included malrotation in 5 patients (22.7%), duodenal atresia in 1 patient (4.5%), duodenal web in 1 patient (4.5%), Meckel diverticulum in 4 patients (18.1%), Morgagni hernia in 1 patient (4.5%) and anal atresia in 3 patients (13.6%). Twelve patients (54.5%) had cardiovascular anomalies such as an atrial septal defect, a ventricular septal defect, patent ductus arteriosus pulmonary hypertension, pulmonary stenosis, tricuspid failure, and mitral valve failure, but none of these anomalies required surgery. Other congenital anomalies included hypothyroid in 1 (4.5%) and cleft limb in 1 (4.5%).
Twelve patients (54.5%) had duodenal obstruction diagnosed by prenatal ultrasound scan. All of the patients presented with vomiting. Bilious vomit or gastric aspirate was observed for 9 patients (40.9%), and nonbilious vomit was observed for the rest of the patients. Plain abdominal films were taken for all patients and showed the typical double bubble with or without distal gas in 20 (90.9%) patients. The 2 remaining patients (9.1%) had only one gas shadow, representing a dilated stomach. Upper gastrointestinal contrast studies were obtained in 6 (27.2%) of the infants to confirm the diagnosis. All of these contrast studies showed a partial duodenal obstruction. Three patients (13.6%) had three or more other anomalies, all of which required additional surgical treatment. The surgical repairs to treat the additional defects included Ladd’s procedure for malrotation (22.7%) and hernia repair (4.5%) for Morgagni hernia. Fourteen of the 22 patients have survived (63.6%). The causes of death of the reaming patients were combinations of sepsis, pneumonia, brain hemorrhage, and cardiac anomaly. Annular pancreas was identified in all 22 patients during surgery. Corrective surgical procedures consisted of duodenoduodenostomy in 9 patients (40.9%), duodenojejunostomy in 9 patients (40.9%), and gastrojejunostomy in 4 patients (18.2%). None of the duodenums were tapered. There were no statistically significant differences in the starting time of enteral feeding among the surgical procedures (p=0.46). The average hospital stay was 24.1±15.5 days (range: 3–64). No statistically significant correlation between the surgical procedure and hospitalization time existed (p=0.45).
Ten patients (45.4%) required postoperative mechanical ventilation, with an average duration of 14.1 days. The mean duration of parenteral nutritional support was 15.2±11.5 days (range: 1–46 days). Patients started enteral feeding and achieved enteral feeding goals at an average of 15.4 days (range: 3–40) and 21.9 days (range: 3–64), respectively. The causes of death of 8 patients (36.3%) were reported such as sepsis, pneumonia, and renal insufficiency. Three of them died because of candida-related infection due to long-term total parenteral nutrition (death on days 24, 46, and 63). One patient died due to renal failure (970 g), and another died due to postoperative pneumonia and septicemia. One patient (1235 g) died during surgery, and one patient died day after surgery for no identifiable reason. One patient born at 28 weeks with pulmonary stenosis and anal atresia did not tolerate enteral feeding and died on day 16 after surgery. There was no statistically significant correlation between the surgical technique and the death rate (p=0.82). The effect of long-term TPN on the death rate was evaluated, and there was no significant correlation between them (p=0.32). Analysis of the gestational week of the patients and the death rate showed no relationship (p=0.7). Evaluation of the data on cardiac anomalies and death rates showed no correlation (p=0.8). No statistically significant correlation was found between the death rates and either gastrointestinal system anomalies or chromosomal anomalies (p=0.8 and p=0.18, respectively).
Discussion
Congenital duodenal obstruction during the newborn period is a relatively common abnormality and may be complete or partial, intrinsic or extrinsic. Intrinsic atresia or stenoses are relatively common. Duodenal obstruction has an incidence of 1 in 7,000 live births and accounts of half of small intestinal atresia [9, 10]. Although infants with complete duodenal atresia present during the neonatal period, duodenal web or stenosis may present later, during the first years of life. Extrinsic obstruction has many causes, including annular pancreas, malrotation, and anterior portal vein, which are frequently associated with duodenal atresia [9]. Annular pancreas is a developmental abnormality that occurs during the early development of the foregut. Annular pancreas is thought to occur when the ventral bud fails to rotate behind the duodenum, leaving pancreatic tissue fully encircling the second portion of the duodenum (Figure 1). Annular pancreas frequently coexists with intrinsic duodenal anomalies, and some cases of annular pancreas are found only incidentally during laparotomy [11]. All of our patients underwent surgery to treat duodenal obstruction, and the diagnosis of annular pancreas was confirmed during surgery.
Figure 1.
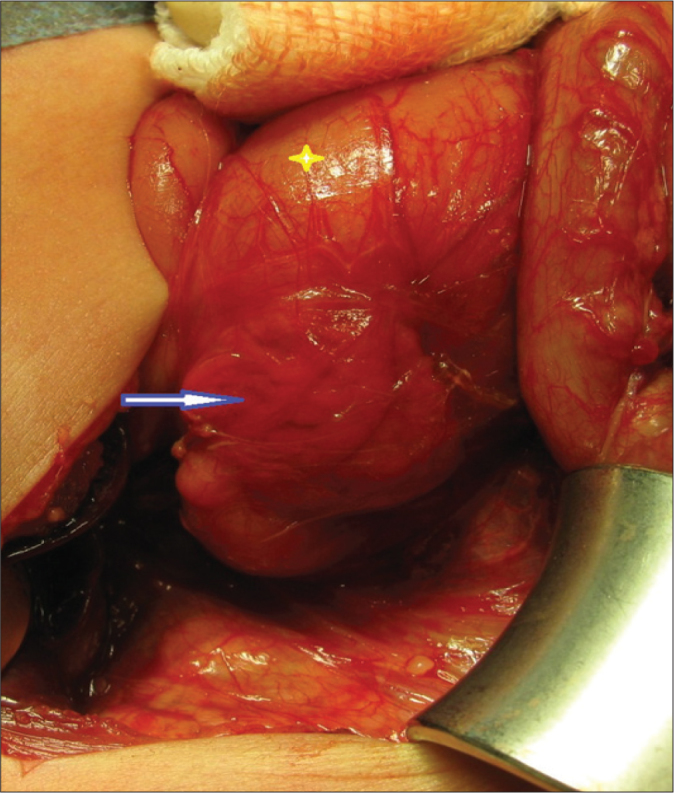
Annular pancreas encircling the second portion of the duodenum (arrow), and the dilated first part of the duodenum (*).
The obstruction was postampullary in nearly 80% of cases and preampullary in 20%. However, our study found that bilious vomit or gastric aspirate was observed in only 9 patients (40.9%). Because polyhydramnios is common, approximately 45% of these infants were born before term. More than half of our patients were born preterm, and half of the patients had low birth weight. These results seem parallel those in the literature. Additional anomalies were seen in 52% of patients. Trisomy 21 was present in approximately 30% of infants with duodenal atresia [9, 12]. We observed nearly the same rates of trisomy 21 among our cases. Other associated anomalies included congenital heart disease, esophageal atresia, anorectal malformations, renal anomalies, and biliary atresia [13]. We found congenital heart disease in 54.5% of our patients, similar to that found in other studies in the literature. On the other hand, congenital gastrointestinal anomalies, including malrotation, duodenal atresia, duodenal web, Meckel diverticulum, Morgagni hernia and anal atresia, were found to be the second most frequent congenital malformation.
Surgical procedures performed to treat duodenal atresia include duodenoduodenostomy, duodenojejunostomy and, less commonly, gastrojejunostomy [14,15]. Our surgical procedures were all duodenoduodenostomies or duodenojejunostomies. Kimura’s diamond-shaped end-to-end anastomosis technique was used for all of our duodenoduodenostomies, and we found statistically significant correlation between the surgical technique and the delay before starting enteral feeding.
Long-term outcomes for most of the patients with duodenal obstruction were excellent. Problems frequently developed for two reasons: abnormalities of gastrointestinal function, and difficulties due to associated structural or chromosomal anomalies [16]. Late gastrointestinal complications included megaduodenum, duodenogastric reflux, gastritis, peptic ulcers and gastroesophageal reflux. Of these, megaduodenum was the most common. Kimura reported no instances of megaduodenum in a series of patients who underwent diamond-shaped anastomosis, although Weber did not demonstrate any difference between the different techniques [11, 14, 17].
We investigated the causes of death of our patients and found that sepsis was the primary cause and resulted from long-term TPN. Megaduodenum may be the reason for dependence on TPN support. We evaluated all of these possibilities but found no statistically significant differences.
Long-term problems may also occur because of other structural or chromosomal anomalies, the most common of which is trisomy 21 [18]. Duodenal atresia and stenosis, intestinal malrotation, and trisomy 21 can be found in combination with annular pancreas at a rate of 10–20%. Relatively high mortality rates for duodenal obstructions associated with trisomy 21 have also been reported [19]. However, that is unlikely in our cases. The effect of trisomy 21 on the death rate of our cases did not seem to correlate with that reported in other studies. This rate may be largely due to the significantly higher incidence of congenital heart anomalies. In later series, decreasing mortality rates have been reported. The overall survival rates of infants with anomalies associated with duodenal obstruction have improved dramatically over the past several decades.
Early postoperative survival after the repair of duodenal atresia has steadily improved over the past 40 years, from approximately 60% to over 90% in practice, owing to improved neonatal and anesthesia management, total parenteral nutrition, and more aggressive treatment of associated anomalies [12]. Our survival rate was 65% forced us to the reasons. All possibilities were investigated with no success. All of our results showed us that the mortality rate can be reduced through improvement of our neonatal care system.
Footnotes
Conflict of interest statement: The authors declare that they have no conflict of interest to the publication of this article.
References
- 1.Merrill JR, Raffensperger JG. Pediatric annular pancreas: Twenty years’ experience. J Pediatr Surg. 1976;11:921–5. doi: 10.1016/s0022-3468(76)80067-x. [DOI] [PubMed] [Google Scholar]
- 2.McCollum MO, Jamieson DH, Webber EM. Annular pancreas and duodenal stenosis. J Pediatr Surg. 2002;37:1776–7. doi: 10.1053/jpsu.2002.36722. [DOI] [PubMed] [Google Scholar]
- 3.Kiernan PD, ReMine SG, Kiernan PC, Remine WH. Annular pancreas: Mayo clinic experience from 1957 to 1976 with review of the literature. Arch Surg. 1980;115:46–50. doi: 10.1001/archsurg.1980.01380010038007. [DOI] [PubMed] [Google Scholar]
- 4.Ladd AP, Madura JA. Congenital duodenal anomalies in the adult. Arch Surg. 2001;136:576–84. doi: 10.1001/archsurg.136.5.576. [DOI] [PubMed] [Google Scholar]
- 5.Sencan A, Mir E, Gunsar C, Akcora B. Symptomatic annular pancreas in newborns. Med Sci Monit. 2002;8:434–7. [PubMed] [Google Scholar]
- 6.Hays DM, Greaney EM, Jr, Hill JT. Annular pancreas as a cause of acute neonatal duodenal obstruction. Ann Surg. 1961;53:103–12. doi: 10.1097/00000658-196101000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kiesewetter WB, Koop CE. Annular pancreas in infancy. Surg. 1954;36:146–59. [PubMed] [Google Scholar]
- 8.Bailey PV, Tracy TF, Jr, Connors RH, Mooney DP, Lewis JE, Weber TR. Congenital duodenal obstruction: A 32-year review. J Pediatr Surg. 1993;28:92–5. doi: 10.1016/s0022-3468(05)80364-1. [DOI] [PubMed] [Google Scholar]
- 9.Grosfeld JL, Rescorla FJ. Duodenal atresia and stenosis: Reassessment of treatment and outcome based on antenatal diagnosis, pathologic variance, and long-term follow-up. World J Surg. 1993;17:301–9. doi: 10.1007/BF01658696. [DOI] [PubMed] [Google Scholar]
- 10.Stauffer UG, Irving I. Duodenal atresia and stenosis. Long term results. Prog Pediatr Surg. 1977;10:49–60. [PubMed] [Google Scholar]
- 11.Weber TR, Lewis J E, Mooney D, Connors R. Duodenal atresia: a comparision of technique of repair. J Pediatr Surg. 1986;21:1133–6. doi: 10.1016/0022-3468(86)90025-4. [DOI] [PubMed] [Google Scholar]
- 12.Keckler SJ, St Peter SD, Spilde TL, Ostlie DJ, Snyder CL. The influence of trisomy 21 on the incidence and severity of congenital heart defects in patients with duodenal atresia. Pediatr Surg Int. 2008;24:921–3. doi: 10.1007/s00383-008-2185-x. [DOI] [PubMed] [Google Scholar]
- 13.Cragan JD, Martin ML, Moore CA. Descriptive epidemiology of small intestinal atresia in Atlanta, Georgia. Teratology. 1993;48:441. doi: 10.1002/tera.1420480508. [DOI] [PubMed] [Google Scholar]
- 14.Kimura K, Mukohara N, Nishijima E, Muraji T, Tsugawa C, Matsumoto Y. Diamond-shape anastomosis for duodenal atresia: an experience with 44 patients over 15 years. J Pediatr Surg. 1990;25:977–9. doi: 10.1016/0022-3468(90)90241-z. [DOI] [PubMed] [Google Scholar]
- 15.Alexander F, DiFiore J, Stallion A. A triangular tapered duodenoplasty for the treatment of congenital duodenal obstruction. J Pediatr Surg. 2002;37:862–4. doi: 10.1053/jpsu.2002.32888. [DOI] [PubMed] [Google Scholar]
- 16.Spigland N, Yazbeck S. Complications associated with surgical treatment of congenital intrinsic duodenal obstruction. J Pediatr Surg. 1990;25:1127–30. doi: 10.1016/0022-3468(90)90746-v. [DOI] [PubMed] [Google Scholar]
- 17.Azhough R, Bayat A, Hashemzadeh S, Khaki AA, Motayagheni N, Tarzamni MK. The combination of annular pancreas and duodenum inversum presenting with delayed gastric emptying, pain, and feeding intolerance. Am J Gastroenterol. 2009;104:1328–9. doi: 10.1038/ajg.2009.68. [DOI] [PubMed] [Google Scholar]
- 18.Mirza B, Ijaz L, Saleem M, Sheikh A. Multiple associated anomalies in a single patient of duodenal atresia: a case report. Cases J. 2008;1:215. doi: 10.1186/1757-1626-1-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mustafawi AR, Hassan ME. Congenital duodenal obstruction in children: a decade’s experience. Eur J Pediatr Surg. 2008;18:93–7. doi: 10.1055/s-2008-1038478. [DOI] [PubMed] [Google Scholar]