

Abstract
OASL1 is a member of the 2’-5’-oligoadenylate synthetase (OAS) family and promotes viral clearance by activating RNase L. OASL1 interacts with the 5’-untranslated region (UTR) of interferon regulatory factor 7 (Irf7) and inhibits its translation. To identify the secondary structure required for OASL1 binding, we examined the 5’-UTR of the Irf7 transcript using “selective 2’-hydroxyl acylation analyzed by primer extension” (SHAPE). SHAPE takes advantage of the selective acylation of residues in single-stranded regions by 1-methyl-7-nitroisatoic anhydride (1M7). We found five major acylation sites located in, or next to, predicted single-stranded regions of the Irf7 5’-UTR. These results demonstrate the involvement of the stem structure of the Irf7 5’-UTR in the regulation of Irf7 translation, mediated by OASL1. [BMB Reports 2014; 47(10): 558-562]
Keywords: Irf7, OASL1, SHAPE, Secondary structure, 1M7
INTRODUCTION
During innate immune responses against invading microorganisms, pattern-recognition receptors recognize conserved pathogen-associated molecular patterns and directly activate signaling cascades, thereby inducing the expression of various inflammatory mediators and type 1 interferons (IFNs) (1, 2). In a previous study, it was established that OASL1, a member of the OAS family, binds to the secondary structure of Irf7 mRNA and inhibits its translation (3). The IRF family contains the major transcription factors required for interferon responses, and these play roles in the cell cycle, apoptosis, oncogene regulation, and hematopoietic cell differentiation (4, 5). Along with IRF3, Irf7 is especially well known as a major regulator of type1 IFN production in response to viral infections (6-8). However, the molecular mechanism behind the OASL1-mediated translational regulation of Irf7, in particular, the cis-element of the Irf7 transcript required for translational regulation, remains unknown.
Recently, many researchers have studied the secondary and tertiary structures of RNAs using various methods, such as the V1 and S1 nucleases or 1-methyl-7-nitroisatoic anhydride (1M7) (9-11). Recent studies have shown that “selective 2’-hydroxyl acylation analyzed by primer extension” (SHAPE) is a powerful method for analyzing single-stranded regions, allowing the prediction of secondary structure (11-14). The importance of RNA secondary structure in modulating the expression level of inflammatory mediators has been illustrated in several cases (15). In a previous study, our group suggested that the secondary structure of mRNA affected the interaction between OASL1 and the Irf7 5’-UTR (3). However, the exact nature of the 5’-UTR required for OASL1 binding has not been established. Here, we investigated the single-stranded regions of the Irf7 5’-UTR that are known to have suppressive effects on translational initiation. We identified five major single-stranded regions of the Irf7 5’-UTR that overlap with the bulged regions, predicted using computer modeling of the secondary RNA structures. These results confirm the requirement for a dsRNA structure for the translational inhibition mediated by OASL1 and identified the cis-element involved in the OASL1-mediated translational regulation of Irf7.
RESULTS
Irf7 5’-UTR has a secondary structure
As demonstrated previously in our laboratory, OASL1 inhibits the translation of Irf7 by binding to the Irf7 5’-UTR (3). Specifically, full suppressive activity was confirmed with the 1-215 fragment of the Irf7 5’-UTR. We used a program to predict the structure of the Irf7 5’-UTR and performed mutation analysis to confirm the requirement for several secondary structures in the translational regulation of Irf7. To further confirm the validity of these secondary structures, we examined the biochemical properties of the Irf7 5’-UTR. The Irf7 5’-UTR was transcribed in vitro and the mobility of the transcripts was confirmed using 2% agarose gel electrophoresis, which revealed several forms of the Irf7 transcript, migrating as rather blurry bands.
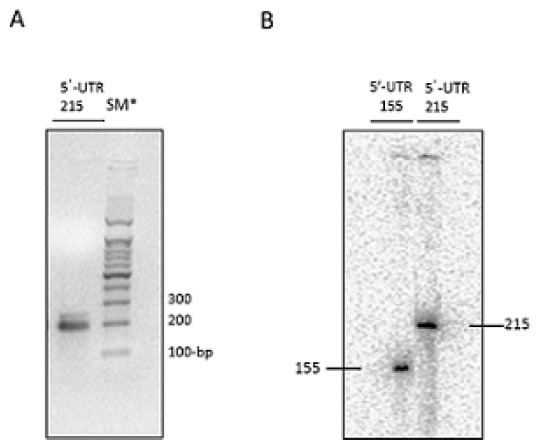
To determine whether the diffuse migrating properties of the UTR were caused by secondary structure, we denatured it at high temperature before electrophoresis. Although the transcript was incubated at 80℃ for up to 10 min, two bands were still seen clearly (Fig. 1A). We next analyzed the transcripts on a 15% denaturing urea-polyacrylamide gel. For this, a cDNA of the Irf7 5’-UTR was synthesized using a [γ-32P]ATP-labeled probe. A cDNA of the exact size of the Irf7 5’-UTR was visible on the denaturing gel (Fig. 1B); a clear and sharp 215-nucleotide (nt) band was seen. These result suggested that the transcripts of the Irf7 5’-UTR may have a secondary structure. To determine the secondary structure of the transcripts in detail, we then analyzed the Irf7 5’-UTR region with SHAPE to identify the singled-stranded regions of the transcripts (14,16-18).
Fig. 1. Irf7 5’-UTR analyzed on a denaturing polyacrylamide gel. pBluescript II SK(+)Irf7 5’-UTR1-215 was linearized using BamH1. Transcripts of the Irf7 5’-UTR1-215 were created using an in vitro transcript assay. (A) The newly generated transcripts appeared as two bands on a 2% agarose gel, even though we denatured them at high temperature, indicating the presence of some secondary structure. (B) A cDNA of the transcript was synthesized using a [γ-32P]ATP-labeled probe and analyzed on a 15% denaturing urea polyacrylamide-bisacrylamide gel. A cDNA of the exact size was clearly seen as a single band in comparison with the pBluescript II SK (+) Irf7 5’-UTR1-155 control cDNA. *SM: size marker.
Five acylation sites were identified on adenosine residues using 1M7
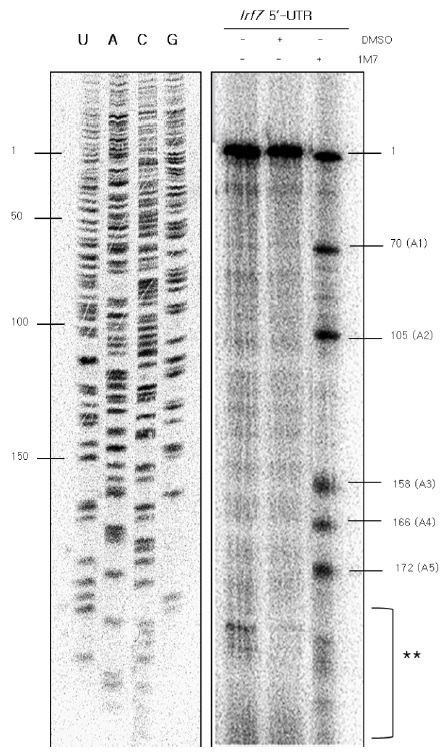
To explore the secondary structure of the Irf7 5’-UTR, we performed SHAPE experiments on the 1-215 fragment of the Irf7 5’-UTR using 1M7 dissolved in DMSO at pH 8.0 under denaturing conditions. 1M7 is known to modify RNA by nucleophilic attack on the 2’-hydroxyl group, causing formation of a 2’-O-adduct, especially in single-stranded regions, loops, or bulges that are conformationally unconstrained or in flexible nucleotide regions (11,19,20). Consequently, 1M7 caused acylation of the single-stranded regions of Irf7 5’-UTR transcripts. cDNAs were synthesized from 1M7-modified mRNA of the Irf7 5’-UTR using a [γ-32P]ATP-labeled probe. Acylation induced breaks in the transcripts and allowed the [γ-32P] ATP-labeled probe to extend only up to where the RNA was acylated. The newly synthesized cDNAs were then analyzed on a 15% urea-acrylamide gel. SHAPE analysis of the Irf7 5’-UTR led to the identification of five major bands (at nucleotide positions 70, 105, 158, 166, and 172), revealing single-stranded regions of the Irf7 5’-UTR (Fig. 2). Our experimental data revealed that single-stranded regions exist at the second and fourth stem structures and the region connecting the second and third stems of the Irf7 5’-UTR, and all five acylation sites were identified on adenosine residues. This is consistent with the observation that adenosine is the most reactive of the four bases (17).
Fig. 2. SHAPE modification led to the identification of five distinct acylation sites in the Irf7 5’-UTR. For SHAPE, the Irf7 5’-UTR transcript was chemically modified using 1M7, dissolved in DMSO at pH 8.0, under denaturing conditions. The Irf7 5’-UTR mRNA was treated with 1 μl of 50 mM 1M7 dissolved in DMSO or 1 μl of DMSO. cDNAs were generated up to the acylation sites at 42℃ for 2 h using a [γ-32P]ATP-labeled probe and then loaded onto a 15 % denaturing urea-acrylamide gel. The single-stranded acylation regions were analyzed using a USB sequencing ladder. The location of the mRNA was deduced from the cDNA. The presence of the SHAPE reagent 1M7, and DMSO of Irf7 5’-UTR are indicated by the ‘+’ in each lane. The first lane was treated with neither DMSO nor 1M7. The second lane was treated with only the solvent, DMSO. The third lane was treated with the SHAPE reagent 1M7. The locations of the nucleotides and the acylation sites are marked on the left and right side of the gel, respectively. The first and second lanes were used as controls for the SHAPE reactions. The sites marked with “1” and double asterisks were excluded from the single-stranded region following SHAPE modification. The site marked “1” represents the size of the whole mRNA, and the asterisk-marked region was also seen in controls. All five single-stranded reactions occurred at adenosine residues.
The secondary structure of the nt 1-215 Irf7 5’-UTR was the same as computed predictions
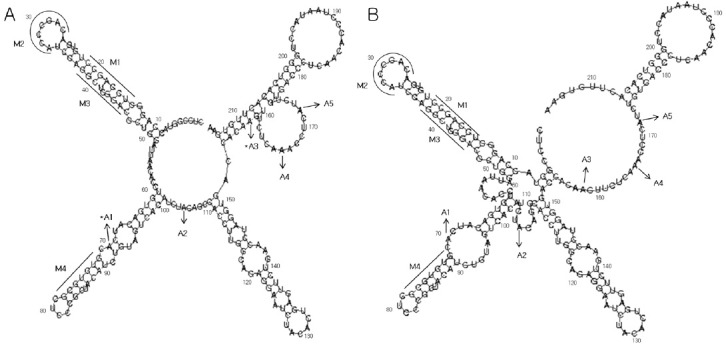
We next used several prediction programs (Vienna fold, Centroidfold, CentroidHomfold, RNA fold, and pknotsRG) to predict the secondary structure of the Irf7 5’-UTR. All of the predictions indicated four stem loops and some bulges. We applied our experimental data to the predictions from Vienna fold using the minimum free energy method (Fig. 3A). Three of the five single-stranded regions matched the predictions from Vienna fold (A2, A4, and A5). These regions were located in the center of the big bulge regions. There were two mismatched areas (A1 and A3), located at the boundaries of predicted bulges. Although these areas were predicted to be a paired region by Vienna fold, the experimental results identified them as single-stranded regions. However, the overall structure matched the computed prediction. Additionally, we compared our current data with previous predictions based on mutation assays. According to our previous report, the inhibition of type 1 IFN translation apparently decreased upon mutation of regions (M1, M3, and M4) from double-stranded to single-stranded (Supplementary Table 1) (3). However, the M2 region did not influence the binding affinity of OASL1. Those regions retained their structure even though the sequence was converted to its complement. Our experimental data confirmed that the three regions (M1, M3, and M4) on the predicted stem structure are not single-stranded (Fig. 3B). Overall, the secondary structure of the Irf7 5’-UTR was found to be the same as the computed predictions, suggesting that the stem-loop structure of Irf7 UTR is a key cis-element for OASL1-mediated translational regulation.
Fig. 3. Most single-stranded regions matched the computer modeling predictions. The secondary structure of Irf7 5’-UTR1- 215 was predicted using Vienna fold, which calculates both minimal free energy and base-pairing possibilities. (A) The plain sequence of the Irf7 5’UTR was applied with no constraint. The experimentally identified single-stranded regions are indicated by a series of acylations (A). The A2, A4, and A5 regions were identified as unpaired regions, confirming the computer predictions; however, regions A1 and A3 did not match the computer predictions. The asterisk indicates the mismatched areas. Mutated regions are indicated with an ‘M’. In M1 and M4, the double-stranded sequences UCCAGCC and GUGUGCG were mutated to single-stranded AGGUCGG and CACACGC, respectively. In M2, the unpaired CAGCCCA was changed to the complementary sequence. M1 and M3 were converted to the complementary sequence but the double-stranded structure was retained. (B) The experimentally identified single-stranded regions were constrained.
DISCUSSION
Irf7, a member of the IRF family, is known as a master regulator of type 1 IFN-dependent immune responses (5,6,8). A previous study in our laboratory found that Irf7 is highly regulated by OASL1, at the mRNA level (3). We previously showed that Irf7 translation is controlled by OASL1 through an interaction in the 5’-UTR (3). In this study, we analyzed the Irf7 5’-UTR to identify the secondary structure required for specific binding of OASL1. Single-stranded regions of the mRNA were modified by SHAPE, and the deduced structure based on the result was compared to the predicted secondary structure from five different structure prediction programs. We found that the identified single-stranded regions mostly matched the structures predicted by the five programs. However, the SHAPE analysis revealed that two regions located next to bulge areas were single stranded. This discrepancy may reflect the physical property of the transcripts under the given reaction conditions. However, these weak interactions may also play regulatory roles in association with other regulatory factors, such as OASL1, which require a specific secondary structure. Together, these data indicate that the secondary structure of the Irf7 5’-UTR appears to assume the predicted structure and that the double-stranded regions of the Irf7 5’-UTR are probably important elements for the inhibition of OASL1-mediated Irf7 translation. However, the exact OASL1 binding site remains to be determined. To define the exact binding sequence of OASL1, it will be necessary to analyze the binding of OASL1 to the Irf7 5’-UTR using an in vitro binding assay and application of dimethyl sulfide footprinting (21), especially of the first and second stem loops. These will contribute to understanding the molecular mechanism underlying OASL1-mediated translational regulation.
MATERIALS AND METHODS
Plasmid Construction and template preparation
The full lengths of cDNA sequence encoding Irf7 5’-UTR 1-215 was subcloned from Metluc2-Irf7 5’-UTR 1-215 into pBluescript II SK (+) vector containing a T7 promoter using restriction enzymes, Hind III and EcoR I. pBluescript II SK (+) Irf7 5’-UTR 1-215 was linearized with BamH I.
In vitro transcription assay
pBluescript II SK (+) Irf7 5’-UTR 1-215 was incubated with T7 polymerase (Enzynomics) and mixture (RNase-free water, rNTPs (Promega), 100 mM DTT (Invitrogen), recombinant RNasin (Promega), 10× T7 polymerase (Enzynomics) buffer) at 37℃ for 1 h to generate mRNA. RQ1 DNase (Promega) was added to the mixture and the incubated was continued at 37℃ for 30 min to remove the templates. The newly generated mRNAs were purified by phenol-chloroform precipitation (RNase-free water 80 μl, phenol:chloroform:isoamyl alcohol 25:24:1, saturated with 10 mM Tris, pH 8.0, 1 mM EDTA (Sigma) 100 μl, 100% ethanol (Sigma) 275 μl, 5 μM NH4Ac (Sigma) 10 μl, 75% ethanol 100 μl).
1M7 synthesis
According to the 1M7 synthesis protocol, 4-nitroisatoic anhydride (4NIA) was dissolved in 60 ml anhydrous dimethylfluoride (DMF) in a 250-ml flask under N2. A mixture of sodium hydride (NaH) was made in 15 ml DMF in a separate 500 ml flask under N2 and stirred. The 4NIA solution was added to the mixture of NaH in DMF slowly and stirred. Subsequently, methyl iodide (MeI) was added and mixed well at room temperature for 4 h. The mixture was poured into ice cold 1 N HCl (100 ml) and filtered by vacuum filtration. The precipitate was rinsed twice with cold water, three times with ether, and dried overnight in an oven (purity 99.9%). Then, the dried 1M7 was dissolved in dimethyl sulfoxide (DMSO) at a final concentration 50 mM.
SHAPE modification
RNA (20 pmol in ½ TE buffer, 6 μl) was heated to denature it at 95℃ for 2 min and quickly cooled on ice for 5 min. Then, 3 μl physiological-like folding buffer containing 333 mM HEPES (Sigma), 333 mM NaCl (Samchun), 33.3 mM MgCl2 (Sigma), pH 8.0, was added and incubated at 37℃ for 20 min. Next, 50 mM 1M7 (1 μl) or DMSO (1 μl), as a control, was added and allowed to react at 37℃ for 70 s and cooled on ice. In prequench reactions, the mixture color changed from bright yellow to dark yellow and the RNAs interacting with 1M7 were degraded by hydrolysis. Modified RNAs were recovered by ethanol precipitation with RNase-free water (90 μl), 5 M NaCl (5 μl), and 100% ethanol (400 μl) at -80℃ for 30 min, then, washed with 75% ethanol and redissolved in 1× TE (10 μl).
End-labeling and primer extension
An oligonucleotide (synthesized by Bioneer) GGA TCC CCC GGG CTG CAG GA was end-labeled with a [γ-32P] ATP using T4 polynucleotide kinase (PNK) and 10× polynucleotide kinase buffer at the 5’-hydroxyl terminus by incubating at 37℃ for 1 h, and was purified with phenol-chloroform at -20℃ for 30 min. Modified RNAs were heated at 65℃ for 5 min to anneal and incubated with end-labeled probe at 42℃ for 2 h. The end-labeled probes were extended up to the modified sites. The extension process was terminated by incubating at 70℃ for 10 min.
Sequencing
pBluescript II SK (+) Irf7 5’-UTR 1-215 was hydrolyzed and used as a template for a sequencing ladder. The sequencing ladder was prepared according to USB’s protocol. Briefly, Sequenase reaction buffer and an appropriate primer (TTC ACA AGT GTG ACC CAG GTA TTA GGG TG) were added to the hydrolyzed DNA and the mixture was incubated at 65℃ for 2 min to denature and cooled at room temperature for 1530 min. Subsequently, dGTP, [α-35S] ATP, 0.1 M DTT, and polymerase were added and the mixture was again incubated at room temperature for 5 min. ddNTPs were added for the termination reaction and incubated at 37℃ for 5 min. To inactivate the extension, stop solution was added and incubated at 75℃ for 2 min. A model S2 sequencing gel electrophoresis apparatus (Gibco BRL) was used to separate the extended cDNA. All sequencing ladders were loaded directly from the incubation at 75℃ and the extended cDNA samples were denatured at 95℃ for 5 min just before loading on a 15% urea-acrylamide- bisacrylamide gel. The samples were separated at 900 V for 12 h and analyzed using the Bio imaging analysis system (Fujifilm Life Sciences).
RNA structure prediction
The secondary structure of the mRNA was predicted using Vienna fold software. Vienna fold predicts both lowest free-energy structures and base pair probabilities from RNA or DNA sequences. The minimum free energy (MFE) method searches for the most energetically stable structure and is calculated by summing the total folding energies. To calculate each base pair in a secondary structure, RNA sequences were subjected to a function e, where e(ri, rj) is the energy of a base pair. If both ri and rj are a pair, but not with each other, ri pairs with rk1 and rj pairs with rk2, where i < k1 < k2 < j. For example, if Ei+1, j = Eij, i is unpaired and if Ei, j-1 = Eij, j is unpaired. The end of a helix, defined by I and j in the algorithm, is considered for exterior loops or branches. Based on this algorithm, our experimental data derived from the SHAPE modification was processed and used to constrain the newly predicted secondary structure.
Acknowledgments
This work was supported by the Global Research Laboratory Program of the National Research Foundation (NRF), funded by the Ministry of Science, ICT & Future Planning (K20705000006-12A0500-00610).
References
- 1.Akira S., Uematsu S., Takeuchi O. Pathogen recognition and innate immunity. Cell. (2006);124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 2.Lee M. S., Kim Y. J. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu. Rev. Biochem. (2007);76:447–480. doi: 10.1146/annurev.biochem.76.060605.122847. [DOI] [PubMed] [Google Scholar]
- 3.Lee M. S., Kim B., Oh G. T., Kim Y. J. OASL1 inhibits translation of the type I interferon-regulating transcription factor Irf7. Nat. Immunol. (2013);14:346–355. doi: 10.1038/ni.2535. [DOI] [PubMed] [Google Scholar]
- 4.Taniguchi T., Ogasawara K., Takaoka A., Tanaka N. IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. (2001);19:623–655. doi: 10.1146/annurev.immunol.19.1.623. [DOI] [PubMed] [Google Scholar]
- 5.Takaoka A., Tamura T., Taniguchi T. Interferon regulatory factor family of transcription factors and regulation of oncogenesis. Cancer Sci. (2008);99:467–478. doi: 10.1111/j.1349-7006.2007.00720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Honda K., Yanai H., Negishi H., Asagiri M., Sato M., Mizutani T., Shimada N., Ohba Y., Takaoka A., Yoshida N., Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. (2005);434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 7.Ning S., Pagano J. S., Barber G. N. Irf7: activation, regulation, modification and function. Genes Immun. (2011);12:399–414. doi: 10.1038/gene.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sato M., Hata N., Asagiri M., Nakaya T., Taniguchi T., Tanaka N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. (1998);441:106–110. doi: 10.1016/S0014-5793(98)01514-2. [DOI] [PubMed] [Google Scholar]
- 9.Kertesz M., Wan Y., Mazor E., Rinn J. L., Nutter R. C., Chang H. Y., Segal E. Genome-wide measurement of RNA secondary structure in yeast. Nature. (2010);467:103–107. doi: 10.1038/nature09322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wan Y., Qu K., Ouyang Z., Chang H. Y. Genome-wide mapping of RNA structure using nuclease digestion and high-throughput sequencing. Nat. Protoc. (2013);8:849–869. doi: 10.1038/nprot.2013.045. [DOI] [PubMed] [Google Scholar]
- 11.Mortimer S. A., Weeks K. M. A fast-acting reagent for accurate analysis of RNA secondary and tertiary structure by SHAPE chemistry. J. Am. Chem. Soc. (2007);129:4144–4145. doi: 10.1021/ja0704028. [DOI] [PubMed] [Google Scholar]
- 12.Lucks J. B., Mortimer S. A., Trapnell C., Luo S., Aviran S., Schroth G. P., Pachter L., Doudna J. A., Arkin A. P. Multiplexed RNA structure characterization with selective 2'-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq). Proc. Natl. Acad. Sci. U. S. A. (2011);108:11063–11068. doi: 10.1073/pnas.1106501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Z., Parisien M., Scheets K., Miller W. A. The cap-binding translation initiation factor, eIF4E, binds a pseudoknot in a viral cap-independent translation element. Structure. (2011);19:868–880. doi: 10.1016/j.str.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Low J. T., Weeks K. M. SHAPE-directed RNA secondary structure prediction. Methods. (2010);52:150–158. doi: 10.1016/j.ymeth.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bevilacqua A., Ceriani M. C., Capaccioli S., Nicolin A. Post-transcriptional regulation of gene expression by degradation of messenger RNAs. J. Cell. Physiol. (2003);195:356–372. doi: 10.1002/jcp.10272. [DOI] [PubMed] [Google Scholar]
- 16.Turner R., Shefer K., Ares M.,, Jr. Safer one-pot synthesis of the 'SHAPE' reagent 1-methyl-7-nitroisatoic anhydride (1m7). RNA. (2013);19:1857–1863. doi: 10.1261/rna.042374.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilkinson K. A., Vasa S. M., Deigan K. E., Mortimer S. A., Giddings M. C., Weeks K. M. Influence of nucleotide identity on ribose 2'-hydroxyl reactivity in RNA. RNA. (2009);15:1314–1321. doi: 10.1261/rna.1536209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deigan K. E., Li T. W., Mathews D. H., Weeks K. M. Accurate SHAPE-directed RNA structure determination. Proc. Natl. Acad. Sci. U. S. A. (2009);106:97–102. doi: 10.1073/pnas.0806929106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merino E. J., Wilkinson K. A., Coughlan J. L., Weeks K. M. RNA structure analysis at single nucleotide resolution by selective 2'-hydroxyl acylation and primer extension (SHAPE). J. Am. Chem. Soc. (2005);127:4223–4231. doi: 10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- 20.Gherghe C. M., Shajani Z., Wilkinson K. A., Varani G., Weeks K. M. Strong correlation between SHAPE chemistry and the generalized NMR order parameter (S2) in RNA. J. Am. Chem. Soc. (2008);130:12244–12245. doi: 10.1021/ja804541s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tijerina P., Mohr S., Russell R. DMS footprinting of structured RNAs and RNA-protein complexes. Nat. Protoc. (2007);2:2608–2623. doi: 10.1038/nprot.2007.380. [DOI] [PMC free article] [PubMed] [Google Scholar]