

Abstract
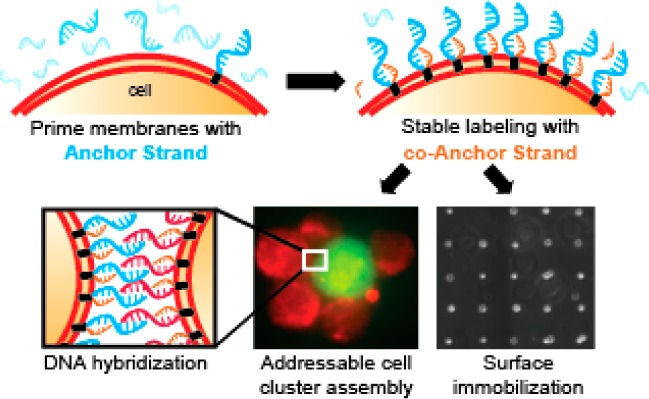
Lipid modifications provide efficient targeting of oligonucleotides to live cell membranes in a range of applications. Targeting efficiency is a function of the rate of lipid DNA insertion into the cell surface and its persistence over time. Here we show that increasing lipid hydrophobicity increases membrane persistence, but decreases the rate of membrane insertion due to the formation of nonproductive aggregates in solution. To ameliorate this effect, we split the net hydrophobicity of the membrane anchor between two complementary oligonucleotides. When prehybridized in solution, doubly anchored molecules also aggregate due to their elevated hydrophobicity. However, when added sequentially to cells, aggregation does not occur so membrane insertion is efficient. Hybridization between the two strands locks the complexes at the cell surface by increasing net hydrophobicity, increasing their total concentration and lifetime, and dramatically improving their utility in a variety of biomedical applications.
Introduction
Lipid-modified oligonucleotides1−3 facilitate uptake of siRNA,4 target DNA nanostructures to lipid bilayers,5 program assembly of 3D microtissues,6,7 enable preparation of live single cell microarrays,8−10 and function as vaccine adjuvants and immunotherapeutics.11,12 These uses are predicated on rapid, efficient, and stable partitioning of these amphiphilic molecules from solution into live cell membranes.13 We recently reported an approach for incorporating dialkylglycerol modified oligonucleotides (DAG) into cell membranes.10 DAG is useful for targeting DNA to the membranes of most cell lines, but suffers when targeting primary or embryonic stem cells (ESCs). Moreover, DAG and other lipid-modified oligonucleotides slowly leave the cell membrane and establish an equilibrium with the surrounding medium.14,15 This loss by re-equilibration limits the ultimate efficiency of incorporation into the bilayer over time.
To improve the concentration of lipid-anchored oligonucleotides in cell membranes, we reasoned that increasing the dialkyl anchor hydrophobicity would increase its thermodynamic stability when inserted into cell membranes.9,14−16 Indeed, previous studies demonstrated that longer lipids are more stable than shorter lipids when reconstituted into synthetic lipid bilayers.3 However, we found that DAG incorporation into live cell membranes (as opposed to synthetic systems) was exquisitely sensitive to alkyl chain length. The addition of even two methylenes completely inhibited partitioning into cell membranes.9 We hypothesized that this was due to a competing self-aggregation reaction17,18 and thus sought an alternative means of introducing greater hydrophobicity to the lipid anchors without aggregation.
Experimental Section
Synthesis of Lipid-Modified Oligonucleotides
Hexadecanoic (Palmitic) acid, octadecanoic (Stearic) acid, icosanoic (Arachidic) acid, docosanoic (Behenic) acid, tetracosanoid (Lignoceric) acid, N,N-diisopropylethylamine (DIPEA), N,N-diisopropylchlorophosphoramidite (DIPC), N,N-dimethylformamide (DMF), methylamine, ammonium hydroxide, and piperidine were obtained from Sigma-Aldrich. HPLC grade acetonitrile, triethylamine, acetic acid, and dichloromethane (DCM) were obtained from Fisher Scientific. Monomethoxytritylamino)hexyl-(2-cyanoethyl)-(N,N-diisopropyl)-phosphoramidite (amine phosphoramidite), standard phosphoramidites, and DNA synthesis reagents were obtained from Azco Biotech. Controlled pore glass (CPG) support, 1-O-dimethoxytrityl-hexyl-disulfide,1′-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite, 10-O-[1-propyl-3-N-carbamoylcholesteryl]-triethylene glycol-1-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite (5′-cholesterol-TEG phosphoramidite), (1-dimethoxytrityloxy-3-O-(N-cholesteryl-3-aminopropyl)-triethylene glycol-glyceryl-2-O-succinoyl-long chain alkylamino-CPG (3′-cholesterol-TEG CPG), and 2-dimethoxytrityloxymethyl-6-fluorenylmethoxycarbonylamino-hexane-1-succinoyl-long chain alkylamino-CPG (3′-amino-modifier C7 CPG), and synthesis columns were obtained from Glen Research. All materials were used as received from manufacturer.
Oligonucleotides were synthesized on an Applied Biosystems Expedite 8909 DNA synthesizer. Amino and cholesterol modified 'DNA strands were synthesized using amine and cholesterol phosphoramidites (100 mM), respectively, using a custom 15 min coupling protocol. For the polythymine regions of the anchor strands (Anch), the capping step was omitted in order to maximize yield. After synthesis of 5′ amino-modified DNA, the MMT protecting group was deprotected manually on the synthesizer by priming alternately with deblock and dry acetonitrile three times and watching for yellow elution. To ensure complete deprotection of the MMT group, the 5′ solid supports were also resuspended in a solution of 20% acetic acid/80% water1 shaking for 1 h at room temperature. The solid support was subsequently washed repeatedly with DMF, DCM, and acetonitrile with acetonitrile as the final wash and then dried with a speedvac system. For the 3′ amino-modified CPG, a solution of 20% piperidine in dimethylformamide was prepared and used to deprotect the CPG support for 10 min at room temperature, followed by DCM and DMF washes with DCM as the final wash. This procedure was repeated twice more to ensure complete deprotection of the FMOC protecting group prior to coupling to the fatty acid. Fatty acid conjugated oligonucleotides were synthesized by coupling the carboxylic acid moiety of the fatty acid to amino modified oligonucleotides with a 3′ or 5′ free amine while on the solid support. The solid support was transferred to an eppendorf tube and resuspended in a solution of dichloromethane containing 200 mM fatty acid, 400 mM DIPEA, and 200 mM DIPC. The eppendorf tubes were sealed with parafilm, crowned with a cap lock, and shaken overnight at room temperature. The next morning, they were washed with DCM and DMF repeatedly and then cleaved off the solid support
Oligonucleotides were cleaved from solid support with a 1:1 mixture of ammonium hydroxide/40% methylamine (AMA) for 1 h at 65 °C with a cap lock followed by evaporation of AMA with a speedvac system. Cleaved oligonucleotides were filtered through 0.2 μm Ultrafree-MC Centrifugal Filter Units (Millipore) to remove any residual CPG support before HPLC purification. Fatty acid modified oligonucleotides were purified from unmodified oligonucleotides by reversed-phase high-performance liquid chromatography (HPLC) using an Agilent 1200 Series HPLC System equipped with a diode array detector (DAD) monitoring at 260 and 300 nm. Purifications used 100 mM triethylamine acetate (TEAA, pH 7) H2O/acetonitrile as a mobile phase on a C8 column (Hypersil Gold, Thermo Scientific) running a gradient between 8 and 95% acetonitrile over 30 min. Pure fractions were collected manually and lyophilized. The resulting powder was then resuspended in distilled water and lyophilized again three more times to remove residual TEAA salts prior to use. Purified FA-modified oligonucleotides were resuspended in distilled water and concentrations were determined by measuring their absorbance at 260 nm on a Thermo-Fischer NanoDrop 2000 series. An aliquot of these stocks was reinjected on the HPLC to ensure >95% purity. If purity was <95%, HPLC purification was repeated. Additionally, select strands were also analyzed by MALDI-TOF (see below). The dialkylglycerol (DAG)-modified oligonucleotides were prepared as previously described.2 Stocks of 250 μM were prepared and from them aliquots of 50 μM were prepared for day-to-day use in order to minimize repeated freeze–thaw cycles.
Cell Lines and Cell Culture
Jurkats were obtained from ATCC (Clone E6–1) ATCC TIB-152 and were grown in suspension using RPMI 1640 media supplemented with 10% fetal bovine serum by volume (UCSF Cell Culture Facility) to a density of 106 cells per mL. The 832–13 pancreatic beta islet cells were obtained from Dr. Tejal Desai (UCSF) at passage 51 and were grown in RPMI 1640 supplemented with 10% fetal bovine serum, 63.7 mg/L penicillin G, 100 mg/L streptomycin SO4, 1 mM sodium pyruvate, and 70 nM 2-mercaptoethanol (UCSF Cell Culture Facility) to approximately 70% confluency on 75 cm2 tissue culture plastic. Dr. Matt Thomson (UCSF) generously provided 46C mouse ES cells. The cells were grown on gelatinized tissue culture plates using N2B27 media supplemented with 1000 U/mL LIF, 3 μM CHIR99021 and 1 μM PD0325901. The cells were lifted from their substrate using accutase and grown to a density of 2–3 x106 cells per cm2. MCF10A cells were kindly provided by Professor Jayanta Debnath (UCSF) and were cultured as previously described.3 Low passage, primary fibroblast, HMEC, and pre-adipocyte cell lines were provided by Jim Garbe (LBNL) and cultured in M87 media supplemented with cholera toxin.
DNA Labeling of Cells and Quantification of Cell Surface Oligonucleotides
For experiments, unless otherwise noted, Jurkat cells were used. Cells were pelleted at 1000 g resuspended in calcium and magnesium-free PBS (UCSF Cell Culture Facility) three times, with a final resuspension volume of 48 μL of PBS per 106 cells. Resuspended cells were labeled with single-stranded DNA by the addition of 1 μL of a 50 μM solution of the anchor strand in water. Cells were gently agitated by gentle vortexing for 5 min at room temperature. Subsequently, 1 μL of a 50 μM solution of the coanchor strand in water was added, bringing both strands to a final concentration of 1 μM. Cells were again gently agitated by slow vortexing for 5 min at room temperature. The cells were then pelleted and resuspended three times in ice-cold PBS to remove unbound or excess oligonucleotides. To quantify the extent of cell surface labeling, cells were incubated with 100 μL of a 20mer complementary 6-FAM modified oligonucleotide (1 μg/mL, Operon), which annealed to the most distal portion of the anchor strand. The strand was incubated for 30–45 min at 4 °C, protected from light. Cells were pelleted and resuspended one time in ice cold PBS before pelleting and resuspending in 100 μL per 106 cells of LIVE/DEAD Fixable Cell Stain (Invitrogen, used per manufacturers instructions) for 15 min at 4 °C protected from light. Cells were washed one last time with ice cold PBS before flow cytometry analysis. Flow cytometry was performed on a FACSCalibur (BD Biosciences, UCSF Laboratory for Cell Analysis) and the data was analyzed using FlowJo software package (Tree Star, Inc.). For stability time course experiments, cells were incubated at 37 °C for the designated amount of time in the presence of serum-free RPMI 1640 before incubating with the fluorescent, complementary oligonucleotide. For the preannealing experiment, a 1 μM solution of C18/C16 and a 0.3 μm solution of C22/C16 fatty acid modified strands in room temperature PBS was prepared and gently agitated for 10 min at room temperature. This solution was used to resuspend the cell pellet after the final wash from media and gently agitated for an additional 10 min at room temperature. This was compared to normal labeling using these same strands at these same concentrations. All reported values are the average of three or more independent measurements, with error bars indicating standard deviations. Graphs were produced using the Prism software package (Graphpad). The heatmap was produced using the R software package (R), specifically using the ggplot2 library.
Measuring Aggregation by Dynamic Light Scattering (DLS)
PBS CMF (UCSF CCF) was filtered by a 0.2 μm vacuum filter. Stock solutions of 250 μM ss-DNA strands were diluted to 1 μM with this filtered PBS prior to transfer to cuvette for measuring by DLS on a Wyatt Technology DynaPro Protein Solutions utilizing the DYNAMICS software package ver 6.10.1.2. Particle size was determined by cumulants analysis. All samples were prepared separately and measured in triplicate.
Surface Preparation for Cell Binding
Lyophilized 5′-amino-modified DNA was resuspended in a buffer of 60 mM sodium citrate, 450 mM sodium chloride, pH = 7.0. DNA was patterned onto aldehyde-silanized glass (Schott) using a micropipette (Figure 4c) or a Nano eNabler (BioForce Nanosciences; Figure 4d,e). Slides were reduced with NaBH4 (Sigma) and passivated with both SigmaCote (Sigma) and Pluronic F108 acid before use. Cells labeled with lipid-DNA were allowed to settle onto patterned glass within a PDMS-based flow cell for 30 min. Flow cells were flushed with ice cold PBS, and only cells hybridized to the surface via DNA were retained. Patterned cells were imaged using phase contrast settings and reconstructed using tiling algorithm (Zen Software, Zeiss). For experiments with mouse embryonic stem cells, no sigmacote was used for surface passivation.
Figure 4.
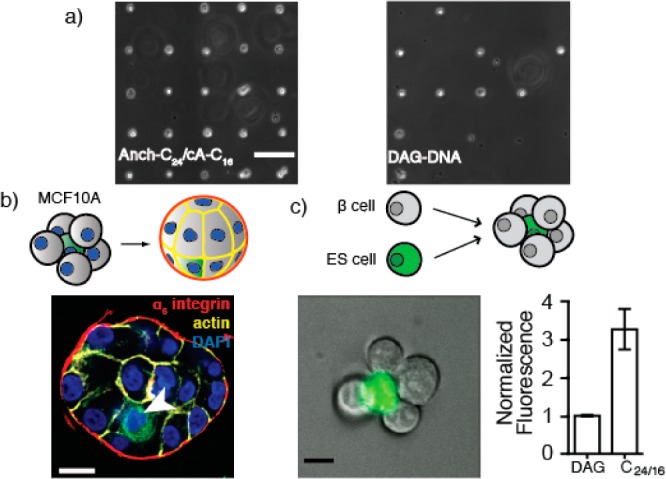
Improved preparation of single cell microarrays and 3D microtissues using stepwise assembly of membrane anchored adhesive oligonucleotides. (a) Single cell microarrays prepared with murine ESCs labeled with Anch100-C24/cA20-C16 or DAG. (b) MCF10A clusters assembled around 1 cell tracker green-stained cell (arrow) establish polarity after 48 h in Matrigel (scale bar is 20 μm). (c) Representative cluster of sorted ESC/β islet cells (scale bar is 10 μm) and labeling efficiency of β islet cells. Error bars are standard deviation of at least three independent measurements.
Programmed Cell Assembly
For quantification of programmed assembly efficiency, CellTracker Green CMFDA and CellTrace Far Red DDAO-SE (Invitrogen) stocks were prepared to a concentration of 10 mM in anhydrous DMSO. Cells were resuspended in 10 μM stain in serum-free media for 30 min at 37 °C followed by 15 min in media supplemented with 10% fetal bovine serum before proceeding through the labeling steps described above. After washing away unreacted DNA, cells were resuspended at 1 × 106 cells/mL. Green cells were mixed with far-red cells at a ratio of 1:60 with 106 cells per 200 μL of ice cold PBS. Mixtures were then agitated at 150 rpm for 10 min in an Ultra-Low Attachment 24-well plate (Corning). This mixture was pelleted and resuspended in ice cold PBS before quantifying via flow cytometry or sorting via fluorescence activated cell sorting (FACS). The labels were reversed to ensure unbiased quantitation. For programmed assemblies of the embryonic stem and pancreatic beta islet cells, CellTracker Green CMFDA was used to stain the ESCs and the islet cells were left unstained. were subsequently labeled with anchor strands 1 and 2, respectively, and assembled as described above. Clusters containing at least 1 green cell were purified from the unassembled cell population using a FACSAria II (UCSF Laboratory for Cell Analysis).
MCF10A Acinus Formation and Imaging
MCF10A cells were cultured as described above. Cells were collected and stained with CellTracker Green CMFDA, as described above or left unstained. The green and unstained cells were subsequently labeled with anchor strands 1 and 2, respectively, and assembled as described above. Clusters containing at least 1 green cell were purified from the unassembled cell population using a FACSAria II (UCSF Laboratory for Cell Analysis) and grown for 48 h in 3D on-top cultures in 8-well chamber slides (Lab-Tek) which were performed as previously described using growth-factor-reduced lrECM lots with protein concentrations between 9 and 11 mg/mL (Matrigel; BD Biosciences) (Debnath et al., 2003). After 48 h, the 3D cultures were fixed with 4% Paraformaldehyde in PBS. The 3D cultures were stained, as previously described by Debnath et al. (2003). Structures were stained with rat anti-human α6-integrin antibodies (Millipore clone NKI-GoH3MAB1378) for the primary and Alexa-568 conjugated goat antirat antibodies (Invitrogen) for the secondary. Alexa-647 conjugated phalloidin (Invitrogen) and 1× DAPI in PBS was used to stain the actin cytoskeleton and nuclei, respectively. Confocal images were acquired on Zeiss Axio Observer Z1 equipped with a Yokogawa spinning disk unit and an EM-CCD camera.
Results and Discussion
Previous reports show that complementary cholesterol-bearing oligonucleotides can be stably targeted to liposomes and supported lipid bilayers via hybridization.19 We envisioned further increasing the hydrophobicity of the membrane anchors to further stabilize duplexes in live cells, rather than artificial lipid bilayers. To prevent aggregation of these more hydrophobic molecules, however, the two strands would need to be added sequentially to cells, rather than as a prehybridized duplex. Under conditions of stepwise addition, a first Anchor strand (Anch) partitions into the lipid bilayer but remains in rapid equilibrium with the medium. A second, co-Anchor (cA) strand is subsequently added and also establishes rapid equilibrium between the lipid bilayer and the medium. However, upon encountering the first strand through diffusion in the phospholipid bilayer, the two strands hybridize, increase the total hydrophobicity of the now doubly anchored duplex and, thus, slowing their exchange with the medium (Scheme 1).
Scheme 1. Stepwise Assembly of Fatty-Acid (FA)-Modified ssDNA into Cell Membranes.

(a) Structure of the Anchor (Anch) and co-Anchor (cA) strands incorporating a lock region for Anch/cA-strand annealing and a polythymine spacer region. A handle region provides adhesion to surfaces and a means of quantifying cell membrane incorporation using complementary fluorescently labeled probes. (b) Model for step-wise assembly of membrane anchored DNA duplexes. FA-DNA molecules insert into the lipid bilayer but remain in rapid equilibrium with the surrounding cell medium. A second, complementary FA-modified oligonucleotide similarly establishes a rapid equilibrium with the cell membrane, but can also hybridize with the first strand in the membrane. The additional FA-anchor in the hybridized duplex alters the equilibrium, locking the co-anchored complex in the membrane. Elevated hydrophobicity can also trigger non-productive aggregate formation in solution.
To explore this strategy, we used fatty acid amides (FA) as more synthetically tractable membrane anchors than previously reported phospholipids or cholesterol.19 Fatty acids are widely commercially available and do not require chemical modification before coupling. Additionally, the conjugation reaction to DNA is not highly water sensitive and requires only one reverse phase purification step after coupling.
Consistent with past studies,13,20 a single FA anchor does not stably label cell membranes when compared to DAG or doubly cholesterol-anchored DNA (Figure S1). For example, a 100 base Anch strand linked to stearic acid (C18) via a 5′ amide (5′-Anch100-C18) did not yield significant DNA incorporation after incubation with cells and washing (Figure 1, column 3). However, addition of a second, 20 base complementary coanchor (cA-) strand linked to palmitic acid (C16) via a 3′ amide (3′-cA20-C16) dramatically increased cell labeling to near that of the DAG and doubly anchored cholesterol (Figure 1, column 6 and Figure S1). No increase was seen upon addition of a noncomplementary 3′-cA20-C16 strand (Figure 1, Column 5), indicating that at least two FA anchors, linked noncovalently through Watson–Crick base pairing in the “lock” region (Scheme 1a), are necessary for stable incorporation.
Figure 1.

Anchor (Anch) and complementary co-Anchor (cA) strands together enhance ssDNA targeting and retention in cell membranes. Fluorescence was measured with flow cytometry and normalized to a C16/C18 DAG-ssDNA control. cA-MM is a 20 base coanchor strand with a DNA sequence noncomplementary to the Anch strand. Error bars are standard deviation of at least three independent measurements.
We found that the number of base pairs in the lock region correlated with initial labeling and retention of oligonucleotides over time, both at 0 and 37 °C. This effect saturated between 15 and 20 bases (Figure S2). Labeling was dose-dependent and occurred without altering cell viability over the examined range of 0.5 to 5 μM (Figure S2). Encouragingly, even these unoptimized molecules were capable of programming cell–cell and cell−surface adhesion of model cell lines with results comparable to DAG when incorporating 60 base polythymine spacers (Figure S3).
These initial findings suggested we could achieve additional improvements in cell membrane incorporation by increasing the length and thus hydrophobicity of FA anchors. We therefore synthesized a series of 5′-Anch100 strands conjugated to saturated FAs between 16 and 24 carbons in length. These Anch strands behaved as predicted when added stepwise to cells in concert with 3′-cA20-C16. Increased lipophilicity of FAs enhanced the labeling efficiency and showed substantial improvement over both DAG and cholesterol linked oligonucleotides (Figure 2A). Anch strands with enhanced hydrophobicity also demonstrated improved retention over time at physiological temperature (Figure S4). Unlike the 5′-Anch100-C18 and 3′-cA20-C16 combination, stepwise addition of more hydrophobic Anch and cA strands was essential for preventing competing aggregation reactions (Figure 2B). Prehybridizing 5′-Anch100-C24 and 3′-cA20-C16 strands led to dramatically reduced cell membrane incorporation compared to prehybridized 5′-Anch100-C18 and 3′-cA20-C16 strands (Figure 2B). Together, these results support the notion that splitting the hydrophobicity of dual-anchored species across two complementary oligonucleotides added stepwise to cells prevents aggregation and improves labeling.
Figure 2.

Lipid hydrophobicity affects cell labeling efficiency of Anch, cA, and prehybridized strands. (a) Membrane incorporation compared to DAG for duplexes with 3′-cA20-C16 strands and Anch strands bearing FA anchors of increasing length. (b) Membrane incorporation of 3′-cA20-C16 and different Anch strands when added stepwise (white bars) or after preannealing (black bars). (c) Heatmap relating average membrane labeling (n = 3) to combinations of Anch and cA strand FA anchor lengths. Error bars are standard deviation of at least three independent measurements.
These data suggested we could achieve further increases in cell labeling and stability by increasing the hydrophobicity of the cA strand in addition to the Anch strand. Surprisingly, increasing the hydrophobicity of the cA strand anchors did not yield additional gains in cell labeling. For instance, stepwise addition of 5′-Anch100-C24 and 3′-cA20-C24, which maximizes hydrophobicity for both strands, actually decreased DNA incorporation when compared to 5′-Anch100-C24/3′-cA20-C16. We investigated this effect by assaying a panel of molecules in which the fatty acid on the Anch and cA strands was varied systematically and independently. We found that increasing hydrophobicity specifically on the coanchor strand decreased labeling (Figure 2C). Indeed, 5′-Anch100-C24/3′-cA20-C16 inserted far more efficiently into cell membranes than 5′-Anch100-C16/3′-cA20-C24 despite containing identical number of phosphodiester bonds and methylene groups.
To explain this trend, we hypothesized that the ratio of anchor hydrophobicity to oligonucleotide length (and thus charge) determines the extent of aggregation. If this were the case, short oligonucleotides would be more prone to aggregation than equivalently modified longer oligonucleotides. To test this notion, we used dynamic light scattering (DLS) to examine the relationship between FA anchor length, oligonucleotide length, and relative aggregation. Both scattered light intensity (Figure 3A) and particle size (Figure S5) correlated with the length of the FA conjugated to the cA strand. In contrast, very little light scattering was observed for any FA conjugated to the 100 base anchor strand (Figure 3B). These results suggested that adding additional bases to the cA strand, increasing its net size and charge, would destabilize aggregates through Coulombic or steric repulsion while simultaneously allowing for increased hydrophobicity of its FA anchor. We therefore synthesized a series of 3′-cA20-C24 strands incorporating an additional 10, 20, or 30 bases at the 5′ end. Consistent with our expectations, DLS revealed an inverse relationship between the number of bases and aggregation (Figure 3B). Moreover, the best of these molecules, 3′-cA50-C24, increased cell labeling in combination with 5′-Anch100-C24 to nearly 7-fold of DAG (Figure 3C). This combination of molecules also showed a dramatic increase in lifetime at the cell surface compared to DAG (Figure 3D). We calculated that the initial rate of decay of these fully optimized strands from the cell surface was nearly 100-fold lower than DAG.
Figure 3.
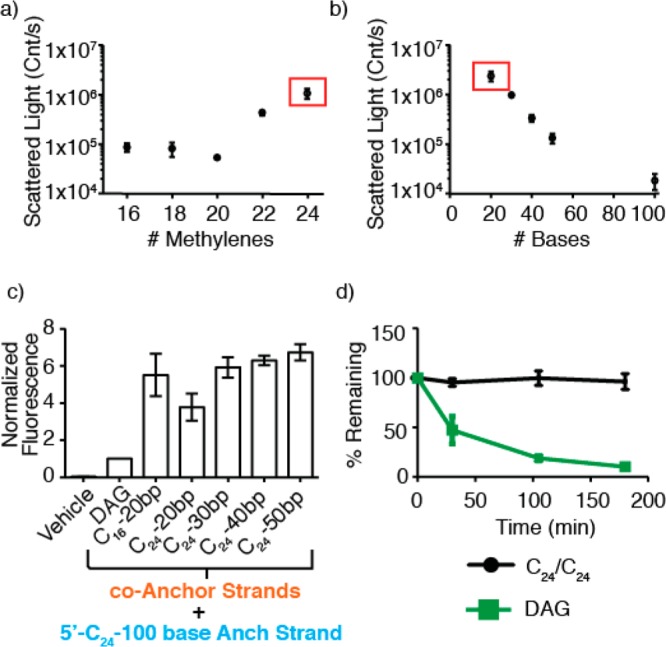
Ratio of FA to oligonucleotide length determines the extent of aggregation. (a) Light scattering from solutions of 3′-cA20 strands as a function of FA anchor length. (b) Light scattering from solutions of 3′-cA-C24 anchored oligonucleotides as a function of the number of DNA bases. The red box indicates the same strand, 3′-cA20-C24. (c) Membrane incorporation compared to DAG for combinations of 3′-cA-C24 with increasing numbers of DNA bases. (d) Incorporation vs time for DAG and 5′-Anch100-C24/3′-cA50-C24-DNA. Error bars are standard deviation of at least three independent measurements.
Given the improved cell labeling enabled by stepwise addition of C24 conjugated oligonucleotides, we investigated whether they could be used for programming cell–cell and cell–surface adhesion for cell types that were inaccessible with DAG. Using even unoptimized 5′-Anch100-C24/3′-cA20-C16, we found efficient adhesion between ESCs and complementary 7 μm diameter spots of DNA patterned on glass surfaces, allowing the preparation of live, single cell microarrays with more than 95% occupancy. By comparison, DAG yielded only 40% binding using identical conditions (Figure 4A). We also used stepwise assembly of FA-anchored DNA to prepare 3D mosaic epithelial tissues from nonmalignant human mammary epithelial cell lines (MCF10A). Aggregates were purified by fluorescence activated cell sorting (FACS) prior to incubation under 3D culture conditions for 48 h in Matrigel. Consistent with previous reports,21,22 aggregates condensed into spherical microtissues with appropriately positioned markers of cell polarity such as α6-integrin (basal) and actin (lateral; Figure 4B). The 5′-Anch100-C24/3′-cA20-C16 combination showed improved labeling compared to DAG in several low passage primary cells and a pancreatic β-cell line (Figures 4C and S6). We therefore used this combination to prepare aggregates of controlled topology from mouse ESCs and pancreatic β-cells (Figure 4C). Previous reports have demonstrated that heterotypic aggregates of this general form can be used to differentiate stem cells into a variety of useful cell types.23−25 Thus, small 3D tissues of this type may find utility in regenerative medicine or basic science research.
Conclusion
In conclusion, stepwise assembly of membrane-anchored oligonucleotides is a modular strategy for targeting DNA to cell membranes with improved efficiency and stability. Insertion of oligonucleotide duplexes into membranes occurs via two FA-anchors with higher net lipophilicity compared to previously reported anchors. Competing self-aggregation is prevented by separating the dual anchors between two molecules that are added sequentially to cells, as well as by balancing the ratio of hydrophobicity to oligonucleotide length. This strategy facilitates new applications such as DNA-mediated adhesion in primary cells, murine ESCs, and pancreatic β cells, cell types that show little to no labeling with DAG. An additional benefit of these molecules is their streamlined synthesis compared to previous methods. We anticipate that the structure/function relationships defined here will prove useful in other applications utilizing lipid-modified oligonucleotides or amphiphiles including vaccine adjuvants, siRNA delivery, and structural DNA nanotechnology.
Acknowledgments
This work was supported by grants from the Department of Defense Breast Cancer Research Program (W81XWH-10-1-1023 and W81XWH-13-1-0221 to Z.J.G.); NICHD (DP2 HD080351-01 to Z.J.G.); The Sidney Kimmel Foundation; and the UCSF Program in Breakthrough Biomedical Research. Z.J.G. is an investigator in the UCSF Center for Systems and Synthetic Biology (NIGMS Systems Biology Center Grant P50 GM081879). This work is partially supported by the Achievement Rewards for College Scientists Foundation Fellowship and the Genentech Foundation Fellowship to S.I.L. The authors thank Michael Todhunter, Noel Jee, Amanda Paulson, Matthew Thomson, Jade McPherson, Michael Broeker, and Allison Doak for technical assistance.
Supporting Information Available
Oligonucleotide sequences, MALDI data, and additional figures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Benkoski J. J.; Höök F. J. Phys. Chem. B 2005, 109, 9773. [DOI] [PubMed] [Google Scholar]
- Yoshina-Ishii C.; Boxer S. G. J. Am. Chem. Soc. 2003, 125, 3696. [DOI] [PubMed] [Google Scholar]
- Tumpane J.; Ljungdahl T.; Wilhelmsson L. M.; Nordén B.; Brown T.; Albinsson B. J. Am. Chem. Soc. 2009, 131, 2831. [DOI] [PubMed] [Google Scholar]
- Kanasty R.; Dorkin J. R.; Vegas A.; Anderson D. Nat. Mater. 2013, 12, 967. [DOI] [PubMed] [Google Scholar]
- Langecker M.; Arnaut V.; Martin T. G.; List J.; Renner S.; Mayer M.; Dietz H.; Simmel F. C. Science 2012, 338, 932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartner Z. J.; Bertozzi C. R. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. S.; Gartner Z. J. Trends Cell Biol. 2012, 22, 683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra R. A.; Douglas E. S.; Mathies R. A.; Bertozzi C. R.; Francis M. B. Angew. Chem., Int. Ed. 2006, 45, 896. [DOI] [PubMed] [Google Scholar]
- Hsiao S. C.; Shum B. J.; Onoe H.; Douglas E. S.; Gartner Z. J.; Mathies R. A.; Bertozzi C. R.; Francis M. B. Langmuir 2009, 25, 6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selden N. S.; Todhunter M. E.; Jee N. Y.; Liu J. S.; Broaders K. E.; Gartner Z. J. J. Am. Chem. Soc. 2012, 134, 765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaar O.; Cazan D.; Klimek L.; Larenas-Linnemann D.; Calderon M. A. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 648. [DOI] [PubMed] [Google Scholar]
- Ishii K.; Akira S. Trends Immunol. 2006, 27, 525. [DOI] [PubMed] [Google Scholar]
- Borisenko G. G.; Zaitseva M. A.; Chuvilin A. N.; Pozmogova G. E. Nucleic Acids Res. 2008, 37, e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Kwong B.; Irvine D. J. Angew. Chem., Int. Ed. 2011, 50, 7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palte M. J.; Raines R. T. J. Am. Chem. Soc. 2012, 134, 6218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvius J. R.; Leventis R. Biochemistry 1993, 32, 13318. [DOI] [PubMed] [Google Scholar]
- Patwa A.; Gissot A.; Bestel I.; Barthélémy P. Chem. Soc. Rev. 2011, 40, 5844. [DOI] [PubMed] [Google Scholar]
- Berti D. Curr. Opin. Colloid Interface Sci. 2006, 11, 74. [Google Scholar]
- Pfeiffer I.; Höök F. J. Am. Chem. Soc. 2004, 126, 10224. [DOI] [PubMed] [Google Scholar]
- Kato K.; Itoh C.; Yasukouchi T.; Nagamune T. Biotechnol. Prog. 2004, 20, 897. [DOI] [PubMed] [Google Scholar]
- Debnath J.; Muthuswamy S. K.; Brugge J. S. Methods 2003, 30, 256. [DOI] [PubMed] [Google Scholar]
- Liu J. S.; Farlow J. T.; Paulson A. K.; Labarge M. A.; Gartner Z. J. Cell Rep. 2012, 2, 1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goerke S. M.; Plaha J.; Hager S.; Strassburg S.; Torio-Padron N.; Stark G. B.; Finkenzeller G. Tissue Eng., Part A 2012, 18, 2395. [DOI] [PubMed] [Google Scholar]
- Saleh F. A.; Whyte M.; Genever P. G. Eur. Cell Mater. 2011, 22, 242. [DOI] [PubMed] [Google Scholar]
- Van Hoof D.; Mendelsohn A. D.; Seerke R.; Desai T. A.; German M. S. Stem Cell Res. 2011, 6, 276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.