

Abstract
Mass spectrometry plays a key role in relative quantitative comparisons of proteins in order to understand their functional role in biological systems upon perturbation. In this review, we review studies that examine different aspects of isobaric labeling-based relative quantification for shotgun proteomic analysis. In particular, we focus on different types of isobaric reagents and their reaction chemistry (e.g., amine-, carbonyl-, and sulfhydryl-reactive). Various factors, such as ratio compression, reporter ion dynamic range, and others, cause an underestimation of changes in relative abundance of proteins across samples, undermining the ability of the isobaric labeling approach to be truly quantitative. These factors that affect quantification and the suggested combinations of experimental design and optimal data acquisition methods to increase the precision and accuracy of the measurements will be discussed. Finally, the extended application of isobaric labeling-based approach in hyperplexing strategy, targeted quantification, and phosphopeptide analysis are also examined.
Keywords: iTRAQ, isobaric tags for relative and absolute quantification, TMT, tandem mass tags, isobaric tags, isobaric labeling, quantitative proteomics, mass spectrometry
1. Introduction
Mass spectrometry (MS) is a powerful tool to assess the relative abundance of proteins among biological samples. Numerous methodologies now support relative quantification measurements, providing a routine means to analyze protein expression patterns and post-translational modification states as a function of biological perturbation. One of the most popular methods for relative quantification through MS is stable isotope labeling of proteins in samples prior to analysis. Labeling can be achieved by the application of combinatorial heavy isotopologues of C, H, N, and O and can be introduced in proteins either by metabolic means or through chemical derivatization processes. In vivo metabolic labeling approaches include techniques such as stable isotope labeling in mammals (SILAM),1 stable isotope labeling by amino acids in cell culture (SILAC),2 and NeuCode (neutron encoding) SILAC.3 The in vitro chemical derivatization processes include techniques such as isotope-coded affinity tags (ICAT),4 dimethyl labeling,5 isobaric mass tags,6,7 and others.8
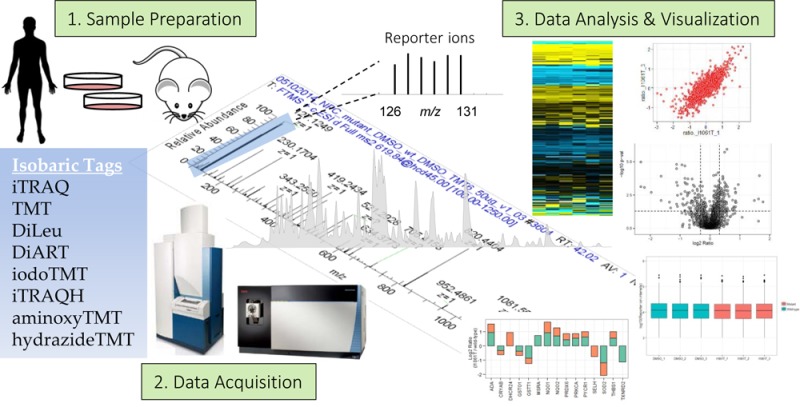
With the exception of isobaric mass tags, stable isotope derivatization methods introduce a small mass difference to identical peptides from two or more samples so that they can be distinguished in the MS1 spectrum. The relative-abundance ratio of peptides is experimentally measured by comparing heavy/light peptide pairs, and then protein levels are inferred from statistical evaluation of the peptide ratios. Isobaric tags, on the other hand, use a different concept for peptide quantification. In isobaric labeling-based quantification, each sample is derivatized with a different isotopic variant of an isobaric mass tag from a set, and then the samples are pooled and analyzed simultaneously in MS. Since the tags are isobaric, peptides labeled with isotopic variants of the tag appear as a single composite peak at the same m/z value in an MS1 scan with identical liquid chromatography (LC) retention time. The fragmentation of the modified precursor ion during MS/MS event generates two types of product ions: (a) reporter ion peaks and (b) peptide fragment ion peaks. The quantification is accomplished by directly correlating the relative intensity of reporter ions to that of the peptide selected for MS/MS fragmentation. The fragment ion peaks observed at higher m/z are specific for peptide amino acid sequence and are used for peptide identifications, which are eventually assigned to the proteins that they represent. Since every tryptic peptide can be labeled in an isobaric labeling method, more than one peptide representing the same protein may be identified, thereby increasing the confidence in both the identification and quantification of the protein. This technology has proved to be successful in numerous experimental contexts for comparative analysis upon perturbation. A general workflow of an isobaric labeling experiment is depicted in Figure 1.
Figure 1.

(a) General workflow of an isobaric labeling experiment. The protocol involves extraction of proteins from cells or tissues followed by reduction, alkylation, and digestion. In the case of TMT 6-plex, up to six samples can be labeled with the six isobaric tags of the reagent. Resulting peptides are pooled at equal concentrations before fractionation and clean up. The TMT-labeled samples are analyzed by LC–MS/MS. (b) In an MS1 scan, same-sequence peptides from the different samples appear as a single unresolved additive precursor ion. Following fragmentation of the precursor ion during MS/MS, the six reporter ions appear as distinct masses between m/z 126–131, and the remainder of the sequence-informative b- and y-ions remains as additive isobaric signals. The reporter ion intensity indicates the relative amount of peptide in the mixture that was labeled with the corresponding reagent.
2. Isobaric Mass Tags
Isobaric mass tags include families of stable isotope chemicals that are used for labeling of peptides. They generate relative quantitative information in an isobaric labeling-based quantification strategy. Isobaric mass tags have identical overall mass but vary in terms of the distribution of heavy isotopes around their structure. The most common isobaric tag is amine-reactive, but tags that react with cysteine residues and carbonyl groups in proteins are also available. The amine specificity of the amine-reactive isobaric mass tags makes most peptides in a sample amenable to this labeling strategy. The tags employ N-hydroxysuccinimide (NHS) chemistry, and the structure consists of three functional groups: an amine-reactive group and an isotopic reporter group (N-methylpiperazine) linked by an isotopic balancer group (carbonyl) for the normalization of the total mass of the tags. The amine-reactive, NHS-ester-activated group reacts with N-terminal amine groups and ε-amine groups of lysine residues to attach the tags to the peptides. The labeling is efficient for all peptides regardless of protein sequence or proteolytic enzyme specificity. The labeling does not occur, however, if the primary amino groups are modified, such as when N-terminal glutamine or glutamic acid forms a ring (pyro-glutamic acid) or if the group is acetylated. The NHS-based isobaric tags may lead to acylation of side chain hydroxyl group of serine, threonine, and tyrosine residues under reaction conditions normally employed for the acylation of primary amines.9 For successful quantification, labeling should be specific to the targeted residues (N-terminal amine and lysyl ε-amine groups in a peptide) and should proceed to completion. Reversal of peptide O-acylation reactions can be achieved by treatment with hydroxylamine that has no disruptive effect on acyl modifications on primary amines.9
The mass normalization group balances the mass difference among the reporter ion groups so that different isotopic variants of the tag have the same mass. The overall mass of reporter and balance components of the molecule are kept constant using differential isotopic enrichment with 13C, 15N, and 18O atoms. The relative intensities of the reporter ion are used to derive quantitative information on the labeled peptides between the samples. Figure 2 shows chemical structure of commercially available isobaric mass tags: tandem mass tag (TMT) and isobaric tags for relative and absolute quantification (iTRAQ).
Figure 2.

(a) (i) Chemical structure of iTRAQ 4-plex reagent.7 The complete molecule consists of a reporter group (based on N-methylpiperazine), a mass balance group (carbonyl), and a peptide-reactive group (NHS ester). The overall mass of the reporter and balance components of the molecule are kept constant using differential isotopic enrichment with 13C, 15N, and 18O atoms. The reporter group ranges in mass from m/z 114–117, whereas the balance group ranges in mass from 28 to 31 Da, such that the combined mass remains constant (145 Da) for each of the four reagents of the iTRAQ 4-plex set. (ii) The tag reacts with peptide N-terminus or ε-amino group of lysine to form an amide linkage that fragments in a similar fashion to that of backbone peptide bonds when subjected to CID. Following fragmentation of the tag amide bond, the balance (carbonyl) moiety is lost as neutral loss, whereas charge is retained by the reporter group. The number in parentheses in the table indicates the number of enriched centers in each section of the molecule.7 (b) Chemical structure of a generic TMT reagent showing the three functional groups: an amine-reactive group that labels the N-terminus and ε-amine group of lysine in peptides, a mass normalization (balance) group that balances mass differences from individual reporter ions to ensure the same overall mass of the reagents, and a reporter group that provides the abundance of a peptide upon MS/MS in individual samples being mixed. The blue dashed lines indicate a cleavable linker that enables the release of the reporter ion from the whole tag upon MS/MS. The TMT reagent family consists of TMTzero, TMTduplex, TMT 6-plex, and TMT 10-plex sets, and each of them is based on the same chemical structure.
2.1. TMT and iTRAQ Isobaric Mass Tags
The application of isobaric tags for simultaneous determination of both the identity and relative abundance of peptide pairs was first demonstrated by Thompson et al. in 2003.6 They synthesized peptides containing a tandem mass tag and showed that this strategy could be used to obtain relative quantification in MS/MS experiment. A year later, Ross et al. published a similar approach using the iTRAQ approach.7 In this study, they demonstrated for the first time the application of isobaric mass tags with 4-fold multiplexing to identify global protein expression trends in a set of isogenic yeast strains. An 8-plex series of iTRAQ reagent performs similarly and increases throughput of analyses by a factor of 2 when compared to that of the 4-plex approach.10 A few year later, Dayon et al.11 showed the increased multiplexing capability of TMT tags and demonstrated its application by using 6-plex TMT reagents in relative quantification of standard protein mixtures at various concentrations. In this study, TMT 6-plex was also used to assess the differential protein abundance in post-mortem cerebrospinal fluid samples after brain injury vs antemortem samples.11
Isobaric reagents are commercially available through vendors such as AB Sciex (Framingham, MA, USA) and Thermo Scientific (Rockford, IL, USA). The iTRAQ reagents available from AB Sciex are set of 4-plex and 8-plex mass tags that can be used to label and derive quantitative information on up to four and eight different biological samples simultaneously. The 4-plex iTRAQ reagents have reporter ion masses at m/z 114–117 and a corresponding balancer group added to accommodate the extra isotopes has masses of 28–31 Da such that they sum to about 145 Da. The 8-plex reagents have reporter ion masses at m/z 113–119 and 121 with a balance group ranging from 24–31 Da. Mass 120 is omitted in iTRAQ 8-plex to avoid contamination from phenylalanine immonium ion (m/z 120.08). Thermo Scientific TMT reagents, available as TMTzero, TMT duplex, TMT 6-plex, and TMT 10-plex, share an identical structure with each other but contain different numbers and combinations of 13C and 15N isotopes in the mass reporter region. The identical structure of TMT reagents facilitates efficient transition from method development using TMTzero or TMT duplex to multiplex quantification using TMT 6-plex or TMT 10-plex. The chemical structure of the TMT tag enables the introduction of five heavy isotopes (13C or 15N) in the reporter group and five heavy isotopes (13C or 15N) in the balancer group to provide six isobaric tags (Figure 3a). Each of the six tags of TMT 6-plex has a specific reporter ion that appears at m/z 126, 127, 128, 129, 130, and 131. TMT 10-plex is an expansion of TMT 6-plex generated by combining current TMT 6-plex reagents with four isotope variants of the tag with 6.32 mDa mass differences between 15N and 13C isotopes.12,13 Even though the mass difference between these reporter ion isotopologues is incredibly small, current generation high-resolution and high mass accuracy analyzers can resolve these ions. The seemingly miniscule difference is sufficient to achieve baseline resolution between the reporter ions when high resolving power is employed (30 K at m/z 400).13 Figure 3b shows the substitution of 15N for 13C to generate new reporter ions that are lighter than the original forms used in TMT 6-plex. In cases where coalescence, fusion of the proximate reporter ion signals into a single measurable entity, phenomenon is observed, the artifact can be completely eliminated by lowering the maximum ion target for MS/MS spectra.14 This modified setting does not result in any losses in identification depth or quantification quality of proteins.14 The high-throughput TMT 10-plex reagent enables concurrent MS analysis and relative quantification of up to 10 different samples derived from cells, tissues, or biological fluids. The higher multiplexing potential also facilitates incorporation of replicates, providing additional statistical validation within any given isobaric labeling experiments.15
Figure 3.

(a) Chemical structure of TMT 6-plex reagents with 13C and 15N heavy isotope positions (blue asterisks). The tags are isobaric, with a different distribution of isotopes between the reporter and mass normalization (balance) groups. (b) The substitution of 15N for 13C to generate new reporter ions that are 6.32 mDa lighter than the original forms used in TMT 6-plex.12 The TMT 6-plex reagents in combination with four isotope variants of the tag with 6.32 mDa mass differences were used to generate TMT 10-plex reagent.
The numbers of identified peptides and proteins in shotgun proteomics experiments have been compared for the three commercially available isobaric mass tags: iTRAQ 4-plex, TMT 6-plex, and iTRAQ 8-plex.16 Even though the number of identified proteins and peptides was largest with iTRAQ 4-plex, followed by TMT 6-plex, and smallest with iTRAQ 8-plex, the precision on the level of peptide–spectrum matches and protein level dynamic range was similar. The discrepancy in peptide identification observed with different n-plex isobaric mass tags was suggested to be due to combination of several factors, such as search algorithms and scoring functions, fragment ions derived from cleavage of the label itself or within the label from precursor ions, or disparate physiochemical properties conferred to the peptides depending on the type of isobaric mass tags used for their derivatization.16 However, in a study by Pottiez et al. on comparison of quantitative measurements of proteins in human plasma samples by iTRAQ 4-plex versus 8-plex reagents, 8-plex tagging provided more consistent ratios than that with 4-plex without compromising protein identification.17 The discrepancies in observations from Pichler et al. and Pottiez et al. could be due to different instruments (LTQ Orbitrap versus MALD-TOF/TOF 4800 platform) and search algorithms (Mascot and Proteome Discoverer software versus ProteinPilot 4.0 with Paragon Algorithm) that were used for the data acquisition and analysis.16,17 Nevertheless, the obvious advantage of 8-plex tagging is that it allows investigation of eight experimental conditions in one analytical experiment. For example, a study of one control and seven experimental conditions can be performed in one 8-plex experiment but would require at least three 4-plex experiments (using the control and up to three experimental samples in each). The three 4-plex experiments would need more instrument time, likely introducing a source of variability, and would be more laborious.
2.2. DiLeu and DiART Isobaric Mass Tags
N,N-Dimethyl leucine (DiLeu) is an isobaric tandem mass tagging reagent that uses isotope-encoded dimethylated leucine as reporters and serves as attractive alternative for iTRAQ and TMT.18 Labeling with DiLeu, however, requires activation of the reagents using 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM)/N-methylmorpholine (NMM) in N,N-dimethylformamide (DMF). Each label can be freshly activated before use. The general structure of DiLeu resembles that of other isobaric mass tags, with an amine-reactive group (triazine ester) targeting the N-terminus and ε-amino group of the lysine side chain of a peptide, a balance group, and a reporter group.18 A mass shift of 145.1 Da is observed for each incorporated label. By using DiLeu isobaric tags, up to four samples can be analyzed simultaneously at greatly reduced cost. The labeling efficiency of DiLeu is comparable to that of the iTRAQ reagents. However, DiLeu-labeled peptides undergo better fragmentation and hence generate higher reporter ion intensities than iTRAQ, thereby offering improved confidence for peptide identification and more reliable quantification.18 Intense reporter ions (dimethylated leucine a1 ion) at m/z 115, 116, 117, and 118 are observed for all pooled samples upon MS/MS. Even though deuterium affects the retention time of small- to intermediate-sized peptides in reversed-phase chromatography,19 the increased polarity of the amine group offsets the small deuterium number difference in 4-plex DiLeu tags.18 Figure 4a shows the chemical structure of a DiLeu tag.
Figure 4.

(a) (i) General structure of dimethyl leucine isobaric (DiLeu) mass tag.18 Reporter ions range from m/z 115–118. (ii) Illustration of formation of new peptide bond at N-terminus or ε-amino group of the lysine side chain and isotope combination of isobaric tags (b) Chemical structure of DiART isobaric reagents.21 Positions containing heavy stable isotopes are illustrated as numbers in the structure, and the table lists the elemental composition of the corresponding numbers. During MS/MS, the DiART-tagged peptides yield reporter ions ranging from m/z 114 to 119.
Deuterium isobaric Amine Reactive Tag (DiART) is another alternative to iTRAQ and TMT for isobaric tagging in quantitative proteomics.20,21 Like iTRAQ or TMT, DiART reagents have three functional groups, an amine-reactive (NHS ester) group for coupling of peptides, a balancer, and a reporter (N,N′-dimethylleucine) with a m/z range of 114–119 (Figure 4b). Up to six samples can be labeled with DiART reagents and analyzed by MS.21 DiART reagents have high isotope purity; hence, unlike that for iTRAQ, TMT, or DiLeu labeling, isotopic impurities correction is not required during data analysis of DiART-labeled samples.21 The performances of DiART and iTRAQ, including their fragmentation mechanisms, the number of identified proteins, and the accuracy of quantification, have been compared.20 Regardless of the peptide sequence, DiART tags generate high-intensity reporter ions compared to those with iTRAQ. Since quantification accuracy is dependent on the intensity of reporter ions,22 as high-intensity reporter ions are less susceptible to underestimation effect,23 DiART labeling quantifies more peptides, including low-abundance ones, and with reliable results.20 While DiLeu uses a nontraditional activation chemistry (DMTMM/NMM in DMF) to label peptides,18,21 DiART uses the same labeling protocol (NHS-ester-based peptide coupling chemistry) as that of TMT and iTRAQ, making it easy for users to switch between the techniques. However, unlike that for iTRAQ or TMT, DiART-labeled samples cannot be analyzed by the HCD-only instrument method due to easy fragmentation of its reporter ions.20 Nevertheless, DiART and DiLeu serve as a cost-effective alternatives to TMT and iTRAQ with comparable labeling efficiency. DiART has been shown to be useful in labeling large quantities of proteins from cell lysates prior to TiO2 enrichment in quantitative phosphoproteomics study.24
2.3. Post-translational Modification- and Cysteine-Specific Isobaric Mass Tags
Isobaric labeling-based quantification can also be used for differential quantification of various protein post-translational modifications. Isobaric mass tags are available that are especially designed to measure relative abundance of modified cysteine residues or carbonylated residues in protein.
2.3.1. Isobaric Reagents for Protein Carbonyl and Glycan Modifications
Carbonylation of proteins is caused by the reactive oxygen and carbonyl species generated as byproducts of lipid oxidation during oxidative stress.25 iTRAQ hydrazide (iTRAQH) is a novel reagent for the selective labeling and relative quantitative analysis of carbonyl groups in proteins.26 iTRAQH was synthesized from iTRAQ and an excess of hydrazine (Figure 5a). iTRAQH reacts with a carbonylated peptide, resulting in the formation of a hydrazone moiety. Consistent with the isobaric labeling approach, peptides labeled with different isotopic variants of iTRAQH reagents are indistinguishable in MS scan. However, the iTRAQH reporter ions in the low m/z region of the MS/MS spectrum provide the relative abundance information on the carbonylated proteins in the samples. The iTRAQH reporter ions have been used as targets for precursor selection in precursor ion scan analysis, which allows selective acquisition of MS/MS spectra of only the carbonylated peptides.26 This eliminates the need for the step involving enrichment of modified peptides prior to LC–MS/MS analysis.
Figure 5.

(a) General structure of iTRAQ hydrazide (iTRAQH) for relative quantitative analysis of carbonylation sites in proteins.26 (b) Chemical structure of the carbonyl-reactive glyco-TMT compounds.27 (Left) Hydrazide reagents; (right) aminoxy reagents. Red asterisks indicate 13C, and blue asterisks, 15N. The table below the compound structures shows isotope codes of the hydrazide- and aminoxy-functionalized glyco-TMT compounds. The carbonyl-reactive tags can be used to quantify a broad range of biologically important molecules including carbohydrates, steroids, or oxidized proteins. (c) Chemical structure of the cysteine-reactive Thermo Scientific iodoTMTzero isobaric mass tag. The iodoTMT reagents are iodoacetyl-activated isobaric mass tags for covalent, irreversible labeling of sulfhydryl (-SH) groups. IodoTMT 6-plex enable measurement of protein and peptide cysteine modifications (S-nitrosylation, oxidation, and disulfide bridges) by multiplex quantitative mass spectrometry. The workflow (not shown in the image) involves derivatization of modified peptides or proteins with the reagent, enrichment of TMT tagged peptide using anti-TMT antibody, and their subsequent elution. The eluent is analyzed by LC–MS/MS to determine the sites of modification and to measure their relative abundance across samples.
On the basis of similar chemistry as that of iTRAQH, the stable isotope-labeled carbonyl-reactive tandem mass tags (glyco-TMTs) have been used for quantification of N-linked glycans.27 Glyco-TMT reagents are derivatives of the original TMT compounds but are functionalized with carbonyl-reactive groups involving either hydrazide chemistry or aminoxy chemistry (Figure 5b). A study reported that aminooxy TMTs outperformed their hydrazide counterparts in labeling efficiency and quantification.27 The glyco-TMT compounds are coded with stable isotopes and enable (i) isobaric quantification in MS/MS spectra and (ii) quantification in MS1 spectra using heavy/light pairs. Isobaric quantification using glyco-TMT can be achieved by using the aminoxy TMT6-128 and TMT6-131 as well as the hydrazide TMT2-126 and TMT2-127 reagents (Figure 5b). The MS1 level quantification is accomplished by the mass difference of 5.0105 Da between the light TMT0 and the heavy TMT6 reagents (Figure 5b) that is sufficient to separate the isotopic patterns of all commonly existing N-glycans. Glycan quantification using heavy and light glycol-TMTs provided more accurate quantification in MS1 spectra over a broad dynamic range compared with that from quantification based on the reporter ions generated in MS/MS spectra.27 Glyco-TMTs with aminooxy-functionalized groups are available commercially from Thermo Scientific (Rockford, IL, USA) as aminoxyTMTzero and aminoxyTMT 6-plex reagents. Labeling with aminoxyTMT reagents involves treating intact proteins or proteolytic digests of proteins extracted from biological specimens with PNGase F/A glycosidases to release N-linked glycans. The free glycans are subsequently purified from protein or peptide matrix and labeled at the reducing end with the aminoxyTMT reagents. The derivatized glycans from individual samples are then combined and analyzed in MS to identify glycoforms in the sample and to quantify reporter ion relative abundance at MS/MS level.
2.3.2. Isobaric Reagents for Tagging Cysteine Residues
Cysteine sulfhydryls in proteins are potential sites of reversible oxidative modification because of the unique redox chemistry of this amino acid.28 S-Nitrosylation is a redox-based protein post-translational modification that occurs in response to nitric oxide signaling and is involved in a wide range of biological processes.28 It involves addition of a nitric oxide (NO) group to a specific cysteine residue of a protein to form S-nitrosothiol. An analytical strategy to enrich and relatively quantify cysteine-containing peptides in complex mixtures has been reported.29 In this strategy, cysteine residues in proteins are first derivatized with N-{2-((2-acryloyl)amino)ethyl-1,3 thiazolidine-4-carboxamide) (ATC) followed by labeling with amine-reactive TMT tags for relative quantification of the targeted peptides after the covalent capture. The workflow involves reduction, derivatization of cysteine residue in protein samples with ATC tag, digestion with trypsin, and differential labeling with TMT tags followed by pooling of the labeled samples. The ATC-derivatized cysteinyl peptides are subsequently isolated on an aldehyde resin through the covalent capture technique and analyzed with LC–MS/MS.
The cysteine-reactive TMT reagents allow measurement of S-nitrosylation occupancy and determination of individual protein thiol reactivity.30,31 However, the disulfide linkage between the (reversible) cysteine-reactive TMT tag and protein thiol group cannot survive the strong reducing conditions normally used during enzymatic digestion for subsequent shotgun proteomic analysis.32 An irreversible cysteine-reactive TMT reagent containing a sulfhydryl-reactive iodoacetyl reactive group called iodoTMT has been developed.32 IodoTMT reagents such as iodoTMTzero and iodoTMT 6-plex are commercially available from Thermo Scientific (Rockford, IL, USA). Each isobaric iodoTMT 6-plex reagent within a set has the same nominal mass and consists of a thiol-reactive iodoacetyl functional group for covalent and irreversible labeling of cysteine, a balancer, and a reporter group. The quantification using iodoTMT tags is achieved by inspection of the reporter ion region in MS/MS spectra. The chemical structure of iodoTMTzero reagent is shown in Figure 5c. An iodoTMT switch assay uses an isobaric set of thiol-reactive iodoTMT 6-plex reagents to specifically detect and quantify protein S-nitrosylation.32,33 The iodoTMT switch assay workflow includes irreversible labeling of S-nitrosylated cysteines followed by enrichment of S-nitrosylated peptides using high-affinity anti-TMT chromatography with competitive elution and finally multiplexed quantification of protein S-nitrosylation via six unique TMT reporter ions.32,33
3. Benefits of Isobaric Labeling-Based Quantification Strategy
Isobaric labeling-based quantification has many advantages compared to other stable isotope labeling techniques, one of which is the ability to perform high-throughput quantification due to sample multiplexing. The ability to combine and analyze several samples within one experiment eliminates the need to compare multiple LC–MS/MS data sets, thereby reducing overall analytical time and run-to-run variation. Moreover, the information replication within LC–MS/MS experimental regimes provides additional statistical validation within any given experiment.15 This is desirable in an analysis where conventional upregulation and downregulation measurements are not nearly as meaningful as obtaining temporal expression patterns of proteins throughout the experimental condition, such as in studies involving different stages of cell differentiation, comparisons of multiple drug treatments, identifications of protein–drug interactions,34 measurement of inhibitor dose response, or time course comparisons.35 When each sample is run separately or with limited multiplexing, as required in label-free, metabolic-labeling and other MS1-based quantification methods, an ion selected for fragmentation on one LC–MS/MS run may not be selected consistently in subsequent runs or spectra of suitable quality may not be acquired. This results in missing observations, affecting identification and quantification. The isobaric labeling strategy, however, is immune to the stochastic nature of data-dependent mass spectrometry because a common precursor ion is fragmented that corresponds to the same peptide species present in all of the labeled samples, yielding quantitative data across samples within an isobaric tagging experiment. Isobaric labeling has been shown to surpass metabolic labeling in quantification precision and reproducibility.36
Isobaric labeling exhibits a wide dynamic range in profiling both high- and low-abundance proteins and proteins with wide array of physiological properties.37 It can be used to identify and quantify proteins across diverse molecular weight and pI ranges, functional categories, and cellular locations.38,39 The isobaric mass tags do not interfere with peptide fragmentation, and the peptide length distribution profile and amino acid content of the isobarically derivatized peptides are similar to those obtained using other MS-based approaches.38 In fact, isobaric tags have been reported to improve the efficiency of MS/MS fragmentation and result in increased signal intensities of native peptides in samples of human parotid saliva that, in general, lack the uniform architecture of tryptic cleavage products, e.g., a basic C-terminal amino acid residue.40
With an MS1-based quantification approach, the co-elution of light and heavy peptides can compromise sensitivity as the ion current is divided between multiple samples during MS analysis. Occurrence of multiple precursor ion species in the MS1 level can also create redundancy in MS/MS scanning events of the same peptide bearing different labels. This results in undersampling of the proteome. It is reported that up to 50% of MS/MS scans acquired during data acquisition can be redundant.41 By contrast, labeling of samples by isobaric mass tags does not increase the sample complexity during chromatographic separation and MS analysis because they are isotope-coded molecules with the same chemical structure and molecular weight, thus eluting at the same chromatographic time and with the same peptide mass. In fact, since differentially labeled but identical peptides from multiple samples are efficaciously merged, an improvement in overall signal-to-noise ratios occurs, allowing good-quality MS/MS data to be acquired from low-copy-number proteins.40,42 Moreover, the sequence informative b- and y-ions in MS/MS spectra also show this summed intensity, which aids sensitivity.43
The in vitro labeling procedure used for isobaric labeling-based quantification strategy is highly efficient and enables this method to be applicable to wide variety of samples such as cultured cells, human tissues and biofluids, and tissues from model animals. This technique has been successfully applied to various biological studies, demonstrating its validity and robustness for quantitative MS-based proteomics.37,42,44−51 Isobaric labeling, especially iTRAQ has been used in identifying and distinguishing protease-generated neo-N termini from N-termini of mature proteins by performing terminal amine isotopic labeling of substrates (TAILS).52,53 After tryptic digestion of iTRAQ-labeled protein samples, N-terminal peptide separation is accomplished using a high-molecular-weight dendritic polyglycerol aldehyde polymer that binds internal tryptic and C-terminal peptides that now have N-terminal alpha amines. The unbound iTRAQ-labeled mature N-terminal and neo-N-terminal peptides and naturally blocked (acetylated, cyclized, and methylated) peptides are recovered by ultrafiltration and analyzed by mass spectrometry. The neo-N-terminal peptides specific to the protease of interest appear only in the protease-treated sample and therefore show a high protease/untreated iTRAQ reporter ion intensity ratio, thus differentiating them from trypsin cleavage products that are present in all samples in equal amounts and therefore have expected iTRAQ ratios of 1.53 The applications of the isobaric labeling strategy have also been extended to studies involving the characterization of post-translational modifications such as phosphorylation41,54−56 and other modifications (discussed in the section above).
4. Instrumentation and Data Acquisition Methods for Isobarically Labeled Samples
Many different mass spectrometers are capable of analyzing isobarically tagged peptides. Initially, isobaric labeling experiments were carried out on MALDI-TOF/TOF57,58 and quadrupole time-of-flight (Q-TOF)7,35 instruments. Quadrupole59 and TOF instruments are capable of detecting low m/z fragment ions in the region where reporter ions are observed. However, the large ion selection window of the TOF/TOF instrument can result in a relatively high background of chemical noise for the reporter groups, compressing the dynamic range of the ratios significantly.58 Quadrupole ion trap geometries generally produce suboptimal results because the reporter ions often lie below the stability limit, as dictated by the precursor peptide mass-to-charge ratio and pseudopotential well parameters used for activation (for example, activation q = 0.23).22 The slow scanning Q-TOF instruments also have less sensitivity for complex mixtures compared to that of linear ion traps.60
Isobaric quantification using standard collision-induced dissociation (CID) conditions is not feasible using ion traps. The “1/3 rule” for ion-trap instruments restricts the analysis of product ions with m/z values less than 25–30% of the precursor ion. This low mass cutoff limitation also applies to hybrid instruments containing an ion-trap for fragmentation, such as the LTQ-FT and the LTQ-Orbitrap.61 This limitation can, in principle, be overcome by pulsed Q dissociation (PQD).62 PQD in the ion trap facilitates detection of low m/z reporter ions, bridging the gap between the linear ion trap with PQD and a quadrupole TOF instrument.60 However, unlike conventional CID spectra, typical PQD spectra are dominated by the unfragmented precursor ion, indicating poor fragmentation efficiency and thus limiting its practical utility for quantification of peptides by iTRAQ or TMT approaches. Nevertheless, Bantscheff et al. and Griffin et al. have shown that by carefully optimizing instrument parameters such as collision energy, activation q, delay time, ion isolation width, number of microscans, repeat count, and number of trapped ions, low m/z fragment ion intensities can be generated that enable accurate peptide quantification.60,63 A combined CID-PQD scan strategy exploits CID for efficient peptide identification and PQD for quantification.49,64
The development of higher energy collision-induced dissociation (HCD) in the LTQ-Orbitrap has also overcome the 1/3 rule limitation. In an ion trap CID is a resonance-based process, whereas HCD is a beam-type CID event that results in a different fragmentation pattern. During HCD, ions are accelerated as they leave the C-trap and then are fragmented in the nitrogen-filled collision cell. The resulting fragments are returned to the C-trap and detected in the Orbitrap mass analyzer. This fragmentation technique allows analysis of the low m/z region of reporter ions in the Orbitrap mass analyzer since there is no mass cutoff for the multipole.65 HCD enables efficient reporter ion generation with high mass accuracy detection, but, in general, it suffers from poor peptide sequence-ion recovery compared to that of the classical ion trap CID analysis. The combined use of CID and HCD for efficient identification and relative quantification of proteins with isobaric tags has been demonstrated.61,66 In this dual-fragmentation method, HCD is used to derive the accurate quantitative information from the reporter ions, whereas CID provides identification of the corresponding peptides. This method alternates MS/MS spectra generated by CID fragmentation with MS/MS spectra obtained from the same precursor ion by HCD fragmentation. Since CID in the ion trap occurs in parallel to acquisition of HCD MS/MS spectra in the Orbitrap, the analysis duty cycle is unaffected. CID and HCD spectra are subsequently combined by merging the peptide sequence-ion m/z range from CID spectra and the reporter ion m/z range from HCD spectra. It should be noted that the extracted intensity values of the reporter ions from each HCD spectrum should be normalized to low ion counts when merging with the respective CID data, otherwise peptide scores can be significantly reduced.61 The CID-HCD method was shown to be superior to HCD alone in terms of sensitivity and ability to identify proteins in complex mixtures.61 However, a recent study has shown that with fine-tuning of the normalized collision energy values on Orbitrap Velos instruments, an HCD-only method can perform better than a CID-HCD dual-fragmentation method.67 This is due to the implementation of the new HCD cell with an axial electric field to push the fragment ions into the C-trap and mounted on Orbitrap XL ETD and Orbitrap Velos instruments that allows an improvement in the analytical precision of the acquired reporter ions.68 In addition, the redundancy in precursor selection in the dual CID-HCD method compared to that for the stand alone HCD method can result in a reduced number of total peptide and protein identifications.67 The use of a stepped HCD scheme in Q Exactive instruments has been shown to enhance the intensity of reporter ions without adversely affecting peptide identifications.69
Another method for analyzing isobaric labeled samples is to use triple-stage mass spectrometry (MS3) in a hybrid ion trap-Orbitrap platform.70 In this approach, a peptide precursor ion is isolated and fragmented with CID-MS/MS to generate a plurality of first-generation product ion species comprising different respective m/z ratios. The most intense product ion in MS/MS scan is then selected for HCD-MS3, yielding quantitative data. This method provides an experimental solution to remove interference, thus eliminating the ratio distortion problem (discussed in the next section). A variant of this method referred to as Multinotch MS371,72 involves selecting and co-isolating two or more of the first-generation product-ion species and fragmenting them to generate a plurality of second-generation fragment ion species including released isobaric tags (Figure 6). The Multinotch MS3 method significantly improves quantitative accuracy and increases the sensitivity of the MS experiment up to n-fold, where n is the number of MS fragments selected and simultaneously isolated.71
Figure 6.
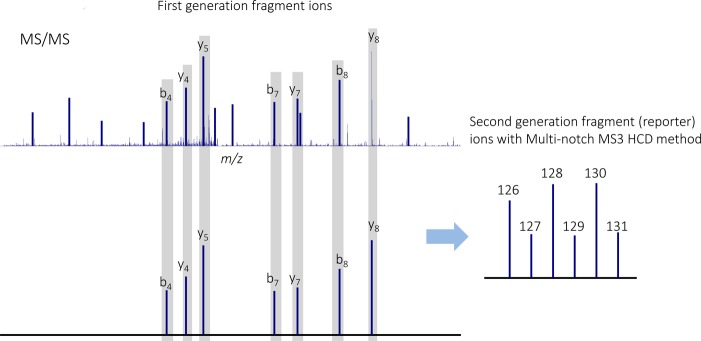
Multinotch MS3 involves selecting and co-isolating multiple MS/MS product ion and fragmenting them to generate a plurality of second-generation fragment ion species including released isobaric tags.71,72 The method increases the sensitivity and quantitative accuracy achieved by isobaric labeling-based quantification approach.
5. Factors Affecting Quantification by Isobaric Labeling: Technical and Bioinformatics Issues
The ratios of the intensity of the reporter ions reflect the relative abundance of the peptides from which they are derived. The integration of the relative quantification data for the peptides allows elucidation of relative protein expression levels. This section discusses the various aspects of data analysis in isobaric labeling-based quantification.
5.1. Evaluation of Labeling Efficiency and Isotope Impurity Correction
Isobaric labeling is usually very efficient; however, when primary amino groups are present elsewhere in the sample, they may interfere with the labeling reaction since they can react with the amine-reactive isobaric mass tags. Hence, proper sample preparation is imperative for the success of an isobaric labeling-based quantification technique and includes either avoiding the use of primary amine-containing buffers such as Tris and ammonium bicarbonate or performing sample cleanup prior to the isobaric labeling reaction.73 To improve detection limits and achieve a reliable estimate of quantification, it is recommended that the labeling efficiency be determined for each isobaric labeling experiment. The labeling efficiency can be ascertained by searching the data separately against protein database using TMT and iTRAQ modifications as variable instead of fixed modifications. Using these parameters, both labeled and unlabeled peptides can be identified and used to calculate labeling efficiency, which is defined as the percent of labeled peptides among all identified peptides. The labeling efficiency can be estimated as
where nti and nki are the number of isobaric tag-labeled N-termini and lysine residues, respectively, and, ntt and nkt are the total number of peptide N-termini and lysine residues, respectively.74 Additionally, due to isotopic contamination in isobaric mass tags, the peaks for each reporter ion will have some contribution from adjacent reporter ions. Hence, prior to data analysis, each of the reporter ion peaks must be corrected to account for isotopic overlap (values reported in the manufacturer’s instruction sheet) in order to achieve accurate quantification. The uncorrected data will appear distorted and confound the observed change in protein expression levels.23 A detailed procedure to calculate true peak areas that account for overlapping isotopic contributions using the reagent purity values provided by the manufacturer is described elsewhere.75
5.2. Ratio Compression and Its Correction
In isobaric labeling-based experiments, accurate ratios can be determined only when a single precursor ion is selected for fragmentation during an MS/MS scan event. It has been observed that the presence of co-eluting peptides within the isolation window used for the selection and subsequent fragmentation of individual peptide ions typically results in an underestimation or compression of actual protein abundance differences in the analyzed samples.23,76,77 This effect is ubiquitous and not dependent only on the instrument used to acquire the data.77 The compression in relative abundance is based on the assumption that the vast majority of proteins in biological studies do not change significantly; therefore, when the peptides from these proteins co-fragment, the reporter ion intensity ratios generated will be less pronounced in terms of fold changes. Precursor ions of similar intensities can produce reporter ions that span over 2 orders of magnitude in intensity.41 This means that very low intensity background ions can significantly contribute to reporter ion signals when they get co-fragmented with a selected precursor ion. Additionally, if the coeluting peptides display a nonequimolar distribution of reporter ions, then the net effect of this co-selection is the unpredictable and context-specific distortion of reporter ion intensities.78 In addition to the distortion in quantification accuracy due to coselection phenomena, the source of quantification error can also be due to presence and interference from artifactual spectral peaks. The reporter ion region in Orbitrap HCD MS/MS spectra contains many signals that are nearly isobaric with reporter ions generated from isobaric mass tags. These signals do not correspond to any plausible chemical compositions and may, in part, be attributed to artifacts related to amplifying and processing the transient signal of the Orbitrap.79 Depending on the mass tolerance used for picking the reporter ion signals, the presence of these nonreporter ion signals may distort the quantification results.
Peptide abundance ratios are calculated by combining data from multiple fractions across MS runs and then averaging across peptides to give an abundance ratio for each parent protein. The measured relative abundance can be influenced by the separation (e.g., SCX) stage in which the MS/MS was acquired, a phenomenon termed as fraction effect.77 Fraction effect for a given peptide is defined as a significant dependence between the measured ratio and the fraction in which the reading was taken. The error within a fraction group for a peptide is smaller than the error between fraction groups and arises from the additional variance from the repeated SCX separation stage.77 The observation of fraction effect could be due to differences in a peptide’s concentration across fractions that contribute to variability in precursor ion intensity measures and subsequent reporter ion peak areas.80 In addition to fraction effect, the measured ratio is also dependent on the precursor ion (i.e., peptide) used to characterize a protein.77,81 The measurement error within a peptide group for a protein was found to be smaller than the error between peptide groups.77 This phenomenon is termed peptide effect. The difference in quantitative value from one peptide to another, even though belonging to the same protein, might result from factors such as post-translational modifications and/or splice variants,80 tryptic digestion artifacts, peptide recovery, and stability.81 Other factors of peptide effect include noise peaks with high signal-to-noise in the reporter ion region,82 sequence of the peptide used for quantification and the possibility of interference from the immonium ion signals in the reporter ion region,23,56 various charge states of the same peptide, and the number of isobaric tags per peptide.73
Since interference due to coisolation is dependent on sample complexity and the number of co-eluting peptides, the ratio compression can be partly alleviated by better fractionation of complex biological samples at the protein or peptide level.83 Ratio compression was observed to be smaller for enriched phosphoproteome samples compared to that for whole proteome samples due to their overall lower sample complexity.41 Another approach involves using an optimized (narrow) MS/MS isolation width setting so that fewer contaminant ions are present during precursor ion activation.76 The high mass resolving power (m/Δm > 15 000) in the reporter ion region also minimizes interference from potential contaminant species that may confound quantification data.22 Delaying peptide selection and fragmentation until the apex of the chromatographic peak during LC–MS/MS analysis has been shown to reduce co-fragmentation by 2-fold.76 With the delayed fragmentation approach, peptides were fragmented with 2.8-fold better signal-to-noise ratios, significantly improving the quantification.76 A targeted mass spectrometric data acquisition methodology with reporter ion-based quantification has been shown to be useful in applications where it is essential to reidentify and requantify a defined set of target proteins in a complex mixture.84 The gas-phase purification85 and MS370,71 methods also eliminate interfering ions in complex mixtures. In Q-TOF instruments, ion mobility (IM) separations have the potential to mitigate quantitative inaccuracies caused by isobaric interference since IM-MS has the ability to separate ions based on charge, m/z, and collision cross section (shape and size).86
The ratio correction can also be achieved by various computational approaches post data acquisition. One of the strategies is to use an algorithm that corrects experimental ratios on the basis of determined peptide interference levels.87 In this method, the measurement for spectrum purity in survey spectra (signal-to-interference measure) was used to improve the accuracy of protein quantification. Signal-to-interference at the time of an MS/MS event is calculated by dividing precursor abundance by the sum of all ion signals observed within the isolation window.76,84 Consequently, values close to one indicate little and values close to zero indicate a high degree of interference caused by co-eluting components. Other informatics approaches include the intensity-based weighed average technique,88 variance-stabilizing normalization,77 and robust statistic-based metric called redescending M-estimator.89 The interference from non-TMT signals can be eliminated by mass difference processing in which TMT reporter ions in HCD spectra are identified via accurate mass differences between TMT reporter ions present within the same tandem mass spectrum instead of applying fixed mass error tolerances for all tandem mass spectra.90 This process leads to unambiguous reporter ion identification and eliminates all non-TMT ions from the spectra. Zhang et al. developed an error model that relates the variance of measured ratios to observed reporter ion intensity and provides a p value, q value, and confidence interval for every peptide identified.22 The identification and exclusion of outlier data, with Grubb’s and Rosner’s tests, that alter or inappropriately skew the average observed expression ratios has shown to result in a more statistically robust estimation of relative protein abundance.82 The ability to consider outlier data, however, can occur only for proteins in which there are more than three MS/MS measurements of protein expression.82 In summary, even though all of the suggested strategies have merit, some techniques only partially remove the problem, and others come with decreased throughput or utilize specialized mass spectrometric instrumentation.
5.3. Reporter Ion Intensity Dynamic Range
Isobaric labeling-based quantification accuracy is also influenced by reporter ion signal intensity and may result in either an underestimation or overestimation of quantification ratio if the signal intensity is outside the detector’s saturation point.91,92 The reporter ions intensities will range between two extremes: the maximum intensity, which corresponds to saturation, and the minimum intensity, which corresponds to the lowest intensity detected. This range is known as the detection limits.89 However, not all reporter ion intensity peaks will lead to accurate relative quantification. The peak intensities of high-abundance peptide ions may be underestimated by a saturation effect of the detector, which is instrument-dependent.92 Nevertheless, high-intensity peptides convey more reliable quantitative information about the protein.23 Larger variances of peptide ratios have been observed for reporter ions of lower intensity93 because the noise associated with low-intensity reporters constitutes a major handicap in determining the statistical significance of the differential expression of a protein.23 Therefore, peptides with higher reporter ion intensities should be given higher weight when used to calculate a protein’s abundance.36 Reporter ion signal intensity can be increased by increasing the MS/MS acquisition duration; however, this comes at the expense of decreased sampling, resulting in fewer protein identifications.93 It is therefore important to estimate the quantification limits of the instrument and the method used in order to assess the reliability of the obtained quantification measurement. This can be achieved by spiking samples with known quantities of reference proteins prior to analysis and confirming the expected protein ratio from the measured reporter ion intensity ratios.89
5.4. Effect of Unique and Shared Peptides in Inferring Protein Ratios
In isobaric labeling, peptide ratios are usually compiled to infer protein ratios. Significant quantification errors arise if a quantified peptide is not unique to its corresponding protein.92 Hence, relative quantification based on shared peptides (i.e., peptides that match multiple proteins or protein isoforms) due to sequence homology should be interpreted with caution.94 For a distinct peptide, its relative abundance ratio is a direct measure of the abundance ratio of its corresponding protein. In contrast, the relative abundance ratio of a shared peptide is a weighted average of the abundance ratios of all its corresponding proteins, with the weighting factors being determined by the absolute abundance of those proteins in the samples.94
Even though isobaric quantification is not dependent on the total number of spectra matching to each protein, a high number of relative abundance ratios obtained from multiple peptide/spectra increase the confidence in the observed protein ratios.89 Both intact protein mass and abundance level influence the reliability of the quantification results since highly abundant proteins generate a larger number of peptides per protein.95 More data, whether from multiple observations per protein or from increasing replication, increases the detection of real signal and reduces false positives.95 Quantitative information derived from proteins identified with a single peptide lacks variance measurements. The identification of so-called one-hit wonders should be filtered intelligently based on the goal of the study.96 These proteins deserve special attention if isobaric labeling is used as a screening tool since potentially important biological information or novel biomarkers may be discarded before they are even considered.
5.5. Estimation of Protein Fold Changes
Fold change has been shown to be a function of protein mass and abundance, with small, low-abundance proteins showing the largest variance.95 A protein is considered to be differentially regulated if the measured fold change exceeds a certain threshold. The actual protein expression level is normally distorted by many factors, with biological variation being the most significant and which ultimately increases the cutoff point.97 The cutoff point that defines significant differential protein regulation upon perturbation can be estimated by including sample replicates in the experiment.73,98 The replicate samples can be technical, experimental, or biological. According to Gan et al.,97 the definition of replicates in terms of isobaric labeling is as follows: a technical replicate will have two identical samples from the same biological source in an isobaric experiment set, whereas a biological replicate will have two distinct biological samples from same condition in an experiment set. The experimental replicate is the actual isobaric experiment replicate, the repetition of the same samples in two or more experimental sets, and they must have the same reference point or control. An illustration of the relationship among technical, experimental, and biological replicates in isobaric labeling experiments is depicted in Figure 7.
Figure 7.
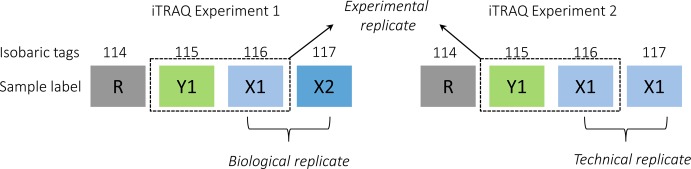
An example defining the relation among technical, experimental, and biological replicates in isobaric labeling (iTRAQ 4-plex in this example) experiments.97 A biological replicate has two distinct biological samples (X1 and X2) from the same condition in an iTRAQ set, whereas a technical replicate has two identical samples (X1 and X1) from the same biological source in an iTRAQ set. An experimental replicate is the repetitive analysis of the setup to assess the variation of the identical sample in two different iTRAQ sets (Y1 and X1 in experiment 1 versus Y1 and X1 in experiment 2). R refers to a reference sample that can be an individual sample or a pooled sample and allows cross-set comparison.
Typically, a technical replicate assesses possible errors contributed from sample preparation, also commonly known as the sample variance. Biological replicates are used to examine the variation of random biological effects. Biological variation is protein-, patient-, and disease-dependent.99 An experimental replicate compares the variation of an identical sample in two different isobaric experiment sets. During analysis of replicate samples, the theoretical relative quantification ratio should be 1:1;99 however, due to associated variations, the observed relative protein ratios might deviate from the theoretical value. The threshold should be chosen such that it encompasses the majority of technical and biological variation among the replicates. Since in isobaric labeling multiple samples are combined and run together, good quantification precision is observed. Hence, the ratio cutoff applied for significant protein change via the isobaric labeling-based quantification approach is lower than the cutoff applied for the label-free quantification approach;100 however, the researcher will need to assess whether such a change is biologically significant.
5.6. Comparison of Multiple Isobaric Labeling Experiments
For comparing biological replicates with isobaric labeling in multiple experimental designs, it is recommended to include a reference sample in each experimental setup. The common reference sample among experiments will allow for cross-set comparison. This can be accomplished by first comparing protein ratios of each sample against its reference within individual experiments and then extending the information among multiple experiments. The reference can be an individual sample or a pooled sample prepared by mixing small aliquots of equal amounts of protein from different individual samples.101 The composition of reference samples does not contribute to missing quantitative values, hence pooling to form a reference sample does not negatively impact the ability to quantitate peptides from comparative individual samples.93,99 The random biological variation in a pooled sample is generally lower, as the biological variation can be normalized by n samples before being introduced into the experiment.97 Pooling provides a representative proteome of all of the samples that are detected in comparative samples and is needed for reliable quantification. It also provides sufficient reference material that can be used in many experiments. Herbrich et al., however, have shown that using a masterpool is counterproductive since the latter is also subjected to experimental noise and can result in highly variable estimates when ratios are calculated.102 According to their study, more precise estimates of protein relative abundance can be obtained by using the available biological data.102 When a reference sample is used, consistency of the reference is necessary throughout the entire experiment, otherwise even small changes to the reference sample are sufficient to alter the proteins that are reported as differentially expressed.93
Regardless of using an individual sample or a pooled sample as a reference, before employing isobaric quantification results for follow-up studies, it is imperative to determine that the data was normalized adequately and the shortlisted protein targets hold merit. Improper normalization might remove some of the biological effects, resulting in attenuated estimates of the protein fold change. Like any other quantification technique, isobaric labeling-based quantification is also biased toward identifying and quantifying a larger percentage of the more abundant proteins, such as ribosomal proteins, heat shock proteins, cytoskeletal proteins, transcription factors, and many others, and often with multiple peptides.103,104 This is mainly due to the fact that their precursor ions have higher signal intensity. The greater signal intensity increases the likelihood that a given peptide will be selected for fragmentation during LC–MS/MS analysis. Since, in most cases, the expression levels of these house-keeping proteins remain unperturbed in related cell types or growth conditions, they can be used as an effective means to determine the reliability of data normalization.104 Normalizing by total intensity is not appropriate when the amount of protein is different in the different quantitative sample such as samples that are enriched for certain proteins by pull-down experiments.
6. Extended Applications of Isobaric Labeling-Based Quantification Strategy
Isobaric labeling experiments can be used for phosphopeptide quantification, and, in cases where the number of samples exceeds the number of isobaric mass tags available for labeling, the throughput can be increased by a hyperplexing method. Isobaric mass tags can also be used for targeted quantification.
6.1. Phosphopeptide Quantification Using Isobaric Mass Tags
Amine-reactive isobaric mass tags have successfully been used in the quantification of post-translational modification such as phosphorylation. Phosphopeptides exist in substoichiometric quantities, and because of the high background of nonphosphorylated peptides in a proteome digest, enrichment of phosphorylated peptides is necessary prior to introduction into the MS. Phosphopeptide enrichment can be performed on isobaric-labeled peptides,24,54,55 or the phosphorylated peptide can be labeled upon enrichment.105 Labeling before enrichment minimizes analytical variations caused by further sample manipulation of individual samples during enrichment, whereas labeling after enrichment might improve the yield of the labeling since the nonphosphorylated peptides would otherwise compete for isobaric reagent and interfere with the complete labeling of phosphopeptides.
During the CID-HCD dual method for the quantification and identification of isobarically tagged phosphopeptides, CID with detection in the linear ion trap provides better sensitivity and can be an advantage for low-abundance precursors such as phosphopeptides. However, quantitative information from the low mass region of subsequent HCD scans may not be available for all such CID scans since HCD scans requires higher ion counts.41 Linke et al. and Wu et al. have examined the optimal fragmentation conditions using the CID-HCD method for iTRAQ-labeled synthetic phosphopeptides in a complex phosphopeptide mix106 and phosphopeptides enriched from cells.105 During CID-MS/MS, the spectra derived from phosphoserine- and phosphothreonine-containing peptides show facile fragmentation of the phosphate group and dominance of neutral phosphate losses from the precursor ions.107 The neutral loss in HCD-MS/MS is much lower, and the sequence-specific fragments are significantly more abundant. With the increasing charge state of the precursor ions, the neutral loss in HCD-MS/MS becomes insignificant and is surpassed by the amide-bond cleavage.105 However, the study of the effect of normalized collision energy on the HCD-MS/MS fragments and reporter ion abundances shows that the HCD identified phosphopeptides and the HCD spectra with reporter ion information are strongly dependent on precursor charge state.105 The 2+ charged precursors are more sensitive to the applied normalized collision energy values than the 3+ charged precursor ions in HCD experiments.106 Thingholm et al. have shown that derivatization with isobaric mass tags significantly increases the average ion charge state of phosphopeptides compared to that of nonlabeled peptides, resulting in a considerable reduction in the number of identified phosphopeptides.108 Interestingly, it was demonstrated that adding a perpendicular flow of ammonia vapor between the needle and the MS orifice in LC–MS/MS analyses reduced the average charge state of isobaric labeled peptides and resulted in an increase in peptide identification. Thus, the application of isobaric labeling strategies for quantitative phosphopeptide analysis requires simultaneous monitoring of peptide backbone dissociation, loss of phosphoryl group, and the generation of reporter ions.
6.2. Hyperplexing with Isobaric Mass Tags
With existing isobaric mass tags, the maximum number of samples that can be combined and analyzed in a single LC–MS/MS experiment is eight in the case of iTRAQ and 10 with TMT. An effort to increase the multiplexing capacity by the combined use of metabolic and isobaric labeling has been demonstrated.109 In this strategy, the mass separation of co-eluting intact peptides with the same sequence in an MS1 scan achieved by duplex (heavy and light) or triplex SILAC labeling was exploited to allow for the simultaneous quantification of multiple sets of TMT 6-plex isobaric labels in a single run. Using a 3 × 6 hyperplexing experiment that enables simultaneous quantification of 18 samples, yeast response to the immunosuppressant drug rapamycin, which inhibits the kinase target of rapamycin (TOR), was monitored by measuring the changes in their protein abundance.109 In this study, three separate cultures of yeast cells grown in light, medium, or heavy SILAC culture medium were treated with 200 mM rapamycin, and samples were removed at 0, 30, 60, 120, and 180 min. A single 120 min sample was taken from parallel cultures treated with DMSO. Equal amounts of peptides from each sample were labeled with 6-plex TMT reagents, mixed, and separated by SCX before LC–MS/MS analysis. The increased multiplexing capacity enabled analyses of multiple biological replicates of a time-course study in the same run, providing the statistical power required to identify significant trends. The hyperplexing technique with combined metabolic labeling and isobaric mass tags can also be extended to 15N-labeled samples. Alternatively, the dimethyl chemical labeling technique can be combined with isobaric mass tags to increase the multiplexing capacity of quantitative proteomics. Theoretically, the combination of iTRAQ 8-plex or TMT 10-plex reagents and triplex SILAC would allow 24 or 30 channels to be monitored simultaneously.
6.3. Targeted Analysis with Isobaric Mass Tags
Proteins that are identified and quantified as differentially expressed can be used for subsequent targeted studies using the isobaric labeling technique to assess reproducibility of the entire procedure and to validate the observed differences in protein expression levels between samples. During biomarker discovery experiments, targeted investigations are necessary to verify proteins with higher variance in additional patient samples or to obtain greater statistical power. For successful targeted analysis, peptides that allow clear protein quantification and are also sufficiently intense should be selected as representative target peptides for validation. Isobaric mass tags are often used for discovery studies to reveal proteins being differentially expressed under any given conditions. However, Stella et al. have shown that isobaric mass tags can be used in combination with multiple reaction monitoring (MRM) for targeted quantification.110 In this study, the instrument monitored the two reporter ions and three transitions for each peptide selected from the target membrane proteins. The relative quantification was achieved by comparing the intensities of the reporter ions generated from the labeled precursor peptide of two samples, wild-type (reporter ion m/z 129) and prion protein(PrP)-knockout (reporter ion m/z 131) cerebellar granule neurons.110 Byers et al. used isotopic versions of TMT reagents for targeted quantification to verify protein regulations observed in a discovery study.111 These isotopic sets of reagents are structurally identical to the isobaric ones but have different numbers of heavy isotopes incorporated and are referred to as light TMT and heavy TMT (Figure 8). The labeling of peptides by these reagents results in an increase in mass of 224 and 229 Da, respectively, per introduced tag.
Figure 8.
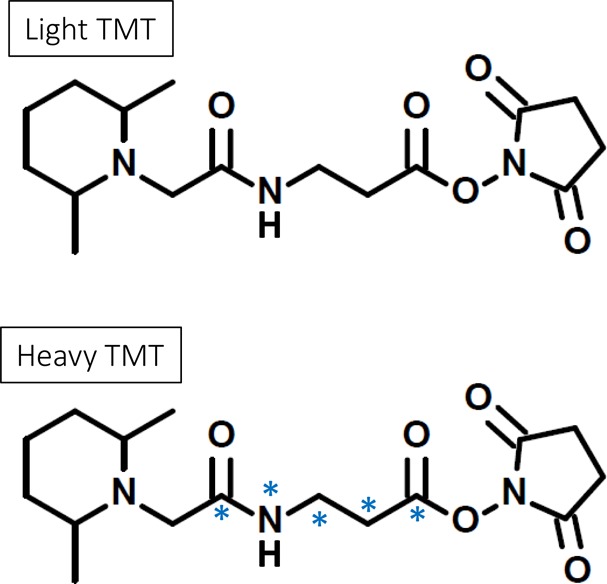
Chemical structure of isotopic reagents, light TMT and heavy TMT, used for targeted quantification.111 The light reagent has no heavy isotope incorporated, whereas the heavy reagent has five heavy isotopes incorporated (4 × 13C and 1 × 15N). Labeling with these reagents introduces mass differences into the peptides from different samples. In targeted experiments, quantification is obtained by structural b and/or y ions generated after collision-induced dissociation.
7. Summary
The isobaric labeling-based quantification technique has developed as a powerful tool for obtaining the relative expression level of proteins in quantitative proteomics studies. Moreover, the ability to multiplex with isobaric mass tags has expanded its applicability to a wide range of sample types. Isobaric mass tags are isotope-coded molecules with the same chemical structure and molecular weight that are used to differentially label peptides without introducing mass difference and sample complexity. The isotopically derivatized peptides display a single peak on an MS spectrum and yield a series of low-mass reporter ions for quantification upon fragmentation in tandem mass spectrometry. However, since peptide quantification ratios are measured to determine protein relative abundance, the variance in peptide ratio measurements will contribute into the protein-level variance, affecting the accuracy of the quantification. Herein, we have reviewed the studies of different aspects of an isobaric labeling-based quantification approach. This includes studies on different types of isobaric reagents and their applications, sources of variation that affect quantification, and the suggested combinations of experimental design and optimal data acquisition methods to increase the precision and accuracy of the measurements. We have also reviewed studies on challenges in data analysis and the proposed solutions for data processing to increase the confidence in the acquired data set.
Acknowledgments
We apologize to authors whose work we could not cite. We would like to thank Drs. Jolene Diedrich, Daniel McClatchy, Aaron Aslanian, and Claire Delahunty for helpful comments and suggestions about the manuscript. Financial support was provided by NIH grant nos. P41 GM103533, R01 HL079442-09, R01 MH067880, and UCLA/NHLBI Proteomics Centers HHSN268201000035C.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Wu C. C.; MacCoss M. J.; Howell K. E.; Matthews D. E.; Yates J. R. III Metabolic labeling of mammalian organisms with stable isotopes for quantitative proteomic analysis. Anal. Chem. 2004, 76, 4951–4959. [DOI] [PubMed] [Google Scholar]
- Ong S. E.; Blagoev B.; Kratchmarova I.; Kristensen D. B.; Steen H.; Pandey A.; Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 2002, 1, 376–386. [DOI] [PubMed] [Google Scholar]
- Rose C. M.; Merrill A. E.; Bailey D. J.; Hebert A. S.; Westphall M. S.; Coon J. J. Neutron encoded labeling for peptide identification. Anal. Chem. 2013, 85, 5129–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gygi S. P.; Rist B.; Gerber S. A.; Turecek F.; Gelb M. H.; Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999, 17, 994–999. [DOI] [PubMed] [Google Scholar]
- Hsu J. L.; Huang S. Y.; Chow N. H.; Chen S. H. Stable-isotope dimethyl labeling for quantitative proteomics. Anal. Chem. 2003, 75, 6843–6852. [DOI] [PubMed] [Google Scholar]
- Thompson A.; Schafer J.; Kuhn K.; Kienle S.; Schwarz J.; Schmidt G.; Neumann T.; Johnstone R.; Mohammed A. K.; Hamon C. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- Ross P. L.; Huang Y. N.; Marchese J. N.; Williamson B.; Parker K.; Hattan S.; Khainovski N.; Pillai S.; Dey S.; Daniels S.; Purkayastha S.; Juhasz P.; Martin S.; Bartlet-Jones M.; He F.; Jacobson A.; Pappin D. J. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 2004, 3, 1154–1169. [DOI] [PubMed] [Google Scholar]
- Yao X.; Freas A.; Ramirez J.; Demirev P. A.; Fenselau C. Proteolytic 18O labeling for comparative proteomics: model studies with two serotypes of adenovirus. Anal. Chem. 2001, 73, 2836–2842. [DOI] [PubMed] [Google Scholar]
- Wiktorowicz J. E.; English R. D.; Wu Z.; Kurosky A. Model studies on iTRAQ modification of peptides: sequence-dependent reaction specificity. J. Proteome Res. 2012, 11, 1512–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe L.; D’Ascenzo M.; Relkin N. R.; Pappin D.; Ross P.; Williamson B.; Guertin S.; Pribil P.; Lee K. H. 8-Plex quantitation of changes in cerebrospinal fluid protein expression in subjects undergoing intravenous immunoglobulin treatment for Alzheimer’s disease. Proteomics 2007, 7, 3651–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayon L.; Hainard A.; Licker V.; Turck N.; Kuhn K.; Hochstrasser D. F.; Burkhard P. R.; Sanchez J. C. Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Anal. Chem. 2008, 80, 2921–2931. [DOI] [PubMed] [Google Scholar]
- McAlister G. C.; Huttlin E. L.; Haas W.; Ting L.; Jedrychowski M. P.; Rogers J. C.; Kuhn K.; Pike I.; Grothe R. A.; Blethrow J. D.; Gygi S. P. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal. Chem. 2012, 84, 7469–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner T.; Becher I.; Sweetman G.; Doce C.; Savitski M. M.; Bantscheff M. High-resolution enabled TMT 8-plexing. Anal. Chem. 2012, 84, 7188–7194. [DOI] [PubMed] [Google Scholar]
- Werner T.; Sweetman G.; Savitski M. F.; Mathieson T.; Bantscheff M.; Savitski M. M. Ion coalescence of neutron encoded TMT 10-plex reporter ions. Anal. Chem. 2014, 86, 3594–3601. [DOI] [PubMed] [Google Scholar]
- Zieske L. R. A perspective on the use of iTRAQ reagent technology for protein complex and profiling studies. J. Exp. Bot. 2006, 57, 1501–1508. [DOI] [PubMed] [Google Scholar]
- Pichler P.; Kocher T.; Holzmann J.; Mazanek M.; Taus T.; Ammerer G.; Mechtler K. Peptide labeling with isobaric tags yields higher identification rates using iTRAQ 4-plex compared to TMT 6-plex and iTRAQ 8-plex on LTQ Orbitrap. Anal. Chem. 2010, 82, 6549–6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottiez G.; Wiederin J.; Fox H. S.; Ciborowski P. Comparison of 4-plex to 8-plex iTRAQ quantitative measurements of proteins in human plasma samples. J. Proteome Res. 2012, 11, 3774–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang F.; Ye H.; Chen R.; Fu Q.; Li L. N,N-Dimethyl leucines as novel isobaric tandem mass tags for quantitative proteomics and peptidomics. Anal. Chem. 2010, 82, 2817–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Sioma C. S.; Wang S.; Regnier F. E. Fractionation of isotopically labeled peptides in quantitative proteomics. Anal. Chem. 2001, 73, 5142–5149. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Wang Q.; Lin L.; Tang Q.; Edwards J. L.; Li S.; Liu S. Comparative evaluation of two isobaric labeling tags, DiART and iTRAQ. Anal. Chem. 2012, 84, 2908–2915. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Wang Y.; Li S. Deuterium isobaric amine-reactive tags for quantitative proteomics. Anal. Chem. 2010, 82, 7588–7595. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Askenazi M.; Jiang J.; Luckey C. J.; Griffin J. D.; Marto J. A. A robust error model for iTRAQ quantification reveals divergent signaling between oncogenic FLT3 mutants in acute myeloid leukemia. Mol. Cell. Proteomics 2010, 9, 780–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ow S. Y.; Salim M.; Noirel J.; Evans C.; Rehman I.; Wright P. C. iTRAQ underestimation in simple and complex mixtures: “the good, the bad and the ugly”. J. Proteome Res. 2009, 8, 5347–5355. [DOI] [PubMed] [Google Scholar]
- Ramsubramaniam N.; Tao F.; Li S.; Marten M. R. Cost-effective isobaric tagging for quantitative phosphoproteomics using DiART reagents. Mol. BioSyst. 2013, 9, 2981–2987. [DOI] [PubMed] [Google Scholar]
- Nystrom T. Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 2005, 24, 1311–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmese A.; De Rosa C.; Chiappetta G.; Marino G.; Amoresano A. Novel method to investigate protein carbonylation by iTRAQ strategy. Anal. Bioanal. Chem. 2012, 404, 1631–1635. [DOI] [PubMed] [Google Scholar]
- Hahne H.; Neubert P.; Kuhn K.; Etienne C.; Bomgarden R.; Rogers J. C.; Kuster B. Carbonyl-reactive tandem mass tags for the proteome-wide quantification of N-linked glycans. Anal. Chem. 2012, 84, 3716–3724. [DOI] [PubMed] [Google Scholar]
- Thomas J. A.; Mallis R. J. Aging and oxidation of reactive protein sulfhydryls. Exp. Gerontol. 2001, 36, 1519–1526. [DOI] [PubMed] [Google Scholar]
- Giron P.; Dayon L.; Turck N.; Hoogland C.; Sanchez J. C. Quantitative analysis of human cerebrospinal fluid proteins using a combination of cysteine tagging and amine-reactive isobaric labeling. J. Proteome Res. 2011, 10, 249–258. [DOI] [PubMed] [Google Scholar]
- Kohr M. J.; Aponte A.; Sun J.; Gucek M.; Steenbergen C.; Murphy E. Measurement of S-nitrosylation occupancy in the myocardium with cysteine-reactive tandem mass tags: short communication. Circ. Res. 2012, 111, 1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray C. I.; Uhrigshardt H.; O’Meally R. N.; Cole R. N.; Van Eyk J. E. Identification and quantification of S-nitrosylation by cysteine reactive tandem mass tag switch assay. Mol. Cell. Proteomics 2012, 11, M111.013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan K. T.; Chen Y. Y.; Pu T. H.; Chao Y. S.; Yang C. Y.; Bomgarden R. D.; Rogers J. C.; Meng T. C.; Khoo K. H. Mass spectrometry-based quantitative proteomics for dissecting multiplexed redox cysteine modifications in nitric oxide-protected cardiomyocyte under hypoxia. Antioxid. Redox Signaling. 2014, 20, 1365–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Z.; Meng F.; Bomgarden R. D.; Viner R. I.; Li J.; Rogers J. C.; Cheng J.; Greenlief C. M.; Cui J.; Lubahn D. B.; Sun G. Y.; Gu Z. Proteomic quantification and site-mapping of S-nitrosylated proteins using isobaric iodoTMT reagents. J. Proteome Res. 2014, 13, 3200–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West G. M.; Tucker C. L.; Xu T.; Park S. K.; Han X.; Yates J. R. III; Fitzgerald M. C. Quantitative proteomics approach for identifying protein-drug interactions in complex mixtures using protein stability measurements. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 9078–9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Wolf-Yadlin A.; Ross P. L.; Pappin D. J.; Rush J.; Lauffenburger D. A.; White F. M. Time-resolved mass spectrometry of tyrosine phosphorylation sites in the epidermal growth factor receptor signaling network reveals dynamic modules. Mol. Cell. Proteomics 2005, 4, 1240–1250. [DOI] [PubMed] [Google Scholar]
- Li Z.; Adams R. M.; Chourey K.; Hurst G. B.; Hettich R. L.; Pan C. Systematic comparison of label-free, metabolic labeling, and isobaric chemical labeling for quantitative proteomics on LTQ Orbitrap Velos. J. Proteome Res. 2012, 11, 1582–1590. [DOI] [PubMed] [Google Scholar]
- Bouchal P.; Roumeliotis T.; Hrstka R.; Nenutil R.; Vojtesek B.; Garbis S. D. Biomarker discovery in low-grade breast cancer using isobaric stable isotope tags and two-dimensional liquid chromatography-tandem mass spectrometry (iTRAQ-2DLC–-MS/MS) based quantitative proteomic analysis. J. Proteome Res. 2009, 8, 362–373. [DOI] [PubMed] [Google Scholar]
- Aggarwal K.; Choe L. H.; Lee K. H. Shotgun proteomics using the iTRAQ isobaric tags. Briefings Funct. Genomics Proteomics 2006, 5, 112–120. [DOI] [PubMed] [Google Scholar]
- Trotter M. W.; Sadowski P. G.; Dunkley T. P.; Groen A. J.; Lilley K. S. Improved sub-cellular resolution via simultaneous analysis of organelle proteomics data across varied experimental conditions. Proteomics 2010, 10, 4213–4219. [DOI] [PubMed] [Google Scholar]
- Hardt M.; Witkowska H. E.; Webb S.; Thomas L. R.; Dixon S. E.; Hall S. C.; Fisher S. J. Assessing the effects of diurnal variation on the composition of human parotid saliva: quantitative analysis of native peptides using iTRAQ reagents. Anal. Chem. 2005, 77, 4947–4954. [DOI] [PubMed] [Google Scholar]
- Mertins P.; Udeshi N. D.; Clauser K. R.; Mani D. R.; Patel J.; Ong S. E.; Jaffe J. D.; Carr S. A. iTRAQ labeling is superior to mTRAQ for quantitative global proteomics and phosphoproteomics. Mol. Cell. Proteomics 2012, 11, M111.014423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S.; Cho K.; Kim J.; Yea K.; Park G.; Lee J.; Ryu S. H.; Kim J.; Kim Y. H. Comparative proteome analysis using amine-reactive isobaric tagging reagents coupled with 2D LC/MS/MS in 3T3-L1 adipocytes following hypoxia or normoxia. Biochem. Biophys. Res. Commun. 2009, 383, 135–140. [DOI] [PubMed] [Google Scholar]
- Unwin R. D. Quantification of proteins by iTRAQ. Methods Mol. Biol. 2010, 658, 205–215. [DOI] [PubMed] [Google Scholar]
- Garbis S. D.; Tyritzis S. I.; Roumeliotis T.; Zerefos P.; Giannopoulou E. G.; Vlahou A.; Kossida S.; Diaz J.; Vourekas S.; Tamvakopoulos C.; Pavlakis K.; Sanoudou D.; Constantinides C. A. Search for potential markers for prostate cancer diagnosis, prognosis and treatment in clinical tissue specimens using amine-specific isobaric tagging (iTRAQ) with two-dimensional liquid chromatography and tandem mass spectrometry. J. Proteome Res. 2008, 7, 3146–3158. [DOI] [PubMed] [Google Scholar]
- Huang Z.; Wang H.; Huang H.; Xia L.; Chen C.; Qiu X.; Chen J.; Chen S.; Liang W.; Huang M.; Lang L.; Zheng Q.; Wu B.; Lai G. iTRAQ-based proteomic profiling of human serum reveals down-regulation of platelet basic protein and apolipoprotein B100 in patients with hematotoxicity induced by chronic occupational benzene exposure. Toxicology 2012, 291, 56–64. [DOI] [PubMed] [Google Scholar]
- Zhou L.; Beuerman R. W.; Chan C. M.; Zhao S. Z.; Li X. R.; Yang H.; Tong L.; Liu S.; Stern M. E.; Tan D. Identification of tear fluid biomarkers in dry eye syndrome using iTRAQ quantitative proteomics. J. Proteome Res. 2009, 8, 4889–4905. [DOI] [PubMed] [Google Scholar]
- Cong Y. S.; Fan E.; Wang E. Simultaneous proteomic profiling of four different growth states of human fibroblasts, using amine-reactive isobaric tagging reagents and tandem mass spectrometry. Mech. Ageing Dev. 2006, 127, 332–343. [DOI] [PubMed] [Google Scholar]
- Zhong J.; Krawczyk S. A.; Chaerkady R.; Huang H.; Goel R.; Bader J. S.; Wong G. W.; Corkey B. E.; Pandey A. Temporal profiling of the secretome during adipogenesis in humans. J. Proteome Res. 2010, 9, 5228–5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien R. N.; Shen Z.; Tachikawa K.; Lee P. A.; Briggs S. P. Quantitative proteome analysis of pluripotent cells by iTRAQ mass tagging reveals post-transcriptional regulation of proteins required for ES cell self-renewal. Mol. Cell. Proteomics 2010, 9, 2238–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch J.; Hansen K. C.; Choi S.; Noh J.; Hirose R.; Roberts J. P.; Matthay M. A.; Burlingame A. L.; Maher J. J.; Niemann C. U. Warm ischemia-induced alterations in oxidative and inflammatory proteins in hepatic Kupffer cells in rats. Mol. Cell. Proteomics 2006, 5, 979–986. [DOI] [PubMed] [Google Scholar]
- Rauniyar N.; Gao B.; McClatchy D. B.; Yates J. R. III Comparison of protein expression ratios observed by sixplex and duplex TMT labeling method. J. Proteome Res. 2013, 12, 1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleifeld O.; Doucet A.; Prudova A.; auf dem Keller U.; Gioia M.; Kizhakkedathu J. N.; Overall C. M. Identifying and quantifying proteolytic events and the natural N terminome by terminal amine isotopic labeling of substrates. Nat. Protoc. 2011, 6, 1578–1611. [DOI] [PubMed] [Google Scholar]
- Prudova A.; auf dem Keller U.; Butler G. S.; Overall C. M. Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Mol. Cell. Proteomics 2010, 9, 894–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P. P.; Kang S. A.; Rameseder J.; Zhang Y.; Ottina K. A.; Lim D.; Peterson T. R.; Choi Y.; Gray N. S.; Yaffe M. B.; Marto J. A.; Sabatini D. M. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 2011, 332, 1317–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai L. K.; Benoist C.; Mathis D.; White F. M. Quantitative phosphoproteomic analysis of T cell receptor signaling in diabetes prone and resistant mice. J. Proteome Res. 2010, 9, 3135–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson C. L.; Dillon R.; Devakumar A.; Shi S. D.; Greig M.; Rogers J. C.; Krastins B.; Rosenblatt M.; Kilmer G.; Major M.; Kaboord B. J.; Sarracino D.; Rezai T.; Prakash A.; Lopez M.; Ji Y.; Priebe W.; Lang F. F.; Colman H.; Conrad C. A. Quantitative phosphoproteomic analysis of the STAT3/IL-6/HIF1alpha signaling network: an initial study in GSC11 glioblastoma stem cells. J. Proteome Res. 2010, 9, 430–443. [DOI] [PubMed] [Google Scholar]
- Wiese S.; Reidegeld K. A.; Meyer H. E.; Warscheid B. Protein labeling by iTRAQ: a new tool for quantitative mass spectrometry in proteome research. Proteomics 2007, 7, 340–350. [DOI] [PubMed] [Google Scholar]
- Keshamouni V. G.; Michailidis G.; Grasso C. S.; Anthwal S.; Strahler J. R.; Walker A.; Arenberg D. A.; Reddy R. C.; Akulapalli S.; Thannickal V. J.; Standiford T. J.; Andrews P. C.; Omenn G. S. Differential protein expression profiling by iTRAQ-2DLC-MS/MS of lung cancer cells undergoing epithelial-mesenchymal transition reveals a migratory/invasive phenotype. J. Proteome Res. 2006, 5, 1143–1154. [DOI] [PubMed] [Google Scholar]
- Wolf-Yadlin A.; Hautaniemi S.; Lauffenburger D. A.; White F. M. Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 5860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin T. J.; Xie H.; Bandhakavi S.; Popko J.; Mohan A.; Carlis J. V.; Higgins L. iTRAQ reagent-based quantitative proteomic analysis on a linear ion trap mass spectrometer. J. Proteome Res. 2007, 6, 4200–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocher T.; Pichler P.; Schutzbier M.; Stingl C.; Kaul A.; Teucher N.; Hasenfuss G.; Penninger J. M.; Mechtler K. High precision quantitative proteomics using iTRAQ on an LTQ Orbitrap: a new mass spectrometric method combining the benefits of all. J. Proteome Res. 2009, 8, 4743–4752. [DOI] [PubMed] [Google Scholar]
- Schwartz J. C.; Syka J. E. P.; Quarmby S. T.. Improving the fundamentals of MSn on 2D linear ion traps: New ion activation and isolation techniques, Proc. 53rd ASMS Conference on Mass Spectrometry and Allied Topics, San Antonio, TX, June 5–9, 2005, oral presentation.
- Bantscheff M.; Boesche M.; Eberhard D.; Matthieson T.; Sweetman G.; Kuster B. Robust and sensitive iTRAQ quantification on an LTQ Orbitrap mass spectrometer. Mol. Cell. Proteomics 2008, 7, 1702–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T.; Gan C. S.; Zhang H.; Zhu Y.; Kon O. L.; Sze S. K. Hybridization of pulsed-Q dissociation and collision-activated dissociation in linear ion trap mass spectrometer for iTRAQ quantitation. J. Proteome Res. 2008, 7, 4831–4840. [DOI] [PubMed] [Google Scholar]
- Olsen J. V.; Macek B.; Lange O.; Makarov A.; Horning S.; Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nat. Methods 2007, 4, 709–712. [DOI] [PubMed] [Google Scholar]
- Dayon L.; Pasquarello C.; Hoogland C.; Sanchez J. C.; Scherl A. Combining low- and high-energy tandem mass spectra for optimized peptide quantification with isobaric tags. J. Proteomics 2010, 73, 769–777. [DOI] [PubMed] [Google Scholar]
- Chiva C.; Sabido E. HCD-only fragmentation method balances peptide identification and quantitation of TMT-labeled samples in hybrid linear ion trap/orbitrap mass spectrometers. J. Proteomics 2014, 96, 263–270. [DOI] [PubMed] [Google Scholar]
- Pichler P.; Kocher T.; Holzmann J.; Mohring T.; Ammerer G.; Mechtler K. Improved precision of iTRAQ and TMT quantification by an axial extraction field in an Orbitrap HCD cell. Anal. Chem. 2011, 83, 1469–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diedrich J. K.; Pinto A. F.; Yates J. R. III Energy dependence of HCD on peptide fragmentation: stepped collisional energy finds the sweet spot. J. Am. Soc. Mass Spectrom. 2013, 24, 1690–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting L.; Rad R.; Gygi S. P.; Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 2011, 8, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senko M. W.Multinotch isolation for ms3 mass analysis. Patent WO/2013/112677, 2013.
- McAlister G. C.; Nusinow D. P.; Jedrychowski M. P.; Wuhr M.; Huttlin E. L.; Erickson B. K.; Rad R.; Haas W.; Gygi S. P. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 2014, 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armenta J. M.; Hoeschele I.; Lazar I. M. Differential protein expression analysis using stable isotope labeling and PQD linear ion trap MS technology. J. Am. Soc. Mass Spectrom. 2009, 20, 1287–1302. [DOI] [PubMed] [Google Scholar]
- Liu T.; Hu J.; Li H. iTRAQ-based shotgun neuroproteomics. Methods Mol. Biol. 2009, 566, 201–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadforth I. P.; Dunkley T. P.; Lilley K. S.; Bessant C. i-Tracker: for quantitative proteomics using iTRAQ. BMC Genomics 2005, 6, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski M. M.; Sweetman G.; Askenazi M.; Marto J. A.; Lang M.; Zinn N.; Bantscheff M. Delayed fragmentation and optimized isolation width settings for improvement of protein identification and accuracy of isobaric mass tag quantification on Orbitrap-type mass spectrometers. Anal. Chem. 2011, 83, 8959–8967. [DOI] [PubMed] [Google Scholar]
- Karp N. A.; Huber W.; Sadowski P. G.; Charles P. D.; Hester S. V.; Lilley K. S. Addressing accuracy and precision issues in iTRAQ quantitation. Mol. Cell. Proteomics 2010, 9, 1885–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoforou A. L.; Lilley K. S. Isobaric tagging approaches in quantitative proteomics: the ups and downs. Anal. Bioanal. Chem. 2012, 404, 1029–1037. [DOI] [PubMed] [Google Scholar]
- Mathur R.; O’Connor P. B. Artifacts in Fourier transform mass spectrometry. Rapid Commun. Mass Spectrom. 2009, 23, 523–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill E. G.; Schwacke J. H.; Comte-Walters S.; Slate E. H.; Oberg A. L.; Eckel-Passow J. E.; Therneau T. M.; Schey K. L. A statistical model for iTRAQ data analysis. J. Proteome Res. 2008, 7, 3091–3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayon L.; Turck N.; Kienle S.; Schulz-Knappe P.; Hochstrasser D. F.; Scherl A.; Sanchez J. C. Isobaric tagging-based selection and quantitation of cerebrospinal fluid tryptic peptides with reporter calibration curves. Anal. Chem. 2010, 82, 848–858. [DOI] [PubMed] [Google Scholar]
- Aggarwal K.; Choe L. H.; Lee K. H. Quantitative analysis of protein expression using amine-specific isobaric tags in Escherichia coli cells expressing rhsA elements. Proteomics 2005, 5, 2297–2308. [DOI] [PubMed] [Google Scholar]
- Ow S. Y.; Salim M.; Noirel J.; Evans C.; Wright P. C. Minimising iTRAQ ratio compression through understanding LC–MS elution dependence and high-resolution HILIC fractionation. Proteomics 2011, 11, 2341–2346. [DOI] [PubMed] [Google Scholar]
- Savitski M. M.; Fischer F.; Mathieson T.; Sweetman G.; Lang M.; Bantscheff M. Targeted data acquisition for improved reproducibility and robustness of proteomic mass spectrometry assays. J. Am. Soc. Mass Spectrom. 2010, 21, 1668–1679. [DOI] [PubMed] [Google Scholar]
- Wenger C. D.; Lee M. V.; Hebert A. S.; McAlister G. C.; Phanstiel D. H.; Westphall M. S.; Coon J. J. Gas-phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nat. Methods 2011, 8, 933–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm R. M.; Lietz C. B.; Li L. Improved isobaric tandem mass tag quantification by ion mobility mass spectrometry. Rapid Commun. Mass Spectrom. 2014, 28, 1051–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski M. M.; Mathieson T.; Zinn N.; Sweetman G.; Doce C.; Becher I.; Pachl F.; Kuster B.; Bantscheff M. Measuring and managing ratio compression for accurate iTRAQ/TMT quantification. J. Proteome Res. 2013, 12, 3586–3598. [DOI] [PubMed] [Google Scholar]
- Onsongo G.; Stone M. D.; Van Riper S. K.; Chilton J.; Wu B.; Higgins L.; Lund T. C.; Carlis J. V.; Griffin T. J. LTQ-iQuant: a freely available software pipeline for automated and accurate protein quantification of isobaric tagged peptide data from LTQ instruments. Proteomics 2010, 10, 3533–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart J. M.; Vaudel M.; Zahedi R. P.; Martens L.; Sickmann A. iTRAQ protein quantification: a quality-controlled workflow. Proteomics 2011, 11, 1125–1134. [DOI] [PubMed] [Google Scholar]
- Pachl F.; Fellenberg K.; Wagner C.; Kuster B. Ultra-high intra-spectrum mass accuracy enables unambiguous identification of fragment reporter ions in isobaric multiplexed quantitative proteomics. Proteomics 2012, 12, 1328–1332. [DOI] [PubMed] [Google Scholar]
- Hu J.; Qian J.; Borisov O.; Pan S.; Li Y.; Liu T.; Deng L.; Wannemacher K.; Kurnellas M.; Patterson C.; Elkabes S.; Li H. Optimized proteomic analysis of a mouse model of cerebellar dysfunction using amine-specific isobaric tags. Proteomics 2006, 6, 4321–4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W. T.; Hung W. N.; Yian Y. H.; Wu K. P.; Han C. L.; Chen Y. R.; Chen Y. J.; Sung T. Y.; Hsu W. L. Multi-Q: a fully automated tool for multiplexed protein quantitation. J. Proteome Res. 2006, 5, 2328–2338. [DOI] [PubMed] [Google Scholar]
- Song X.; Bandow J.; Sherman J.; Baker J. D.; Brown P. W.; McDowell M. T.; Molloy M. P. iTRAQ experimental design for plasma biomarker discovery. J. Proteome Res. 2008, 7, 2952–2958. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii A. I.; Aebersold R. Interpretation of shotgun proteomic data: the protein inference problem. Mol. Cell. Proteomics 2005, 4, 1419–1440. [DOI] [PubMed] [Google Scholar]
- Mahoney D. W.; Therneau T. M.; Heppelmann C. J.; Higgins L.; Benson L. M.; Zenka R. M.; Jagtap P.; Nelsestuen G. L.; Bergen H. R.; Oberg A. L. Relative quantification: characterization of bias, variability and fold changes in mass spectrometry data from iTRAQ-labeled peptides. J. Proteome Res. 2011, 10, 4325–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veenstra T. D.; Conrads T. P.; Issaq H. J. What to do with “one-hit wonders”?. Electrophoresis 2004, 25, 1278–1279. [DOI] [PubMed] [Google Scholar]
- Gan C. S.; Chong P. K.; Pham T. K.; Wright P. C. Technical, experimental, and biological variations in isobaric tags for relative and absolute quantitation (iTRAQ). J. Proteome Res. 2007, 6, 821–827. [DOI] [PubMed] [Google Scholar]
- Glen A.; Gan C. S.; Hamdy F. C.; Eaton C. L.; Cross S. S.; Catto J. W.; Wright P. C.; Rehman I. iTRAQ-facilitated proteomic analysis of human prostate cancer cells identifies proteins associated with progression. J. Proteome Res. 2008, 7, 897–907. [DOI] [PubMed] [Google Scholar]
- Zhou C.; Simpson K. L.; Lancashire L. J.; Walker M. J.; Dawson M. J.; Unwin R. D.; Rembielak A.; Price P.; West C.; Dive C.; Whetton A. D. Statistical considerations of optimal study design for human plasma proteomics and biomarker discovery. J. Proteome Res. 2012, 11, 2103–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Alvarez S.; Hicks L. M. Comprehensive comparison of iTRAQ and label-free LC-based quantitative proteomics approaches using two Chlamydomonas reinhardtii strains of interest for biofuels engineering. J. Proteome Res. 2012, 11, 487–501. [DOI] [PubMed] [Google Scholar]
- Romero R.; Kusanovic J. P.; Gotsch F.; Erez O.; Vaisbuch E.; Mazaki-Tovi S.; Moser A.; Tam S.; Leszyk J.; Master S. R.; Juhasz P.; Pacora P.; Ogge G.; Gomez R.; Yoon B. H.; Yeo L.; Hassan S. S.; Rogers W. T. Isobaric labeling and tandem mass spectrometry: a novel approach for profiling and quantifying proteins differentially expressed in amniotic fluid in preterm labor with and without intra-amniotic infection/inflammation. J. Matern.-Fetal Neonat. Med. 2010, 23, 261–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbrich S. M.; Cole R. N.; West K. P. Jr.; Schulze K.; Yager J. D.; Groopman J. D.; Christian P.; Wu L.; O’Meally R. N.; May D. H.; McIntosh M. W.; Ruczinski I. Statistical inference from multiple iTRAQ experiments without using common reference standards. J. Proteome Res. 2013, 12, 594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshi B. An integrated approach to mapping the proteome of the human bone marrow stromal cell. Proteomics 2006, 6, 5169–5182. [DOI] [PubMed] [Google Scholar]
- Lund T. C.; Anderson L. B.; McCullar V.; Higgins L.; Yun G. H.; Grzywacz B.; Verneris M. R.; Miller J. S. iTRAQ is a useful method to screen for membrane-bound proteins differentially expressed in human natural killer cell types. J. Proteome Res. 2007, 6, 644–653. [DOI] [PubMed] [Google Scholar]
- Wu J.; Warren P.; Shakey Q.; Sousa E.; Hill A.; Ryan T. E.; He T. Integrating titania enrichment, iTRAQ labeling, and Orbitrap CID-HCD for global identification and quantitative analysis of phosphopeptides. Proteomics 2010, 10, 2224–2234. [DOI] [PubMed] [Google Scholar]
- Linke D.; Hung C. W.; Cassidy L.; Tholey A. Optimized fragmentation conditions for iTRAQ-labeled phosphopeptides. J. Proteome Res. 2013, 12, 2755–2763. [DOI] [PubMed] [Google Scholar]
- Nilsson C. L. Advances in quantitative phosphoproteomics. Anal. Chem. 2012, 84, 735–746. [DOI] [PubMed] [Google Scholar]
- Thingholm T. E.; Palmisano G.; Kjeldsen F.; Larsen M. R. Undesirable charge-enhancement of isobaric tagged phosphopeptides leads to reduced identification efficiency. J. Proteome Res. 2010, 9, 4045–4052. [DOI] [PubMed] [Google Scholar]
- Dephoure N.; Gygi S. P. Hyperplexing: a method for higher-order multiplexed quantitative proteomics provides a map of the dynamic response to rapamycin in yeast. Sci. Signaling 2012, 5, rs2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella R.; Cifani P.; Peggion C.; Hansson K.; Lazzari C.; Bendz M.; Levander F.; Sorgato M. C.; Bertoli A.; James P. Relative quantification of membrane proteins in wild-type and prion protein (PrP)-knockout cerebellar granule neurons. J. Proteome Res. 2012, 11, 523–536. [DOI] [PubMed] [Google Scholar]
- Byers H. L.; Campbell J.; van Ulsen P.; Tommassen J.; Ward M. A.; Schulz-Knappe P.; Prinz T.; Kuhn K. Candidate verification of iron-regulated Neisseria meningitidis proteins using isotopic versions of tandem mass tags (TMT) and single reaction monitoring. J. Proteomics 2009, 73, 231–239. [DOI] [PubMed] [Google Scholar]