

Abstract
Mass spectrometry (MS) is a powerful tool to accurately identify and quantify histone post-translational modifications (PTMs). High-resolution mass analyzers have been regarded as essential for these PTM analyses because the mass accuracy afforded is sufficient to differentiate trimethylation versus acetylation (42.0470 and 42.0106 Da, respectively), whereas lower-resolution mass analyzers cannot. Noting this limitation, we sought to determine whether lower-resolution detectors are nonetheless adequate for histone PTM analysis by comparing the low-resolution LTQ Velos Pro with the high-resolution LTQ-Orbitrap Velos Pro. We first determined that the optimal scan mode on the LTQ Velos Pro is the Enhanced scan mode with respect to apparent resolution, number of MS and MS/MS scans per run, and reproducibility of label-free quantifications. We next compared the performance of the LTQ Velos Pro to the LTQ-Orbitrap Velos Pro using the same criteria for comparison, and we found that the main difference is that the LTQ-Orbitrap Velos Pro is able to resolve the difference between acetylation and trimethylation while the LTQ Velos Pro cannot. However, using heavy isotope labeled synthetic peptide standards and retention time information enables confident assignment of these modifications and comparable quantification between the instruments. Therefore, lower-resolution instruments can confidently be utilized for histone PTM analysis.
Keywords: mass spectrometry, histone, PTM, quantification, stable isotope, epigenetics, modification, proteomics, chromatin
Introduction
In eukaryotes, the basic repeating unit of chromatin is the nucleosome, consisting of 147 base pairs of DNA wrapped around an octamer of histone proteins. The octamer contains two copies of each of the core histones, H2A, H2B, H3, and H4.1 Histone proteins are dynamically post-translationally modified to regulate a wide array of nuclear processes, including transcription, DNA damage repair, and signaling pathways.2 Most of this post-translational modification (PTM) occurs on the N-terminal histone tails that protrude from the nucleosomal surface.3 Of the core histones, H3 and H4 undergo the most extensive modifications
Mass spectrometry (MS) has emerged as a powerful tool to characterize histone PTMs as it can accurately identify and quantify all histone PTM profiles in an unbiased manner.2,4 MS can also identify novel modifications as well as multiple modifications on a single peptide.5,6
Most histone PTM analysis by MS has been conducted on high-resolution mass analyzers, such as Orbitraps, because they provide sufficient mass accuracy to differentiate two very common, nearly isobaric PTMs: acetylation and trimethylation, 42.0106 and 42.0470 Da, respectively. For example, the ultrahigh-resolution Fourier transform ion cyclotron resonance mass spectrometer (FTICR–MS) has a resolving power (>106) that can differentiate peptides with almost identical mass (Δm/z < 0.001), and can therefore easily discriminate trimethylation and acetylation based on mass alone.8 Lower-resolution mass analyzers, such as linear ion traps, are unable to resolve this small mass difference.9
However, other techniques can be used to differentiate these modifications on lower-resolution mass analyzers, such as MS/MS spectra in combination with retention time information9−13 or synthetic peptide coelution information.14 The Hunt lab demonstrated that the H3 9–17 AA peptide bearing K14ac or K9me3 eluted 17 min apart based on a 240 min gradient (0 to 100% B), suggesting that relative retention time can be used to distinguish the modifications.15 Subsequently, the Freitas group utilized this shift in retention time to demonstrate that trimethylated and acetylated peptides can be distinguished on low-resolution mass spectrometers based on their relative retention times. They validated their peak assignments by running the same sample on a high-resolution mass spectrometer to resolve the mass difference on the MS1 level and also performed MS/MS for further validation.9
Heavy labeling methods have also been used to differentiate nearly isobaric PTMs. One way to accomplish this task is to use heavy isotope labeled methyl and/or acetyl donors so that the modifications are no longer isobaric.16−19 However, it is relatively time-consuming to wait for the light modifications to turn over. Another option is to spike in heavy labeled synthetic peptides, which will then coelute with the modified peptide of interest, but will be distinguishable based on its unique mass. Lee et al. used synthetic peptide coelution to determine that a potential new modification on a histone peptide (H2AT15ac) was falsely assigned.14
Neutral losses can also be used to lend confidence to identity assignments for isobaric peptides. Trimethylated peptides undergo signature neutral losses during collision induced dissociation (CID) fragmentation, and the resulting mass shifts observed in MS/MS spectra can be used to validate methylation sites.11,21 Fragmentation of acetylated peptides does not result in neutral losses but does produce immonium ions at m/z 143.1 which can be used to verify the presence of acetylated resides.11,13
Since lower-resolution instruments are less expensive, easier to maintain, and somewhat more ubiquitous compared to high-resolution instruments, we aimed to determine if lower-resolution mass analyzers are as capable of robust and accurate histone PTM identification and quantification as high-resolution mass analyzers using several of the above-described techniques. To this end, we compared the performance of a low-resolution linear ion trap (LTQ Velos Pro) and a high-resolution ion trap–Orbitrap hybrid instrument (LTQ-Orbitrap Velos Pro) in comprehensive PTM analysis of histones, one of the most extensively modified proteins in eurkaryotes. Most modifications have a large enough mass difference to be resolved on lower-resolution mass analyzers, and so one of the biggest challenges when using low-resolution detectors for histone PTM analysis is differentiating trimethylation from acetylation.
We first determined the optimal scan mode on the LTQ Velos Pro based on three criteria: (1) reproducibility of relative peptide abundance measurements, (2) resolution of peptides in higher charge states (e.g., +2 and +3 charges), and (3) the number of MS1 and MS2 scans obtained per run. From this analysis, we determined that Enhanced scan mode has the optimal trade-off between resolution and number of scans, while providing very consistent quantification. We then compared the performance of the LTQ Velos Pro on Enhanced scan mode to the Orbitrap Velos Pro using the same criteria listed above as well as mass accuracy. The Orbitrap Velos Pro was able to resolve the mass difference between acetylation and trimethylation, while the LTQ Velos Pro could not. However, these modifications could be accurately quantified on the LTQ Velos Pro using either retention time information or stable isotope-labeled synthetic peptide spike-in standards. As such, we conclude that while high-resolution mass analyzers are ideally suited for histone PTM analysis, low-resolution detectors are also adequate.
Experimental Section
Cell Culture/Harvesting/Nuclei Isolation/Histone Extraction/Separation
HeLa S3 cells were gown in suspension as described previously23 and harvested using standard protocols.24 Nuclei were isolated as previously described.24 Histones were extracted using a salt extraction followed by an acid extraction. Nuclei were resuspended in 0.4 M NaCl buffer containing 1 mM DTT, 0.3 mM AEBSF, and 10 mM sodium butyrate (10:1 buffer:pellet by volume) and were incubated at 4 °C with shaking for 30 min. The nuclei were pelleted at 3000g for 5 min at 4 °C, and the supernatant was removed by decanting. The pellet was resuspended in 2.5 M NaCl containing 1 mM DTT, 0.3 mM AEBSF, and 10 mM sodium butyrate (5:1 by volume). An equal volume of cold 0.4 N H2SO4 was added slowly, and the nuclei were incubated at 4 °C with shaking for 2 h. The nuclei were pelleted at 3400g for 5 min, and proteins were precipitated from the supernatant with TCA as previously described.24 When needed, offline reverse-phase high-performance liquid chromatography (RP-HPLC) was used to further purify histone proteins as described previously.24
Preparation of Histones for MS
Acid-extracted total histones or RP-HPLC-purified histones were chemically derivatized with propionic anhydride and digested using trypsin (Promega; substrate/protease ratio of 20:1) as described previously.24 Histone peptides were subsequently desalted for MS analysis using homemade C18 STAGE tips as described previously.24
Preparation of Synthetic Peptide Standards
The synthetic peptide library was created exactly as described by Lin et al.25 Briefly, 93 synthetic tryptic histone peptides containing heavy-labeled amino acids were synthesized containing the most common PTM profiles. The peptides were purified by RP-HPLC and combined to a final concentration of 0.27 pmol/μL/peptide in water. They were subsequently propionylated and desalted as described.25 The synthetic peptide library was added to a total acid-extracted histone sample in a ratio of 100 fmol of synthetic peptides:1 μg of histone.
Liquid Chromatography Mass Spectrometry
LTQ Scan Mode Comparison Studies
A fused silica microcapillary column (75 μm i.d.) was packed in-house with Reprosil-pur C18 resin (3 μm, Dr. Maisch GmbH). The column was fitted to a commercial fused silica emitter with a 10 μm tip (New Objective). Small aliquots (1.5 μg) of a single histone H4 sample were loaded onto the column by an Eksigent NanoLC AS-2 autosampler. Peptides were separated using an Eksigent NanoLC 2D Plus system HPLC across a 76 min multistep gradient: 2% buffer B for 1 min (A, 0.1% formic acid in water; B, 0.1% formic acid in acetonitrile), 2% to 30% B in 55 min, 30% to 98% B in 15 min, 98% B for 10 min, 98% B to 2% B in 30 s, 2% B for 9.5 min (equilibration) at a constant flow of 250 nL/min. The HPLC was coupled to a linear quadrupole ion trap (LTQ Velos Pro, Thermo Scientific) mass spectrometer operating in Zoom, Enhanced, Normal, or Turbo scan modes (see Table 1 for scan rate information). A full MS spectrum was obtained followed by 6 data-dependent MS/MS acquisitions for the top six most abundant ions using CID fragmentation (collision energy of 40, activation Q of 0.25, and activation time of 10 ms). Three technical replicates were obtained for each scan mode.
Table 1. Scan Rate Information for Linear Ion Trap Scan Modes.
| scan mode | scan rate (Da/s) | MS/MS per duty cycle | av MS1 per runa | av MS2 per runa | rel no. of scans per run |
|---|---|---|---|---|---|
| Zoom | 1,111 | 6 | 1,825.3 | 10,952 | 0.81 |
| Enhanced | 5,000 | 6 | 2,179.5 | 13,077 | 0.97 |
| Normal | 16,666 | 6 | 2,256.7 | 13,540 | 1.00 |
| Turbo | 125,000 | 6 | 2,373.3 | 14,240 | 1.05 |
Based on a 76 min gradient (2% B for 1 min; 2% to 30% B in 55 min; 30 to 98% B in 15 min; 98% B for 10 min; 98% to 2% B in 30 s; 2% B for 9.5 min) at 250 nL/min flow rate using an Eksigent NanoLC Ultra loading pump.
LTQ Velos Pro/LTQ-Orbitrap Velos Pro Comparison Studies
Aliquots (1.5 μg) of the same acid-extracted histone sample with synthetic peptides were run on two separate mass spectrometers: a linear ion trap (LTQ Velos Pro) and a hybrid linear ion trap–Orbitrap (Orbitrap Velos Pro). The same 75 μm i.d. column was used on both instruments (packed in-house with 3 μm C18 resin) fitted with a fused silica emitter with a 10 μm tip (New Objective). The sample was loaded using an Eksigent NanoLC 2D Plus system HPLC for the LTQ instrument while a Thermo Easy nLC 1000 was used to load the samples for the hybrid LTQ-Orbitrap instrument. The same multistep gradient was used for both instruments, consisting of 2% B for 1 min, 2 to 30% B in 40 min, 30% to 98% B in 15 min, 98% B for 10 min, with the exception that the Eksigent NanoLC gradient had an additional 10 min equilibration step at 2% B. Data acquisition for both instruments was broken into 3 segments, 14, 26, and 16 min long, respectively. In the first segment, a full MS was obtained followed by 9 data dependent MS/MS acquisitions of the top nine most abundant ions from the full MS. In the second segment, a full MS was obtained, followed by 5 targeted MS/MS acquisitions (m/z: 528.30, 570.84, 754.93, 761.94, and 768.95) and 5 data dependent MS/MS acquisitions of the top 5 most abundant ions from the full MS. In the third segment, a full MS was acquired followed by 10 data dependent MS/MS acquisitions of the top 10 most abundant ions. All full MS acquisitions for the Orbitrap Velos Pro were obtained in the Orbitrap in profile mode (resolution: 60,000 at m/z 400), while MS/MS acquisitions were obtained in the ion trap. The ion trap of both instruments was operated under Enhanced scan mode. CID fragmentation was used to fragment ions (collision energy of 40, activation Q of 0.25, activation time of 10 ms). Three technical replicates were obtained for each instrument.
Data Analysis
We used an in-house developed algorithm to identify and quantify PTMs for the LTQ-Orbitrap Velos Pro data, as described in Wu et al.26 The algorithm uses MS, MS/MS, and retention time information to accurately identify and provide relative abundances of PTM profiles for tryptic histone peptides. Relative abundance is obtained by integrating the single ion chromatogram of a particular modified peptide in all occupied charge states and dividing it by the total ion current for that peptide in all of its modified forms. The relative abundances of three technical replicates were averaged to obtain the final estimation of relative abundance. All LTQ data were manually quantified because they are not compatible with the aforementioned algorithm. Monoisotopic raw abundance of each peptide was manually extracted from Xcalibur Qual Browser, based on the area underneath extracted ion chromatograms at the full MS level. All detectable charge states were considered, typically [M + H]+, [M + 2H]+2, and [M + 3H]+3 as previously described.24
Results and Discussion
Comparison of LTQ Scan Modes
The LTQ Velos Pro mass spectrometer can operate in four different scan modes that have varying scan rates: Turbo, Normal, Enhanced, and Zoom (rates listed in Table 1). The scan rate affects several important properties, including reproducibility of peptide abundance measurements, resolution, and number of scans collected per run. The optimal scan mode will vary depending on the specific application, and, as such, we sought to determine which scan mode is optimal for histone PTM analysis based on their ability to resolve doubly and triply charged histone peptides while still allowing for the maximal number of scans and consistent quantification. To our knowledge, this study represents the first comprehensive analysis of different scan modes for histone PTM analysis on a low-resolution mass spectrometer.
Three technical replicates of a single histone H4 sample were analyzed for this experiment. The histone sample was purified from HeLa cells treated with butyrate, a histone deacetylase inhibitor, therefore containing more pronounced and combinatorial acetylation profiles. The sample was derivatized using our propionic anhydride methodology as it increases the hydrophobicity of the histone peptides, allowing for better retention on C18 resin, and also prevents trypsin cleavage after lysine. To reduce variance due to instrument setup, the sample replicates were run using identical chromatographic gradients (delivered by different HPLCs) and the same analytical column within 2 days.
Since small changes in PTM abundances can have large biological implications, it is important to run samples for PTM analysis on an instrument that generates highly reproducible results. Therefore, any observed differences can be attributed to biological phenomena rather than technical variances. As such, we investigated the reproducibility of measuring the relative abundances of modifications for a typical example peptide from histone H4 (amino acids 4–17; sequence: GKGGKGLGKGGAKR), for each scan mode. Relative abundances were obtained by integrating the single ion chromatogram (SIC) in MS1 spectra of a particularly modified peptide in all occupied charge states and normalizing the signal by dividing it by the total ion current of that peptide in all of its modified forms, as we and others have performed for many years now.5,28 The relative abundance values obtained on each scan mode were found to have a fairly small standard deviation, indicating that each mode produces highly reproducible abundance measurements (Figure 1). However, some scan modes produce different relative peptide abundance measurements compared to the other scan modes, with Turbo scan mode being the most variable. This variability indicates that Turbo scan mode may lead to inaccuracies in abundance measurements, possibly due to the presence of multiple species in a given m/z range that cannot be resolved. The tri- and tetra-acetylated peptides have larger standard deviations than the other modified forms of the peptide, which is more common for low-abundance peptides.
Figure 1.
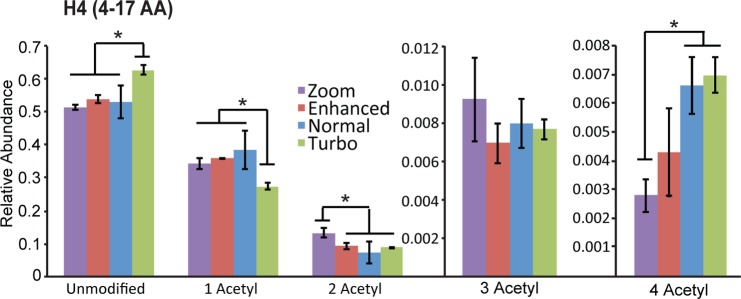
Reproducibility of relative peptide abundance measurements for H4(4–17AA) peptides on each LTQ Velos Pro scan mode. The relative abundance of each modified form of the peptide was calculated from three technical replicates of the same purified H4 sample. Error bars represent standard deviation from average relative peptide abundances. *P < 0.05.
Both resolution and scan rate are important factors affecting accuracy in peptide and PTM identification. Obtaining adequate resolution is vital for identifying and quantifying the PTM profiles present in a sample. If the resolution is too low, the charge state of the peptides cannot be determined, and the identity cannot be confidently assigned. Scan rate largely affects the resolution of an instrument. Ion trap mass analyzers scan and record the m/z range by increasing the radio frequency (rf) voltage applied to the electrode over time to eject peptide ions of increasing m/z from the trap to be detected. Fast scan rates result in poor resolution because the ions of a given m/z do not have enough time to fully eject before the rf increases, causing peak widening.29
The resolution obtained in the four different LTQ Velos Pro scan modes is shown in Figure 2A for an example peptide, diacetylated H4 4–17+2 ([M + 2H]2+ = 761.939). The fastest scan rate, Turbo, is completely unresolved, and the isotopes cannot be distinguished at all. However, the Zoom, Enhanced, and Normal scan modes have sufficient resolution to reliably assign charge states and are therefore amenable to this type of analysis.
Figure 2.

Resolution of diacetylated H4 4–17 on each LTQ Velos Pro scan mode. Mass spectra of (A) doubly charged ([M + 2H]2+ = 761.939) and (B) triply charged ([M + 3H]3+ = 508.295) peptides are displayed. The resolution (Δm/m) of the monoisotopic peak is indicated. (C) Resolution of the peptides from panels A and B as a function of scan rate.
As the charge state increases, it becomes more difficult to resolve the isotopes of a given peptide. Propionylated tryptic histone peptides generally occupy lower charge states (+1 to +3). Figure 2B displays the resolution of the triply charged example peptide ([M + 3H]3+ = 508.295). Zoom and Enhanced modes can resolve the charge state, while Normal and Turbo scan modes do not fully resolve the peptide. Therefore, Zoom and Enhanced scan modes are better options for histone PTM analysis on a low-resolution mass spectrometer. Figure 2C illustrates the relationship between scan rate and resolution for the example peptide in both charge states.
One important note is that scan rate affects the amount of data that can be collected during an MS analysis. Zoom has the best resolution, but is also the slowest scan rate and therefore does not collect as many full MS and MS/MS spectra as Enhanced scan mode (Table 1). Since some modified peptides have low abundances (e.g., K18me1, K23me1), it may be advantageous to sacrifice higher resolution for an increased number of scans. As such, we recommend using Enhanced mode for analyzing histone PTMs as it has sufficient resolution to resolve isotopes for double and triply charged peptides, allows reproducible quantification, and allows for almost 20% more MS2 scans compared to Zoom mode.
Comparison of LTQ and Orbitrap Performance
High-resolution mass analyzers have been at the forefront in histone PTM analysis, despite the fact that low-resolution mass analyzers are less expensive and generally more accessible. One of the main reasons for using a high-resolution detector is that it can resolve the mass difference between trimethylation and acetylation modifications (42.0470 and 42.0106 Da, respectively), which occur at high frequency on histone proteins. However, it is still possible to differentiate these PTMs on a low-resolution detector using a combination of retention time and MS/MS information.9 Furthermore, Krey et al. demonstrated that the low-resolution LTQ and LTQ Velos Pro mass spectrometers can quantify protein abundance as accurately as high-resolution Orbitrap mass spectrometers over a range of protein concentrations.30 As such, low-resolution mass analyzers could potentially be useful in analyzing histone PTM profiles, however to our knowledge no side-by-side comparison between low- and high-resolution mass analyzers has been published yet for comprehensive analysis of histones, which contain one of the most complicated PTM profiles of any eukaryotic protein. We therefore compared the performance of the LTQ and Orbitrap mass analyzers to determine if low-resolution mass analyzers are adequate for this type of complicated analysis.
To conduct this comparison, we used a single sample containing total acid-extracted histones from butyrate-treated HeLa cells, which were subsequently propionylated and digested with trypsin. Propionylated, heavy isotope-labeled synthetic peptides were spiked in the HeLa histone peptides to aid in identification of endogenous histone peptides during data analysis, as will be described in more detail later. We analyzed three technical replicates of this sample on each instrument and compared the resulting reproducibility of relative peptide abundance values, resolution, and mass accuracy. The LTQ Velos Pro was operated in Enhanced scan mode as this was determined to be optimal for histone PTM analysis. The runs were conducted within 3 days on the same analytical column, instrument method, and gradient to reduce differences due to instrument setup.
One drawback of using the Orbitrap Velos Pro mass analyzer is that the scan rate is inherently slower than that of the LTQ Velos Pro mass analyzer. Table 2 compares scan rate information for the LTQ Velos Pro and the Orbitrap Velos Pro. The scan numbers refer to the number of scans collected in the first three segments (see Experimental Section). The Orbitrap Velos Pro collected an average of 873 full MS scans and 8,784 MS/MS scans, while the LTQ Velos Pro collected 1,189 and 11,703, respectively (Table 2). Thus, while high-resolution detectors allow for better mass accuracy and consequently more facilitated data analysis, low-resolution detectors collect a larger amount of information even when operated on one of the slowest scan modes. This trade-off of reduced accuracy for increased MS and MS/MS scans could be desirable for the identification and quantification of rare or low-level PTMs.
Table 2. Scan Rate Information for Ion Trap and Orbitrap Mass Analyzersa.
| detector | av MS1 per run | av MS2 per run | rel no. of scans per run |
|---|---|---|---|
| ion trap (Enhanced) | 1,189 | 11,703 | 1.33 |
| Orbitrap | 873 | 8,784 | 1.00 |
Ion trap chromatography conditions are the same as listed in Table 1. The Orbitrap data is based on a 66 min gradient that omits the final equilibration step (2% B for 1 min; 2% to 30% B in 55 min; 30 to 98% B in 15 min; 98% B for 10 min) at 250 nL/min flow rate using a Thermo Easy NanoLC HPLC.
As mentioned previously, it is important that the instrument used for PTM analysis generate highly reproducible abundance measurements. As such, we compared the reproducibility of calculated relative peptide abundances obtained on the LTQ Velos Pro and the Orbitrap Velos Pro instruments based on three technical replicates each. We only analyzed H3 and H4 because they are the most modified histone proteins and are therefore the most challenging to analyze. The results, as shown in Figure 3, demonstrate that the standard deviation in relative abundance measurements is very small on both instruments, and results are very comparable between both instruments (P = 0.09; paired t test comparing standard deviations between the two instruments), indicating that both instruments are highly reproducible.
Figure 3.
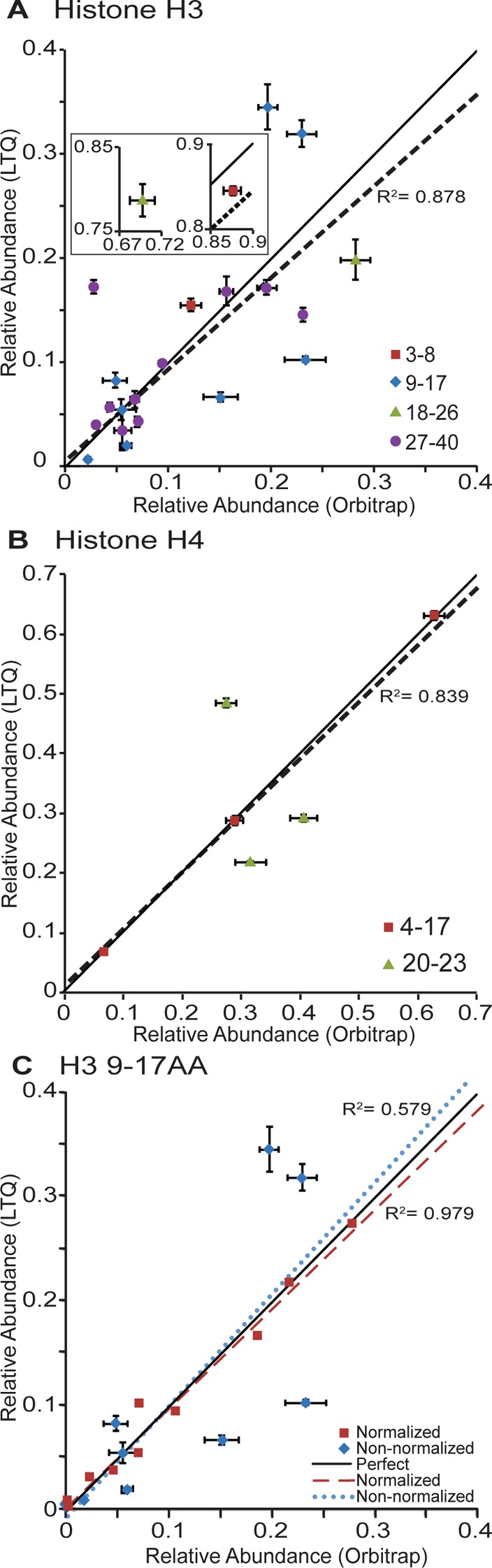
Reproducibility of relative peptide abundance measurements obtained on the LTQ Velos Pro and the Orbitrap Velos Pro. (A, B) Each point represents a particular modified form of either (A) an H3 peptide or (B) an H4 peptide, while the color of the point indicates the identity of the peptide. The black line indicates perfect correlation between relative peptide abundance values on the two instruments. The dashed line is a linear regression for all data points on the plot (Pearson, (A) R2 = 0.878, (B) R2 = 0.839; Spearman, (A) R2 = 0.853, (B) R2 = 0.883). The modified peptides shown in the figure include the following: (A) 3–8, unmodified, K4me1; 9–17, unmodified, K9me1, K9me1K14ac, K9me3K14ac, K9me2, K9me3, K9 or K14ac, K9me2K14ac; 18–26, unmodified, K18ac or K23ac; 27–40, unmodified, K36me1, K36me2, K27me1, K27me2, K27me3, K27me2K36me1, K27me1K36me2, K27me1K36me3, K27me1K36me1; (B) 4–17, unmodified, monoacetylated, and diacetylated; 20–23, unmodified, K20me1, K20me2. (C) Normalization of endogenous relative peptide abundances to synthetic peptide standards. Each point represents a particular modified form of the H3 9–17 peptide, listed in panel B. The blue points represent the data before normalization as shown in panel B, and the red points represent the data after normalization to the synthetic peptide standards. The solid black line represents perfect correlation. The dashed blue line is a linear regression fit of the data before normalization (R2 = 0.579), and the red dotted line is a linear regression fit of the data after normalization (R2 = 0.979).
Since the same sample was analyzed on both instruments, we would expect the calculated relative abundance measurements on each instrument to be similar. The results indicate that while in general this is true, some of these values differ between the two instruments (Figure 3A,B, as evidenced by deviation from the black line that indicates perfect correlation). However, a linear regression fit to all data points for H3 or H4 peptides demonstrates a high degree of correlation between the values obtained on each instrument (Pearson, R2 = 0.878 for H3 and R2 = 0.834 for H4; Spearman, R2 = 0.853 for H3 and R2 = 0.883 for H4), indicating that the instruments produce highly similar abundance measurements. It is also important to note that the measurements are relative, so an error in the abundance of one modified form of a peptide affects the relative abundance measurement of all other forms of that peptide due to the way the data is normalized.
Although the correlation of peptide abundance measurements between the two instruments is relatively high overall, some peptides do not correlate well. The peptide with the lowest correlation between instruments was the H3 9–17 peptide (Pearson: R2 = 0.579). Low correlation is likely a result of the major differences in how the instruments collect data. There were no common features between modifications or peptides that did not correlate well. To correct for differences in instrument data acquisition, the relative peptide abundance measurements of the endogenous peptides can be normalized to those of the synthetic peptide standards, which are present in equal concentrations. To normalize the data, a normalization factor was calculated for each form of the peptide by dividing the expected relative abundance measurements of the synthetic peptides by the observed relative abundance measurement of the synthetic peptides. The average raw abundance values for the endogenous peptides were then multiplied by the respective normalization factor, and the relative peptide abundances were calculated from these corrected values. Our group has previously used this method to correct for differences in ionization efficiencies between differently modified histone peptides during quantification.25 Figure 3C plots the relative abundances of the H3 9–17 peptides before and after normalization to the synthetic peptide standards. After normalization, the abundance measurements obtained on the two instruments are very highly correlated (Pearson: R2 = 0.979), indicating that the LTQ Velos Pro measures histone PTM abundances as accurately as the Orbitrap Velos Pro.
We calculated the average mass accuracy of the LTQ Velos Pro and the Orbitrap Velos Pro based on two example peptides, H3 18–26 and H4 4–17, in different modified forms to gauge what mass tolerance can be used to identify peptides. The mass accuracy for the LTQ Velos Pro ranges from 53.0 to 193 ppm, with an average of 129 ppm, and the mass accuracy for the Orbitrap Velos Pro ranges from 0.00 to 6.62 ppm, with an average of 2.01 ppm (Table 3). Using a mass tolerance of 10 ppm, the Orbitrap Velos Pro can differentiate trimethylation and acetylation, while the LTQ Velos Pro, using a mass tolerance of 150 ppm, cannot, as has been noted by others.9,11 This is demonstrated in Figure 4, which shows the chromatograms of different modified forms of the histone H3 9–17 peptide. For the Orbitrap Velos Pro, the trimethylated and acetylated peptide (row 4 and 5, respectively) have a large enough mass difference under the given mass tolerance to be unequivocally assigned to the single peak, while the LTQ Velos Pro cannot resolve these peptides. The resolution of the LTQ Velos Pro is high enough to differentiate all of the other common PTMs, such as mono- and dimethylation and multiply modified peptides such as K9me1K14ac (Figure 4, rows 1–3).
Table 3. Mass Accuracy Information of Example Peptides for Linear Ion Trap and Orbitrap.
| linear ion trap |
Orbitrap |
||||||
|---|---|---|---|---|---|---|---|
| peptide | modifications | obsd m/z | theor m/z | error Δppm | obsd m/z | theor m/z | error Δppm |
| H3 18–26 | none | 577.93 | 577.85 | 138 | 577.849 | 577.849 | 0.00 |
| K23me1 | 584.90 | 584.86 | 120 | 584.857 | 584.857 | 0.00 | |
| K18me1 | 584.93 | 584.86 | 120 | 584.857 | 584.857 | 0.00 | |
| K18ac or K23ac | 570.93 | 570.84 | 158 | 570.842 | 570.841 | 1.75 | |
| K18acK23ac | 563.93 | 563.83 | 177 | 563.832 | 563.833 | 1.18 | |
| H4 4–17 | none | 776.10 | 775.95 | 193 | 775.955 | 775.955 | 0.00 |
| 1 Ac | 769.02 | 768.95 | 91.0 | 768.948 | 768.947 | 1.30 | |
| 2 Ac | 762.02 | 761.94 | 105 | 761.934 | 761.939 | 6.56 | |
| 3 Ac | 754.97 | 754.93 | 53.0 | 754.926 | 754.931 | 6.62 | |
| 4 Ac | 748.02 | 747.92 | 134 | 747.924 | 747.922 | 2.67 | |
| average | 129 | average | 2.01 | ||||
Figure 4.
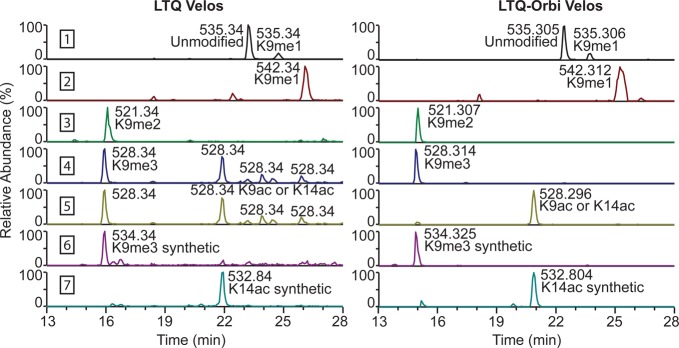
Chromatographic information for H3 9–17 obtained on LTQ Velos Pro or LTQ-Orbitrap Velos Pro. The mass tolerance used for selection is 150 ppm for the LTQ Velos Pro and 10 ppm for the LTQ-Orbitrap Velos Pro. In each row, labeled 1 to 7, the mass of the peptide bearing a specific modification was specified. Rows 1 to 5 represent endogenous peptides while rows 6 and 7 represent heavy-labeled synthetic peptides.
The acetylated and trimethylated peptides can still be distinguished on the LTQ, however, by using relative retention time information and high-resolution MS/MS data as has been noted by the Freitas group.9 However, this group did not derivatize their samples with propionic anhydride and did not use this technique to quantify peptides, and so we decided to apply this method to quantify histone PTMs for the instrument comparison. Synthetic peptides eliminate the need to perform high-resolution MS to validate peak assignments. The retention time of acetylated peptides is later than their corresponding methylated peptides because acetyl groups are more hydrophobic than trimethyl groups (Figure 4, row 4 versus 5).9 Since the peptides are propionylated in this study, the di- and trimethylated peptides are more hydrophilic than the unmodified peptide. Di- and trimethylation prevent propionylation so K9 is propionylated in the unmodified peptide and is not propionylated in the di- and trimethylated peptides. Since propionyl groups are more hydrophobic than di- and trimethylation, the unmodified peptide is more hydrophobic and elutes later (Figure 4, row 3 and 4 compared to 1).
Heavy-labeled synthetic peptides bearing a trimethyl or acetyl group at the residue of interest can also be used to validate peak assignments. Similar methods have been described by others, but there have been no studies using heavy-labeled synthetic peptides to differentiate nearly isobaric PTMs.16−19 The synthetic peptides have the same retention time as the unlabeled endogenous histone peptides containing the same modifications, as 13C or 15N isotopes do not influence retention on C18 columns, but impart a unique mass.31 Therefore, we can determine which ambiguous peak contains a trimethylated or acetylated peptide by seeing which coelutes with the synthetic peptide bearing the respective modification. In Figure 4, synthetic peptides were used to differentiate trimethylated and acetylated peptides. Rows 6 and 7 show the chromatograms for the H3 9–17 synthetic peptides bearing K9me3 or K14ac, respectively. The K14ac synthetic peptide (row 7) elutes under the K14ac peptide peak in the endogenous trace (row 5), and the K9me3 synthetic peptide (row 6) elutes under the corresponding K9me3 peak in the endogenous trace (row 4). Therefore, low-resolution detectors can be used to accurately quantify and identify histone PTM profiles when supplemented with synthetic peptides.
The results of the current study show for the first time that low-resolution mass analyzers, such as the LTQ Velos Pro, are adequate for comprehensive PTM analysis although the data analysis is more easily facilitated on high-resolution instruments. The reproducibility of obtained relative peptide abundances is equally high on the LTQ Velos Pro as the Orbitrap Velos Pro. Although the LTQ Velos Pro is not able to resolve the small mass difference between acetylation and trimethylation, other orthogonal lines of evidence can be used to accurately identify the peak containing each modified peptide. As such, high-resolution does not seem to be absolutely required for histone PTM analysis.
Acknowledgments
B.A.G. gratefully acknowledges funding from an NIH Innovator grant (DP2OD007447) from the Office of the Director, NIH Grant R01GM110174, and the National Science Foundation Early Faculty CAREER award.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Luger K.; Mäder A. W.; Richmond R. K.; Sargent D. F.; Richmond T. J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [DOI] [PubMed] [Google Scholar]
- Britton L.-M. P.; Gonzales-Cope M.; Zee B. M.; Garcia B. A. Breaking the histone code with quantitative mass spectrometry. Expert Rev. Proteomics 2011, 8, 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove M. S. Histone proteomics and the epigenetic regulation of nucleosome mobility. Expert Rev. Proteomics 2007, 4, 465–478. [DOI] [PubMed] [Google Scholar]
- Karch K. R.; Black B. E.; Garcia B. A. Identification and interrogation of combinatorial histone modifications. Front. Epigenomics Epigenet. 2013, 4, 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young N. L.; DiMaggio P. A.; Plazas-Mayorca M. D.; Baliban R. C.; Floudas C. A.; Garcia B. A. High Throughput Characterization of Combinatorial Histone Codes. Mol. Cell. Proteomics 2009, 8, 2266–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M. R.; Trelle M. B.; Thingholm T. E.; Jensen O. N. Analysis of posttranslational modifications of proteins by tandem mass spectrometry. BioTechniques 2006, 40, 790–798. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Eugeni E. E.; Parthun M. R.; Freitas M. A. Identification of novel histone post-translational modifications by peptide mass fingerprinting. Chromosoma 2003, 112, 77–86. [DOI] [PubMed] [Google Scholar]
- Yang L.; Tu S.; Ren C.; Bulloch E. M. M.; Liao C.-L.; Tsai M.-D.; Freitas M. A. Unambiguous determination of isobaric histone modifications by reversed-phase retention time and high-mass accuracy. Anal. Biochem. 2010, 396, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klammer A. A.; Yi X.; MacCoss M. J.; Noble W. S. Improving Tandem Mass Spectrum Identification Using Peptide Retention Time Prediction across Diverse Chromatography Conditions. Anal. Chem. 2007, 79, 6111–6118. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Yau P. M.; Chandrasekhar B.; New R.; Kondrat R.; Imai B. S.; Bradbury M. E. Differentiation between peptides containing acetylated or tri-methylated lysines by mass spectrometry: An application for determining lysine 9 acetylation and methylation of histone H3. PROTEOMICS 2004, 4, 1–10. [DOI] [PubMed] [Google Scholar]
- Falick A. M.; Hines W. M.; Medzihradszky K. F.; Baldwin M. A.; Gibson B. W. Low-mass ions produced from peptides by high-energy collision-induced dissociation in tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 1993, 4, 882–893. [DOI] [PubMed] [Google Scholar]
- Kim J. Y.; Kim K. W.; Kwon H. J.; Lee D. W.; Yoo J. S. Probing Lysine Acetylation with a Modification-Specific Marker Ion Using High-Performance Liquid Chromatography/Electrospray-Mass Spectrometry with Collision-Induced Dissociation. Anal. Chem. 2002, 74, 5443–5449. [DOI] [PubMed] [Google Scholar]
- Lee S.; Tan M.; Dai L.; Kwon O. K.; Yang J. S.; Zhao Y.; Chen Y. MS/MS of Synthetic Peptide Is Not Sufficient to Confirm New Types of Protein Modifications. J. Proteome Res. 2013, 12, 1007–1013. [DOI] [PubMed] [Google Scholar]
- Syka J. E. P.; Marto J. A.; Bai D. L.; Horning S.; Senko M. W.; Schwartz J. C.; Ueberheide B.; Garcia B.; Busby S.; Muratore T.; et al. Novel Linear Quadrupole Ion Trap/FT Mass Spectrometer: Performance Characterization and Use in the Comparative Analysis of Histone H3 Post-translational Modifications. J. Proteome Res. 2004, 3, 621–626. [DOI] [PubMed] [Google Scholar]
- Zee B. M.; Levin R. S.; Dimaggio P. A.; Garcia B. A. Global turnover of histone post-translational modifications and variants in human cells. Epigenet. Chromatin 2010, 3, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X.-J.; Zee B. M.; Garcia B. A. Heavy methyl-SILAC labeling coupled with liquid chromatography and high-resolution mass spectrometry to study the dynamics of site-specific histone methylation. Methods Mol. Biol. 2013, 977, 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. M.; Gafken P. R.; Zhang Z.; Gottschling D. E.; Smith J. B.; Smith D. L. Mass spectrometric quantification of acetylation at specific lysines within the amino-terminal tail of histone H4. Anal. Biochem. 2003, 316, 23–33. [DOI] [PubMed] [Google Scholar]
- Ren C.; Liu S.; Ghoshal K.; Hsu P.-H.; Jacob S. T.; Marcucci G.; Freitas M. A. Simultaneous metabolic labeling of cells with multiple amino acids: Localization and dynamics of histone acetylation and methylation. Proteomics: Clin. Appl. 2007, 1, 130–142. [DOI] [PubMed] [Google Scholar]
- Afjehi-Sadat L.; Garcia B. A. Comprehending dynamic protein methylation with mass spectrometry. Curr. Opin. Chem. Biol. 2013, 17, 12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C. E.; Kelleher N. L.; Mizzen C. A. Mass Spectrometric Characterization of Human Histone H3: A Bird’s Eye View. J. Proteome Res. 2006, 5, 240–247. [DOI] [PubMed] [Google Scholar]
- Lin S.; Garcia B. A. Examining Histone Posttranslational Modification Patterns by High Resolution Mass Spectrometry. Methods Enzymol. 2012, 512, 3–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.; Wein S.; Gonzales-Cope M.; Otte G. L.; Yuan Z.-F.; Afjehi-sadat L.; Maile T.; Berger S. L.; Rush J.; Lill J. R.; et al. Stable Isotope labeled histone peptide library for histone post-translational modification and variant quantification by mass spectrometry. Mol. Cell. Proteomics 2014, mcp.O113.036459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; DiMaggio P. A.; Perlman D. H.; Zakian V. A.; Garcia B. A. Novel Phosphorylation Sites in the S. cerevisiae Cdc13 Protein Reveal New Targets for Telomere Length Regulation. J. Proteome Res. 2013, 12, 316–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton L.-M. P.; Newhart A.; Bhanu N. V.; Sridharan R.; Gonzales-Cope M.; Plath K.; Janicki S. M.; Garcia B. A. Initial characterization of histone H3 serine 10 O-acetylation. Epigenetics 2013, 8, 1101–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong P. S. H.; Cooks R. G. Ion Trap Mass Spectrometry. Curr. Sep. 1997, 16, 85–92. [Google Scholar]
- Krey J. F.; Wilmarth P. A.; Shin J.-B.; Klimek J.; Sherman N. E.; Jeffery E. D.; Choi D.; David L. L.; Barr-Gillespie P. G. Accurate Label-Free Protein Quantitation with High- and Low-Resolution Mass Spectrometers. J. Proteome Res. 2014, 1321034–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi E. C.; Li X.; Cooke K.; Lee H.; Raught B.; Page A.; Aneliunas V.; Hieter P.; Goodlett D. R.; Aebersold R. Increased quantitative proteome coverage with 13C/12C-based, acid-cleavable isotope-coded affinity tag reagent and modified data acquisition scheme. Proteomics 2005, 5, 380–387. [DOI] [PubMed] [Google Scholar]