

Abstract
Despite decades of research, therapy for diseases caused by abnormal protein folding and aggregation (amyloidoses) is limited to treatment of symptoms and provides only temporary and moderate relief to sufferers. The failure in developing successful disease-modifying drugs for amyloidoses stems from the nature of the targets for such drugs – primarily oligomers of amyloidogenic proteins, which are distinct from traditional targets, such as enzymes or receptors. The oligomers are metastable, do not have well-defined structures, and exist in dynamically changing mixtures. Therefore, inhibiting the formation and toxicity of these oligomers likely will require out-of-the-box thinking and novel strategies. We review here the development of a strategy based on targeting the combination of hydrophobic and electrostatic interactions that are key to the assembly and toxicity of amyloidogenic proteins using lysine (K)-specific “molecular tweezers” (MTs). Our discussion includes a survey of the literature demonstrating the important role of K residues in the assembly and toxicity of amyloidogenic proteins and the development of a lead MT derivative called CLR01, from an inhibitor of protein aggregation in vitro to a drug candidate showing effective amelioration of disease symptoms in animal models of Alzheimer’s and Parkinson’s diseases.
Keywords: Amyloid, Alzheimer’s disease, Parkinson’s disease, inhibitor, oligomer, lysine, molecular tweezers
INTRODUCTION
Aberrant protein folding and aggregation cause over 30 human diseases called amyloidoses, for which currently there is no cure [1]. Some of the most devastating amyloidoses are those that affect the central nervous system (CNS), such as Alzheimer’s disease (AD) and Parkinson’s disease (PD). In each disease, one or more proteins self-assemble into toxic oligomers that disrupt cellular function and communication, and proceed to form insoluble amyloid aggregates characterized by fibrillar morphology and cross-β structure [2, 3]. An attractive strategy for preventing and treating amyloidoses is inhibition or modulation of the self-assembly process in a manner that would prevent formation of the toxic oligomers and fibrils [4].
Of the different amyloidoses, research on AD has been the most prominent and in many cases has led the way towards discoveries in other diseases. Nonetheless, to date, there is no disease-modifying therapy for AD or other amyloidoses. The failure of AD clinical trials in the last decade [5, 6] might be partially attributed to the common methods of drug discovery, namely large-scale compound screens or empirically identified leads. Though high-throughput screens generally are efficient and leads discovered empirically, particularly from food sources, tend to be safe, lack of understanding of the mechanism of action of the compounds selected by either strategy may result in pursuing nonviable leads and unpredictable complications. The necessity for better mechanistic understanding has been underscored in recent years by studies showing that many inhibitors of protein aggregation are promiscuous in their inhibition, a behavior attributable to the inhibitors forming colloids around protein fibrils and thereby sequestering them rather than actually disrupting the assembly process [7, 8].
Complicating matters further, inhibition of fibril formation or dissociation of existing fibrils may yield both toxic [9, 10], or non-toxic oligomers, depending on the particular proteins and inhibitors studied [4]. Large-scale, high-throughput screens based on identifying binding interactions are best suited for compounds that fit tightly in a small, deep pocket and bind to the target with affinity similar to, or higher than, the natural ligand or substrate. However, in the case of amyloid, stable structure in the fibrils is achieved as a sum of numerous weak interactions spread across large, flat areas, where it would be difficult for a small molecule to compete effectively [11]. In addition, because of the high flexibility of amyloids, even molecules that bind with relative high affinity can be accom-modated without sufficiently disrupting the amyloid lattice [12]. Inhibiting oligomer formation and toxicity is even more difficult because the structures of oligomers not only are unknown, but also are constantly changing. These challenges in drug discovery and development specifically for protein aggregation may be circumvented by using rational approaches to the problem, considering the weakest links of the target(s) and the fundamental interactions within and among the individual building blocks necessary for abnormal self-assembly and toxicity. Because the structures of the most toxic species — the metastable oligomers of amyloidogenic proteins — are poorly understood [13-15], continued research to-wards better characterization of these structures is vital. At the same time, the tremendous magnitude of the financial and psychological burden AD and other amyloidoses create for individuals and societies makes the search for disease-modifying intervention highly urgent. Therefore, scientists and physicians do not have the luxury of waiting for complete structural details to be deciphered and must act now to create the best possible solutions to the problem using currently available information.
To rationally approach treatment, we must first understand the biological basis of the target diseases. The genome of a living organism may encode >100,000 proteins, all of which must adopt particular three-dimensional structures, which are encoded by their amino acid sequences and sometimes require particular environments, as in the case of naturally unfolded proteins [16, 17], to carry out their biological function [18]. Given that self-association of polypeptides can be induced in many unrelated proteins, the cross-β secondary structure of amyloids has been hypothesized to be a primordial, default structure of polypeptides [18]. Moreover, Dobson went as far as proposing that potentially any protein might form amyloid given the appropriate conditions [19]. This view has been debated in light of evidence that particular sequences may be required for amyloid formation [20]. Nonetheless, in view of the high abundance of such sequences in biologically active proteins, it might be surprising that only a few dozen diseases of protein aggregation are known [1].
Remarkably, the shared, common structural characteristics of the amyloid deposits found in all amyloidoses are independent of the amino acid sequence of the proteins that comprise them [21-23]. The proteins involved in amyloidoses can be divided into natively structured and natively unfolded proteins. For the first group, which includes proteins such as prion protein, transthyretin, Cu/Zn-superoxide dismutase 1, and β2-microglobulin, amyloid formation requires partial unfolding, leading to formation of metastable, toxic oligomers and subsequently, β-sheet-rich fibrils. Proteins in the second group, including amyloid-β protein (Aβ), tau, α-synuclein (α-syn), and islet amyloid polypeptide (IAPP), are thought to undergo partial folding to create similar metastable structures leading to self-assembly and toxicity [4].
Aberrant protein self-assembly involves formation of multiple oligomeric structures, ranging from dimers to protofibrillar structures, most of which have been reported to be toxic [13]. Most likely, different mechanisms of oligomerization and fibrillization act in concert, and the contribution of each depends largely on the experimental and environmental conditions, as well as on the particular protein under study. A common view of amyloid formation is as a nucleation-dependent polymerization reaction. The nucleation step has a high-energy barrier and therefore is the rate-limiting step. Following nucleation, a relatively rapid fibril elongation process takes place [24].
Common molecular interactions mediate the aberrant self-assembly process, including backbone and side-chain hydrogen bonds complemented by hydrophobic and electrostatic interactions involving side chains of particular amino acids. The role of hydro-phobic interactions is well known. For example, a major driving force of aggregation in the case of naturally folded proteins is exposure of hydrophobic regions that are buried in the native structure, due to partial unfolding of the protein, followed by abnormal sequestering of the exposed side chains through uncontrolled intermolecular interactions leading to aggregation. In comparison, the contribution of electrostatic interactions is less well recognized, though multiple studies have indicated their importance in amyloid assembly, specifically in fibril morphology [25], the size of oligomers [26], and the relative amount of α-helix or β-sheet secondary structures involved in amyloid formation [27]. For example, many familial AD-linked mutations within the Aβ-encoding region of the app gene lead to an increase in the positive charge of Aβ sequence and to enhanced aggregation kinetics [28, 29]. Based on these realizations, we reasoned that in the absence of common primary structures or of higher-order structures of amyloidogenic proteins prior to their self-assembly, a strategy aimed at inhibiting formation of the toxic assemblies should focus on the common, fundamental molecular interactions that drive the aberrant self-assembly reaction — the combination of hydrophobic and electrostatic interactions.
Unique in their ability to participate in both hydrophobic and electrostatic interactions among the twenty proteinogenic amino acids are K residues. Due to this unique feature, K residues play a prominent role in protein folding and biological processes [30]. For example, ubiquitination at K residues marks proteins for proteasomal degradation and acetylation of histones at K residues regulates transcription.
K residues also play an important role in the assembly and toxicity of amyloidogenic proteins. In the microtubule-associated protein tau, aberrant post-translational modifications of K residues are frequently found in disease [31]. In addition to normal ubiquitination of specific K residues to mark the protein for degradation, ubiquitination of other K residues has been found in the microtu bule-binding domain of soluble paired helical filaments (PHF), impairing tau binding and microtubule integrity [32]. In particular, ubiquitination of K6 within PHF (using numbering for the longest, 441-residue tau isoform) is thought to inhibit ubiquitin-dependent protein degradation suggesting an explanation for the failure of the 26S ubiquitin-proteasome system (UPS) in the presence of neurofibrillary tangles [32].
K residues also are involved in motifs that regulate tau phosphorylation. Normal phosphorylation of tau at S, Y, or T residues regulates the ability of tau to promote microtubule assembly, whereas abnormal hyperphosphorylation facilitates the polymerization of tau into PHF [33]. One of the kinases involved in tau phosphorylation, the microtubule affinity-regulating kinase (MARK), targets S residues in K-X-G-S motifs [34]. Phosphorylation by MARK induces dissociation of tau from microtubules and prevents its degradation [35]. It has been suggested that phosphorylation by MARK and the related kinase, PAR-1, may be a prerequisite for the subsequent action of cyclin-dependent kinase 5 and glycogen synthase kinase 3β [36], leading to the pathologically hyperphosphory-lated tau.
Recently, mono-methylation of K residues in tau has been identified as another possible post-translational modification that pre-dominantly co-localizes with neurofibrillary tangles [37]. Acetylation of K residues in tau, including at K280 within the microtubule-binding domain, has been shown to inhibit tau function by impairing tau-microtubule interactions and promoting pathological tau aggregation [38]. Immunohistochemical and biochemical studies of brains from patients with AD and related tauopathies and from tau transgenic mice showed that acetylated-tau pathology was specifically associated with insoluble, thioflavin-S-positive tau aggregates [38]. Moreover, deletion of K280 has been linked with both frontotemporal dementia [39] and Alzheimer’s disease [40]. This deletion has been shown to lead to a highly fibrillogenic tau variant [41]. The deletion reduces inclusion of exon 10 in the mapt cognate gene, which leads to a lower levels of the 4-repeat tau isoform and thus has an effect opposite to many other disease causing tau mutations. K280 deletion possibly causes pathology by facilitating an extended state that allows the propagation of β-structure downstream of the fibrillogenic hexapeptide, V306QIVYK311 within the third repeat [42]. This hexapeptide can be targeted to inhibit fibrilization as shown by Li et al. [43] who used oleocanthal extracted from olive oil and found that it inhibited tau fibrilization by forming an adduct with K311.
In Aβ40, site-specific modification at the only two K residues in the sequence of the protein, K16 and K28, by cholesterol oxidation products, resulted in enhanced aggregation kinetics relative to the unmodified protein [44]. An early structural element observed in the Aβ monomer is a turn in region 21–30 [45]. This turn is stabilized by a salt-bridge between the side chains of D23 or E22 and K28 [45-52] and has been suggested to be key in the nucleation of Aβ assembly [45]. Supporting this view, Sciaretta et al. [53] have shown that Aβ40 containing a lactam cross-link between D23 and K28 forms amyloid fibrils substantially faster than wild-type (WT) Aβ40, with no detectable lag phase in the fibrillization process. In agreement with the hypothesis of the prominent role of this turn region in further assembly, the turn also has been found in the fibril structure of Aβ40 and Aβ42 [54-57].
A suggested mechanism by which amyloidogenic protein assemblies cause toxicity is disruption of membrane integrity. This could result from direct distortion of physical properties of membranes [58, 59], which may result in apoptosis [60], increased membrane permeabilization and conductivity [61], or formation of non-specific channels [62] and disruption of calcium, and/or other electrolyte homeostasis [63-68]. Many studies point to a combination of electrostatic and hydrophobic interactions, potentially in volving “lysine snorkeling” [69], as the driving force behind the membrane disruption by amyloidogenic proteins.
Yoshiike et al. [70] suggested that the surface composition of different morphologies of amyloid fibrils, specifically clusters of positive charge, is the cause of the observed range of fibril toxicity. Through chemical modification or amino acid substitution in Aβ, they showed that changes in the surface structures of Aβ fibrils led to changes in properties responsible for electrostatic and/or hydrophobic interactions and could be manipulated to suppress Aβ toxicity [70]. Thus, in both oligomers and fibrils, electrostatic attraction between positively charged K residues and negatively charged membrane phospholipid head groups, together with hydrophobic interactions between the K butylene and lipid hydrocarbon chains presumably contribute to the toxic effect of amyloidogenic protein assemblies [65, 67, 70, 71].
Recently, the binding sites of five toxicity inhibitors, Congo red, Myricetin, melatonin, nicotine, and curcumin on Aβ were explored computationally and then tested in vitro to gain insight into the surface components of aggregates that contribute to the biological effects [72]. All five molecules were found to dock at or near K28, supporting the importance of this residue in Aβ assembly and toxicity. The other K residue in Aβ, K16, resides next to the central hydrophobic cluster (CHC, residues 17–21), which is known to be important in regulating Aβ fibrillogenesis [73-75]. K16 itself has been reported to be solvent-exposed and thus not participate directly in Aβ self-assembly but rather be available for interaction with cell membranes or potential inhibitors [76-78]. The triple substitution R5A, K16A, and K28A in Aβ resulted in significant loss of Aβ40 fibril toxicity in human embryonic kidney cells [70], presumably due to removal of the positively charged residues that could interact with, and disrupt, the cell membrane. These examples demonstrate the prominent role K residues play in the assembly and toxicity of the proteins involved in AD and likely in other amyloid-related diseases. Below we discuss our recent work on the role of K residues in Aβ and the use of K-specific molecular tweezers (MTs) as broad-spectrum inhibitors of Aβ and other amyloidogenic protein toxicity.
K RESIDUES ARE IMPORTANT DETERMINANTS OF Aβ STRUCTURE, ASSEMBLY KINETICS, AND TOXICITY
To gain insight into the role of K residues in Aβ assembly and toxicity, each of the two K residues was individually substituted by A and the effect of the substitution on secondary structure, morphology, and toxicity in cell culture of both Aβ40 and Aβ42 was studied [79]. Circular dichroism (CD) spectroscopy was used to monitor the initial secondary structure and the temporal change of secondary structure in six Aβ alloforms: WT Aβ40, [K16A]Aβ40, [K28A]Aβ40, WT Aβ42, [K16A]Aβ42, and [K28A]Aβ42. The spectra subsequently were deconvoluted to allow for quantitative comparison of the content of the α-helix, β-sheet, and statistical coil structures in the substituted analogues relative to the WT peptides, during their self-assembly [79].
Comparing the initial and final α-helix, β-sheet, and statistical coil content of WT Aβ40 to the two Aβ40 analogues containing K→A substitutions, Sinha et. al. found that initially, the three analogues had similar content of each conformational element — predominantly statistical coil and a small contribution of β-sheet. Following incubation under conditions that promote fibril formation, the conformational transition of the α-substituted Aβ analogues were substantially slower than those of their WT counterparts. The slowest transition was of [K16A]Aβ42, which did not reach completion at the final time point measured – 9 days, compared to the transition of WT Aβ42, which was complete in 24 hours. Interestingly, following incubation, [K16A]Aβ40 showed a higher β-sheet and lower statistical coil content than WT Aβ40. In fact, the β-sheet content of [K16A]Aβ40 fibrils was as high as that of Aβ42, which is more amyloidogenic, and typically its fibrils display higher β-sheet content than Aβ40 fibrils [79].
The conformational change in the case of the K→A Aβ42 analogues was substantially more variable compared to the Aβ40 analogues. The initial α-helix content of the Aβ42 analogues was minimal and similar to each other. However, over time, in WT Aβ42 and [K28A]Aβ42 the α-helix content decreased to zero and the β-sheet content increased, whereas in [K16A]Aβ42, the α-helix content increased and became the predominant secondary structure element with concomitant decrease of β-sheet content to zero. This is a highly unusual behavior and we are unaware of any other substitution that promotes such a high increase in α-helix content in an Aβ42 analogue.
Though [K28A]Aβ42 followed a conformational transition similar to that of WT Aβ42, it had substantially lower final β-sheet content, lower than all the Aβ40 analogues, and a corresponding high final statistical coil content, the highest of all the six Aβ alloforms. Thus, both K16 and K28 were found to be important determinants in Aβ42 assembly into β-sheet-rich fibrils, supporting the central role K residues play in this process.
Using the same samples characterized by CD spectroscopy, the effect of the K→A substitutions on Aβ morphology was analyzed by electron microscopy (EM). All six alloforms displayed quasiglobular or amorphous morphology immediately following dissolution and progressed to form fibrillar structures [79]. Aβ40, Aβ42 and their K16A analogues all showed long, unbranched fibrils. However, corresponding to its unusual behavior measured by CD spectroscopy, [K16A]Aβ42 also was characterized by abundant globular structures that appeared to “decorate” the fibrils. Presumably, within these structures [K16A]Aβ42 was in the predominantly α-helical conformation observed by CD spectroscopy. Interestingly, both [K28A]Aβ40 and [K28A]Aβ42 produced thicker, shorter, branched fibrils and a wider distribution of fibril diameters compared to the WT and K16A analogues, suggesting that the K28A substitution induced a higher nucleation rate relative to the analogues containing the native K28 residue. As the overall conformational transition kinetics in both [K28A]Aβ40 and [K28A]Aβ42 was slower than in their WT counterparts, the data indicate a large decrease in the elongation rates of the K28A analogues. These observations are in agreement with the important role of the electrostatic and hydrophobic interactions involving K28, which stabilize a turn in the Aβ(21–30) region [45].
The stark contrast between the structural transitions of [K16A]Aβ40 and [K16A]Aβ42, namely the high final β-sheet content of [K16A]Aβ40 versus the very low β-sheet content of [K16A]Aβ42, suggests an important interplay between the C-terminus and the CHC region, which is affected differently in Aβ40 and Aβ42 by the adjacent, charged K16. In Aβ40, this residue appears to inhibit early folding that would lead to high β-sheet content, whereas in Aβ42, K16 seems to facilitate β-sheet formation. It is interesting to compare and contrast these results with the study by Usui et al., in which K16 in Aβ40 was modified by the addition of cholesterol oxidation products leading to increased aggregation kinetics and toxicity [44]. The increase in β-sheet content correlates with the removal of the positive charge of K16, and/or a general increase in hydrophobicity in this region, yet the different modifications of K16 by Sinha et al. and Usui et al. led to opposite effects on the assembly kinetics and toxicity of the resulting peptides.
Modeling studies suggest that in Aβ40, because the C-terminus is shorter and less hydrophobic than in Aβ42, the N-terminus competes with the C-terminus for interaction with the CHC [26, 80, 81]. Thus, the C-terminus–CHC interactions are a higher component in early Aβ42 folding versus Aβ40 [81] and consequently, perturbation of K16 would be predicted to affect Aβ42 more than Aβ40. Possibly, in Aβ40 K16 promotes electrostatic N-terminus–CHC interactions (e.g., with D1, E3, D7), and thus the substitution of K16 by A shifts the scales towards increased C-terminus–CHC leading to increased β-sheet content. In the case of Aβ42, where the C-terminus–CHC interaction is predominant, conceivably K16 facilitates formation of an intermediate state necessary for the transition into β-sheet and the absence of such stabilization explains the substantial decrease in β-sheet content and slow conformational transition in [K16A]Aβ42. Alternatively, the obligatory α-helical intermediate in the conformational transition of Aβ [82] may be stabilized by the K16A substitution in Aβ42.
To assess the impact of the A substitutions on Aβ-induced toxicity, the six alloforms were incubated with differentiated rat pheochromocytoma (PC-12) cells and the lactate-dehydrogenase release (LDH) assay was used to measure cell viability [79]. Unexpectedly, despite the relatively minor influence of the K16A substitution on Aβ40 assembly, [K16A]Aβ40 was not toxic to the cells even at a concentration as high as 100 μM ((Fig. 1), reprinted with permission from Sinha et al., ACS Chem. Neurosci 2012; 3: 473-81. Copyright (2012) American Chemical Society [79]). Previously, Ono et al. found that the level of toxicity of small Aβ40 oligomers correlated with β-sheet content [83]. Thus, it was highly surprising that even though the β-sheet content in [K16A]Aβ40 was higher than that of WT Aβ40 and similar to the WT Aβ42 levels, no toxicity was observed with this isoform. These data strongly suggest direct involvement of K16 in Aβ-mediated toxicity, presumably through interaction with the cell membrane. In support of this inter-pretation of the data, [K16A]Aβ42 also showed substantially reduced toxicity relative to WT Aβ42 (Fig. 1) despite the distinct effects of the K16A substitution on Aβ40 and Aβ42 conformation.
Fig. (1). Toxicity of Aβ40, Aβ42, and their α-substituted analogues.
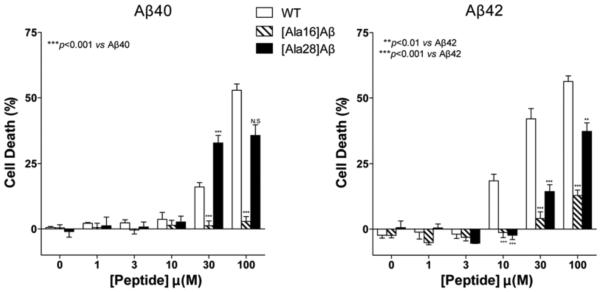
Aβ40, Aβ42, and their respective K→A analogues were incubated with differentiated PC-12 cells for 48 h and cell death was measured using the lactate dehydrogenase (LDH) release assay. The results are an average of four to six experiments and are presented as mean ± SEM. Statistical significance was calculated by one-way ANOVA.
The K28A substitution also led to reduced toxicity of the resulting Aβ40 and Aβ42 analogues relative to the WT counterparts, but the reduction in toxicity was substantially smaller than for the K16A analogues. In the case of the [K28A]Aβ analogues, the decreased toxicity correlated with major perturbation of the conformational transition and assembly process. Therefore, a conservative interpretation of the data is that substitution of K28 by A disrupts the turn in Aβ(21–30) leading to formation of oligomers and fibrils with different structures from those formed by the WT peptides. These structures are still toxic, but to a lesser extent than those of WT Aβ40 and Aβ42.
Based on the surprising findings that substitution of K16 by A led to a dramatic loss of toxicity in both Aβ40 and Aβ42, we predict that mutations in app leading to this or similar substitutions may be found to be protective from AD, similar to the recently discovered protective mutation that causes an A2T substitution in Aβ [84]. Interestingly, a new kindred has been discovered recently in which a heterozygous app mutation leading to a K16N substitution causes familial AD [85]. Ostensibly, this might be perceived as contradicting our hypothesis that K16 is directly involved in mediating Aβ toxicity. However, in support of our interpretation, [K16N]Aβ42 was found to have reduced toxicity compared to WT Aβ42. Apparently, the early onset familial AD associated with this mutation correlates with formation of highly toxic mixed oligomers of WT and K16N Aβ and possibly by increased total Aβ concentration due to perturbation of the α-secretase cleavage site and/or reduced Aβ clearance [85].
Recently, our group discovered that deletion of K1 in IAPP, a highly amyloidogenic peptide hormone whose self-assembly is closely associated with pancreatic β-cell death in type-2 diabetes, causes substantial decrease in the peptide’s toxicity [86]. These findings provide additional support for an important role of K residues in mediating toxicity of amyloidogenic proteins. We hope that the data presented here will prompt researchers in the field to explore the role of K residues in the structure and toxicity of additional amyloidosis-associated proteins.
The data discussed above suggest that disruption of molecular interactions involving K residues may lead to modulation of Aβ assembly and inhibition of Aβ toxicity. Moreover, because K residues are common to almost all proteins, this strategy may be applied to inhibition of the toxicity of amyloidogenic proteins in general, not just Aβ. At the same time, this very argument raises the question whether the strategy can be applied specifically to inhibition of toxic amyloidogenic proteins without interfering with normal protein folding and function. In the following sections we describe the application of MTs, which bind non-covalently with moderate affinity to K residues and achieve this very goal, inhibiting the assembly and toxicity of amyloidogenic proteins without interfering with normal physiology.
MOLECULAR TWEEZERS: ARTIFICIAL K RECEPTORS
The MTs developed by Drs. Klärner and Schrader at University of Duisburg-Essen, Germany, are small molecules that act as selective, artificial K receptors. MTs bind with Kd = 10–20 μM to K residues and with ~10-times lower affinity to R residues in peptides and proteins (Fig. 2) [87-89]. MTs have a horseshoe-shaped structure composed of two hydrocarbon arms capable of hydrophobic interactions with the alkyl side chains of K residues, which get threaded through the central cavity [87, 89]. At their bridgehead, MTs have negatively charged groups, e.g. phosphates, which form ionic interactions with the positively charged ammonium or guanidinium groups of K and R, respectively. Binding of the MT derivative termed CLR01 ((Fig. 2B), reprinted with permission from Sinha et al., J. Am. Chem. Soc. 133:16958-69. Copyright (2011) American Chemical Society [88]) to K16 and K28, and to a lower extent to R5, in Aβ was confirmed by solution-state, 2D-NMR and electron-capture dissociation mass spectrometry [88]. Because MTs utilize the same types of interactions, hydrophobic and electrostatic, found in early Aβ assembly and presumably K-mediated interaction of Aβ with cell membranes, following the report on the first Kspecific MTs [87], Gal Bitan hypothesized that competition by MTs for these interactions would inhibit Aβ assembly and decrease Aβ-induced toxicity [88].
Fig. (2). Molecular structures of MTs CLR01 and CLR03.
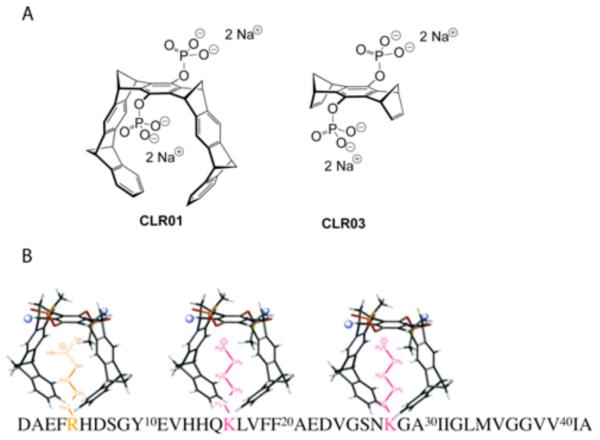
A) The hydrocarbon arms of CLR01 participate in hydrophobic interactions and the phosphate groups at the bridgehead participate in electrostatic interactions. CLR03 lacks the hydrocarbon arms and thus cannot participate in hydrophobic interactions and is used as a negative control. B) Illustration of 3 molecules of CLR01 interacting with the K and R residues of Aβ42. The binding to R is ~10-times weaker than to K [87].
The micromolar affinity of MTs for K may seem at first to be a shortcoming for a drug candidate. However, this actually appears to be the key feature of the ability of MTs to affect the abnormal folding and assembly of amyloidogenic proteins without disrupting normal physiology. Because of their moderate affinity and labile binding, MTs disrupt only the weak interactions involved in aberrant self-assembly but not the stable structures or high-affinity interactions of normal proteins, which are tightly controlled and have been shaped by millions of years of evolution. Thus, MTs are process-specific, rather than protein-specific because they disrupt the abnormal folding and assembly of multiple amyloidogenic proteins, but not the normal folding and function of stable proteins.
The data presented below have been obtained using the MT derivatives CLR01 and CLR03 (Fig. 2A). CLR03 differs from CLR01 in that it has a truncated hydrocarbon skeleton. Consequently, it can form electrostatic, but not hydrophobic interactions, is not expected to bind specifically to K residues, and has been used as a negative control. In multiple experiments CLR03 was found to have minimal or no effect on the assembly or toxicity of the proteins under study, highlighting the functional importance of the hydrophobic side arms of MTs for interacting with K residues.
MOLECULAR TWEEZERS ARE BROAD-SPECTRUM ASSEMBLY AND TOXICITY INHIBITORS IN VITRO
To test the hypothesis that perturbation of molecular interactions involving K residues would disrupt amyloidogenic assembly, the effect of CLR01 on formation of β-sheet-rich fibrils was analyzed in nine different proteins that were either naturally unstructured (Aβ, tau, α-syn, IAPP, calcitonin, and PrP(106–126)) or structured (insulin, β2-microglobulin, and transthyretin). The percentage of K residues in these proteins ranges from 2.7–10.7% (Table 1, reprinted with permission from Sinha et al., J. Am. Chem. Soc. 133:16958-69. Copyright (2011) American Chemical Society [88]). Each protein was incubated under aggregation-promoting conditions in the absence or presence of CLR01 or CLR03. The aggregation reaction was followed by thioflavin T (ThT) fluorescence [90] and EM, except in the cases of TTR and PrP(106–126), which did not show sufficient ThT binding upon fibril formation and therefore their aggregation was monitored by the increase in turbidity at α = 360 nm [91, 92].
Table 1.
Amyloidogenic proteins used by Sinha et al. [88].
| Protein | Length (aa) |
Lys | % Lys | Arg | % Arg | Associated Disease | Reference |
|---|---|---|---|---|---|---|---|
| Aβ40, Aβ42 | 40, 42 | 2 | 5.0, 4.8 | 1 | 2.5, 2.4 | AD | [130] |
| Tau (embryonic) |
352 | 37 | 10.5 | 14 | 4.0 | AD, tauopathies | [130] |
| α-Syn | 140 | 15 | 10.7 | 0 | 0.0 | PD, synucleinopathies | [131] |
| IAPP | 37 | 1 | 2.7 | 1 | 2.7 | Type-2 diabetes | [132] |
| Calcitonin | 32 | 1 | 3.1 | 0 | 0.0 | Medullary Carcinoma of the Thyroid | [133] |
| Insulin | 51 | 2 | 3.9 | 1 | 2.0 | Injection-related nodular amyloidosis | [134] |
| β2-Microglobulin | 99 | 8 | 8.1 | 5 | 5.1 | Dialysis-related amyloidosis | [135] |
| Transthyretin | 147 | 8 | 5.4 | 5 | 3.4 | Senile systemic amyloidosis, Familial amyloid polyneuropathy |
[136, 137] |
| PrP(106-126) | 21 | 2 | 9.5 | 0 | 0.0 | [138] |
The protein concentration in each case was selected to enable aggregation within a reasonable time frame, ranging from <2 hours for the most amyloidogenic peptide, IAPP, to over a week for the slowest protein, α-syn. In most cases, CLR01 was found to inhibit the aggregation completely at a 1:1 concentration ratio and in some cases at sub-stoichiometric concentrations (less CLR01 than protein), suggesting inhibition of both the nucleation and elongation steps [88]. Interestingly, sub-stoichiometric inhibition was found in the cases of calcitonin and IAPP, both of which are hormones sharing some structural similarity, such as a disulfide-bridge constrained 6/7-residue region in their N-terminus. In agreement with the ThT fluorescence/turbidity measurements, morphological examination of all proteins studied showed formation of amorphous aggregates in the presence of CLR01, as opposed to the typical amyloid fibrils formed in the absence of MTs or in the presence of CLR03 [88]. The only exception to the rule was PrP(106–126), whose aggregation was not inhibited by CLR01, presumably because the two K residues in this peptide are located away from the amyloidogenic sequence that mediates its aggregation.
In follow-up experiments, the capability of CLR01 to inhibit the toxicity induced by amyloidogenic proteins was tested. The proteins were incubated first under conditions that promote oligomerization and then added exogenously to cultured cells. Differentiated PC-12 cells were used to study Aβ, α-syn, calcitonin, β2-microglobulin, and transthyretin, whereas rat insulinoma (RIN5fm) cells were used to characterize IAPP and insulin. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay was used for measurement of cell viability in all cases [88]. In addition, because α-syn is predominantly an intracellular protein, CLR01 inhibition of endogenous α-syn expressed in HEK 293 cells also was tested [93].
Because MTs potentially can bind to any exposed K and at sufficiently high concentration likely would disrupt cellular processes, it was important to determine first whether a sufficient window existed between CLR01 concentrations that reduced cell viability and those needed for inhibition of toxicity caused by amyloidogenic proteins. CLR01 was found to increase cell viability by 5–15% relative to control cells at concentrations up to 200 μM, whereas at 400 μM, it caused a 10–20% decrease in cell viability [88]. Therefore, CLR01 concentration was kept below 400 μM in all subsequent experiments. The results are summarized in Table 2 (reprinted with permission from Sinha et al., J. Am. Chem. Soc. 133:16958-69. Copyright (2011) American Chemical Society [88]). Because the proteins were added exogenously in these assays, the CLR01 concentration at half-maximal inhibition (IC50) depended on the concentration of the respective protein. The CLR01:protein stoichiometry, estimated as the ratio between the concentration of the protein used and the IC50, varied, though in most cases it was in the same order of magnitude. Because the endogenous concentrations of most of the proteins used are in the nM range, the data suggested that nM concentrations of CLR01, several orders of magnitude below toxic concentrations, might inhibit aggregation of these proteins in vivo. In support of this view, though the IC50 of CLR01 for inhibition of 20 μM exogenously added α-syn was 3–4 μM, 1 μM of CLR01 was sufficient for complete inhibition of cell death induced by expression of endogenous α-syn in HEK 293 cells [93].
Table 2.
CLR01 inhibits toxicity of amyloidogenic proteins (from Sinha et al. [88]).
| Protein | Concentration in toxicity assay (μM) | Cells | IC50 (μM) | IC50:Conc. |
|---|---|---|---|---|
| Aβ40 | 20 | PC-12 | 14±11 | 0.7 |
| Aβ42 | 10 | PC-12 | 52±18 | 5.2 |
| α-Syn | 20 | PC-12 | 3±1 | 0.15 |
| IAPP | 0.01 | RIN5fm | 6±3 | 600 |
| Calcitonin | 15 | PC-12 | 28±4 | 1.9 |
| Insulin | 5 | RIN5fm | 13±2 | 2.8 |
| β2-Microglobulin | 10 | PC-12 | 28±6 | 2.8 |
| Transthyretin | 1 | PC-12 | 54±19 | 54 |
CLR01 REMODELS Aβ AND α-SYN OLIGOMERS INTO NON-TOXIC STRUCTURES AND DISSOCIATES PREFORMED FIBRILS
Due to the increasing evidence that soluble oligomers of Aβ and other amyloidogenic proteins are the most toxic structural species involved in amyloidoses [13, 15, 94], Sinha et al. [88] explored how CLR01 affected Aβ42 oligomerization using dot blots with the oligomer-specific antibody, A11 [95]. In the absence of MTs, A11 immunoreactivity was observed immediately and continued to increase over several days. In contrast, Aβ42 samples incubated in the presence of CLR01 never showed A11 reactivity suggesting that the reaction of CLR01 with Aβ was fast and induced structural changes precluding formation of the toxic oligomers recognized by A11. Interestingly, dynamic light scattering (DLS) experiments showed that oligomers formed by 10 μM Aβ42 in the absence or presence of equimolar CLR01 had essentially the same sizes, though their abundance increased relative to Aβ42 alone [88]. SDS-PAGE and native-PAGE/Western blot analyses of 200 nM Aβ42 prepared in neurobasal cell culture medium or artificial cerebrospinal fluid for electrophysiologic experiments did not show a difference in assembly size between preparations in the absence or presence of CLR01 [96]. Thus, the structural changes CLR01 induces in Aβ42 oligomers are relatively small and cannot be discerned by low-resolution methods, such as DLS, EM, or PAGE/Western blot. Nonetheless, these structural changes have two important outcomes — the oligomers formed in the presence of CLR01 are non-toxic, and binding of CLR01 prevents fibril formation.
The effect of CLR01 also was assessed on oligomerization of αsyn. Samples of 20 μM α-syn were incubated for 12 days and aliquots were analyzed daily using SDS-PAGE and native-PAGE [93]. α-Syn migrated in several bands likely corresponding to monomer, low molecular weight oligomers, and a smear of higher molecular weight assemblies. Similarly to the data observed with Aβ42, in the presence of CLR01 there was no difference in the size of the α-syn oligomers prepared in the absence or presence of CLR01 detected by SDS-PAGE or native-PAGE yet the abundance of high-molecular weight species increased in the presence of CLR01 [93]. Based on the data observed for Aβ42 and α-syn, we hypothesize that upon interaction with CLR01, amyloidogenic proteins self-assemble into oligomers of similar size to those formed in the absence of the MT, but are not toxic. The increase in abundance of such oligomers in the presence of CLR01 is attributed to stabilization of these structures and prevention of further aggregation and fibril formation.
If MTs are to be developed as therapeutic drugs, it is important to examine whether they can not only prevent protein aggregation but also disassemble pre-formed fibrils, as those are expected to exist in the affected tissues of patients. Therefore, the capability of CLR01 to dissociate fibrils of Aβ ((Fig. 3A,B), adapted with permission from Sinha et al., J. Am. Chem. Soc. 133:16958-69. Copyright (2011) American Chemical Society [88]), α-syn ((Fig. 3C)), reproduced with permission from reference [93]), or IAPP [86] was examined. Each protein was allowed to aggregate and form immature or mature fibrils and then incubated further with 10-fold excess CLR01. In all cases, ThT fluorescence measurements showed that CLR01 caused steady dissociation of the present fibrils. EM images revealed remodeling of the assembly state first from long and unbranched, to shorter, branched fibrils and then to amorphous aggregates. The putative mechanism by which the excess CLR01 effected fibril dissociation is by shifting the equilibrium between the fibrillar and soluble states of the proteins and preventing reassociation of monomers with the fibrils. In vivo, this is predicted to allow clearance of the offending proteins by various mechanisms, such as the proteasome, lysosome, and/or specific and non-specific proteolytic enzymes, which fail to clear these proteins in their aggregated, fibrillar forms.
Fig. (3). CLR01 disaggregates Aβ and α-syn fibrils in vitro.

Disaggregation of A) Aβ40, B) Aβ42, or C) a-syn fibrils by CLR01 was initiated in each case at two time points, the first when immature fibrils formed and the second when fibrils had a chance to mature and consolidate for at least 2 weeks. The reactions were monitored using ThT fluorescence. Electron micrographs were obtained periodically and show the morphology of each protein at the indicated time points.
COMPARISON OF CLR01 WITH ASSEMBLY INHIBITORS CURRENTLY IN CLINICAL TRIALS
Approximately fifty treatments are in phase-2 or 3 clinical trials for AD, of which 10–20% target Aβ assembly directly or indirectly [97, 98]. Examples of drug candidates in this category include ELND-005, Sunphenon, Tramiprosate/Alzhemed, and PBT2, of which the first three compounds originated from natural sources. Tramiprosate is a glycosaminoglycan mimetic that binds to soluble Aβ and prevents fibrillization [99]. Though it was found to be effective in preclinical animal studies, Tramiprosate failed in clinicaltrials, potentially due to methodological issues, and has been now rebranded as a natural memory-protecting agent called Vivimind (http://www.canadadrugcenter.com/Health-Canada-Approves-VIVIMIND.asp). PBT2 is a 2nd-generation clioquinol derivative and similar to clioquinol, it is a chelating agent for Zn2+ and Cu2+, which are thought to induce brain Aβ aggregation. PBT2 has shown positive results in phase-2 clinical trials and has yet to progress to phase-3 trials (http://finance.yahoo.com/news/pranas-pbt2-clinical-trials-cited-172631353.html).
ELND-005 is a natural sugar derivative, scyllo-inositol (SI), which has good oral bioavailability and crosses the blood-brain barrier via inositol transporters. Though the mechanism by which SI interacts with Aβ is not understood, the compound has been reported to promote both dissociation of Aβ aggregates and modulation of the aggregation process into formation of relatively large, presumably non-toxic assemblies [100-102]. Studies of SI in transgenic mouse models of AD showed reduction in soluble and insoluble Aβ, plaque burden, and synaptic loss, and improved spatial memory [100]. Phase-2 clinical trials for ELND-005 have been completed with mixed results, of which 9 deaths in the high-dose groups caused significant safety concerns and cognitive and functional co-primary endpoints did not achieve statistical significance [103].
Sunphenon is the commercial name of (–)-epigallocatechin-3-gallate (EGCG), a polyphenol from green tea thought to function by many modalities, including anti-aggregation, scavenging of reactive oxygen species and other antioxidant activities, activation of cell signaling cascades, such as the protein kinase C pathway, and Fe3+ chelation [98, 104]. Though EGCG has relatively poor bioavailability, it has been reported to inhibit Aβ and α-syn assembly and toxicity, presumably by binding to the unfolded proteins and preventing conversion to toxic oligomers [105]. Peripheral administration of EGCG decreased Aβ plaque load in mice [106]. A phase-2/3 clinical trial pursued by Charite University, Berlin, Germany currently is recruiting patients.
In contrast to SI and EGCG, for which the mechanistic basis of the interaction with Aβ or other amyloidogenic proteins is not known, the binding of CLR01 to these proteins occurs at K, and to a lower extent, R residues and the basis for the binding has been wellcharacterized. Therefore, it was of interest to compare the capability of these three compounds to inhibit the aggregation, oligomerization, and toxicity of Aβ.
Three-fold excess of either EGCG or CLR01 were found to inhibit completely the formation of β-sheet-rich fibrils in Aβ42 [107]. In contrast, surprisingly, 10-fold excess of SI showed only week inhibition. ThT fluorescence in samples of Aβ42 incubated in the presence of 10-fold excess SI increased steadily with no apparent lag phase, though the rate of increase was slower than that of Aβ42 alone, suggesting that SI might facilitate nucleation, and attenuate elongation of Aβ42 fibrils, similarly to the effect of the K28A substitution discussed above.
The effect of the three inhibitors on Aβ42 oligomerization was assessed using a the oligomer-specific antibody A11 [95], as described above. Aβ42 incubated in the presence of CLR01 or EGCG did not show A11 immunoreactivity, whereas in agreement with the weak inhibition observed in ThT fluorescence experiments, Aβ42 incubated in the presence of SI showed similar A11 immunoreactivity to control samples of Aβ42 alone. The data suggested a similar, quick remodeling of Aβ42 assemblies by both EGCG and CLR01, but not SI, to a conformation that is not recognized by A11.
The effect of the three compounds on Aβ-induced toxicity was compared in differentiated PC-12 cells, primary hippocampal neurons, and mixed primary hippocampal neuronal/glial cultures using the LDH-release assay ((Fig. 4), reprinted with permission from Sinha et al., ACS Chem Neurosci. 3:451-8. Copyright (2012) American Chemical Society [107]). Again, in agreement with the inhibition of aggregation and oligomerization, both CLR01 and EGCG were potent inhibitors of cell death [107], whereas SI showed weak or no inhibition in the different cell cultures.
Fig. (4). Comparison of inhibition of Aβ42-induced cell death by SI, EGCG, and CLR01.
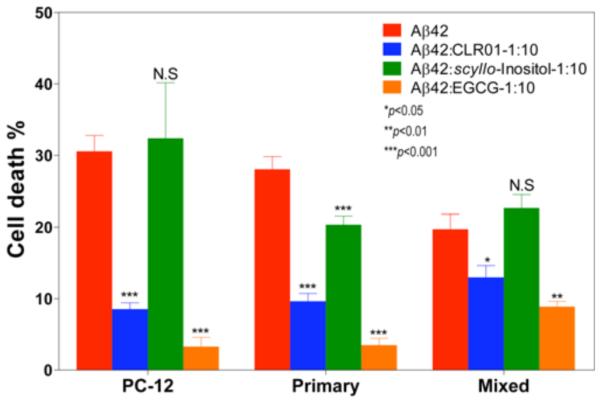
Ten μM Aβ42 was added to differentiated PC-12 cells, primary rat hippocampal neurons, or primary rat hippocampal neurons mixed with glial cells in the absence or presence of 10-fold excess of each inhibitor. Cells were incubated with the peptide:inhibitor mixtures for 48 h and cell death was measured using the LDH release assay. The data are presented as mean ± SEM for 3 independent experiments. *p<0.05, **p<0.01, ***p<0.001 compared to the Aβ42 in each group (one-way ANOVA).
Interestingly, solution-state NMR investigation of the interaction of EGCG with Aβ40 revealed only weak contacts between the polyphenol and Aβ monomers, and could not identify a binding site, in contrast to the clear binding sites identified for CLR01 in similar experiments [107]. These findings suggest that EGCG binds to Aβ at later assembly stages than CLR01. A recent solid-state NMR study has suggested that EGCG binding interferes particularly with aromatic interactions in the CHC region of Aβ [108], yet how this interaction relates to the inhibition of Aβ oligomerinduced toxicity remains to be elucidated.
CLR01 PROTECTS SYNAPTIC STRUCTURE AND FUNCTION FROM Aβ42-INDUCED TOXICITY
To evaluate the potential of MTs as disease-modifying therapy in AD, the ability of CLR01 to prevent synapse loss was evaluated, because synapse loss is a strong anatomical correlate of cognitive deficits in AD [109, 110]. In the presence of CLR01, dendritic spine density of rat primary hippocampal neurons treated with Aβ42 was four-times higher than in the absence of CLR01 [96], demonstrating that the compound protected not only neuronal viability but also synaptic integrity. In subsequent electrophysiologic experiments, excitatory post-synaptic currents induced by stimuli mimicking action potentials were rescued to baseline level by CLR01 when a mixture of Aβ42 and CLR01 was incubated with rat primary hippocampal neurons as compared to ~50% decreased amplitude of these currents in the presence of Aβ42 alone. In similar experiments, both the amplitude and frequency of spontaneous miniature excitatory post-synaptic currents were rescued to baseline levels in the presence of CLR01 [96]. Importantly, CLR01 also was found to protect neurons against Aβ42-induced inhibition of long-term potentiation (LTP), a cellular correlate of learning and memory ((Fig. 5), reprinted with permission from Attar et al. [96]). In mouse hippocampal brain slices, co-application of a freshly prepared mixture of Aβ42 with CLR01 significantly mitigated the level of Aβ-induced LTP inhibition. Furthermore, pre-incubation of Aβ42 with CLR01 rescued LTP to an even higher extent — ~80% of baseline levels. In agreement with the in vitro data discussed above, these results suggest that CLR01 rapidly remodels Aβ42 into a non-toxic form.
Fig. (5). CLR01 attenuates Aβ42-induced inhibition of long-term potentiation at CA3–CA1 synapses.

A) Time course of field excitatory postsynaptic potential (fEPSP) amplitudes before and after high-frequency stimulation (HFS, indicated by arrow) in slices treated for 20 min with vehicle (black circles), 200 nM Aβ42 (grey circles), or 200 nM Aβ42 + 2 μM CLR01 pre-incubated for 1 h prior to application (white circles). Results are expressed as percentages of baseline fEPSP amplitude (=100%). Insets show representative traces of fEPSP at baseline (dotted lines) and during the last 5 min of LTP recording (solid lines). B) Bar graph showing mean LTP changes measured during the last 5 min of recording following slice exposure to vehicle or different combinations of 200 nM Aβ42, 2 μM CLR01 and 2 μM CLR03. **p < 0.001; *p < 0.05 using Student’s unpaired t-test.
CLR01 PROTECTS ZEBRAFISH FROM α-SYN TOXICITY
Zebrafish (ZF) embryos have been emerging as efficient models for investigating amyloid disease mechanisms, and for screening and evaluating drug candidates [111-113]. For example, a recent study has examined methylene blue as an inhibitor of tau and polyglutamine-induced toxicity in ZF, and unfortunately, did not find beneficial effects of the drug in this model [114].
CLR01’s ability to inhibit α-syn-induced toxicity in vivo was tested in a novel ZF model that expresses human wild type α-syn as a fusion protein with red fluorescent protein (DsRed) under a neuronal promoter starting at ~12 hours post fertilization (hpf). Following expression of the fusion protein, α-syn and DsRed are cleaved apart rapidly intracellularly to release native α-syn. This results in robust α-syn expression and DsRed fluorescence in surviving embryos, but the survival rate of embryos expressing these proteins is about half that of embryos expressing DsRed alone, illustrating the toxic effect of the α-syn expression. Moreover, ~80% of surviving embryos expressing α-syn show various degrees of deformation resulting from extensive neuronal apoptosis and nearly 100% of the embryos are paralyzed. Consequently, all deformed and most nondeformed embryos die within 10 days post fertilization.
Addition of CLR01 to the water environment of the ZF embryos at 8 hpf caused a dramatic increase in ZF survival ((Fig. 6), adapted with permission from Prabhudesai et al. [93]) and improved the phenotype in a dose-dependent manner [93]. At 10 days post fertilization, nearly half of the higher dosed group remained alive and overall, CLR01 increased survival at 10 days post fertilization by 13-fold. CLR01 also reduced apoptosis levels to control conditions, i.e., apoptosis levels in CLR01-treated ZF expressing α-syn and DsRed were the same as those in ZF expressing DsRed alone.
Fig. (6). CLR01 ameliorates α-syn neurotoxicity in zebrafish (ZF).
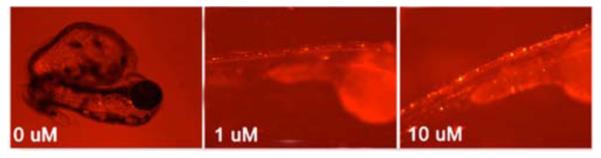
ZF embryos were treated with CLR01 at 8 hpf and were monitored for abnormal appearance and survival. Bright-field and fluorescent overlay images were taken at 72 hpf. For quantitative analysis, see [93].
Immunohistochemistry revealed abundant α-syn-immunoreactive small clumps in DsRed-positive neurons of untreated ZF ((Fig. 7A,B), reprinted with permission from Prabhudesai et al. [93]). In contrast, ZF treated with CLR01 also showed α-syn immunoreactivity but the protein was soluble and dispersed homogeneously together with DsRed in the cytoplasm (Fig. 7C-E). The concentration level of α-syn measured by Western blot decreased by ~80% in treated ZF (Fig. 7F), without changes in levels of DsRed or α-syn expression, as assessed by reverse-transcriptase polymerase chain reaction [93].
Fig. (7). CLR01 prevents α-syn aggregation and proteasome inhibition.

(a–e) ZF embryos expressing α-syn-DsRed (72 hpf) were subjected to immunohistochemistry. Green represents anti-a-syn antibody binding, red is DsRed, and blue is 4',6-diamidino-2-phenylindole-stained nuclei. (a, b) Representative neurons in untreated ZF. (c, d) CLR01-treated embryos. (e) Merged image of panels (c and d). (f) α-Syn expression inhibits the 26S ubiquitin-proteasome system (UPS) in ZF embryos. Embryos were lysed and proteins subjected to WB analysis (10 embryos per condition, N = 4). Lane 1, DsRed control; lane 2, untreated ZF expressing α-syn-DsRed; lane 3, α-syn-DsRed expressing ZF treated with CLR01; lane 4, α-syn-DsRed expressing ZF treated with CLR01 and lactacystin; lane 5, untreated wild-type embryos. Optical densities for α-syn (green bars) and DsRed (red bars) were normalized to actin and expressed as the percentage of untreated controls (*p < 0.0002 using Student’s t-test).
Many proteins that aggregate in neurodegenerative disorders have been shown to inhibit protein degradation [115]. To assess whether this was the case here and whether it was related to the decrease in α-syn concentration levels upon CLR01 treatment, the UPS was inhibited in treated ZF using lactacystin (Lac). Upon treatment with both CLR01 and Lac, α-syn was still soluble, rather than clumped, in the neurons of the treated ZF, yet α-syn concentration remained at the same level as in untreated ZF (Fig. 7F), demonstrating that the UPS was largely responsible for the 80% decrease in α-syn concentration levels observed in CLR01-treated ZF in the absence of Lac [93]. The data suggest that the UPS is inhibited by α-syn assemblies and that by remodeling α-syn into a soluble form, CLR01 allows rapid clearance of the protein by the UPS. The data are not only encouraging and supporting further exploration of the use of CLR01 for PD therapy, but also suggest a plausible mechanism by which CLR01 works in vivo — keeping the offending protein in a soluble, non-toxic form and thereby facilitating its rapid clearance.
Importantly, the data support the process-specific mechanism of MTs because the putative labile binding of CLR01 to the K residues in α-syn efficiently inhibited the protein’s aggregation but not its ubiquitination, which occurs at K residues and is required for UPS-mediated degradation.
CLR01 PROTECTS TRANSGENIC MICE FROM AD-LIKE PATHOLOGY WITH NO APPARENT TOXICITY
Utilizing a tritium-labeled analogue, the blood-brain barrier permeability of CLR01 was found to be ~2% one-hour following intravenous injection [96]. This is quite a remarkable result for a negatively charged compound, such as CLR01, which suggests that the compound enters the brain using a mechanism other than passive diffusion. Interestingly, the same permeability was observed in both WT and triple-transgenic mice [116] at 12-months of age. CLR01 also was found to be 100% stable in both mouse and human, liver microsome and plasma preparations in vitro for onehour.
The capability of CLR01 to affect AD-related brain pathology was tested in the same triple-transgenic mice used for the bloodbrain barrier studies. These mice express human mutant forms of amyloid β-protein precursor, presenilin-1, and tau, and conse-quently develop Aβ plaques and neurofibrillary tangles in AD-relevant brain regions (hippocampus, cortex, amygdala) and deficits in synaptic plasticity [116]. Development of AD-like phenotype is relatively slow in these mice compared to many other AD mouse models [117, 118]. Though memory deficits have been observed by one group at 2–4 months of age [119, 120] and by certain other studies beginning at 6–8 months of age [121-124], most studies did not find deficits in this model until older ages [125-127].
Fourteen-month-old mice were treated for 28 days with 40 μg/kg/day of CLR01 administered using subcutaneously implanted pumps. Brain immunohistochemistry revealed a significant decrease in amyloid plaque load in mice treated with CLR01 compared to vehicle-treated mice, in the hippocampus, entorhinal, perirhinal, and piriform cortices, and in the amygdala ((Fig. 8), reprinted with permission from Attar et al. [96]). Similar decreases were observed also in levels of neurofibrillary tangles and of hippocampal and cortical microglia. Interestingly, the study confirmed a previous report by Hirata-Fukae et al. [128] who found that female triple-transgenic mice had substantially higher Aβ levels, yet similar hyperphosphorylated tau levels, when compared to male mice. Importantly, no signs of toxicity were observed in the treated mice by any of the criteria employed, including weight loss or mortality [96]. Furthermore, video-assisted monitoring of mouse activity found no differences upon treatment in velocity, path shape or mobility or the habituation rates of these measures, which previously have been implicated as signs of drug-induced central nervous system toxicity [129].
Fig. (8). CLR01 decreases amyloid β-protein and hyperphosphorylated-tau deposition and ameliorates microgliosis in transgenic mouse brain.

Tripletransgenic mice were treated with 40 μg/kg/day CLR01 or vehicle. A, D, G, I) Vehicle-treated transgenic mouse hippocampus. B, E, H, J) CLR01-treated transgenic mouse hippocampus. A, B) transgenic mouse brain stained with monoclonal antibody (mAb) 6E10 showing amyloid plaque deposition. C) % Aβ burden was quantified by calculating the total 6E10-stained area divided by the total area measured (Hippo – hippocampus, Ent – entorhinal, Peri – perirhinal, Pir – piriform cortices, Amyg – amygdala). D, E) transgenic mouse brain showing mAb AT8-positive neurofibrillary tangles in the CA1 region. F) % Aggregated hyperphosphorylated tau load was quantified by calculating the total AT8-stained area divided by the total area. G, H) transgenic mouse brain stained with mAb HT7 for total tau. I, J) transgenic mouse brain showing Iba1-positive microglia in the subiculum and CA1 region. K) Number of stained microglia in a 1.14 mm2 area of hippocampus per treatment condition. The scale bar in panel B applies to both panels A and B. The scale bar in panel J applies to panels D–J. *p < 0.05, **p < 0.01, ***p < 0.001 compared to vehicle-treated mice using linear mixed effects models for panels C and F and Student’s unpaired t-test for panel K.
SUMMARY AND FUTURE DIRECTIONS FOR CLR01
To date, K-specific MTs have been shown to inhibit the assembly and toxicity of thirteen different disease related amyloidogenic proteins ([88] and unpublished results). CLR01, the lead MT, has the ability to disassemble fibrils and prevent formation of toxic oligomers of both Aβ and α-syn. In cell culture, CLR01 prevents cell death at levels comparable to EGCG, a compound currently in clinical trials, and prevents Aβ-induced retraction of dendritic spines, inhibition of basal synaptic transmission, and long-term potentiation. In vivo, CLR01 inhibits deformation and early death in a ZF model of α-syn toxicity by keeping α-syn soluble and restoring α-syn degradation by the proteasome. Importantly, CLR01 has been shown recently to reduce brain Aβ and hyperphosphorylated tau levels in a mouse model of AD without any indications of toxicity. Thus far, multiple experiments in various systems have produced positive data supporting further development of MTs in general, and CLR01 in particular, as therapeutic agents for AD, PD, and other amyloid-related diseases. These are exciting and somewhat surprising results for small molecules that act using a heretoforeunexplored, process-specific mechanism, binding to virtually any exposed K residue with micromolar affinity.
Following these initial studies, many questions remain to be answered. Further experiments need to be done to assess the optimal dose and duration of treatment in mice and the effect of CLR01 on cognitive/behavioral deficits. Though toxic effects have not been observed at the doses administered to date, the therapeutic window needs to be determined. In addition, the impact of CLR01 on normal protein folding and aggregation, for example in blood clotting, needs to be explored in more detail. Future studies also will include additional exploration of structure–activity relationships in MTs and drug formulation for optimal blood–brain barrier permeability and oral bioavailability.
CONCLUSIONS
We present a novel approach to the problem of aberrant protein folding and self-assembly into toxic oligomers and aggregates, which underlies over 30 cureless human diseases. The approach is based on targeting K residues as key elements participating in the combination of hydrophobic and electrostatic interactions that are central in the aberrant assembly process. Our findings demonstrate that K residues are highly important mediators of both assembly and toxicity of Aβ and IAPP, supporting the rationale behind this strategy, and suggest that K residues likely play similar roles in the aggregation and toxicity of other amyloidogenic proteins.
Rather than screening molecules for their ability to break the amyloid cross-β structure, or exploring empirically found neu-traceuticals, the application of MTs as broad-spectrum inhibitors of amyloid proteins’ toxicity is based on targeting the fundamental molecular interactions involved in nascent oligomer formation. To our knowledge, CLR01 is the first example of an inhibitor selected using a rational approach that uses a process-specific, rather than a protein-specific, mode of action. Using process-specific inhibitors is highly attractive for developing disease-modifying therapy for amyloidoses because the inhibitors act only on the pathologic aggregation while sparing normal physiological processes (e.g., production of the offending proteins). The labile, moderate-affinity binding of CLR01 to K residues is the key to selectively blocking the weak interactions involved in amyloidogenesis without disrupting structurally stable proteins and their normal physiological functions.
MTs provide a unique, mechanism-based strategy for development of disease-modifying drugs for amyloidoses. Additional study of these molecules in vivo is necessary for moving forward from pre-clinical to clinical trials for AD and related diseases. Pursuing these studies is supported by the data outlined here, including the inhibitory effect of CLR01 on the oligomerization, aggregation, and toxicity of multiple amyloidogenic proteins, the amelioration of disease phenotype in animal models of AD and PD, and the favorable pharmacological profile of CLR01.
ACKNOWLEDGEMENTS
This work was supported by the Jim Easton Consortium for Alzheimer’s Drug Discovery and Biomarker Development at UCLA, RJG Foundation grant 20095024, a grant from the Cure Alzheimer’s Fund, UCLA Alzheimer’s Disease Research Center Pilot Award (from NIH/NIA P50AG016570), UCLA Clinical and Translational Science Institute (NIH/NCRR UL1TR000124), and an Individual Pre-doctoral National Research Service Award 1F31AG037283 (to AA).
ABBREVIATIONS
- AD
Alzheimer’s disease
- Aβ
Amyloid β-protein
- CD
Circular dichroism
- CHC
Central hydrophobic cluster
- CNS
Central nervous system
- DLS
Dynamic light scattering
- EGCG
(–)-epigallocatechin-3-gallate
- EM
Electron microscopy
- fEPSP
Field excitatory postsynaptic potential
- HFS
High-frequency stimulation
- hpf
Hours post fertilization
- IAPP
Islet amyloid polypeptide
- Lac
Lactacystin
- LDH
Lactate dehydrogenase
- LTP
Long-term potentiation
- mAb
Monoclonal antibody
- MARK
Microtubule affinity-regulating kinase
- MT
Molecular tweezer
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide
- PD
Parkinson’s disease
- PHF
Paired helical filaments
- SI
scyllo-inositol
- α-syn
α-synuclein
- ThT
Thioflavin T
- UPS
26S ubiquitin-proteasome system
- WT
Wild type
- ZF
Zebrafish
Footnotes
CONFLICT OF INTEREST
Gal Bitan is a co-author and co-inventor of a patent application entitled “Molecular Tweezers for the Treatment of Amyloid-Related Diseases” (2009), International Patent Application No. PCT/US2010/026419, USA Patent Application No. 13/203,962, European Patent Application 10 708 075.6. Gal Bitan also is a cofounder and a shareholder of Clear Therapeutics, Inc.
Aida Attar has no conflict of interest to disclose.
REFERENCES
- [1].Buxbaum J. The amyloidoses. Mt Sinai J Med. 1996;63:16–23. [PubMed] [Google Scholar]
- [2].Merlini G, Westermark P. The systemic amyloidoses: clearer understanding of the molecular mechanisms offers hope for more effective therapies. J Intern Med. 2004;255:159–78. doi: 10.1046/j.1365-2796.2003.01262.x. [DOI] [PubMed] [Google Scholar]
- [3].Rahimi F, Bitan G. The structure and function of fibrillar and oligomeric assemblies of amyloidogenic proteins. In: Rahimi F, Bitan G, editors. Pre-fibrillar amyloidogenic protein assemblies— common cytotoxins underlying degenerative diseases. Springer Science+Media B.V.; Dordrecht: 2012. pp. 1–36. [Google Scholar]
- [4].Liu T, Bitan G. Modulating self-assembly of amyloidogenic proteins as a therapeutic approach for neurodegenerative diseases: strategies and mechanisms. ChemMedChem. 2012;7:359–74. doi: 10.1002/cmdc.201100585. [DOI] [PubMed] [Google Scholar]
- [5].Grill JD, Cummings JL. Current therapeutic targets for the treatment of Alzheimer's disease. Expert Rev Neurother. 2010;10:711–28. doi: 10.1586/ern.10.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mullard A. Sting of Alzheimer's failures offset by upcoming prevention trials. Nat Rev Drug Discov. 2012;11:657–60. doi: 10.1038/nrd3842. [DOI] [PubMed] [Google Scholar]
- [7].Feng BY, Toyama BH, Wille H, et al. Small-molecule aggregates inhibit amyloid polymerization. Nat Chem Biol. 2008;4:197–9. doi: 10.1038/nchembio.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45:1712–22. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- [9].Martins IC, Kuperstein I, Wilkinson H, et al. Lipids revert inert Aβ amyloid fibrils to neurotoxic protofibrils that affect learning in mice. EMBO J. 2008;27:224–33. doi: 10.1038/sj.emboj.7601953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sharma AK, Pavlova ST, Kim J, et al. Bifunctional compounds for controlling metal-mediated aggregation of the Aβ42 peptide. J Am Chem Soc. 2012;134:6625–36. doi: 10.1021/ja210588m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Roberts BE, Shorter J. Escaping amyloid fate. Nat Struct Mol Biol. 2008;15:544–6. doi: 10.1038/nsmb0608-544. [DOI] [PubMed] [Google Scholar]
- [12].Landau M, Sawaya MR, Faull KF, et al. Towards a pharmacophore for amyloid. PLoS Biol. 2011;9:e1001080. doi: 10.1371/journal.pbio.1001080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rahimi F, Shanmugam A, Bitan G. Structure-function relationships of pre-fibrillar protein assemblies in Alzheimer's disease and related disorders. Curr Alzheimer Res. 2008;5:319–41. doi: 10.2174/156720508784533358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat Rev Mol Cell Biol. 2007;8:101–12. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- [15].Fandrich M. Oligomeric intermediates in amyloid formation: structure determination and mechanisms of toxicity. J Mol Biol. 2012;421:427–40. doi: 10.1016/j.jmb.2012.01.006. [DOI] [PubMed] [Google Scholar]
- [16].Das S, Mukhopadhyay D. Intrinsically unstructured proteins and neurodegenerative diseases: conformational promiscuity at its best. IUBMB life. 2011;63:478–88. doi: 10.1002/iub.498. [DOI] [PubMed] [Google Scholar]
- [17].Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- [18].Dobson CM. The structural basis of protein folding and its links with human disease. Philos Trans R Soc Lond B Biol Sci. 2001;356:133–45. doi: 10.1098/rstb.2000.0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–90. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- [20].Goldschmidt L, Teng PK, Riek R, Eisenberg D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc Natl Acad Sci USA. 2010;107:3487–92. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Makin OS, Serpell LC. Structures for amyloid fibrils. FEBS J. 2005;272:5950–61. doi: 10.1111/j.1742-4658.2005.05025.x. [DOI] [PubMed] [Google Scholar]
- [22].Dobson CM. Principles of protein folding, misfolding and aggregation. Semin Cell Dev Biol. 2004;15:3–16. doi: 10.1016/j.semcdb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- [23].Pastor MT, Kummerer N, Schubert V, et al. myloid toxicity is independent of polypeptide sequence, length and chirality. J Mol Biol. 2008;375:695–707. doi: 10.1016/j.jmb.2007.08.012. [DOI] [PubMed] [Google Scholar]
- [24].Jarrett JT, Lansbury PT., Jr. Seeding "one-dimensional crystallization" of amyloid: A pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993;73:1055–8. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- [25].Marshall KE, Morris KL, Charlton D, et al. Hydrophobic, aromatic, and electrostatic interactions play a central role in amyloid fibril formation and stability. Biochemistry. 2011;50:2061–71. doi: 10.1021/bi101936c. [DOI] [PubMed] [Google Scholar]
- [26].Yun S, Urbanc B, Cruz L, Bitan G, Teplow DB, Stanley HE. Role of electrostatic interactions in amyloid β-protein (Aβ) oligomer formation: a discrete molecular dynamics study. Biophys J. 2007;92:4064–77. doi: 10.1529/biophysj.106.097766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yanagi K, Ashizaki M, Yagi H, Sakurai K, Lee YH, Goto Y. Hex-afluoroisopropanol induces amyloid fibrils of islet amyloid polypeptide by enhancing both hydrophobic and electrostatic interactions. J Biol Chem. 2011;286:23959–66. doi: 10.1074/jbc.M111.226688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li H, Monien BH, Fradinger EA, Urbanc B, Bitan G. Biophysical characterization of Aβ42 C-terminal fragments: inhibitors of Aβ42 neurotoxicity. Biochemistry. 2010;49:1259–67. doi: 10.1021/bi902075h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Betts V, Leissring MA, Dolios G, Wang R, Selkoe DJ, Walsh DM. Aggregation and catabolism of disease-associated intra-Aβ mutations: reduced proteolysis of AβA21G by neprilysin. Neurobiol Dis. 2008;31:442–50. doi: 10.1016/j.nbd.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Freiman RN, Tjian R. Regulating the regulators: lysine modifications make their mark. Cell. 2003;112:11–7. doi: 10.1016/s0092-8674(02)01278-3. [DOI] [PubMed] [Google Scholar]
- [31].Gendron TF, Petrucelli L. The role of tau in neurodegeneration. Mol Neurodegener. 2009;4:13. doi: 10.1186/1750-1326-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cripps D, Thomas SN, Jeng Y, Yang F, Davies P, Yang AJ. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J Biol Chem. 2006;281:10825–38. doi: 10.1074/jbc.M512786200. [DOI] [PubMed] [Google Scholar]
- [33].Utton MA, Vandecandelaere A, Wagner U, et al. Phosphorylation of tau by glycogen synthase kinase 3β affects the ability of tau to promote microtubule self-assembly. Biochem J. 1997;323:741–7. doi: 10.1042/bj3230741. Pt 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Drewes G, Ebneth A, Preuss U, Mandelkow EM, Mandelkow E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell. 1997;89:297–308. doi: 10.1016/s0092-8674(00)80208-1. [DOI] [PubMed] [Google Scholar]
- [35].Dickey CA, Kamal A, Lundgren K, et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. 2007;117:648–58. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Nishimura I, Yang Y, Lu B. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell. 2004;116:671–82. doi: 10.1016/s0092-8674(04)00170-9. [DOI] [PubMed] [Google Scholar]
- [37].Thomas SN, Funk KE, Wan Y, et al. Dual modification of Alzheimer's disease PHF-tau protein by lysine methylation and ubiquitylation: a mass spectrometry approach. Acta Neuropathol (Berl) 2012;123:105–17. doi: 10.1007/s00401-011-0893-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cohen TJ, Guo JL, Hurtado DE, et al. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2011;2:252. doi: 10.1038/ncomms1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rizzu P, Van Swieten JC, Joosse M, et al. High prevalence. of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet. 1999;64:414–21. doi: 10.1086/302256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Momeni P, Pittman A, Lashley T, et al. Clinical and pathological features of an Alzheimer's disease patient with the MAPT βK280 mutation. Neurobiol Aging. 2009;30:388–93. doi: 10.1016/j.neurobiolaging.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Barghorn S, Zheng-Fischhofer Q, Ackmann M, et al. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry. 2000;39:11714–21. doi: 10.1021/bi000850r. [DOI] [PubMed] [Google Scholar]
- [42].Huang A, Stultz CM. The effect of a βK280 mutation on the un-folded state of a microtubule-binding repeat in Tau. PLoS Comp Biol. 2008;4:e1000155. doi: 10.1371/journal.pcbi.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Li W, Sperry JB, Crowe A, Trojanowski JQ, Smith AB, 3rd, Lee VM. Inhibition of tau fibrillization by oleocanthal via reaction with the amino groups of tau. J Neurochem. 2009;110:1339–51. doi: 10.1111/j.1471-4159.2009.06224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Usui K, Hulleman JD, Paulsson JF, Siegel SJ, Powers ET, Kelly JW. Site-specific modification of Alzheimer's peptides by cholesterol oxidation products enhances aggregation energetics and neurotoxicity. Proc Natl Acad Sci USA. 2009;106:18563–8. doi: 10.1073/pnas.0804758106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lazo ND, Grant MA, Condron MC, Rigby AC, Teplow DB. On the nucleation of amyloid β-protein monomer folding. Protein Sci. 2005;14:1581–96. doi: 10.1110/ps.041292205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tarus B, Thirumalai D, Straub JE. Structures and free-energy landscapes of the wild type and mutants of the Aβ21-30 peptide are determined by an interplay between intrapeptide electrostatic and hydrophobic interactions. J Mol Biol. 2008;379:815–29. doi: 10.1016/j.jmb.2008.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Murray MM, Krone MG, Bernstein SL, et al. Amyloid β-Protein: Experiment and Theory on the 21-30 Fragment. J Phys Chem B. 2009 doi: 10.1021/jp808384x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Fawzi NL, Phillips AH, Ruscio JZ, Doucleff M, Wemmer DE, Head-Gordon T. Structure and dynamics of the Aβ(21-30) peptide from the interplay of NMR experiments and molecular simulations. J Am Chem Soc. 2008;130:6145–58. doi: 10.1021/ja710366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cruz L, Urbanc B, Borreguero JM, Lazo ND, Teplow DB, Stanley HE. Solvent and mutation effects on the nucleation of amyloid β-protein folding. Proc Natl Acad Sci USA. 2005;102:18258–63. doi: 10.1073/pnas.0509276102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Chen W, Mousseau N, Derreumaux P. The conformations of the amyloid-β (21-30) fragment can be described by three families in solution. J Chem Phys. 2006;125 doi: 10.1063/1.2337628. 084911. [DOI] [PubMed] [Google Scholar]
- [51].Borreguero JM, Urbanc B, Lazo ND, Buldyrev SV, Teplow DB, Stanley HE. Folding events in the 21-30 region of amyloid β-protein (Aβ) studied in silico. Proc Natl Acad Sci USA. 2005;102:6015–20. doi: 10.1073/pnas.0502006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Baumketner A, Bernstein SL, Wyttenbach T, et al. Structure of the 21-30 fragment of amyloid β-protein. Protein Sci. 2006;15:1239–47. doi: 10.1110/ps.062076806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sciarretta KL, Gordon DJ, Petkova AT, Tycko R, Meredith SC. Aβ40-Lactam(D23/K28) models a conformation highly favorable for nucleation of amyloid. Biochemistry. 2005;44:6003–14. doi: 10.1021/bi0474867. [DOI] [PubMed] [Google Scholar]
- [54].Petkova AT, Ishii Y, Balbach JJ, et al. A structural model for Alzheimer's β-amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci USA. 2002;99:16742–7. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Petkova AT, Yau WM, Tycko R. Experimental constraints on quaternary structure in Alzheimer's β-amyloid fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer's β-amyloid fibrils. Science. 2005;307:262–5. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- [57].Lührs T, Ritter C, Adrian M, et al. 3D structure of Alzheimer's amyloid-β(1-42) fibrils. Proc Natl Acad Sci USA. 2005;102:17342–7. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].McLaurin J, Chakrabartty A. Characterization of the interactions of Alzheimer β-amyloid peptides with phospholipid membranes. Eur J Biochem. 1997;245:355–63. doi: 10.1111/j.1432-1033.1997.t01-2-00355.x. [DOI] [PubMed] [Google Scholar]
- [59].Waschuk SA, Elton EA, Darabie AA, Fraser PE, McLaurin J. Cellular membrane composition defines Aβ-lipid interactions. J Biol Chem. 2001;276:33561–8. doi: 10.1074/jbc.M103598200. [DOI] [PubMed] [Google Scholar]
- [60].Demeester N, Baier G, Enzinger C, et al. Apoptosis induced in neuronal cells by C-terminal amyloid β-fragments is correlated with their aggregation properties in phospholipid membranes. Mol Membr Biol. 2000;17:219–28. doi: 10.1080/09687680010015838. [DOI] [PubMed] [Google Scholar]
- [61].Kayed R, Sokolov Y, Edmonds B, et al. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein mis-folding diseases. J Biol Chem. 2004 doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- [62].Kagan BL, Azimov R, Azimova R. Amyloid peptide channels. J Membr Biol. 2004;202:1–10. doi: 10.1007/s00232-004-0709-4. [DOI] [PubMed] [Google Scholar]
- [63].Kawahara M, Kuroda Y, Arispe N, Rojas E. Alzheimer's β-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J Biol Chem. 2000;275:14077–83. doi: 10.1074/jbc.275.19.14077. [DOI] [PubMed] [Google Scholar]
- [64].Choo-Smith LP, Garzon-Rodriguez W, Glabe CG, Surewicz WK. Acceleration Of Amyloid Fibril Formation By Specific Binding Of Aβ-(1-40) Peptide to Ganglioside-Containing Membrane Vesicles. J Biol Chem. 1997;272:22987–90. doi: 10.1074/jbc.272.37.22987. [DOI] [PubMed] [Google Scholar]
- [65].Bokvist M, Lindstrom F, Watts A, Grobner G. Two types of Alzheimer's β-amyloid (1-40) peptide membrane interactions: aggregation preventing transmembrane anchoring versus accelerated surface fibril formation. J Mol Biol. 2004;335:1039–49. doi: 10.1016/j.jmb.2003.11.046. [DOI] [PubMed] [Google Scholar]
- [66].Wang CN, Chi CW, Lin YL, Chen CF, Shiao YJ. The neuroprotective effects of phytoestrogens on amyloid β protein-induced toxicity are mediated by abrogating the activation of caspase cascade in rat cortical neurons. J Biol Chem. 2001;276:5287–95. doi: 10.1074/jbc.M006406200. [DOI] [PubMed] [Google Scholar]
- [67].Chauhan A, Ray I, Chauhan VP. Interaction of amyloid β-protein with anionic phospholipids: possible involvement of Lys28 and C-terminus aliphatic amino acids. Neurochem Res. 2000;25:423–9. doi: 10.1023/a:1007509608440. [DOI] [PubMed] [Google Scholar]
- [68].McLaurin J, Yang D, Yip CM, Fraser PE. Review: modulating factors in amyloid-β fibril formation. J Struct Biol. 2000;130:259–70. doi: 10.1006/jsbi.2000.4289. [DOI] [PubMed] [Google Scholar]
- [69].Gebhardt M, Henkes LM, Tayefeh S, et al. Relevance of lysine snorkeling in the outer transmembrane domain of small viral potassium ion channels. Biochemistry. 2012;51:5571–9. doi: 10.1021/bi3006016. [DOI] [PubMed] [Google Scholar]
- [70].Yoshiike Y, Akagi T, Takashima A. Surface structure of amyloid-β fibrils contributes to cytotoxicity. Biochemistry. 2007;46:9805–12. doi: 10.1021/bi700455c. [DOI] [PubMed] [Google Scholar]
- [71].Hertel C, Terzi E, Hauser N, Jakob-Rotne R, Seelig J, Kemp JA. Inhibition of the electrostatic interaction between β-amyloid peptide and membranes prevents β-amyloid-induced toxicity. Proc Natl Acad Sci USA. 1997;94:9412–6. doi: 10.1073/pnas.94.17.9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Keshet B, Gray JJ, Good TA. Structurally distinct toxicity inhibitors bind at common loci on β-amyloid fibril. Protein Sci. 2010;19:2291–304. doi: 10.1002/pro.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].de Groot NS, Aviles FX, Vendrell J, Ventura S. Mutagenesis of the central hydrophobic cluster in Aβ42 Alzheimer's peptide. Side-chain properties correlate with aggregation propensities. FEBS J. 2006;273:658–68. doi: 10.1111/j.1742-4658.2005.05102.x. [DOI] [PubMed] [Google Scholar]
- [74].Esler WP, Stimson ER, Ghilardi JR, et al. Point substitution in the central hydrophobic cluster of a human β-amyloid congener disrupts peptide folding and abolishes plaque competence. Biochemistry. 1996;35:13914–21. doi: 10.1021/bi961302+. [DOI] [PubMed] [Google Scholar]
- [75].Yan Y, Liu J, McCallum SA, Yang D, Wang C. Methyl dynamics of the amyloid-β peptides Aβ40 and Aβ42. Biochem Biophys Res Commun. 2007;362:410–4. doi: 10.1016/j.bbrc.2007.07.198. [DOI] [PubMed] [Google Scholar]
- [76].Zhang SS, Casey N, Lee JP. Residual structure in the Alzheimer’s disease peptide Probing the origin of a central hydrophobic cluster. Fold Des. 1998;3:413–22. doi: 10.1016/S1359-0278(98)00054-6. [DOI] [PubMed] [Google Scholar]
- [77].Zhang S, Iwata K, Lachenmann MJ, et al. The Alzheimer's peptide Aβ adopts a collapsed coil structure in water. J Struct Biol. 2000;130:130–41. doi: 10.1006/jsbi.2000.4288. [DOI] [PubMed] [Google Scholar]
- [78].Chen Z, Krause G, Reif B. Structure and orientation of peptide inhibitors bound to β-amyloid fibrils. J Mol Biol. 2005;354:760–76. doi: 10.1016/j.jmb.2005.09.055. [DOI] [PubMed] [Google Scholar]
- [79].Sinha S, Lopes DH, Bitan G. A Key Role for Lysine Residues in Amyloid β-Protein Folding, Assembly, and Toxicity. ACS Chem Neurosci. 2012;3:473–81. doi: 10.1021/cn3000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Urbanc B, Cruz L, Yun S, et al. In silico study of amyloid β-protein folding and oligomerization. Proc Natl Acad Sci USA. 2004;101:17345–50. doi: 10.1073/pnas.0408153101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Yang M, Teplow DB. Amyloid β-protein monomer folding: free-energy surfaces reveal alloform-specific differences. J Mol Biol. 2008;384:450–64. doi: 10.1016/j.jmb.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Kirkitadze MD, Condron MM, Teplow DB. Identification and characterization of key kinetic intermediates in amyloid β-protein fibrillogenesis. J Mol Biol. 2001;312:1103–19. doi: 10.1006/jmbi.2001.4970. [DOI] [PubMed] [Google Scholar]
- [83].Ono K, Condron MM, Teplow DB. Structure-neurotoxicity relationships of amyloid β-protein oligomers. Proc Natl Acad Sci USA. 2009;106:14745–50. doi: 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Jonsson T, Atwal JK, Steinberg S, et al. A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature. 2012;488:96–9. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- [85].Kaden D, Harmeier A, Weise C, et al. Novel APP/Aβ mutation K16N produces highly toxic heteromeric Aβ oligomers. EMBO Mol Med. 2012;4:647–59. doi: 10.1002/emmm.201200239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Lopes DHJ, Attar A, Du Z, et al. The molecular tweezer CLR01 inhibits islet amyloid polypeptide assembly and toxicity via an unexpected mechanism: Submitted for publication [Google Scholar]
- [87].Fokkens M, Schrader T, Klärner FG. A molecular tweezer for lysine and arginine. J Am Chem Soc. 2005;127:14415–21. doi: 10.1021/ja052806a. [DOI] [PubMed] [Google Scholar]
- [88].Sinha S, Lopes DH, Du Z, et al. Lysine-specific molecular tweezers are broad-spectrum inhibitors of assembly and toxicity of amyloid proteins. J Am Chem Soc. 2011;133:16958–69. doi: 10.1021/ja206279b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Talbiersky P, Bastkowski F, Klärner FG, Schrader T. Molecular clip and tweezer introduce new mechanisms of enzyme inhibition. J Am Chem Soc. 2008;130:9824–8. doi: 10.1021/ja801441j. [DOI] [PubMed] [Google Scholar]
- [90].LeVine H., 3rd Quantification of β-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999;309:274–84. doi: 10.1016/s0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- [91].Kanapathipillai M, Ku SH, Girigoswami K, Park CB. Small stress molecules inhibit aggregation and neurotoxicity of prion peptide 106-126. Biochem Biophys Res Commun. 2008;365:808–13. doi: 10.1016/j.bbrc.2007.11.074. [DOI] [PubMed] [Google Scholar]
- [92].Wang SS, Wen WS. Examining the influence of ultraviolet C irradiation on recombinant human γD-crystallin. Mol Vis. 2010;16:2777–90. [PMC free article] [PubMed] [Google Scholar]
- [93].Prabhudesai S, Sinha S, Attar A, et al. A novel "Molecular Tweezer" inhibitor of α-synuclein neurotoxicity in vitro and in vivo. Neurotherapeutics. 2012;9:464–76. doi: 10.1007/s13311-012-0105-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Straub JE, Thirumalai D. Principles governing oligomer formation in amyloidogenic peptides. Curr Opin Struct Biol. 2010;20:187–95. doi: 10.1016/j.sbi.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Kayed R, Head E, Thompson JL, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–9. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- [96].Attar A, Ripoli C, Ricardi E, et al. Protection of primary neurons and mouse brain from Alzheimer’s pathology by molecular tweezers. Brain. 2012;135:3735–48. doi: 10.1093/brain/aws289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M. Alzheimer's disease: clinical trials and drug development. Lancet Neurol. 2010;9:702–16. doi: 10.1016/S1474-4422(10)70119-8. [DOI] [PubMed] [Google Scholar]
- [98].Amijee H, Scopes DI. The quest for small molecules as amyloid inhibiting therapies for Alzheimer's disease. J Alzheimers Dis. 2009;17:33–47. doi: 10.3233/JAD-2009-1044. [DOI] [PubMed] [Google Scholar]
- [99].Gervais F, Paquette J, Morissette C, et al. Targeting soluble Aβ peptide with Tramiprosate for the treatment of brain amyloidosis. Neurobiol Aging. 2007;28:537–47. doi: 10.1016/j.neurobiolaging.2006.02.015. [DOI] [PubMed] [Google Scholar]
- [100].McLaurin J, Kierstead ME, Brown ME, et al. Cyclohexanehexol inhibitors of Aβ aggregation prevent and reverse Alzheimer phenotype in a mouse model. Nat Med. 2006;12:801–8. doi: 10.1038/nm1423. [DOI] [PubMed] [Google Scholar]
- [101].Fenili D, Brown M, Rappaport R, McLaurin J. Properties of scylloinositol as a therapeutic treatment of AD-like pathology. J Mol Med. 2007;85:603–11. doi: 10.1007/s00109-007-0156-7. [DOI] [PubMed] [Google Scholar]
- [102].Shaw JE, Chio J, Dasgupta S, et al. Aβ(1-42) assembly in the presence of scyllo-inositol derivatives: identification of an oxime link-age as important for the development of assembly inhibitors. ACS Chem Neurosci. 2012;3:167–77. doi: 10.1021/cn2000926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Salloway S, Sperling R, Keren R, et al. A phase 2 randomized trial of ELND005, scyllo-inositol, in mild to moderate Alzheimer disease. Neurology. 2011;77:1253–62. doi: 10.1212/WNL.0b013e3182309fa5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Mandel SA, Amit T, Kalfon L, Reznichenko L, Weinreb O, Youdim MB. Cell signaling pathways and iron chelation in the neurorestorative activity of green tea polyphenols: special reference to epigallocatechin gallate (EGCG) J Alzheimers Dis. 2008;15:211–22. doi: 10.3233/jad-2008-15207. [DOI] [PubMed] [Google Scholar]
- [105].Ehrnhoefer DE, Bieschke J, Boeddrich A, et al. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol Biol. 2008;15:558–66. doi: 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- [106].Rezai-Zadeh K, Shytle D, Sun N, et al. Green tea epigallocatechin-3-gallate (EGCG) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in Alzheimer transgenic mice. J Neurosci. 2005;25:8807–14. doi: 10.1523/JNEUROSCI.1521-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Sinha S, Du Z, Maiti P, et al. Comparison of three amyloid assembly inhibitors: the sugar scyllo-inositol, the polyphenol epigallocatechin gallate, and the molecular tweezer CLR01. ACS Chem Neurosci. 2012;3:451–8. doi: 10.1021/cn200133x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Lopez del Amo JM, Fink U, Dasari M, et al. Structural properties of EGCG-induced, nontoxic Alzheimer's disease Aβ oligomers. J Mol Biol. 2012;421:517–24. doi: 10.1016/j.jmb.2012.01.013. [DOI] [PubMed] [Google Scholar]
- [109].DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–64. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- [110].Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–80. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- [111].Gama Sosa MA, De Gasperi R, Elder GA. Modeling human neurodegenerative diseases in transgenic systems. Hum Genet. 2012;131:535–63. doi: 10.1007/s00439-011-1119-1. [DOI] [PubMed] [Google Scholar]
- [112].Xia W. Exploring Alzheimer's disease in zebrafish. J Alzheimers Dis. 2010;20:981–90. doi: 10.3233/JAD-2010-1412. [DOI] [PubMed] [Google Scholar]
- [113].Paquet D, Schmid B, Haass C. Transgenic zebrafish as a novel animal model to study tauopathies and other neurodegenerative disorders in vivo. Neuro-degenerative diseases. 2010;7:99–102. doi: 10.1159/000285515. [DOI] [PubMed] [Google Scholar]
- [114].van Bebber F, Paquet D, Hruscha A, Schmid B, Haass C. Methylene blue fails to inhibit Tau and polyglutamine protein dependent toxicity in zebrafish. Neurobiol Dis. 2010;39:265–71. doi: 10.1016/j.nbd.2010.03.023. [DOI] [PubMed] [Google Scholar]
- [115].Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–5. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- [116].Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron. 2003;39:409–21. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- [117].Kitazawa M, Medeiros R, Laferla FM. Transgenic mouse models of Alzheimer disease: developing a better model as a tool for therapeutic interventions. Curr Pharm Des. 2012;18:1131–47. doi: 10.2174/138161212799315786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Lithner CU, Hedberg MM, Nordberg A. Transgenic mice as a model for Alzheimer's disease. Curr Alzheimer Res. 2011;8:818–31. doi: 10.2174/156720511798192736. [DOI] [PubMed] [Google Scholar]
- [119].Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic Interactions between Aβ, tau, and α-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci. 2010;30:7281–9. doi: 10.1523/JNEUROSCI.0490-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Clinton LK, Billings LM, Green KN, et al. Age-dependent sexual dimorphism in cognition and stress response in the 3xTg-AD mice. Neurobiol Dis. 2007;28:76–82. doi: 10.1016/j.nbd.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Aβ causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–88. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- [122].Medina DX, Caccamo A, Oddo S. Methylene blue reduces Aβ levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 2011;21:140–9. doi: 10.1111/j.1750-3639.2010.00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Caccamo A, Maldonado MA, Bokov AF, Majumder S, Oddo S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2010;107:22687–92. doi: 10.1073/pnas.1012851108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].McKee AC, Carreras I, Hossain L, et al. Ibuprofen reduces Aβ, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Res. 2008;1207:225–36. doi: 10.1016/j.brainres.2008.01.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Gulinello M, Gertner M, Mendoza G, et al. Validation of a 2-day water maze protocol in mice. Behav Brain Res. 2009;196:220–7. doi: 10.1016/j.bbr.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Halagappa VK, Guo Z, Pearson M, et al. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer's disease. Neurobiol Dis. 2007;26:212–20. doi: 10.1016/j.nbd.2006.12.019. [DOI] [PubMed] [Google Scholar]
- [127].Nelson RL, Guo Z, Halagappa VM, et al. Prophylactic treatment with paroxetine ameliorates behavioral deficits and retards the development of amyloid and tau pathologies in 3xTgAD mice. Exp Neurol. 2007;205:166–76. doi: 10.1016/j.expneurol.2007.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Hirata-Fukae C, Li HF, Hoe HS, et al. Females exhibit more extensive amyloid, but not tau, pathology in an Alzheimer transgenic model. Brain Res. 2008;1216:92–103. doi: 10.1016/j.brainres.2008.03.079. [DOI] [PubMed] [Google Scholar]
- [129].Hess EJ, Albers LJ, Le H, Creese I. Effects of chronic SCH23390 treatment on the biochemical and behavioral properties of D1 and D2 dopamine receptors: potentiated behavioral responses to a D2 dopamine agonist after selective D1 dopamine receptor upregulation. J Pharmacol Exp Ther. 1986;238:846–54. [PubMed] [Google Scholar]
- [130].Goedert M, Spillantini MG. A century of Alzheimer's disease. Science. 2006;314:777–81. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- [131].Venda LL, Cragg SJ, Buchman VL, Wade-Martins R. α-Synuclein and dopamine at the crossroads of Parkinson's disease. Trends Neurosci. 2010;33:559–68. doi: 10.1016/j.tins.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Clark A, Charge SB, Badman MK, MacArthur DA, de Koning EJ. Islet amyloid polypeptide: actions and role in the pathogenesis of diabetes. Biochem Soc Trans. 1996;24:594–9. doi: 10.1042/bst0240594. [DOI] [PubMed] [Google Scholar]
- [133].Melvin KE, Miller HH, Tashjian AH., Jr. Early diagnosis of medullary carcinoma of the thyroid gland by means of calcitonin assay. N Engl J Med. 1971;285:1115–20. doi: 10.1056/NEJM197111112852004. [DOI] [PubMed] [Google Scholar]
- [134].Endo JO, Rocken C, Lamb S, Harris RM, Bowen AR. Nodular amyloidosis in a diabetic patient with frequent hypoglycemia: sequelae of repeatedly injecting insulin without site rotation. J Am Acad Dermatol. 2010;63:e113–4. doi: 10.1016/j.jaad.2010.03.001. [DOI] [PubMed] [Google Scholar]
- [135].Floege J, Ehlerding G. β-2-microglobulin-associated amyloidosis. Nephron. 1996;72:9–26. doi: 10.1159/000188801. [DOI] [PubMed] [Google Scholar]
- [136].Joao Saraiva M, Mendes Sousa M, Cardoso I, Fernandes R. Familial amyloidotic polyneuropathy: protein aggregation in the peripheral nervous system. J Mol Neurosci. 2004;23:35–40. doi: 10.1385/jmn:23:1-2:035. [DOI] [PubMed] [Google Scholar]
- [137].Plante-Bordeneuve V, Said G. Transthyretin related familial amyloid polyneuropathy. Curr Opin Neurol. 2000;13:569–73. doi: 10.1097/00019052-200010000-00011. [DOI] [PubMed] [Google Scholar]
- [138].Vassallo N. Properties and pathogenicity of prion-derived peptides. Protein Peptide Lett. 2009;16:230–8. doi: 10.2174/092986609787601651. [DOI] [PubMed] [Google Scholar]