

Abstract
RNA polymerase II carboxyl-terminal domain (RNAPII CTD) phosphatases are responsible for the dephosphorylation of the C-terminal domain of the small subunit of RNAPII in eukaryotes. Recently, we demonstrated the identification of several interacting partners with human small CTD phosphatase1 (hSCP1) and the substrate specificity to delineate an appearance of the dephosphorylation catalyzed by SCP1. In this study, using the established cells for inducibly expressing hSCP1 proteins, we monitored the modification of β-O-linked N-acetylglucosamine (O-GlcNAc). O-GlcNAcylation is one of the most common post-translational modifications (PTMs). To gain insight into the PTM of hSCP1, we used the Western blot, immunoprecipitation, succinylayed wheat germ agglutininprecipitation, liquid chromatography-mass spectrometry analyses, and site-directed mutagenesis and identified the Ser41 residue of hSCP1 as the O-GlcNAc modification site. These results suggest that hSCP1 may be an O-GlcNAcylated protein in vivo, and its N-terminus may function a possible role in the PTM, providing a scaffold for binding the protein(s). [BMB Reports 2014; 47(10): 593-598]
Keywords: CTD phosphatase SCP1, Inducible mammalian expression system, O-GlcNAc, Post-translational modification
INTRODUCTION
RNA polymerase II (RNAPII) is a key complex multisubunit enzymatic component in the transcription apparatus for the synthesis of mRNAs and many noncoding RNAs and for its regulation 1), 2). Eukaryotic RNAPII has several (up to 52 in mammals) conserved tandem heptad repeats (Y1S2P3T4S5P6S7, namely C-terminal domain (CTD)) at its C-terminus that is not present in other RNAPs (3). The phosphorylation of S2 and S5 of this repeat along with proline cis/trans isomerization and glycosylation creates a code that seemingly can be read to mediate a number of processes 4), 5) and is thought to act as a scaffold to coordinate the binding of proteins involved in the different phases of transcription and couples the transcription with other nuclear process. Indeed, the phosphorylation status of this repeat is a hallmark for different transcriptional sdtages of the RNAPII complexes (6). Thus, the enzymes responsible for the phosphorylation status of these serine (Sr) residues play pivotal roles in the regulation of transcription and related processes (7). These events are largely mediated by the participation of the proline-directed kinases such as cyclin-dependent kinases. Moreover, several phosphatases have been implicated for the removal of phosphates from the CTD, thereby mediating transitions in the transcription cycle (8, 9). A systematic approach investigating the genome-wide distribution of the CTD modifications indicated a considerable crosstalk between the CTD kinases and phosphatases, suggesting that the transcription operates in a uniform mode at virtually all the genes (10).
In higher eukaryotes, small CTD phosphatases (SCPs) with activities preferential for phosphoryl-Ser5 (PS5) were identified, containing a catalytic domain (FCPH domain) with Mg+2-binding DXDX(T/V) signature motif but lack a breast cancer protein related C-terminal domain (11,12). The SCPs are related to the catalytic subunit of FCP1, the first discovered CTD phosphatase, which is highly conserved, essential enzyme for dephosphorylating the CTD of RNAPII and preferential for phosphorylating PS2. These are also transcriptional regulators for gene silencing activities in neuronal genes and phase regulation in the cell cycle (13,14). The catalytic mechanisms of the SCPs and the structural basis for their CTD specificity are well understood (15,16); however, the identity of physiological regulatory mechanisms explaining the biological activities of the SCPs has remained mainly elusive.
The β-O-linked N-acetylglucosamine (O-GlcNAc) modification (O-GlcNAcylation) has been proposed to act as a nutrient sensor and changes in response to many signals, including the morphogens, cell cycle, development, extracellular glucose levels, and numerous forms of cellular stress (17,18). This modification of Ser and threonine (Thr) residues of various nuclear, mitochondrial, and cytoplasmic proteins is one of a number of post-translational modifications (PTMs) thought to mediate cellular function, and a variety of proteins can be O-GlcNAcylated (19). The O-GlcNAcylation is highly dynamic and interestingly regulates protein function in a manner analogous to the phosphorylation of protein by changing the localization of proteins, regulating protein-protein or protein-DNA interactions, altering the half-life of proteins, and regulating the activity of proteins (20). Taken together, it is well established that the O-GlcNAcylation is a key PTM employed by the cells and animals to rapidly respond for the survival against stress.
Previously, we demonstrated the identification of several interacting partners with hSCP1 and the substrate specificity to delineate an appearance of the dephosphorylation reaction catalyzed by SCP1 phosphatase using a specific inducible expressing system of the human SCP1 (hSCP1) (21). SCP1 protein subjected to diverse PTMs in spite of the known O-GlcNAcylation of RNAPII (5)., particularly CTD, has not been reported. In this study, we interrogated the PTMs into this SCP1 as a CTD phosphatase and report hSCP1 as a target for the O-GlcNAcylation. The O-GlcNAcylated hSCP1 peptides were analyzed by mass spectrometry using the established NIH/3T3 cells expressing hSCP1 proteins under the tight control by doxycycline. Moreover, the intrinsically disordered N-terminal portion of SCP1 may provide a scaffold for binding the protein(s) involved in the PTM such as the O-GlcNAcylation.
RESULTS AND DISCUSSION
Establishment of NIH/3T3 cell lines for hSCP1
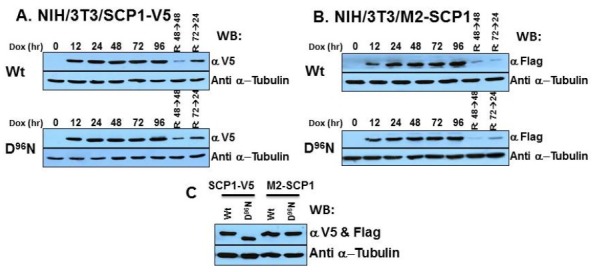
The active site of the CTD-phosphatases was characterized by the signature motif ψψψDXDX(T/V)ψψ, where ψ is a hydrophobic residue. The D96 in the hSCP1 has been shown to be catalytically pivotal. The hSCP1 was tagged either with the Flag sequence at its N-terminus (5'-region) or V5 epitope sequence at its C-terminus (3'-region) and then subcloned into pTRE-IRES-EGFP mammalian inducible plasmid (21-23). After the transfections of wild type (Wt) and D96N mutant hSCP1 in pTRE-IRES-EGFP vector placing together pEF1α-Tet, several candidates were primarily chosen based on their antibiotic selection (puromycin, 10 μg/ml) and microscopic appearance for the EGFP monitored into the IRES-driven EGFP expression in order for the expression to be tracked by fluorescence (Fig. 1) and finally selected using the Western blot analysis with α-Flag and α-DYKDDDDK antibodies or α-V5 epitope antibodies in all the NIH/3T3 cells (Fig. 2). The selected and used expression clones in this study were shown to be >99% expression of the EGFP whenever they were induced by 2 μg/ml doxycycline as an inducer (Fig. 1C). An increase in the hSCP1 expression was only shown in response to doxycycline depending on the induction time (Fig. 2). However, after the treatment of doxycycline for the indicated periods of time, when the cells were replated on new culture dishes with fresh medium without the inducer, the hSCP1 finally reverted to no expression. Moreover, the immunoprecipitation analyses with 2 mg of the total cell lysates with appropriate antibodies did not show any positive signal from the noninduced cell lines. Thus, its expression was under the tight control of doxycycline and is a suitable, unique, and useful system for characterizing hSCP1. The established hSCP1 (Wt and D96N) inducible NIH/3T3 cells were designated as NIH/3T3/M2-hSCP1 and NIH/3T3/hSCP1-V5 for the N-terminal Flag-tagged hSCP1 and C-terminal V5-epiotpe tagged hSCP1, respectively. To confirm the enzymatic activities of the prepared recombinant hSCP1, the Mg2+-dependent phosphatase activities of the immunocomplexes from the induced expressed hSCP1 (Wt) from the NIH/3T3 cells were measured by the enzymatic activity against the artificial substrate, para-nitrophenyl phosphate (pNPP) (21), whereas those from the mutant hSCP1 (D96N) inducible cell lines did not show any activity. This is in a very good agreement with the previous reports. The preparation of the enzyme source for the SCP1 in the mammalian system is not reported. The production of a full-length version of recombinant SCP1 in the functional form under native conditions was not possible, primarily because of solubility issues (24). Furthermore, even purifying the full-length form of the hSCP1 from E. coli expression system has also not been possible previously for another reason, because the protein consistently underwent proteolysis along the N-terminus during the expression (16). The N-terminus was predicted to be structurally disordered, overcoming the biggest challenge of its solubility, because of the presence of the disordered N-terminus. The full-length SCP1 could only be purified under denaturing conditions followed by refolding. However, it had very little enzymatic activity remaining, and the N-terminus-truncated SCP1 purified under native conditions was functional (25). In this study, as shown in Fig. 2C, only mutant hSCP1(D96N) from NIH/3T3/ hSCP1(D96N)-V5 cells had a slightly faster electrophoretic mobility than hSCP1 from NIH/3T3/hSCP1(Wt)-V5 and NIH/3T3/M2-hSCP1(Wt), and even hSCP1 from NIH/3T3/M2- hSCP1(D96N) cells. Hence, this truncation is likely because of specific proteolytic cleavage of the full-length hSCP1 and lability of the hSCP1(D96N) protein toward the proteolytic cleavage. However, the formation of the truncated version of hSCP1 was abolished, when the hSCP1 from NIH/3T3/ M2-hSCP1 cells was masked with the Flag-tag sequence in its N-terminal (Fig. 2C, lane 4). All together, these data indicate that our C-terminal tagged hSCP1(D96N) in the inducibly expressing cells (NIH/3T3/hSCP1-V5) was expressed as a truncated version of the hSCP1 protein at its N-terminal region. In fact, several reports have demonstrated that the N-terminus of the SCP1 is predicted to be structurally disordered and play a role of the intrinsically disordered N-terminus in the protein-protein interactions (26-28). Our data demonstrate that the mammalian hSCP1 inducible cell lines are suitable, useful, and effective tools for dissecting the cellular and physiological functions of each hSCP1 in vivo.
Fig. 1. Microscopic assays for the EGFP expression in hSCP1 inducible cells. Images of hSCP1(Wt and D96N)-induced NIH/3T3 cells in the absence (A) or presence (B) (Phase contrast image) and (C) of 2 μg/ml doxycycline for 72 h were taken using a Leika M205FA stereomicroscope (Leika Microsystems, Wetzler, Germany) equipped with the LAS software for fluorescence imaging.

Fig. 2. Establishment of hSCP1-expressing inducible NIH/3T3 cells ((A) NIH/3T3/hSCP1-V5 and (B) NIH/3T3/M2-hSCP1). The expression levels of hSCP1 (Wt and D96N) treated with or without the inducer for the indicated periods of time were monitored by Western blot analyses with the appropriate antibodies. For reverted cells, the cells were treated with the inducer for the indicated periods, and after the replacement with fresh medium, the cells were allowed to grow for the indicated periods of time without the inducer. (C) For the comparison of molecular sizes of hSCP1 (Wt and D96N) from NIH/3T3/hSCP1-V5 and NIH/3T3/M2-hSCP1 cells, equal amounts (30 μg) of total cell lysates from the abovementioned cell lines were analyzed by immunoblotting with a mixture of α-DYKDDDDK and α-V5 antibodies and detected with HRP-conjugated secondary antibodies.
hSCP1 modification by O-GlcNAcylation
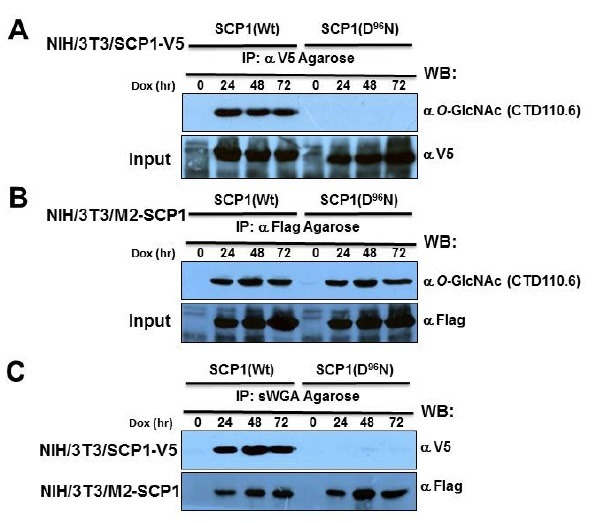
The usefulness of our inducible protein expressing cell systems to validate the proteome profile changes, dissect the signaling network, and monitor the PTM, particularly phosphorylation has been previously (22,23,29,30) considered. In this study, we first addressed the question whether the hSCP1 is a target substrate of O-GlcNAc transferase (OGT). For this, immunologically confirmed epitope tagged hSCP1s (Wt and D96N) at different induction time points (0, 24, 48, 72 h) were used in the presence of Thiamet-G as an inhibitor of O-GlcNAcase, immunoprecipitated with either the α-Flag or α-V5 epitope antibody-conjugated Agarose beads and analyzed for the O-GlcNAcylation by Western blot analysis using α-O-GlcNAc antibody (CTD110.6) at each induction time point. As shown in Figs. 3A and 3B, hSCP1 except the truncated hSCP1(D96N) from NIH/3T3/hSCP1-V5, lacking the N-terminal region, was modified by the O-GlcNAc. Most proteins undergo PTMs, which might be important for their function. Moreover, in many proteins, some regions are partially unstructured or even native folded under the physiological conditions. Furthermore, the cell lysates from each hSCP1-expressed cells were incubated with sWGA Agarose, a lectin that specifically interacts with terminal GlcNAc moieties. The sWGA precipitates were resolved by SDS-PAGE and Western blotting using either α-Flag or α-V5 epitope antibody. Fig. 3C shows that also hSCP1 except the truncated hSCP1 (D96N) from NIH/3T3/hSCP1-V5 was detected in the sWGA precipitates, indicating that they carry O-GlcNAc moiety in their N-terminus and is in a very good agreement with the previous immunoprecipitation result.
Fig. 3. Human SCP1 as a substrate target for O-GlcNAcylation. The immunologically defined epitope-tagged hSCP1s (Wt and D96N) were induced in NIH/3T3/hSCP1-V5 (A) and NIH/3T3/M2-hSCP1 (B) cells for the indicated periods and hSCP1 from equal amount of total cell lysate (2 mg) were immunoprecipitated with either α-Flag antibody or α-V5 epitope antibody. O-GlcNAcylated hSCP1 was detected using the α-O-GlcNAc antibody. For reprobing the blots, the blots were washed, incubated with BlotFresh Western Blot Stripping Reagent, and remonitored the input hSCP1 proteins. (B) At 72 h of the postinduction, total cell lysates (2 mg) from the noninduced and induced cells for the indicated periods were incubated with sWGA Agarose beads. The affinity-purified hSCP1 in the precipitate was resolved onto SDS-PAGE and probed with either α-DYKDDDDK or α-V5 epitope antibody.
Conformational disorder is proposed to be important for binding diversity in the protein-protein interactions (26-28). In many in vitro studies of the recombinant SCP1, the inability to produce an active full-length version of the SCP1 under native conditions necessitated the use of a truncated form, but the results regarding its protein-protein interaction were inconclusive (15,16). In this study, an active full-length hSCP1 in mammalian system was successfully produced and shown to possess in vivo O-GlcNAcylation, thus demonstrating a pivotal role of its N-terminus in events such as PTMs and proteolysis, which might provide a specific location for the protein-protein interaction platform.
Ser41 residue in hSCP1 as an O-GlcNAcylation site
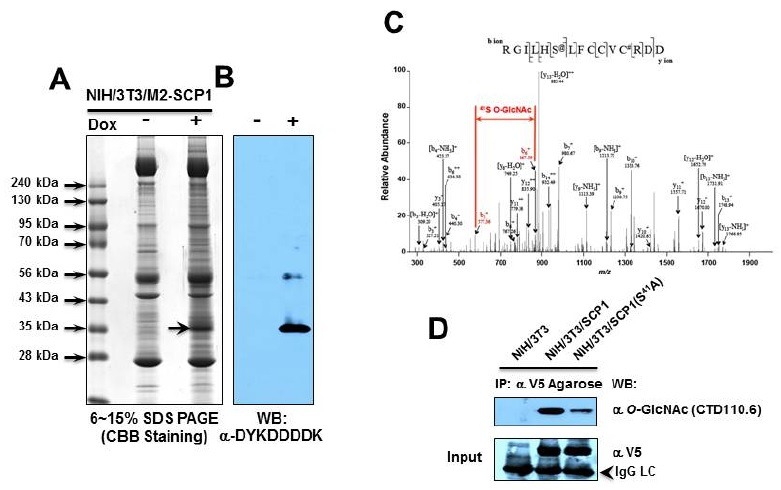
To determine the O-GlcNAcylation site of hSCP1, 20 mg of total cell lysate from NIH/3T3/M2-hSCP1 for Flag-tagged hSCP1(Wt) with the lysis buffer A, described in “Materials and Methods” section, was immunoprecipitated with the α-Flag antibody-conjugated Agarose beads. The immunoprecipitates were resolved onto 6-15% gradient sodium dodecyl sulphate--polyacrylamide gel electrophoresis (SDS-PAGE) and visualized by Coomassie Brilliant Blue G250 (Fig. 4A). After confirming the recombinant hSCP1 of the immunoprecipitate (1/30 volume) with α-DYKDDDDK antibody (Fig. 4B), the excised hSCP1 band was digested with Glu-C. The extracted peptides were analyzed using Q-TOF MS, and the O-GlcNAc- modified peptide was identified by calculating the difference of 203.2. Only one O-GlcNAc-modified peptide of hSCP1 corresponding to residue R36GILHS41LFCCVCRDD50 was identified, and the Q-TOF spectrum and sequencing results of the GlcNAc-modified peptide corresponding to residues 36-50 are shown in Fig. 4C. The expected increase in mass by the O-GlcNAc modification was 203.2 Da. To assess the O-GlcNAcylation site at S41, a full length hSCP1 (S41A) tagged with V5-epitope at its C-terminal region was created. Either hSCP1 (S41A) or hSCP1 (Wt) in pcDNA3 vector was then transfected into NIH/3T3 cells in the presence of 10 μM Thiamet-G. Alterations in the level of O-GlcNAcylation were monitored by immunoprecipitation with V5-Agarose beads and Western blot analysis. The O-GlcNAcylation level of hSCP1 (S41A) proteins was significantly reduced relative to the hSCP1 (Wt) transfected cells (Fig. 4D). Because of the presence of an internal Ser residue in this peptide, we concluded that S41 was modified by the O-GlcNAc.
Fig. 4. Determination of O-GlcNAcylation site on hSCP1. (A) The Flag-tagged hSCP1 (Wt) from NIH/3T3/M2-hSCP1 cells (20 mg of total cell lysate) was subjected to the immunoprecipitation with α-Flag-Agarose. The immunoprecipitates were subjected to 6-15% SDS-PAGE and stained with CBB. Arrow indicates hSCP1. (B) A small portion of immunoprecipitate was subjected to Western blot analysis with α-DYKDDDDK antibody. (C) The Q-TOF spectrum and the sequencing results of a GlcNAc-modified peptide corresponding to residues 36-50 are shown. S41 residue in hSCP1 is an O-GlcNAcylation site. The expected increase in mass by the O-GlcNAc modification is 203.2 Da. (D) Whole cell lysates of NIH/3T3 transfected with V5-tagged hSCP1 (empty vector (pcDNA3), Wt and S41A) were immunoprecipitated with V5-epitope Agarose beads and characterized by Western blot analysis with α O-GlcNAc antibody. The protein input was monitored with α-V5 antibody after stripping the blot. Arrow head indicates IgG light chain.
Despite its importance in coordinately regulating phosphorylation and dephosphorylation of the CTD in the transcription and mRNA processing, the mechanism of the regulation of SCP1 phosphatase remains largely undefined. It is not clear whether the PTMs of the SCP1 phosphatases by enzymatic reactions are essential for their pathway. The PTMs create a highly dynamic relay system, responding to signaling events without requiring de novo protein synthesis and offer a platform for signaling pathway components involved in the regulation of the signaling system, hence providing alternative opportunities for targeting the specific signaling pathway.
In this study, we established clonal cell lines in which the expressions of hSCP1(Wt) and dominant negative hSCP1 (D96N) are under the tight control of an antibiotic, doxycycline as an inducer. Moreover, we demonstrate that hSCP1 was modified with O-GlcNAcylation in NIH/3T3 cells and identified an O-GlcNAcylated peptide from the hSCP1, and S41 located near the N-terminal region the O-GlcNAcylation site. These experiments may shed some light on not only the successful production of the functional full-length hSCP1 in the mammalian system, but also the future evaluation of the functional role of its N-terminus in the protein-protein interaction. Although the functional role of the O-GlcNAcylation on the N-terminal region of SCP1 such as the role of O-GlcNAcylated RNAPII CTD remains speculative, accumulating evidences indicate that the reversible PTMs such as O-GlcNAcylation are mostly used to modulate the protein stability, allowing catalytic functions of the ordered proteins and is frequently used for the signaling activities of intrinsically disordered proteins. We believe that a novel O-GlcNAcylated protein, hSCP1, identified in this study could function as a valuable resource for a further study of the role of RNAPII CTD phosphatases, a newly emerging family of phosphatases.
MATERIALS AND METHODS
Cell culture and establishment of inducible hSCP1-expressing cell lines
NIH/3T3 cells were purchased from ATCC (Bethesda, MD, USA) and maintained in high glucose (25 mM) DMEM (Life Technologies, Burlington, ON, USA) supplemented with 10% bovine serum and antibiotics (Life Technologies) in a humidified incubator at 37℃ with 5% CO2 as recommended by the manufacturer’s instruction. Tetracycline-inducible cell lines for the specific hSCP1 protein expression were established as previously described (23,29,30). The expressed hSCP1s (Wt and D96N mutant) were tagged with V5 epitope and Flag sequence at the C- and N-terminal regions of the hSCP1 cDNA, respectively. For O-GlcNAcase inhibition, the established expressing cells were cultured in the presence of 10 μM Thiamet-G (Cayman Chemical, Ann Arbor, MI, USA) during the induction time.
Immunoblot analysis, immunoprecipitation assays, and sWGA lectin precipitation
The cells were washed with ice-cold PBS and lysed using lysis buffer A (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, and 1% Triton X-100 containing protease inhibitor cocktails (Roche, Manheim, Germany) for particularly Flag tagged hSCP1 as per manufacturer’s recommendation or conventional RIPA buffer containing protease inhibitors. The procedures for Western blot analysis and immunoprecipitation assays were performed as described previously (21,29). The anti-V5 epitope antibody-conjugated Agarose beads and α-Flag antibody-conjugates were purchased from Sigma-Aldrich. Primary antibodies used are as follows: mouse α-Flag (M2 clone, Sigma-Aldrich), rat α-DYKDDDDK (BioLegend, San Diego, CA, USA), mouse α-V5 epitope (Santa Cruz Biotech., Santa Cruz, CA, USA), mouse α-O-GlcNAc (CTD110.6 clone, Covance, Prinston, NJ, USA), and rabbit a α-tubulin (AbClon, Seoul, Korea). Succinylated wheat germ agglutinin (sWGA) lectin provides a convenient method for enriching and detecting O-GlcNAc-modified proteins. Precipitation with sWGA Agarose (Vector, Burlingame, CA, USA) was performed as previously described (19). The detection of α-tubulin as a primary antibody was used as an internal control. For reprobing the blots, the blots were washed in 1X TBS to remove the chemiluminescent substrate and incubated with BlotFresh Western Blot Stripping Reagent (SignaGen Laboratories, Gaithersburg, MD, USA) according to the manufacturer’s instruction.
Phosphatase assays
The hSCP1-catalyzed dephosphorylation of the phosphorylated substrate was performed as previously described (21).
Site-directed mutagenesis and transient expression of SCP1
Ser41 of the hSCP1 cDNA was mutagenized to Ala using a QuikChangeTM site-directed mutagenesis kit (Stratagene, La Jolla, CA). The sequence change was verified using an ABI Prism sequencing kit (Applied Biosystems, Foster City, CA, USA). Transfections for the transient expression were performed using FuGENE 6 Reagent (Roche) according to the manufacturer’s instruction. 3 μg of cDNA construct in pcDNA3 was used for all the transfections.
Mass spectrometric analysis
The specific protein bands from the CBB-stained gel of the immunoprecipitated complexes (20 mg of total cell lysate) were digested with endoproteinase Glu-C (Promega, Madison, WI, USA) at 37℃ according to the manufacturer’s instruction. The resulting peptides were extracted using 50% acetonitrile and dried in a vacuum evaporator for further analysis. Mass spectrometric analysis was performed using a QSTAR Pulsar Q-TOF MS (Applied Biosystems) equipped with a nanoelectrospray ion source (Protana, Odense, Denmark) as described previously (21,29).
Acknowledgments
This study was supported by grants from the National Research Foundation of Korea: 2010-0024199 for YYB and 2013R1A1 A2009145 for JHK.
References
- 1.Eicks D., Geyer M. The RNA polymerase II carboxyl-terminal domain (CTD) code. Chem. Rev. (2013);113:8456–8490. doi: 10.1021/cr400071f. [DOI] [PubMed] [Google Scholar]
- 2.Proudfoot N. J., Furger A., Dye M. J. Integrating mRNA processing with transcription. Cell. (2002);108:501–512. doi: 10.1016/S0092-8674(02)00617-7. [DOI] [PubMed] [Google Scholar]
- 3.Palancade B., Bensaude O. Investigating RNA polymerase II carboxyl-terminal domain (CTD) phosphorylation. Eur. J. Biochem. (2003);270:3859–3870. doi: 10.1046/j.1432-1033.2003.03794.x. [DOI] [PubMed] [Google Scholar]
- 4.Buratowski S. The CTD code. Nat. Struct. Biol. (2003);10:679–680. doi: 10.1038/nsb0903-679. [DOI] [PubMed] [Google Scholar]
- 5.Kelly W. G., Dahmus M. E., Hart G. W. RNA polymerase II is a glycoprotein: Modification of the COOH-terminal domain by O-GlcNAc. J. Biol. Chem. (1993);268:10416–10424. [PubMed] [Google Scholar]
- 6.Phatnani H. P., Greenleaf A. L. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. (2006);20:2922–2936. doi: 10.1101/gad.1477006. [DOI] [PubMed] [Google Scholar]
- 7.Chapman R. D., Heidemann M., Albert T. K., Mailhammer R., Flatley A., Meisterernst M., Kremmer E., Eicks D. Transcribing RNA polymerase II is phosphorylated at CTD residue serine-7. Science. (2007);318:1780–1782. doi: 10.1126/science.1145977. [DOI] [PubMed] [Google Scholar]
- 8.Lin P. S., Marshall N. F., Dahmus M. E. CTD phosphatase: role in RNA polymerase II cycling and the regulation of transcription elongation. Prog. Nucleic Acid Res. Mol. Biol. (2002);72:333–365. doi: 10.1016/s0079-6603(02)72074-6. [DOI] [PubMed] [Google Scholar]
- 9.Cho E. J., Kobor M. S., Kim M., Buratowski S. Opposing effects of Ctk1 kinase and Fcp1 phosphatase at Ser2 of the RNA polymerase II C-terminal domain. Genes Dev. (2001);15:3319–3329. doi: 10.1101/gad.935901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bataille A. R., Jeronimo C., Jacques P. É., Laramee L., Fortin M. E., Forest A., Bergeron M., Hanes S. D., Robert F. A universal RNA polymerase II CTD cycle is orchestrated by complex interplays between kinase, phosphatase, and isomerase enzymes along genes. Mol. Cell. (2012);45:158–170. doi: 10.1016/j.molcel.2011.11.024. [DOI] [PubMed] [Google Scholar]
- 11.Koiwa H., Hausmann S., Bang W. Y., Ueda A., Kondo N., Hiraguri A., Fukuhara T., Bahk J. D., Yun D. J., Bressan R. A., Hasegawa P. M., Shuman S. Arabidopsis C-terminal domain phosphatase-like 1 and 2 are essential Ser-5-specific C-terminal domain phosphatases. Proc. Natl. Acad. Sci. U.S.A. (2004);101:14539–14544. doi: 10.1073/pnas.0403174101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harikrishna R. R., Kim H., Noh K., Kim Y. J. Diverse roles of RNA polymerase II C-terminal domain phosphatase SCP1. BMB Rep. (2014);47:192–196. doi: 10.5483/BMBRep.2014.47.4.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeo M., Lee S. K., Lee B., Ruiz E. C., Pfaff S. L., Gill G. N. Small CTD phosphatases function in silencing neuronal gene expression. Science. (2005);307:596–600. doi: 10.1126/science.1100801. [DOI] [PubMed] [Google Scholar]
- 14.Fabian-Marwedel T., Umeda M., Sauter M. The rice cyclin-dependent kinase-activating kinase R2 regulates S-phase progression. Plant Cell. (2002);14:197–200. doi: 10.1105/tpc.010386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y., Kim Y., Genoud N., Gao J., Kelley J. W., Pfaff S. L., Gill G. N., Dixon J. E., Noel J. P. Determinants for dephosphorylation of the RNA polymerase II C-terminal domain by Scp1. Mol. Cell. (2006);24:759–770. doi: 10.1016/j.molcel.2006.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamenski T., Heilmeier S., Meinhart A., Cramer P. Structure and mechanism of RNA polymerase II CTD phosphatases. Mol. Cell. (2004);15:399–407. doi: 10.1016/j.molcel.2004.06.035. [DOI] [PubMed] [Google Scholar]
- 17.Yang W. H., Kim J. E., Nam H. W., Ju J. W., Kim H. S., Kim Y. S., Cho J. W. Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat. Cell Biol. (2006);8:1074–1083. doi: 10.1038/ncb1470. [DOI] [PubMed] [Google Scholar]
- 18.Zachara N. E., Molina H., Wong K. Y., Pandey A., Hart G. W. The dynamic stress-induced “O-Glcnacome” highlights functions for O-GlcNAc in regulating DNA damage/ repair and other cellular pathways. Amino Acids. (2011);40:793–808. doi: 10.1007/s00726-010-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zachara N. E., Vosseller K., Hart G. W. Detection and analysis of proteins modified by O-linked N-acetylglucosamine. Curr. Protoc. Protein Sci. (2011);66:12.8.1–12.8.33. doi: 10.1002/0471140864.ps1208s66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hart G. W., Housley M. P., Slawson C. Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. (2007);446:1017–1022. doi: 10.1038/nature05815. [DOI] [PubMed] [Google Scholar]
- 21.Kim Y. J., Bahk Y. Y. A study of substrate specificity for a CTD phosphatase, SCP1, by proteomics screening of binding partners. Biochem. Biophys. Res. Comm. (2014);448:189–124. doi: 10.1016/j.bbrc.2014.04.089. [DOI] [PubMed] [Google Scholar]
- 22.Kim S., Lee Y. Z., Kim Y. S., Bahk Y. Y. A proteomic approach for protein-profiling the oncogenic ras induced transformation (H-, K-, and N-ras) in NIH/3T3 mouse embryonic fibroblasts. Proteomics. (2008);8:3082–3093. doi: 10.1002/pmic.200800106. [DOI] [PubMed] [Google Scholar]
- 23.Bahk Y. Y., Cho I.-H., Kim T. S. A cross-talk between oncogenic Ras and tumor suppressor PTEN through FAK Tyr861 phosphorylation in NIH/3T3 mouse embryonic fibroblasts. Biochem. Biophys. Res. Comm. (2008);377:1199–1204. doi: 10.1016/j.bbrc.2008.10.157. [DOI] [PubMed] [Google Scholar]
- 24.Jaramillo-Tatis S., Bamm V. V., Vassall K. A., Harauz G. Over-expression in E. coli and purification of functional full-length murine small C-terminal domain phosphatase (SCP1, or Golli-interacting protein). Protein Expr. Purif. 2014 doi: 10.1016/j.pep.2010.05.013.. [DOI] [PubMed] [Google Scholar]
- 25.Zhang M., Liu J., Kim Y., Dixon J. E., Pfaff S. L., Gill G. N., Noel J. P., Zhang Y. Structural and functional analysis of the phosphoryl transfer reaction mediated by the human small C-terminal domain phosphatase, SCP1. Protein Sci. (2010);19:974–986. doi: 10.1002/pro.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uversky V. N. The most important thing is the tail: multitudinous functionalities of intrinsically disordered protein termini. FEBS Lett. (2013);587:1891–1901. doi: 10.1016/j.febslet.2013.04.042. [DOI] [PubMed] [Google Scholar]
- 27.Uversky V. N., Gillespie J. R., Fink A. L. Why are natively unfolded proteins unstructured under physiologic conditions. Proteins. (2000);41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::AID-PROT130>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 28.Kriwacki R. W., Hengst L., Tennant L., Reed S. I., Wright P. E. Structural studies of p21Waf1/Cip1/ Sdi1 in the free and Cdk2-bound state: conformational disorder mediates binding diversity. Proc. Natl. Acad. Sci. U.S.A. (1996);93:11504–11509. doi: 10.1073/pnas.93.21.11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bahk Y. Y., Lee J., Cho I.-H., Lee H.-W. An analysis of an interactome for apoptosis factor, Ei24/PIG8, using the inducible expression system and shotgun proteomics. J. Proteome Res. (2010);9:5270–5283. doi: 10.1021/pr100552y. [DOI] [PubMed] [Google Scholar]
- 30.Lim Y., Han I., Jeon J., Park H., Bahk Y. Y., Oh E.S. Phosphorylation of focal adhesion kinase at tyrosine 861 is crucial for Ras transformation of fibroblasts. J. Biol. Chem. (2004);279:29060–29065. doi: 10.1074/jbc.M401183200. [DOI] [PubMed] [Google Scholar]