

Abstract

Protein biomarker discovery and validation in current omics era are vital for healthcare professionals to improve diagnosis, detect cancers at an early stage, identify the likelihood of cancer recurrence, stratify stages with differential survival outcomes, and monitor therapeutic responses. The success of such biomarkers would have a huge impact on how we improve the diagnosis and treatment of patients and alleviate the financial burden of healthcare systems. In the past, the genomics community (mostly through large-scale, deep genomic sequencing technologies) has been steadily improving our understanding of the molecular basis of disease, with a number of biomarker panels already authorized by the U.S. Food and Drug Administration (FDA) for clinical use (e.g., MammaPrint, two recently cleared devices using next-generation sequencing platforms to detect DNA changes in the cystic fibrosis transmembrane conductance regulator (CFTR) gene). Clinical proteomics, on the other hand, albeit its ability to delineate the functional units of a cell, more likely driving the phenotypic differences of a disease (i.e., proteins and protein–protein interaction networks and signaling pathways underlying the disease), “staggers” to make a significant impact with only an average ∼1.5 protein biomarkers per year approved by the FDA over the past 15–20 years. This statistic itself raises the concern that major roadblocks have been impeding an efficient transition of protein marker candidates in biomarker development despite major technological advances in proteomics in recent years.
Keywords: quantitative targeted proteomics, protein biomarker, multiplex proteomics assays, MRM−MS, immunoassays
The National Cancer Institute’s Clinical Proteomic Technologies for Cancer Initiative (NCI-CPTC) that launched the Clinical Proteomic Tumor Analysis Consortium – (CPTAC) has tackled many issues that plagued the clinical proteomics field to ensure a more streamlined transition from research to the clinic by (1) introducing “verification” between discovery and validation using analytically robust, multiplexed targeted proteomic technologies; (2) developing open-access regulatory science presubmission documents (mock 510k) with the FDA on how to appropriately validate multiplex proteomic assays and thus creating a framework to inform the proteomics community regarding analytical validation requirements sought by all international regulatory agencies; (3) generating and well-characterizing renewable affinity reagents for assay validation for the community; and (4) implementing data metrics for proteomic data sets to be more transparent and accessible by the public.
Overview
Advances and Challenges in Clinical Proteomics
In the postgenomic era, great technological and informatics progresses have been made to characterize thousands to tens of thousands of proteins in complex biological systems or clinical specimens by modern proteomic approaches, such as mass spectrometry (MS), immunohistochemistry, multiplexed immunological assays, and protein microarrays. Differential proteomic analyses of high-quality clinical human cancer biospecimens (e.g., tissues and biofluids) allow comparison between normal and diseased samples or between patient samples from different disease stages or diseases themselves. These preclinical studies have shown promise in identifying protein candidates responsible for or derived from the biology of disease by means of their differential protein expression level, altered post-translational modifications (PTMs), or isoform pattern. In particular, advances in protein science such as removal of high and moderate abundance proteins in blood and multidimensional chromatographic separation of complex mixtures of proteins,1,2 in addition to leaps in MS,3−5 have enabled the detection of low abundance proteins indicative of the disease in these biological and clinical samples. In reality, proteomics has now been widely applied across a variety of scientific disciplines, ranging from elucidating the molecular pathogenesis of disease, discovering and characterizing novel drug targets, to uncovering diagnostic, predictive, and prognostic biomarker candidates.6−8
Discovery proteomics often begins with differential (semiquantitative) analysis of the detectable proteome inventories between normal and disease states that are indicative of the disease pathophysiology. PTMs including phosphorylation, glycosylation, acetylation, and ubiquitination (to name but a few) have also stirred up much interest in this field for many years, as changes in phosphorylation, for instance, have been demonstrated to be involved in disease pathology and also been targeted for therapeutic development, such as kinase inhibitors. In addition to MS-based protein discovery studies, approaches such as nucleic-acid-programmable protein array9 and flow cytometry10 to quantify proteins have also furthered the understanding of the molecular mechanisms of diseases. In summary, clinical proteomics has improved significantly in the past couple of decades in the area of technology development/standardization and bioinformatics to enable confident identification of molecular disease signatures. As these instrumentation and technologies become more affordable with certain MS platforms already well-entrenched in research or clinical laboratories, clinical proteomics has a great future ahead for improving disease diagnosis, prognosis, and prediction of therapeutic outcome, as demonstrated by two recently FDA-cleared MS-based assays for microbe detection using Vitek MS Clinical Microbiology System (BioMérieux). However, its full potential has not been realized because the rate of FDA-approved protein blood biomarkers in the USA has been stagnant over the past 15–20 years for all diseases,11 in sharp contrast with over a thousand claimed biomarker candidates in the scientific literature for cancer alone. This discrepancy inevitably raises the question in the research community: What causes the congestion between discovery (using deep proteomic technologies) and validation (using more analytically stringent platforms)?
Over the years, through many discussions, the scientific community has identified several major barriers potentially attributable to this discrepancy, including: (1) a lack of high-quality, well-annotated biospecimens due to inappropriate biospecimen accrual, storage, and processing (preanalytical variables) critical for any studies; (2) measurement inconsistency and a lack of reproducibility within/across proteomic platforms (analytical); (3) difficulty in verifying biomarker candidates before large-scale clinical trials using immunoassays; (4) a lack of knowledge by the research community regarding the analytical evaluation criteria required by the regulatory agency for these distinct processes to progress through the approval pipeline; (5) a lack of publicly accessible, high-quality affinity reagents, reference materials, and data sets for data mining, hypotheses generation, and experimental validation prior to clinical validation; (6) a lack of data analysis and visualization tools for large-scale omics data sets (e.g., proteogenomics) in the research stage and of lockdown of software during subsequent validation studies; and (7) a lack of appropriate statistical and experimental study design, involving statistically rigorous clinical samples for early stage biomarker discovery and verification studies and biospecimen selection bias or collection without a specific clinical question in mind (Figure 1). If proteomics is to successfully penetrate clinical chemistry, diagnostics, and therapeutics, the implementation of standards and metrics will be required in the translational path to ensure that observed changes are reflective of true disease biology instead of preanalytical or workflow variability, followed by proper large-scale validation. As a result, an improved understanding of the challenges by translational researchers and strategies in each stage of a proteomics pipeline is fundamental for accelerating the pace of biomarker development and facilitating the implementation of novel clinical tests. Herein, this article focuses on several considerations of the regulatory review criteria necessary for analytical validation of MS-based assays.
Figure 1.

National Cancer Institute—Clinical Proteomic Technologies for Cancer Initiative’s restructured biomarker development pipeline. The incorporation of a verification step based on MRM–MS or iMRM–MS multiplexed assays into the current biomarker development pipeline between discovery and validation is beneficial for streamlining the transition of protein discovery candidates to large-scale clinical trials commonly validated by immunoassays.
Incorporating Verification into the Biomarker Pipeline Using Targeted Proteomics
Traditional discovery proteomics using modern MS workflows (as previously described) usually goes directly to a clinical validation stage where large cohorts of patients are evaluated using well-established, validated methodologies (such as enzyme-linked immunosorbent assays (ELISAs)) prior to clinical implementation. In addition, many published biomarker studies in the literature were often stuck in the discovery stage without any downstream confirmation at all, presumably one of the contributing factors for failure in biomarker development. This pipeline, however, is not the most efficient in transitioning candidates because immunoassays, the current gold standard in clinical validation for protein markers, are low-throughput, costly, and time-consuming to develop and implement. This is due to the fact that affinity reagents for novel protein candidates do not currently exist in many cases, especially for PTM markers, or are difficult to develop and that it is difficult to multiplex protein targets in complex matrices without having significant interferences and cross-reactivity. Additional problems include inaccurate measurements resulting from the antigen epitopes being masked by protein–protein interaction or other mechanisms or being lost by processing in some cases. These limitations of immunoassays have spurred the development of alternative approaches for more sensitive, accurate, and target-descriptive and cost-effective assays. For example, recent advances in peptide-based targeted MS proteomic technologies called multiple reaction monitoring MS (MRM–MS) and protein microarrays have enabled researchers to apply such “bridge technologies” for analytically validating and triaging candidates from discovery in tissues and biofluids.
To enable more efficient transition of candidates, the National Cancer Institute (NCI) launched the Clinical Proteomic Technologies for Cancer Initiative (NCI-CPTC) in 2006 (http://proteomics.cancer.gov) to address just such a gap in the conventional biomarker pipeline. Five years later, CPTC investigators have made significant progress in developing quantitative proteomic workflows to verify protein biomarker candidates by incorporating technology and data analysis standards, generating well-characterized reagents using standardized protocols for the research community (affinity and reference materials), designing statistically powered biomarker research studies and an open-access policy for proteomics data.12−15 This standardized multistage proteomic workflow with “verification” in the middle serves as decision points that enable researchers to accurately and reproducibly identify large numbers of proteins.8,16 In this pipeline, verification is to triage “biomarker candidates” from discovery proteomics using analytically robust, multiplexed targeted assays to confirm findings on statistically sufficient number of samples annotated with clinical data (ideally on an orthogonal cohort).16 If proteins successfully pass verification stage, they are considered verified biomarkers that will feed into clinical validation studies such as clinical trials. This streamlined pipeline is currently used in the Clinical Proteomic Tumor Analysis Consortium’s (CPTAC) proteomic studies of genomically characterized colorectal, breast, and ovarian tumors by the Cancer Genome Atlas to create a comprehensive human proteogenomic atlas for the research community to further the understanding of cancer on the molecular level.
Verification Using Multiple Reaction Monitoring Mass Spectrometry Methodologies in Preclinical Research
A main verification technology currently applied by the proteomics community (including CPTC researchers) is MRM–MS, a methodology commonly used in clinical reference laboratories to measure small molecules in plasma and urine, such as drug metabolites,17,18 and for newborn screening. It was only recently that MRM–MS was applied in preclinical research studies to measure large numbers of candidate peptides in patient samples.19,20 MRM–MS provides a cost-effective way (with an approximate lead development time of 3 months) to measure the abundance of a specific peptide candidate or a PTM site and examine the correlation between any changes in its abundance with the presence or stage of a given disease.
Briefly, MRM assay development starts with precursor (parent) peptide ions determined from empirical MS data, database mining of peptide libraries, or bioinformatic predictions to develop the optimal peptide candidates and three to five transitions to monitor. On a typical triple quadrupole mass spectrometer (TQMS), peptide quantitation as a surrogate for protein measurement is made by measuring the intensities of the product (daughter) ions during collision. To accurately quantitate these target peptides, chemically identical, heavy isotope-labeled peptides of known amounts serving as internal standards are often spiked into the biological matrices of interest. This sole attribute makes MRM–MS a desirable alternative to immunoassays, making it a complementary approach to existing methodologies. Additionally, beneficial attributes include a faster assay development timeline, lower cost, high multiplexing capability (up to 150plex), potential for higher specificity using MS detectors as the secondary antibodies, and unique advantages for PTMs, mutations, and splice variants of any given protein. Conversely, MRM–MS assays as compared with immunoassays have several disadvantages including (i) fairly complex selection processes for determining target precursor peptide ions as surrogates,21 for example, synthesizing recombinant or in vitro protein standards to aid in the selection of suitable surrogate peptides, although community-based databases such as SRMAtlas and CPTAC assay portal provide such information to streamline these selection processes for the research community; (ii) unit resolution of TQMS, making it difficult to resolve complex components in samples (i.e., causing interferences); (iii) analytical issues due to additional sample preparation, such as proteolysis; and (iv) insufficient sensitivity of direct MRM–MS assays (at best in the range of μg protein/mL), thus requiring an immunoenrichment step, or immuno-MRM assay (iMRM–MS), such as stable isotope standards and capture by anti-peptide antibodies (SISCAPA) or i-MALDI (immuno-matrix-assisted laser desorption ionization).22,23 These “enhanced” versions of MRM assays require the development of antipeptide antibodies to elevate sensitivity detection limits by as much as 103 to 104 fold over direct MRM–MS assays.22 As a result, iMRM–MS assays in their current stage of development are comparably sensitive to many ELISAs, with the advantages of lower sample consumption, ease of multiplexing, and ability to detect and avoid interferences, and are equally applicable to complex biofluid matrices (e.g., cells, plasma, serum, CSF, urine, and other biofluids). Immuno-MRM assays have demonstrated its ability to routinely measure proteins in the low ng protein/mL level from 30 μL of plasma, with the best antibodies achieving pg protein/mL plasma detection. The sensitivity of iMRM assays can be further increased by increasing the volume of input biospecimen to generate reproducible results, especially with automation. Taking advantage of the multiplexing capability of MRM–MS, next-generation proteomic assays can quantitate tens or hundreds of proteins involved in an entire biological pathway (the so-called thematic panels, such as DNA damage pathway or receptor tyrosine kinase pathway) or a set of protein biomarkers from samples of interest in just a single run. Immuno-MALDI, the complement of LC–iMRM–MS assays where peptides bound to the antibody beads are spotted directly on a MALDI plate and eluted from the beads by the MALDI matrix (e.g., α-cyano-4-hydroxycinnamic acid (CHCA, alpha-cyano), is easy to use and very high-throughput.24,25 Whereas the quantitation of the peptide with its peak height or using iMALDI is determined from an MS1 spectrum, peptide identities are confirmed with MS/MS or in the “MRM mode” on an MALDI-MS/MS instrument. As compared with ELISA that measures intact proteins, analytical validation processes for MRM–MS and iMRM–MS need to be better established, especially when previous studies have demonstrated that values of protein concentrations measured by ELISA and MRM–MS methodologies differed. For example, an MRM–MS method provided concentration values for C-reactive protein in human plasma approximately ten times the concentration measured by the ELISA method.26 Such differences are perhaps not surprising because differences between different ELISA instruments/platforms using the same methodology have been observed for protein analytes. The comparison and calibration of MRM–MS assays to ELISAs for protein analytes could be made by establishing new reference ranges for the same analytes in the same matrices from a sufficient number of patient samples under study in the preclinical verification stage and subsequently for a large-scale clinical validation study as required by the FDA to demonstrate the assay’s analytical validity.
To implement universally accepted performance metrics to assist the proteomics community with analytical requirement for different tiers of proteomic assays, NCI-CPTC recently organized a workshop with representatives from multiple communities to develop best practice (fit-for-purpose) guideline documents for what information must be provided to journals to qualify/analytically validate targeted assays in clinical proteomics.27 The CPTAC Assay Portal that serves as a public repository of well-characterized MS-based targeted proteomic assays was released (http://assays.cancer.gov) to complement the outputs/metrics from this workshop.
Understanding Regulatory Requirements for MRM–MS Assay Validation for the Clinic
In general, The Centers for Medicare & Medicaid Services (CMS) through Clinical Laboratory Improvement Amendments (CLIA) established in 1988 regulates clinical laboratories by implementing quality-testing standards to ensure the accuracy, reliability, and timeliness of patient test results (http://www.cms.hhs.gov/clia). The Food and Drug Administration (FDA) regulates commercially marketed in vitro diagnostic assays (IVDs). In fact, categorization under CLIA regulations based on their technical complexity and requirements for lab personnel training is the responsibility of the FDA. CLIA categories encompass waived tests, tests of moderate complexity, and tests of high complexity. Device authorization by the US FDA depends on the ability of the assay sponsor to provide analytical and clinical data demonstrating that the device performance meets claimed intended use based on its risk (Class I, Class II, and Class III).28 De novo mechanism is currently available to expedite the review process. A premarket submission document required by the regulatory agency includes (1) a device’s intended use and (2) a description of the device covering the instrument and all components, requisite standards and reagents, and analytical and clinical studies that evaluate the performance of the device for its intended use. For novel markers, both analytical detection of any analyte and the significance of the measurement for clinical management of patients need to be demonstrated. The analytical performance of a device (assay) should be described in terms of accuracy, precision, and performance around the cutoff points, specificity, sensitivity, and linearity, limit of detection, and limit of quantitation. Furthermore, appropriate internal/external controls and calibrators used in the assay need to be provided in any assay submission. The clinical performance of an assay can be demonstrated through either clinical data or credible published data supporting the intended clinical use in certain cases.29
Instrumentation Requirements
Historically, MS platforms have been classified as Class I low-risk platforms, requiring only general controls and exempt from 510(k) processes, unless their intended use is for the diagnosis, monitoring, and screening of neoplastic diseases, cardiovascular diseases, and diabetes. Despite this, MS instruments under any 510(k) exemption are still required to follow standard GMP guidelines (http://www.fda.gov/medicaldevices/deviceregulationandguidance/postmarketrequirements/qualitysystemsregulations/ucm230127.htm) in addition to being registered and listed. Recently, MS manufacturers have begun to take the steps to register their instruments. However, the FDA view of regulations differentiates between old-generation MS instrumentation under FDA’s exempt regulation and more complex modern MS instrumentation. This is clearly evidenced by the most recent FDA public workshop discussing the role of proteomics in the clinic with focus on MS (Proteomics in the Clinic on June 13, 2014; http://www.fda.gov/MedicalDevices/NewsEvents/WorkshopsConferences/ucm392858.htm). Specifically, instrumentation (LC, MS, and all components in between) used to run a multiplex assay is defined by the classification (risk) of that assay submitted to the FDA for that particular intended use. For instance, a Class III high-risk assay using MS would involve the evaluation of that particular MS instrument as a part of a Class III assay for that intended use. Consequently, it is critical that the research and clinical chemistry communities understand these regulatory frameworks. Navigating any regulatory process is, however, a huge challenge and is further intensified when proteomic technologies brought under review as validated biomarker discovery platforms are relatively recent considerations for the FDA and are constantly evolving, similar to the genomics world. Fortunately, early interactions between assay submitter with the FDA are highly encouraged by the regulatory agency to help the sponsor better understand all FDA requirements, regulations, and guidance documents while allowing the FDA reviewers to familiarize themselves with these emerging technologies.
Currently, multiplexed MRM–MS is defined by the FDA as a series of simultaneous MS measurements concerning one or more peptide transitions (as representative of the peptides and by inference as protein surrogates) via a common process of sample preparation, measurement (i.e., a single high-performance liquid chromatography injection), and data interpretation. As previously discussed, LC–MS instrument design from different manufacturers and interfaced HPLC systems could be vastly different, for example, microflow versus nanoflow, orthogonal electrospray versus direct electrospray). This could potentially cause nonequivalent analytical performance of an assay on different instruments. To address this, the NCI-CPTC network conducted a multicenter study that demonstrated the reproducibility of direct MRM–MS assays on instrument platforms and across different laboratories.30 Ultimately, the highest coefficient of variation was ≤22% using a single transition of MRM for all peptides except for one, even in the most complex scenario where each center had to prepare their own samples including trypsin digestion at each individual lab sites. This result for research-grade MRM assays is very encouraging and has built foundations for future improvements in analytical reproducibility using standardized protocols and reference materials and may further be improved by incorporating more streamlined sample preparation procedures. While CPTAC’s multicenter study to measure seven proteins in plasma was not intended for clinical use, we envisioned that automation combined with user-friendly software should reduce complex workflows, analytical variability between instrumentation platforms and laboratories, and current requirement for specialists to perform these assays.
To obtain regulatory authorization, one has to demonstrate equivalent analytical performance on different MS instrument platforms, after which independent regulatory evaluations of the same analyte(s) on other instruments may become simplified or unnecessary.29,31 For example, providing data on a specific MS platform in conjunction with any other instrumentation as a part of the assay system (e.g., LC, etc.) could be a part of the initial submission to the FDA for a diagnostic assay (with an intended use). If performance is deemed adequate, an approval of an assay is granted first only on that specific instrument used in evaluating performance. This establishes a device predicate, which can be followed by subsequent submissions with any changes to the predicate, for example, adding another instrument platform to streamline the complicated path to regulatory authorization. For MRM technologies, one can envision that regulatory authorization can be obtained on the most commonly used TQMS (a predicate) to measure one single protein analyte with a low-risk intended use (preferably cleared on an immunoassay platform previously). This clearance would make this TQMS instrument manufactured under quality systems available for clinical laboratory testing, followed by the addition of other TQMS platforms for the same intended use. Regulatory evaluation of instrument platforms should encompass all components and accessories (e.g., nanoflow columns, trap columns, silica tubings, electrospray source, and predictive software if needed). Even the slightest changes to a predicate would have to be re-examined by the assay sponsor prior to submissions to ensure the safety and effectiveness of the modified platform. Upon accomplishing that, other analytes or kits can be cleared on the same platform by providing assay-specific components of the instrumentation without the requirement for additional information specific for the platform.31 However, each analyte in the assay panel has to be individually validated.
Software Considerations: In Vitro Diagnostic Multivariate Index Assay
An in vitro diagnostic multivariate index assay (IVDMIA) software combines the values of multiple variables from multiple measurements using an interpretation function to generate a single, patient-specific result (e.g., a classifier, index or score) whose derivation is nontransparent by end users. Therefore, it becomes very complicated for individual users to validate such scores. The FDA currently has a guidance document to help assay sponsors understand the regulatory framework for an IVDMIA submission (http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm071455.pdf). Furthermore, the research community has assessed current practices on classifiers and composite scores using DNA microarray-based predictive models,32 which could be extrapolated to proteomics. Indeed, we have some success stories in the development and implementation of IVDMIA using proteomic results recently. OVA1 test (Vermillion) is one example of a FDA-cleared IVDMIA using individually measured values of five proteins in blood to derive a patient-specific score. The intended use for this IVDMIA is to help a physician evaluate the likelihood that an ovarian adnexal mass is malignant or benign prior to a planned surgery. The Risk of Ovarian Malignancy Algorithm (ROMA) developed by Fujirebio Diagnostics, another FDA-cleared qualitative serum test combining the results of HE4, CA125, and menopausal status into a single numerical score, is intended to aid in the assessment of whether a premenopausal or postmenopausal woman with an ovarian adnexal mass is at high or low likelihood of finding ovarian cancer malignancy upon surgery. There are authorized IVDMIAs using genomic results as well, such as AlloMap Molecular Expression Testing using quantitative RT-PCR to assess the gene expression profile of RNA isolated from peripheral blood mononuclear cells, which aids in the identification of heart transplant recipients with stable allograft function who have a low probability of moderate/severe acute cellular rejection (ACR) at the time of testing in combination with standard clinical assessment.
What Can Be Learned from the NCI-CPTC’s Mock 510(k) Assay Submission?
With the ever evolving nature of LC–MS instrument platforms, establishing a standardized consensus list of evaluation criteria for complex proteomic tests should help and provide accurate test results for clinical use. To this regard, the NCI-CPTC decided to go beyond the interlaboratory study and collaborate with the FDA and other stakeholders at a joint workshop to discuss analytical evaluation requirements to turn research-grade assays into clinical grade (Figure 2). Consequently, a mock 510(k) premarket submission with mock up data33 was developed by CPTAC investigators to educate the research community on requisite FDA regulatory processes for assay authorization using iMRM–MS. This enabled the publication of otherwise proprietary information involved in assay submission with FDA review comments, serving as a first-of-its-kind regulatory cliffs notes for clinical proteomics community. This multiplex iMRM–MS assay (PepCa10) measures a total of 10 tryptic peptides from 5 plasma proteins relevant to cancer, yielding a single qualitative result intended to determine whether a female patient with Breast Imaging-Reporting And Data System (BI-RADS) category 4 requires subsequent breast biopsy.33 In this assay, researchers specifically addressed the need to include controls (e.g., concatamers) for the assessment of variability of tryptic digestion of proteins in different samples from published literature34 and for evaluating analytical recovery of immunoaffinity-enriched peptides during sample preparation before it can be considered for use in the clinic.35 A concatamer, a heavy-isotope-labeled, in vitro synthetic protein standard with repeated copies of amino acid sequences for monitoring peptide targets used in this submission, could be used as an internal control to assess trypsin digestion variability and efficiency. Alternatively, a heavy-isotope-labeled intact recombinant protein standard resembling the entire amino acid sequence of the measured protein target (not presented in the mock submission) can be used to gauge analytical variability. Subsequently, each of the 10 peptide analytes in PepCa10 was measured by a TQMS via a ratio in which the peak area of the analyte peptide (unlabeled) was divided by that of its respective internal standard peptide (heavy-isotope-labeled) added immediately prior to peptide quantitation in the MS (providing a standard against which to measure analyte recovery). If there are any changes in the relative amount of the two peptides for a given protein in a cancer patient plasma compared with normal patient plasma, it could indicate the presence of diagnostic alterations in the parent protein or suggest the effect of single nucleotide polymorphisms (SNPs) or PTMs present, and one needs to understand why.
Figure 2.
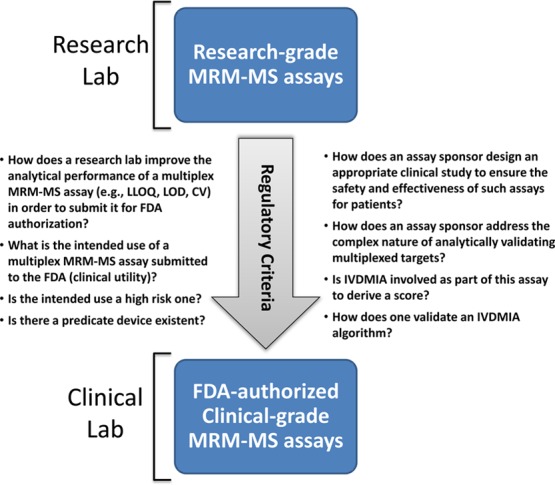
Role of regulatory framework in analytically validating multiplex MS-based assays. The NCI-CPTC’s mock 510(k) presubmission documents publicly accessible by the research community presents a regulatory framework for understanding the evaluation criteria required by the FDA to successfully move a research-grade MRM–MS (or iMRM–MS) assay into the clinic.
For a panel of multiple protein analytes measured by their proteotypic peptides, the FDA requires that all analytes in the panel meet analytical performance criteria as previously illustrated, especially their performance data around the cutoff points. In particular, this type of assays should address cross-reactivity or interference of analytes within and outside the assay panel in a similar way to immunoassays. Furthermore, the FDA requires the demonstration of the “overall measured protein concentration” from representative proteotypic peptides from the same parent protein to be reproducible in multiple measurements. If one of the peptides has consistently been shown as an outlier during replicate measurements, the assay sponsor needs to fully understand why this has occurred (e.g., an outlier could conceivably stem from PTMs, interferences from other proteins, and SNPs), what effect this outliner would have on the entire assay, and whether this outlier would be readily identified and mitigated by the assay quality control system.
Finally, manufacturing LC–MS instruments under GMP guidelines as a quality system should help reduce operator-specific bias, requirements for highly specialized expertise and analytical variability. If and when interpretive IVDMIA software is incorporated at the backend of analytical measurements, as presented in this mock submission to obtain a patient-specific score, the FDA generally requires that algorithms used for data and interpretation of final results be predefined and locked down before analyzing clinical study data. Any alteration of the algorithm to better fit the data poststudy is usually deemed inappropriate.
Present and Future of MS-Based Clinical Proteomics
Although there are currently no FDA-authorized direct MRM or iMRM–MS-based proteomic assays available, clinical proteomics has made headway in the regulatory arena. The first MALDI-TOF-based MS system (VITEK MS) has been cleared by the FDA for rapid identification of disease-causing bacterial and yeast infection and shows great promise of MS-based clinical proteomics applications. Using one device, the assay submitted by bioMérieux is able to identify almost 200 different microorganisms (including the timely identification of pathogenic microorganisms) even though the device is not based on an MRM-specific MS platform and not all of the analytes are necessarily proteins. To gain FDA clearance, bioMérieux conducted a multicenter study consisting of 7068 clinical isolates on their VITEK MALDI MS platform, after which performance accuracy was compared with the gold standard 16S ribosomal RNA gene sequencing for several categories of pathogens including Gram positive aerobes, fastidious Gram negative bacteria, and various yeast. It was demonstrated that the overall accuracy of VITEK MS for these organisms was 93.6% compared with nucleic acid sequencing (a predicate) clearly allowing efficacious, improved clinical decision regarding those specific infectious diseases. In addition, a peptide-based iMRM–MS assay for thyroglobulin36 developed by ARUP Laboratories and Quest Diagnostics could potentially overcome existing interference problems in current immunoassays from circulating autoantibodies in 20% of the patient population. Lab-based iMRM–MS assays have been developed for similar rationales to measure parathyroid hormone in blood37 and total pepsin/pepsinogen in saliva.38 We firmly believe that such progress in clinical proteomics will pave the way for greater success by accumulating and sharing knowledge and experience in understanding validation criteria for the regulatory authorization of multiplex MS-based assays.
Commonly Referenced Clinical Laboratory Standards Institutes’ Documents for Assay Validation
Clinical Laboratory Standards Institutes (CLSI) (http://www.clsi.org) is an organization that intends to develop global consensus standards and guidelines for healthcare testing. CLSI documents provide helpful information to the assay sponsors and the regulatory agency for preparing and reviewing premarket submissions. Because of the rigorous nature of CLSI document review prior to publication, these documents have been approved by consensus of many stakeholders in particular areas, followed by public review procedures for new and revised standards documents, for example, relevant to MS is C57 (Draft 2)—Mass Spectrometry for Androgen and Estrogen Measurements in Serum (http://clsi.org/standards/documents-for-public-review/). The FDA can either fully or partially recognize CLSI documents as standards, and compliance may be accepted as evidence of fulfillment of certain FDA analytical requirements. A commonly referenced document, EP-17A, Vol. 24, No. 34, Protocols for Determination of Limits of Detection and Limits of Quantitation; Approved Guideline, for example, is often cited in assay submissions for analytical performance of a test. While CLSI documents on multiplex MRM–MS-based proteomic assays currently do not exist, general guidance could be extrapolated from the nucleic-acid-based multiplex world, for example, MM-17A (Verification and Validation of Multiplex Nucleic Acid Assays; Approved Guideline), and from MS, for example, NBS04-A (Newborn Screening by Tandem Mass Spectrometry; Approved Guideline) and C50-A (Mass Spectrometry in the Clinical Laboratory: General Principles and Guidance; Approved Guideline).
Conclusions
The National Cancer Institute’s Clinical Proteomic Technologies for Cancer initiative (NCI-CPTC) and other international efforts (e.g., HUPO’s Proteomics Standards Initiative) have helped to bridge the gap between biomarker discovery and large-scale clinical validation by introducing “verification” using multiplex-targeted proteomic technologies. Improved understanding of regulatory framework by the clinical proteomics community of the analytical review requirements of a multiplex protein assay by the FDA for its intended use is fundamentally important in the process of successfully validating marker candidates in every stage of a biomarker pipeline to reach its clinical utility.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Whiteaker J. R.; Zhang H.; Eng J. K.; Fang R.; Piening B. D.; Feng L. C.; et al. Head-to-head comparison of serum fractionation techniques. J. Proteome Res. 2007, 62828–836. [DOI] [PubMed] [Google Scholar]
- Ernoult E.; Bourreau A.; Gamelin E.; Guette C. A proteomic approach for plasma biomarker discovery with iTRAQ labeling and OFFGEL fractionation. J. Biomed. Biotechnol. 2010, 2010, 927917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.; Whiteaker J. R.; Paulovich A. G. The evolving role of mass spectrometry in cancer biomarker discovery. Cancer Biol. Ther. 2009, 8121083–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener M. C.; Sachs J. R.; Deyanova E. G.; Yates N. A. Differential mass spectrometry: a label-free LC-MS method for finding significant differences in complex peptide and protein mixtures. Anal. Chem. 2004, 76206085–6096. [DOI] [PubMed] [Google Scholar]
- Geiger T.; Cox J.; Ostasiewicz P.; Wisniewski J. R.; Mann M. Super-SILAC mix for quantitative proteomics of human tumor tissue. Nat. Methods 2010, 75383–385. [DOI] [PubMed] [Google Scholar]
- García-Foncillas J.; Bandrés E.; Zárate R.; Remírez N. Proteomic analysis in cancer research: potential application in clinical use. Clin. Transl. Oncol. 2006, 84250–261. [DOI] [PubMed] [Google Scholar]
- An H. J.; Lebrilla C. B. A glycomics approach to the discovery of potential cancer biomarkers. Methods Mol. Biol. 2010, 600, 199–213. [DOI] [PubMed] [Google Scholar]
- Rifai N.; Gillette M. A.; Carr S. A. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat. Biotechnol. 2006, 24, 971–983. [DOI] [PubMed] [Google Scholar]
- Anderson K. S.; Sibani S.; Wallstrom G.; Qiu J.; Mendoza E. A.; Raphael J.; et al. Protein microarray signature of autoantibody biomarkers for the early detection of breast cancer. J. Proteome Res. 2011, 10185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishhan V. V.; Khan I. H.; Luciw P. A. Multiplexed microbead immunoassays by flow cytometry for molecular profiling: Basic concepts and proteomics applications. Crit. Rev. Biotechnol. 2009, 29129–43. [DOI] [PubMed] [Google Scholar]
- Anderson N. L. The clinical plasma proteome: a survey of clinical assays for proteins in plasma and serum. Clin. Chem. 2010, 562177–185. [DOI] [PubMed] [Google Scholar]
- Paulovich A. G.; Billheimer D.; Ham A. J.; Vega-Montoto L.; Rudnick P. A.; Tabb D. L.; et al. Interlaboratory study characterizing a yeast performance standard for benchmarking LC-MS platform performance. Mol. Cell. Proteomics 2010, 92242–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez H.; Snyder M.; Uhlén M.; Andrews P.; Beavis R.; Borchers C.; et al. Recommendations from the 2008 International Summit on Proteomics Data Release and Sharing Policy: the Amsterdam principles. J. Proteome Res. 2009, 873689–3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloriam D. E.; Orchard S.; Bertinetti D.; Björling E.; Bongcam-Rudloff E.; Borrebaeck C. A.; et al. A community standard format for the representation of protein affinity reagents. Mol. Cell. Proteomics. 2010, 911–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skates S. J.; Gillette M. A.; Labaer J.; Carr S. A.; Anderson L.; Liebler D. C.; et al. Statistical design for biospecimen cohort size in proteomics-based biomarker discovery and verification studies. J. Proteome Res. 2013, 12125383–5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteaker J. R.; Lin C.; Kennedy J.; Hou L.; Trute M.; Sokal I.; et al. A targeted proteomics-based pipeline for verification of biomarkers in plasma.. Nat. Biotechnol. 2011, 297625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.; Zhang J.; Zhang L.; Liu W.; Weisel C. P. Selective detection of monohydroxy metabolites of polycyclic aromatic hydrocarbons in urine using liquid chromatography/triple quadrupole tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2004, 18192299–2308. [DOI] [PubMed] [Google Scholar]
- Xu X.; Roman J. M.; Issaq H. J.; Keefer L. K.; Veenstra T. D.; Ziegler R. G. Quantitative measurement of endogenous estrogens and estrogen metabolites in human serum by liquid chromatography-tandem mass spectrometry. Anal. Chem. 2007, 79207813–7821. [DOI] [PubMed] [Google Scholar]
- Kuhn E.; Wu J.; Karl J.; Liao H.; Zolg W.; Guild B. Quantification of C-reactive protein in the serum of patients with rheumatoid arthritis using multiple reaction monitoring mass spectrometry and 13C-labeled peptide standards. Proteomics 2004, 441175–1186. [DOI] [PubMed] [Google Scholar]
- Kiyonami R.; Schoen A.; Prakash A.; Peterman S.; Zabrouskov V.; Picotti P.; et al. Increased selectivity, analytical precision, and throughput in targeted proteomics. Mol. Cell. Proteomics. 2011, 102M110.002931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stergachis A. B.; MacLean B.; Lee K.; Stamatoyannopoulos J. A.; MacCoss M. J. Rapid empirical discovery of optimal peptides for targeted proteomics. Nat. Methods 2011, 8121041–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn E.; Addona T.; Keshishian H.; Burgess M.; Mani D. R.; Lee R. T.; et al. Developing multiplexed assays for troponin I and interleukin-33 in plasma by peptide immunoaffinity enrichment and targeted mass spectrometry. Clin. Chem. 2009, 5561108–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson N. L.; Anderson N. G.; Haines L. R.; Hardie D. B.; Olafson R. W.; Pearson T. W. Mass spectrometric quantitation of peptides and proteins using stable isotope standards and capture by anti-peptide antibodies (SISCAPA). J. Proteome Res. 2004, 32235–244. [DOI] [PubMed] [Google Scholar]
- Reid J. D.; Holmes D. T.; Mason D. R.; Shah B.; Borchers C. H. Towards the development of an immuno MALDI (iMALDI) mass spectrometry assay for the diagnosis of hypertension. J. Am. Soc. Mass Spectrom. 2010, 21101680–1686. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Parker C. E.; Fuller J. R.; Kawula T. H.; Borchers C. H. An immunoaffinity tandem mass spectrometry (iMALDI) assay for detection of Francisella tularensis. Anal. Chim. Acta 2007, 605170–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams D. K.; Muddiman D. C. Absolute quantification of C-reactive protein in human plasma derived from patients with epithelial ovarian cancer utilizing protein cleavage isotope dilution mass spectrometry. J. Proteome Res. 2009, 821085–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr S. A.; Abbatiello S. E.; Ackermann B. L.; Borchers C.; Domon B.; Deutsch E. W.; Grant R. P.; Hoofnagle A. N.; H Uumlttenhain R.; Koomen J. M.; Liebler D. C.; Liu T.; Maclean B.; Mani D. R.; Mansfield E.; Neubert H.; Paulovich A. G.; Reiter L.; Vitek O.; Aebersold R.; Anderson L.; Bethem R.; Blonder J.; Boja E.; Botelho J.; Boyne M.; Bradshaw R. A.; Burlingame A. L.; Chan D.; Keshishian H.; Kuhn E.; Kinsinger C.; Lee J.; Lee S. W.; Moritz R.; Oses-Prieto J.; Rifai N.; Ritchie J.; Rodriguez H.; Srinivas P. R.; Townsend R. R.; Van Eyk J.; Whiteley G.; Wiita A.; Weintraub S. Targeted Peptide Measurements in Biology and Medicine: Best Practices for Mass Spectrometry-based Assay Development Using a Fit-for-Purpose Approach. Mol. Cell. Proteomics. 2014, 133907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield E.; O’Leary T. J.; Gutman S. I. Food and Drug Administration regulation of in vitro diagnostic devices. J. Mol. Diagn. 2005, 712–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boja E. S.; Rodriguez H. The path to clinical proteomics research: integration of proteomics, genomics, clinical laboratory and regulatory science. Korean J. Lab. Med. 2011, 31261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addona T. A.; Abbatiello S. E.; Schilling B.; Skates S. J.; Mani D. R.; Bunk D. M.; et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 2009, 277633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boja E. S.; Jortani S. A.; Ritchie J.; Hoofnagle A. N.; Težak Ž; Mansfield E.; et al. The journey to regulation of protein-based multiplex quantitative assays. Clin. Chem. 2011, 574560–567. [DOI] [PubMed] [Google Scholar]
- Shi L.; Reid L. H.; Jones W. D.; Shippy R.; Warrington J. A.; Baker S. C.; et al. The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nat. Biotechnol. 2010, 288827–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnier F. E.; Skates S. J.; Mesri M.; Rodriguez H.; Tezak Z.; Kondratovich M. V.; et al. Protein-based multiplex assays: mock presubmissions to the US Food and Drug Administration. Clin. Chem. 2010, 562165–171. [DOI] [PubMed] [Google Scholar]
- Hoofnagle A. N. Peptide lost and found: internal standards and the mass spectrometric quantification of peptides. Clin. Chem. 2010, 56101515–1517. [DOI] [PubMed] [Google Scholar]
- Ciccimaro E.; Hanks S. K.; Yu K. H.; Blair I. A. Absolute quantification of phosphorylation on the kinase activation loop of cellular focal adhesion kinase by stable isotope dilution liquid chromatography/mass spectrometry. Anal. Chem. 2009, 8193304–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnir M. M.; Rockwood A. L.; Roberts W. L.; Abraham D.; Hoofnagle A. N.; Meikle A. W. Measurement of thyroglobulin by liquid chromatography-tandem mass spectrometry in serum and plasma in the presence of antithyroglobulin autoantibodies. Clin. Chem. 2013, 596982–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grebe S. K. G.; Singh R. J. J. LC-MS/MS in the clinical laboratory—where to from here?. Clin. Biochem. Rev. 2011, 32, 5–31. [PMC free article] [PubMed] [Google Scholar]
- Neubert H.; Gale J.; Muirhead D. Online high-flow peptide immunoaffinity enrichment and nanoflow LC-MS/MS: assay development for total salivary pepsin/pepsinogen. Clin. Chem. 2010, 56, 1413–1423. [DOI] [PubMed] [Google Scholar]