

Abstract
In the face of the global epidemic of metabolic syndrome (MetS) and its strong association with the increasing rate of cardiovascular morbidity and mortality, it is critical to detect MetS at an early stage in the clinical setting to implement preventive intervention long before the complications arise. Lipopolysaccharide, the cell wall component of Gram-negative bacteria produced from diet-disrupted gut microbiota, has been shown to induce metabolic endotoxemia, chronic low-grade inflammation, and ultimately insulin resistance. Therefore, ameliorating the inflammation and insulin resistance underlying MetS by gut microbiota-targeted, dietary intervention has gained increasing attention. In this review, we propose using dynamic monitoring of a set of translational biomarkers related with the etiological role of gut microbiota, including lipopolysaccharide binding protein (LBP), C-reactive protein (CRP), fasting insulin, and homeostasis model assessment of insulin resistance (HOMA-IR), for early detection and prevention of MetS via nutritional modulation. LBP initiates the recognition and monomerization of lipopolysaccharide and amplifies host immune responses, linking the gut-derived antigen load and inflammation indicated by the plasma levels of CRP. Fasting plasma insulin and HOMA-IR are measured to evaluate insulin sensitivity that is damaged by pro-inflammatory cytokines. The dynamic monitoring of these biomarkers in high-risk populations may provide translational methods for the quantitative and dynamic evaluation of dysbiosis-induced insulin resistance and the effectiveness of dietary treatment for MetS.
Keywords: biomarkers, gut microbiota, insulin resistance, metabolic syndrome, trajectory analysis
Introduction
Metabolic syndrome (MetS), a cluster of multiple cardiovascular risk factors, is one of the most significant public health challenges worldwide (Alberti et al., 2005). Although the clinical criteria of MetS have been defined differently by various organizations, all have come to an agreement on the core components: insulin resistance, central obesity, hyperglycemia, dyslipidemia, and hypertension (Kassi et al., 2011). The International Diabetes Federation estimated that 20–25% of the world's adult population have MetS (Alberti et al., 2006), but many are unaware that they are suffering from this syndrome. People with MetS are 3–5 times more likely to have heart attack/stroke or type 2 diabetes compared with unaffected individuals (Stern et al., 2004; Ärnlöv et al., 2010; Grundy, 2012). The rise in comorbid chronic diseases accounts for much of the clinical and economic burden of MetS to individuals, their families, and society (Wild & Byrne, 2006; Scholze et al., 2010). Early detection and intensive management of MetS, to reduce the long-term risk of cardiovascular disease and diabetes, are now possible and bring appreciable benefits (WHO, 2005; Després et al., 2008). Because of the urgent need for strategies to prevent MetS, there has been increased interest in exploring translational methods for early diagnosis and prevention with biochemical or genetic markers.
Although the molecular mechanisms underlying MetS are not fully understood, the presence of MetS is commonly associated with inflammation (Haffner, 2006), insulin resistance (Lann & LeRoith, 2007), endothelial dysfunction (Hajer et al., 2007), renal dysfunction (Servais et al., 2008), oxidative stress (Onat et al., 2006), disturbed hemostasis (Mansfield et al., 1996), and neurohormonal activation (Prasad & Quyyumi, 2004; Olsen et al., 2005). Several biological markers of these physiologic and pathological phenomena have been proposed as risk factors for MetS and its associated complications, such as white blood cell count, high-sensitivity C-reactive protein (hs-CRP), homeostasis model assessment insulin resistance index (HOMA-IR), homocysteine, cystatin C, uric acid, plasminogen activator inhibitor-1, fibrinogen, aldosterone, renin, and B-type natriuretic peptide (Lee et al., 2009). These biomarkers are readily measured in clinical practice, and altered levels could serve as diagnostic criteria to guide clinical management. However, generally, they are applied individually or independently, and it is less clear whether variations of biomarker levels are pathogenetically related to the incidence of MetS. In addition, although impaired fasting glycemia or impaired glucose tolerance can predict the risk of developing diabetes, it can only indicate an individual's glycemic state at a single point in time (Stern et al., 2002; Meigs et al., 2003). It is most likely that the risk of developing diabetes is present at the stage when glucose concentration is still at the ‘normal range’ (Rydén et al., 2007). Therefore, monitoring early biomarker profiles that participate in the pathogenesis of MetS will help identify patients at risk and allow them to take effective preventive interventions.
Low-grade, chronic activation of the innate immune system is regarded as an important underlying condition for the pathogenesis of obesity and associated metabolic disorders (Wellen & Hotamisligil, 2005). The process might partly depend on the immunomodulatory effects exerted by dietary compounds in the gut and beyond (Zeyda & Stulnig, 2007). Particularly in recent years, gut microbiota shaped by day-to-day dietary changes is considered a possible causative factor of metabolic disorders, as well as a therapeutic target (Cani & Delzenne, 2011; Vrieze et al., 2012; Everard et al., 2013; Kimura et al., 2013; Zhao, 2013). Modulating gut microbiota by designed dietary schemes has become a promising strategy to help manage obesity and metabolic abnormalities (Sanz et al., 2010). In the current review, we propose that the measurement of longitudinal changes in gut microbiota-based, translational biomarkers could be applied to monitor insulin sensitivity alterations during MetS development and progression. This multimarker approach includes lipopolysaccharide binding protein (LBP), CRP, fasting insulin, and HOMA-IR, all of which represent key links to the potential causal pathway from unhealthy diet to initiation of insulin resistance and MetS (Fig. 1): diet-induced gut microbiota dysbiosis, namely, an increase in opportunistic pathogens and a decrease in gut barrier protectors (Zhang et al., 2010), leads to increased gut permeability, increased lipopolysaccharide in the bloodstream to provoke low-grade inflammation, which can increase insulin resistance. An effective intervention would reverse this pathway, as indicated by these biomarkers. This new biomarker profile could not only be used for the early detection of insulin resistance, but also for assessing the degree of success in treating MetS.
Fig 1.
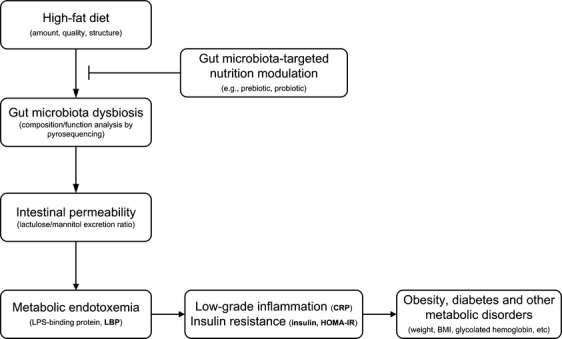
The biomarkers represent key links to the potential causal pathway from high-fat diet to initiation of metabolic endotoxemia, insulin resistance, and MetS and the restoration of dysbiosis by gut microbiota-targeted nutritional approach can abolish obesity-associated metabolic features.
Gut microbiota, inflammation, and insulin resistance
Insulin resistance, in which the physiologic concentration of insulin cannot produce a blood glucose lowering effect, is the most accepted hypothesis to describe the pathophysiology of MetS (Eckel et al., 2005). This pathological phenomenon is often seen in obesity, stress, infection, or severe illness. Simultaneously, obesity and metabolic abnormalities are closely associated with chronic low-grade inflammation characterized by abnormal cytokines production and activation of inflammatory signaling pathways (Wellen & Hotamisligil, 2005). Increasing evidence directly implicates the abundance of pro-inflammatory cytokines in insulin resistance in the liver, muscle, and adipose tissue (Neuschwander-Tetri & Caldwell, 2003), which have multifaceted effects on insulin resistance susceptibility genes, including those regulating lipid synthesis, gluconeogensis, and adipogenesis (Kim, 2010). Increased levels of inflammatory cytokines, such as tumor necrosis factor-α and interleukin-6, can facilitate serine phosphorylation of insulin receptor substrate-I, contributing to insulin resistance (Reaven, 1988). Thus, inflammation suppresses insulin-signaling pathways and makes the human body less responsive to insulin, increasing the risk for insulin resistance (Xu et al., 2003; Cai et al., 2005). Therefore, low-grade chronic inflammation is viewed as an important underlying condition for the pathogenesis of MetS and a possible target of prevention and/or therapy.
Diet is considered the key determinant influencing the overall chronic inflammatory response (Bulló et al., 2007); however, the primary mediator(s) between diet and inflammation remain to be elucidated. Some dietary compounds in high-fat meals, such as the saturated fatty acids frequently consumed in substantial quantities by obese individuals, have been demonstrated to bind to Toll-like receptor 4 (TLR4) to activate nuclear factor kappa-B, leading to over-expression of pro-inflammatory cytokines. These increases are completely absent after a meal rich in fiber and fruit (Ghanim et al., 2009).
Gut microbiota, consisted of trillions of microorganisms (c. 1 × 1013–1 × 1014, biomass > 1 kg) and modulated mainly by diet (Scott et al., 2012), has recently been recognized as a primary mediator for human health (Cani & Delzenne, 2009; Ley, 2010; Monteiro & Azevedo, 2010; Kelly et al., 2012; Everard & Cani, 2013). Current evidences support that alterations in composition and/or metabolic activity of gut microbiota play pivotal role in the pathogenesis of obesity and related disorders (Musso et al., 2011). Lipopolysaccharide, the cell wall component of Gram-negative bacteria living in the gut, has been identified as an important factor for inducing this metabolically triggered chronic inflammation associated with obesity (Cani et al., 2007a). High-fat diet-disrupted gut microbiota, with the increase in the proportion of lipopolysaccharide producers and the decrease in intestinal barrier protectors Bifidobacterium spp., released lipopolysaccharide into the host bloodstream through a partially impaired gut barrier to act as a primary mediator for inflammation, leading to insulin resistance and obesity (namely metabolic endotoxemia) (Cani et al., 2007a). The plasma concentration of lipopolysaccharide increased 2–3 times in high-fat diet-fed obese mice, comparable to what has been found in human subjects with MetS (Cani et al., 2007a; Creely et al., 2007). Subcutaneous injection of lipopolysaccharide in otherwise lean and healthy mice fed on normal chow evoked systemic inflammation and eventually induced insulin resistance and obesity, whereas knock-out of CD14, a co-receptor of TLR4, abolished these responses to lipopolysaccharide treatment (Cani et al., 2007a). Supporting normal levels of bifidobacteria by adding oligofructose maintained a gut barrier less permeable to lipopolysaccharide and prevented high-fat diet-fed mice from developing insulin resistance and obesity (Cani et al., 2007b). In human subjects, increased lipopolysaccharide content was also associated with an increased BMI and high-fat diet (Erridge et al., 2007; Lajunen et al., 2008). Thus, metabolic endotoxemia induced by intestinal dysbiosis has been proposed as a possible contributor to the inflammatory state associated with MetS.
Specific bacteria, which are positively associated with MetS, have been identified. In our previous work, we found that the endotoxin-producing Enterobacter decreased in relative abundance from 35% of a morbidly obese volunteer's gut bacteria to non-detectable, during which time the volunteer lost 51.4 kg of 174.8 kg initial weight and recovered from hyperglycemia and hypertension after 23 weeks on the whole grains, traditional Chinese medicinal foods, and prebiotics diet (WTP diet) (Fei & Zhao, 2013). The strain Enterobacter cloacae B29 isolated from the volunteer's gut induced germfree C57BL/6J mice to develop obesity and insulin resistance on a high-fat diet, suggesting that the endotoxin-producing bacterium may be a causative factor in the development of obesity in its human host (Fei & Zhao, 2013). After the Enterobacter population reduced to almost non-detectible, the human host started to reduce endotoxin load in his serum, alleviate his inflammation and recover from insulin resistance and other metabolic deteriorations. In B29-induced obese mice, we observed increased endotoxin load in the serum, increased systemic and local inflammation and significantly increased insulin resistance (Fei & Zhao, 2013). Thus, there seems to be a causal path between endotoxin producers in the gut and obesity/insulin resistance disease endpoints.
Gut bacteria, which are negatively correlated with MetS, have also been identified. Akkermansia muciniphila, an obligate mucin degrader that grows almost fully dependent on the mucin (Derrien et al., 2004), inversely correlates with body weight (Santacruz et al., 2010; Karlsson et al., 2012) and type 1 diabetes (Hansen et al., 2012), and increases by c. 100-fold with prebiotic (oligofructose) treatment (Everard et al., 2011). Very recently, Everard et al. (2013) demonstrated that the abundance of A. muciniphila decreased in genetic and high-fat-fed obese and type 2 diabetic mice, which was restored and correlated with the improved metabolic profile after prebiotics administration. In addition, introduction of A. muciniphila by daily oral gavage reversed high-fat diet-induced metabolic disorders, indicating the potential application of A. muciniphila in prevention or treatment of obesity and related complications.
MetS can thus be driven by structurally aberrant gut microbiota with increased pathobionts such as E. cloacae B29 and decreased gut barrier protectors such as A. muciniphila, which may have been perturbed by host gene defect (Vijay-Kumar et al., 2010), dietary change (Turnbaugh et al., 2008), antibiotics administration (Ajslev et al., 2011), or gastrointestinal tract illnesses/surgery (Liou et al., 2013).
Gut microbiota-targeted nutritional modulation in the management of MetS
Due to its key role in the initiation, propagation, and development of insulin resistance and obesity, which is associated with increased risks for other metabolic abnormalities, the pro-inflammatory state is a possible target of prevention and/or therapy for MetS (Hotamisligil et al., 1993; Cai et al., 2005). Directly inhibiting the inflammation by pharmacological interventions, such as inhibitors of inflammatory kinases and agonists of relevant transcription factors/cytokines, prevents insulin resistance (Moller & Berger, 2003; Kaneto et al., 2004). But the problem is that by blocking the action of individual inflammatory mediators, other redundant components may be sufficient to continue propagating the inflammatory signal (Wellen & Hotamisligil, 2005). On the other hand, weakening or even inhibiting inflammation in the first place may compromise the immune response, leaving the system vulnerable to subsequent injury or infection (Gregor & Hotamisligil, 2011). Therefore, the most effective anti-inflammatory therapy strategies should address the root causes of inflammation, which include metabolic endotoxemia (Monteiro & Azevedo, 2010; Gregor & Hotamisligil, 2011; Moreira et al., 2012).
Diet modification is an integral part of lifestyle intervention to reduce MetS risk factors (Grundy et al., 2006; Bulló et al., 2007). Numerous studies of weight loss have demonstrated that improvement in metabolic parameters due to dietary intervention is often associated with a decrease in circulating levels of inflammatory markers (Imayama et al., 2012; Fayh et al., 2013; Nicklas et al., 2013). However, not all dietary weight loss interventions lead to reduced inflammation; for example, inflammatory markers increased in overweight children after they effectively lost weight on a low carbohydrate, high-fat diet (Alvarez et al., 2009). Therefore, when choosing an intervention for weight management, priority should be given to schemes that can decrease the inflammatory tone, eventually decreasing the risk of progression to metabolic diseases, not just reducing the body weight temporarily.
The homeostasis of gut microbiota depends on host physiology and environmental conditions; moreover, it also relies on day-to-day dietary changes (Turnbaugh et al., 2009). The composition and/or activity of gut microbiota is a factor that characterizes obese vs. lean individuals, and diabetic vs. non diabetic patients (Bäckhed et al., 2004; Caesar et al., 2010; Qin et al., 2012; Everard et al., 2013; Karlsson et al., 2013). More importantly, the changes associated with the above disorders in gut microbes can be reversed by nutritional intervention (Ley et al., 2006; Cani et al., 2007b; Zhang et al., 2012a; Everard et al., 2013). Because of the dominating role of diet in shaping the composition and gene transcription network of gut microbiota (Sonnenburg et al., 2005; Zhang et al., 2010), modulation of gut microbiota by nutrients with prebiotic properties is a promising strategy for managing obesity and metabolic diseases (Delzenne & Cani, 2010; Sanz et al., 2010; Delzenne et al., 2011). This dietary scheme should not only meet human nutritional needs, but also balance gut microbiota. A diet protecting against MetS should be rich in whole grains, fruits, vegetables, lean meats, and fish, and low-fat or fat-free dairy products, and avoid processed foods (Bulló et al., 2007).
Following the encouraging results from experimental models aimed at affecting gut microbiota, both probiotic (live microorganisms beneficial to the host organism, such as Bifidobacterium and Lactobacillus spp.) (An et al., 2011; Chen et al., 2011; Fåk & Bäckhed, 2012) and prebiotic (non-digestible food ingredients that stimulate the growth and/or activity of probiotic, such as oligofructose, galactooligosaccharide, etc.) (Everard et al., 2011; Neyrinck et al., 2012) approaches were effective in managing the metabolic diseases associated with obesity. A few clinical studies using dietary interventions to manipulate gut microbiota and host metabolism have succeeded in linking the intervention to beneficial phenotypic changes (Cani et al., 2009; Parnell & Reimer, 2009; Bays et al., 2012). Targeted approaches, such as fluorescence in situ hybridization and real-time PCR, have been used to evaluate the gut microbiota in overweight or obese patients after dietary treatment (Musso et al., 2010). Weight loss was associated with an increase in Bacteroides fragilis and Lactobacillus and a decrease in Bifidobacterium longum and Clostridium coccoides in overweight adolescents after 10 weeks on a calorie restriction diet (Santacruz et al., 2009). Ley et al. (2006) found that the relative proportion of Bacteroidetes over Firmicutes increased in obese subjects after weight reduction on low carbohydrate or low fat diets, indicating that modulating gut microbiota can be an effective means for weight management. However, it remains obscure how these reported changes may lead to weight loss or improved metabolic health (Everard et al., 2013; Liou et al., 2013). Our recent study showed that after a 23-weeks WTP dietary intervention (9 weeks strict intervention followed by a 14-week maintenance period), 89 central obese volunteers (BMI ≥ 28 kg m−2) lost 5.79 ± 4.64 kg (6.62 ± 4.94%) weight. The incidence of MetS decreased from 60.67% (baseline) to 31.46% (9 weeks later) and 29.21% (23 weeks later), in addition to the improvement in insulin sensitivity, lipid profiles, and blood pressure. Plasma endotoxin load as LBP was also significantly reduced, with concomitant decrease in CRP, tumor necrosis factor-α, interleukin-6, and an increase in adiponectin, indicating a significant alleviation of the inflammatory condition. Pyrosequencing of fecal samples showed that phylotypes related to endotoxin-producing opportunistic pathogens of Enterobacteriaceae and Desulfovibrionaceae were reduced significantly, while those related to gut barrier-protecting bacteria of Bifidobacteriaceae increased. These results suggest that modulation of the gut microbiota via dietary intervention may enhance the intestinal barrier integrity, reduce circulating antigen load, and ultimately ameliorate the inflammation, insulin resistance, and metabolic phenotypes (Xiao et al., 2013). This also indicates that there is a possible causal linkage from dietary alteration of gut microbiota to reduced endotoxin load in serum and alleviation of inflammation in human body.
Gut microbiota-based host biomarkers to prevent MetS
Gut microbiota dysbiosis (mainly caused by high-fat/energy diet) affects host gut barrier permeability, increases endotoxin load, and evokes inflammation, which lead to downstream metabolic consequences (Cox & Blaser, 2013). Manipulating the gut microbiota composition with diet becomes a promising ‘pharmaco-nutritional’ approach to reverse the dysbiosis and host metabolic disorders (Cani & Delzenne, 2011; Kovatcheva-Datchary & Arora, 2013). However, assessing qualitative and quantitative alterations of gut microbiota is still not feasible in the conventional healthcare system. Developing a set of host biomarkers related to changes in gut microbiota should be an alternative for MetS prevention and management at the population level. Here, we propose a set of biomarkers for the dietary management of MetS in a public health project: diet modulates the composition of gut microbiota to suppress opportunistic pathogens and promote gut barrier protectors and decreases gut permeability, thus alleviating metabolic endotoxemia (LBP reduces) and inflammatory tone (CRP reduces), and improving insulin sensitivity (fasting insulin and HOMA-IR decrease).
LBP
Various toxins produced by particular members of gut microbiota may enter the bloodstream via enterohepatic circulations or the impaired gut barrier to affect host immunity and metabolism (Manco et al., 2010; Zhao & Shen, 2010). Experimental data indicated that lipopolysaccharide, derived from Gram-negative bacteria in the gut, plays a key role in driving systemic inflammation, insulin resistance, and fat mass development (Cani et al., 2007a). However, because of its short half-life, low concentration, and high susceptibility to interfering substances (Novitsky, 1998), the utility of lipopolysaccharide detection via the Limulus lysate assay is limited in routine clinical setting and large-scale studies. The development of more reliable methods for lipopolysaccharide detection in blood samples is needed. Furthermore, individual endotoxin preparations from various Gram-negative bacteria vary widely in their capacity to mediate activation of alternative pathways (Morrison & Ryan, 1987; Erridge et al., 2002; Coats et al., 2011; Matsuura, 2013). The lipopolysaccharide produced from Escherichia coli, Salmonella minnesota, and other Enterobacteriaceae has potent inflammation-inducing capacity, usually nearly 100- to 1000-fold higher than the lipopolysaccharide from B. fragilis (Lindberg et al., 1990; Erridge et al., 2002; Hakansson & Molin, 2011). The total amount of bacterial lipopolysaccharide from different sources varies dramatically in the capacity to lead to metabolic endotoxemia and the inflammatory response.
LBP is an acute-phase protein mainly produced in the liver that circulates in the blood, which can be conveniently detected by commercial ELISA kits. LBP initiates the recognition of lipopolysaccharide and amplifies host immune responses to lipopolysaccharide (Schumann et al., 1990), which indicates the amount of effective lipopolysaccharide and could be a reliable biomarker linking lipopolysaccharide load and the induced innate immune response (Lepper et al., 2007). LBP also binds to other bacterial compounds, including glycolipids of spirochetes, lipoteichoic acid, lipomannan of mycobacteria, at least two types of lipopeptides, and elements of the pneumococcal cell wall to modulate their ability to stimulate the innate immune system and therefore can be a more general indicator of the exogenous antigen load in the host (Schröder & Schumann, 2005; Cesaro et al., 2011).
Circulating LBP concentration is significantly increased in non-alcoholic fatty liver disease patients (Guerra et al., 2007), glucose-intolerant men (Gubern et al., 2006), and coronary artery disease patients (Lepper et al., 2007), and becomes an inflammatory marker associated with obesity-related insulin resistance (Moreno-Navarrete et al., 2012). Sun et al. (2010) found that increased circulating LBP is associated with obesity, MetS, and type-2 diabetes in apparently healthy Chinese people in a cross-sectional cohort study. These findings suggest a potential association between LBP and metabolic disorders, and the possibility of using LBP as a biomarker for early detection, diagnosis, and progression of MetS. Furthermore, our previous work found that increased serum LBP levels in Wistar rats after high-fat diet was essentially prevented by berberine co-administration, which accompanied with significant reductions in bacterial diversity and the total bacterial population resulting in a decrease in the free antigen load in the host (Zhang et al., 2012b). Another report showed that C57BL/6J mice with 30% calorie restriction had higher abundance of gut barrier-protecting bacteria, such as Lactobacillus and Bifidobacterium, but markedly reduced opportunistic pathogens, such as Enterobacteraceae and Streptococcaceae. These changes in the gut microbiota were also concomitant with significantly reduced serum levels of LBP (Zhang et al., 2013).
CRP
Metabolic endotoxemia, evoked by antigens that originate from gut microbiota (e.g. lipopolysaccharide, peptidoglycans, and flagellin) through the dysfunction intestinal barrier, induces chronic inflammation (Cani et al., 2007a, 2008; Everard & Cani, 2013). CRP, a stable downstream biomarker of inflammation, is the most widely measured acute-phase reactant responding to tissue injury, infection, and inflammation in clinical practice (Pepys & Baltz, 1983). A chronically increased level of CRP is associated with obesity (Hiura et al., 2003; Piéroni et al., 2003), atherosclerosis (Libby, 2002), and MetS (Fröhlich et al., 2000). In patients with MetS, CRP increases linearly with the number of abnormalities that comprise this syndrome (Pradhan et al., 2001; Devaraj et al., 2004). Therefore, a significant reduction in CRP levels through weight loss could reduce the risk of cardiovascular disease and other obesity-associated chronic diseases. Large studies have been conducted to investigate the association of weight loss by a dietary approach and CRP levels (Dietrich & Jialal, 2005). Heilbronn et al. (2001) found a 26% reduction in CRP plasma concentrations in 83 healthy obese women who performed 12 weeks of energy restriction using a low-fat diet with an average weight loss of 7.9 kg. The weight loss achieved by dietary programs ranged from 3 to 15 kg and was accompanied by a 7–48% reduction in CRP levels (Dietrich & Jialal, 2005). Gut microbiota alterations in obese subjects are associated with local and systemic inflammation, for example, plasma CRP was also increased in these subjects (P = 0.0005) and correlated with the Bacteroidetes/Firmicutes ratio (r = −0.41, P = 0.03) (Verdam et al., 2013). Brignardello et al. (2010) observed an inverse correlation between CRP concentrations and G + C abundance, suggesting that bacterial populations with high DNA GC contents may modulate inflammatory processes in the host. High-cocoa flavanol intervention in healthy human volunteers significantly reduced CRP concentrations, which correlated with the amounts of specific bacteria (Bifidobacteria: r = −0.438, P < 0.05; Lactobacilli: r = −0.492, P < 0.01) (Tzounis et al., 2011). The CRP level, which is downstream of LBP in the metabolic endotoxemia causal pathway, can thus be employed as a biomarker for inflammation in managing MetS (Fig. 1).
Fasting insulin
Insulin is a hormone central to regulating carbohydrate and fat metabolism, which causes cells in the liver, muscle, and fat tissue to take up glucose from the blood. Fasting plasma insulin (FPI) concentration is one of the most practical ways to estimate insulin resistance from the clinical perspective (Monzillo & Hamdy, 2003). Fasting insulin has been used in population-based studies, high values of which reflect the presence of insulin resistance (Bo et al., 2012; Gagnon et al., 2012; Zuo et al., 2013). There is a good correlation between FPI and insulin sensitivity derived from the hyperinsulinemic euglycemic clamp (Yeni-Komshian et al., 2000). Despite some limitations, such as the pulsatile mode of insulin secretion (Seino et al., 2011) and the lack of established standards for insulin assays (Staten et al., 2010; Kalathil et al., 2013), FPI level can still be an important biomarker for monitoring the trend of increasing insulin resistance.
HOMA-IR
Because fasting insulin does not provide an accurate evaluation of insulin sensitivity in MetS risk populations per se, this biochemical parameter has been incorporated in a formula for better estimation of insulin sensitivity. The HOMA is another method used to quantify insulin sensitivity and islet β-cell function (Matthews et al., 1985). HOMA is calculated from FPI and fasting plasma glucose (FPG), using the following mathematic formula: HOMA-IR = FPI × FPG/22.5. HOMA-IR correlates well with the glucose disposal rate derived from the hyperinsulinemic euglycemic clamp (Emoto et al., 1999; Katsuki et al., 2001), a low value of which indicates high insulin sensitivity. Therefore, HOMA-IR is most useful for the evaluation of insulin sensitivity in euglycemic individuals and in persons with mild diabetes for large population-based studies that require a simple method to assess insulin sensitivity. As it has become increasingly evident that gut microbiota plays a significant role in the development of insulin resistance (Delzenne & Cani, 2011; Shen et al., 2012; Vrieze et al., 2012), insulin sensitivity indicators, fasting insulin, and HOMA-IR index were widely used in the nutritional modulation of gut microbiota in the context of obesity and insulin resistance (Gao et al., 2009; Dewulf et al., 2013; Vulevic et al., 2013).
Trajectory analysis
Developmental trajectories of individuals entering MetS or in the process of interventions are poorly understood (Franco et al., 2009). The trend in an individual person is more important than a single point value that stays at the ‘normal range’. Tabák et al. (2009) analyzed data from a prospective occupational cohort study of 6538 British civil servants without diabetes mellitus as the baseline. During a median follow-up period of 9.7 years, 505 diabetes cases were diagnosed. In this group, a linear increase in fasting glucose was followed by a steep quadratic increase starting 3 years before the diagnosis of diabetes, and HOMA insulin sensitivity decreased steeply during the 5 years before diagnosis. This suggests that a description of biomarker trajectories leading to diabetes diagnosis could contribute to more-accurate risk prediction models that use repeated measures available to patients through regular checkups.
In fact, except for LBP, the biomarkers we propose have been widely used in clinical practice. However, both doctors and patients regard these biomarkers as diagnostics and pay more attention to the exact value of the biomarkers relative to the normal range at a single time point. To support a preventive public health program, these biomarkers should be continuously monitored through regular checkups. A trajectory for each biomarker in each individual over a long period of time should be used to monitor the increase in disease risk long before real disease symptoms become manifest. An ‘inflection point’ in the curve could indicate increased risks of illness, even though the exact value is still below the diagnostic threshold. A continued increase in LBP, CRP, and HOMA-IR in an individual is a strong indication that the endotoxin level is increasing in the bloodstream and that the immune system is responding by increasing inflammation, leading to increased insulin resistance. Conversely, continued decrease in these biomarkers in an individual over a period of dietary intervention may be a good indication that the intervention is effectively reducing disease risk. Due to the fact that these parameters can be routinely checked in community clinics, an eHealth database could be established for the local population to construct each individual's health trajectory for the predictive, preventative, preemptive, and personalized management of metabolic diseases.
Conclusion
The nutritional modulation strategy for gut microbiota causes changes in biomarker profiles indicative of metabolic endotoxemia, systemic inflammation, and reduced insulin resistance in MetS patients. Monitoring the trajectories of biomarker profiles will provide quantitative, dynamic, and translational methods aimed at the root cause for evaluating dysbiosis-induced insulin resistance, as well as the degree of success in preventing MetS by dietary treatment. Prospective studies in different populations are needed to further confirm the association between the gut microbiota-based biomarker panel and the development of MetS, and to evaluate the success of applying trajectory analyses of related biomarkers in managing MetS for the prevention of metabolic diseases.
Acknowledgments
The authors would like to acknowledge support from the National Natural Science Foundation of China Program Grant 30730005 and 973 Program Grant 2007CB513002. The authors have no conflict of interest to declare.
References
- Ajslev T, Andersen C, Gamborg M, Sørensen T. Jess T. Childhood overweight after establishment of the gut microbiota: the role of delivery mode, pre-pregnancy weight and early administration of antibiotics. Int J Obes (Lond) 2011;35:522–529. doi: 10.1038/ijo.2011.27. [DOI] [PubMed] [Google Scholar]
- Alberti KGMM, Zimmet P. Shaw J. The metabolic syndrome—a new worldwide definition. Lancet. 2005;366:1059–1062. doi: 10.1016/S0140-6736(05)67402-8. [DOI] [PubMed] [Google Scholar]
- Alberti KGMM, Zimmet P. Shaw J. Metabolic syndrome—a new world-wide definition. A consensus statement from the International Diabetes Federation. Diabet Med. 2006;23:469–480. doi: 10.1111/j.1464-5491.2006.01858.x. [DOI] [PubMed] [Google Scholar]
- Alvarez JA, Higgins PB, Oster RA, Fernandez JR, Darnell BE. Gower BA. Fasting and postprandial markers of inflammation in lean and overweight children. Am J Clin Nutr. 2009;89:1138–1144. doi: 10.3945/ajcn.2008.26926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An HM, Park SY, Lee DK, Kim JR, Cha MK, Lee SW, Lim HT, Kim KJ. Ha NJ. Antiobesity and lipid-lowering effects of Bifidobacterium spp. in high fat diet-induced obese rats. Lipids Health Dis. 2011;10:116. doi: 10.1186/1476-511X-10-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ärnlöv J, Ingelsson E, Sundström J. Lind L. Impact of body mass index and the metabolic syndrome on the risk of cardiovascular disease and death in middle-aged men. Circulation. 2010;121:230–236. doi: 10.1161/CIRCULATIONAHA.109.887521. [DOI] [PubMed] [Google Scholar]
- Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF. Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. P Natl Acad Sci USA. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bays H, Evans JL, Maki KC, Evans M, Maquet V, Cooper R. Anderson JW. Chitin-glucan fiber effects on oxidized low-density lipoprotein: a randomized controlled trial. Eur J Clin Nutr. 2012;67:2–7. doi: 10.1038/ejcn.2012.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bo S, Musso G, Gambino R, Villois P, Gentile L, Durazzo M, Cavallo-Perin P. Cassader M. Prognostic implications for insulin-sensitive and insulin-resistant normal-weight and obese individuals from a population-based cohort. Am J Clin Nutr. 2012;96:962–969. doi: 10.3945/ajcn.112.040006. [DOI] [PubMed] [Google Scholar]
- Brignardello J, Morales P, Diaz E, Romero J, Brunser O. Gotteland M. Pilot study: alterations of intestinal microbiota in obese humans are not associated with colonic inflammation or disturbances of barrier function. Aliment Pharmacol Ther. 2010;32:1307–1314. doi: 10.1111/j.1365-2036.2010.04475.x. [DOI] [PubMed] [Google Scholar]
- Bulló M, Casas-Agustench P, Amigó-Correig P, Aranceta J. Salas-Salvadó J. Inflammation, obesity and comorbidities: the role of diet. Public Health Nutr. 2007;10:1164–1172. doi: 10.1017/S1368980007000663. [DOI] [PubMed] [Google Scholar]
- Caesar R, Fåk F. Bäckhed F. Effects of gut microbiota on obesity and atherosclerosis via modulation of inflammation and lipid metabolism. J Intern Med. 2010;268:320–328. doi: 10.1111/j.1365-2796.2010.02270.x. [DOI] [PubMed] [Google Scholar]
- Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J. Shoelson SE. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat Med. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cani PD. Delzenne NM. Interplay between obesity and associated metabolic disorders: new insights into the gut microbiota. Curr Opin Pharmacol. 2009;9:737–743. doi: 10.1016/j.coph.2009.06.016. [DOI] [PubMed] [Google Scholar]
- Cani PD. Delzenne NM. The gut microbiome as therapeutic target. Pharmacol Ther. 2011;130:202–212. doi: 10.1016/j.pharmthera.2011.01.012. [DOI] [PubMed] [Google Scholar]
- Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007a;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- Cani PD, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM, Gibson GR. Delzenne NM. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia. 2007b;50:2374–2383. doi: 10.1007/s00125-007-0791-0. [DOI] [PubMed] [Google Scholar]
- Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM. Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- Cani PD, Lecourt E, Dewulf EM, Sohet FM, Pachikian BD, Naslain D, De Backer F, Neyrinck AM. Delzenne NM. Gut microbiota fermentation of prebiotics increases satietogenic and incretin gut peptide production with consequences for appetite sensation and glucose response after a meal. Am J Clin Nutr. 2009;90:1236–1243. doi: 10.3945/ajcn.2009.28095. [DOI] [PubMed] [Google Scholar]
- Cesaro C, Tiso A, Del Prete A, Cariello R, Tuccillo C, Cotticelli G, del Vecchio Blanco C. Loguercio C. Gut microbiota and probiotics in chronic liver diseases. Dig Liver Dis. 2011;43:431–438. doi: 10.1016/j.dld.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Chen JJ, Wang R, Li X-F. Wang R-L. Bifidobacterium longum supplementation improved high-fat-fed-induced metabolic syndrome and promoted intestinal Reg I gene expression. Exp Biol Med (Maywood) 2011;236:823–831. doi: 10.1258/ebm.2011.010399. [DOI] [PubMed] [Google Scholar]
- Coats SR, Berezow AB, To TT, Jain S, Bainbridge BW, Banani KP. Darveau RP. The lipid A phosphate position determines differential host Toll-like receptor 4 responses to phylogenetically related symbiotic and pathogenic bacteria. Infect Immun. 2011;79:203–210. doi: 10.1128/IAI.00937-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox LM. Blaser MJ. Pathways in microbe-induced obesity. Cell Metab. 2013;17:883–894. doi: 10.1016/j.cmet.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creely SJ, McTernan PG, Kusminski CM, Fisher M, Da Silva NF, Khanolkar M, Evans M, Harte AL. Kumar S. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am J Physiol Endocrinol Metab. 2007;292:E740–E747. doi: 10.1152/ajpendo.00302.2006. [DOI] [PubMed] [Google Scholar]
- Delzenne NM. Cani PD. Nutritional modulation of gut microbiota in the context of obesity and insulin resistance: potential interest of prebiotics. Int Dairy J. 2010;20:277–280. [Google Scholar]
- Delzenne NM. Cani PD. Gut microbiota and the pathogenesis of insulin resistance. Curr Diab Rep. 2011;11:154–159. doi: 10.1007/s11892-011-0191-1. [DOI] [PubMed] [Google Scholar]
- Delzenne NM, Neyrinck AM. Cani PD. Modulation of the gut microbiota by nutrients with prebiotic properties: consequences for host health in the context of obesity and metabolic syndrome. Microb Cell Fact. 2011;10:S10. doi: 10.1186/1475-2859-10-S1-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien M, Vaughan EE, Plugge CM. de Vos WM. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol. 2004;54:1469–1476. doi: 10.1099/ijs.0.02873-0. [DOI] [PubMed] [Google Scholar]
- Després JP, Poirier P, Bergeron J, Tremblay A, Lemieux I. Alméras N. From individual risk factors and the metabolic syndrome to global cardiometabolic risk. Eur Heart J Suppl. 2008;10:B24–B33. [Google Scholar]
- Devaraj S, Rosenson RS. Jialal I. Metabolic syndrome: an appraisal of the pro-inflammatory and procoagulant status. Endocrinol Metab Clin North Am. 2004;33:431–453. doi: 10.1016/j.ecl.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Dewulf EM, Cani PD, Claus SP, Fuentes S, Puylaert PG, Neyrinck AM, Bindels LB, de Vos WM, Gibson GR. Thissen J-P. Insight into the prebiotic concept: lessons from an exploratory, double blind intervention study with inulin-type fructans in obese women. Gut. 2013;62:1112–1121. doi: 10.1136/gutjnl-2012-303304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich M. Jialal I. The effect of weight loss on a stable biomarker of inflammation, C-reactive protein. Nutr Rev. 2005;63:22–28. doi: 10.1111/j.1753-4887.2005.tb00107.x. [DOI] [PubMed] [Google Scholar]
- Eckel RH, Grundy SM. Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:1415–1428. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- Emoto M, Nishizawa Y, Maekawa K, Hiura Y, Kanda H, Kawagishi T, Shoji T, Okuno Y. Morii H. Homeostasis model assessment as a clinical index of insulin resistance in type 2 diabetic patients treated with sulfonylureas. Diabetes Care. 1999;22:818–822. doi: 10.2337/diacare.22.5.818. [DOI] [PubMed] [Google Scholar]
- Erridge C, Bennett-Guerrero E. Poxton IR. Structure and function of lipopolysaccharides. Microbes Infect. 2002;4:837–851. doi: 10.1016/s1286-4579(02)01604-0. [DOI] [PubMed] [Google Scholar]
- Erridge C, Attina T, Spickett CM. Webb DJ. A high-fat meal induces low-grade endotoxemia: evidence of a novel mechanism of postprandial inflammation. Am J Clin Nutr. 2007;86:1286–1292. doi: 10.1093/ajcn/86.5.1286. [DOI] [PubMed] [Google Scholar]
- Everard A. Cani PD. Diabetes, obesity and gut microbiota. Best Pract Res Clin Gastroenterol. 2013;27:73–83. doi: 10.1016/j.bpg.2013.03.007. [DOI] [PubMed] [Google Scholar]
- Everard A, Lazarevic V, Derrien M, Girard M, Muccioli GG, Neyrinck AM, Possemiers S, Van Holle A, François P. de Vos WM. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes. 2011;60:2775–2786. doi: 10.2337/db11-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG. Delzenne NM. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. P Natl Acad Sci USA. 2013;110:9066–9071. doi: 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fåk F. Bäckhed F. Lactobacillus reuteri prevents diet-induced obesity, but not atherosclerosis, in a strain dependent fashion in Apoe−/− mice. PLoS One. 2012;7:e46837. doi: 10.1371/journal.pone.0046837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayh APT, Lopes AL, da Silva AMV, Reischak-Oliveira Á. Friedman R. Effects of 5% weight loss through diet or diet plus exercise on cardiovascular parameters of obese: a randomized clinical trial. Eur J Nutr. 2013;52:1443–1450. doi: 10.1007/s00394-012-0450-1. [DOI] [PubMed] [Google Scholar]
- Fei N. Zhao L. An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J. 2013;7:880–884. doi: 10.1038/ismej.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco OH, Massaro JM, Civil J, Cobain MR, O'Malley B. D'Agostino RB. Trajectories of entering the metabolic syndrome. Circulation. 2009;120:1943–1950. doi: 10.1161/CIRCULATIONAHA.109.855817. [DOI] [PubMed] [Google Scholar]
- Fröhlich M, Imhof A, Berg G, Hutchinson WL, Pepys MB, Boeing H, Muche R, Brenner H. Koenig W. Association between C-reactive protein and features of the metabolic syndrome. Diabetes Care. 2000;23:1835–1839. doi: 10.2337/diacare.23.12.1835. [DOI] [PubMed] [Google Scholar]
- Gagnon C, Lu ZX, Magliano DJ, Dunstan DW, Shaw JE, Zimmet PZ, Sikaris K, Ebeling PR. Daly RM. Low serum 25-hydroxyvitamin D is associated with increased risk of the development of the metabolic syndrome at five years: results from a national, population-based prospective study (The Australian Diabetes, Obesity and Lifestyle Study: AusDiab) J Clin Endocrinol Metab. 2012;97:1953–1961. doi: 10.1210/jc.2011-3187. [DOI] [PubMed] [Google Scholar]
- Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, Cefalu WT. Ye J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. 2009;58:1509–1517. doi: 10.2337/db08-1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanim H, Abuaysheh S, Sia CL, Korzeniewski K, Chaudhuri A, Fernandez-Real JM. Dandona P. Increase in plasma endotoxin concentrations and the expression of toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal. Diabetes Care. 2009;32:2281–2287. doi: 10.2337/dc09-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregor M. Hotamisligil G. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- Grundy SM. Pre-diabetes, metabolic syndrome, and cardiovascular risk. J Am Coll Cardiol. 2012;59:635–643. doi: 10.1016/j.jacc.2011.08.080. [DOI] [PubMed] [Google Scholar]
- Grundy SM, Cleeman JI, Daniels SR, et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute scientific statement. Curr Opin Cardiol. 2006;21:1–6. doi: 10.1097/01.hco.0000200416.65370.a0. [DOI] [PubMed] [Google Scholar]
- Gubern C, Lopez-Bermejo A, Biarnes J, Vendrell J, Ricart W. Fernandez-Real JM. Natural antibiotics and insulin sensitivity: the role of bactericidal/permeability-increasing protein. Diabetes. 2006;55:216–224. [PubMed] [Google Scholar]
- Guerra RA, Casafont F, Crespo J, Cayón A, Mayorga M, Estebanez A, Fernadez-Escalante J. Pons-Romero F. Lipopolysaccharide-binding protein plasma levels and liver TNF-alpha gene expression in obese patients: evidence for the potential role of endotoxin in the pathogenesis of non-alcoholic steatohepatitis. Obes Surg. 2007;17:1374–1380. doi: 10.1007/s11695-007-9243-7. [DOI] [PubMed] [Google Scholar]
- Haffner SM. The metabolic syndrome: inflammation, diabetes mellitus, and cardiovascular disease. Am J Cardiol. 2006;97:3–11. doi: 10.1016/j.amjcard.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Hajer GR, Van Der Graaf Y, Olijhoek JK, Verhaar MC. Visseren FLJ. Levels of homocysteine are increased in metabolic syndrome patients but are not associated with an increased cardiovascular risk, in contrast to patients without the metabolic syndrome. Heart. 2007;93:216–220. doi: 10.1136/hrt.2006.093971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakansson A. Molin G. Gut microbiota and inflammation. Nutrients. 2011;3:637–682. doi: 10.3390/nu3060637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen CHF, Krych L, Nielsen DS, Vogensen FK, Hansen LH, Sørensen SJ, Buschard K. Hansen A. Early life treatment with vancomycin propagates Akkermansia muciniphila and reduces diabetes incidence in the NOD mouse. Diabetologia. 2012;55:2285–2294. doi: 10.1007/s00125-012-2564-7. [DOI] [PubMed] [Google Scholar]
- Heilbronn LK, Noakes M. Clifton PM. Energy restriction and weight loss on very-low-fat diets reduce C-reactive protein concentrations in obese, healthy women. Arterioscler Thromb Vasc Biol. 2001;21:968–970. doi: 10.1161/01.atv.21.6.968. [DOI] [PubMed] [Google Scholar]
- Hiura M, Kikuchi T, Nagasaki K. Uchiyama M. Elevation of serum C-reactive protein levels is associated with obesity in boys. Hypertens Res. 2003;26:541–546. doi: 10.1291/hypres.26.541. [DOI] [PubMed] [Google Scholar]
- Hotamisligil G, Shargill N. Spiegelman B. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- Imayama I, Ulrich CM, Alfano CM, Wang C, Xiao L, Wener MH, Campbell KL, Duggan C, Foster-Schubert KE. Kong A. Effects of a caloric restriction weight loss diet and exercise on inflammatory biomarkers in overweight/obese postmenopausal women: a randomized controlled trial. Cancer Res. 2012;72:2314–2326. doi: 10.1158/0008-5472.CAN-11-3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalathil S, Napier C, Pattman S, Wark G, Abouglila K. James R. Variable characteristics with insulin assays. Pract Diab. 2013;30:118–120. [Google Scholar]
- Kaneto H, Nakatani Y, Miyatsuka T, Kawamori D, Matsuoka T, Matsuhisa M, Kajimoto Y, Ichijo H, Yamasaki Y. Hori M. Possible novel therapy for diabetes with cell-permeable JNK-inhibitory peptide. Nat Med. 2004;10:1128–1132. doi: 10.1038/nm1111. [DOI] [PubMed] [Google Scholar]
- Karlsson CL, Önnerfält J, Xu J, Molin G, Ahrné S. Thorngren-Jerneck K. The microbiota of the gut in preschool children with normal and excessive body weight. Obesity. 2012;20:2257–2261. doi: 10.1038/oby.2012.110. [DOI] [PubMed] [Google Scholar]
- Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, Nielsen J. Bäckhed F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 2013;498:99–103. doi: 10.1038/nature12198. [DOI] [PubMed] [Google Scholar]
- Kassi E, Pervanidou P, Kaltsas G. Chrousos G. Metabolic syndrome: definitions and controversies. BMC Med. 2011;9:48. doi: 10.1186/1741-7015-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuki A, Sumida Y, Gabazza EC, Murashima S, Furuta M, Araki-Sasaki R, Hori Y, Yano Y. Adachi Y. Homeostasis model assessment is a reliable indicator of insulin resistance during follow-up of patients with type 2 diabetes. Diabetes Care. 2001;24:362–365. doi: 10.2337/diacare.24.2.362. [DOI] [PubMed] [Google Scholar]
- Kelly CJ, Colgan SP. Frank DN. Of microbes and meals: the health consequences of dietary endotoxemia. Nutr Clin Pract. 2012;27:215–225. doi: 10.1177/0884533611434934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E. Insulin resistance at the crossroads of metabolic syndrome: systemic analysis using microarrays. Biotechnol J. 2010;5:919–929. doi: 10.1002/biot.201000048. [DOI] [PubMed] [Google Scholar]
- Kimura I, Ozawa K, Inoue D, Imamura T, Kimura K, Maeda T, Terasawa K, Kashihara D, Hirano K. Tani T. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun. 2013;4:1829. doi: 10.1038/ncomms2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovatcheva-Datchary P. Arora T. Nutrition, the gut microbiome and the metabolic syndrome. Best Pract Res Clin Gastroenterol. 2013;27:59–72. doi: 10.1016/j.bpg.2013.03.017. [DOI] [PubMed] [Google Scholar]
- Lajunen T, Vikatmaa P, Bloigu A, Ikonen T, Lepäntalo M, Pussinen PJ, Saikku P. Leinonen M. Chlamydial LPS and high-sensitivity CRP levels in serum are associated with an elevated body mass index in patients with cardiovascular disease. Innate Immun. 2008;14:375–382. doi: 10.1177/1753425908099172. [DOI] [PubMed] [Google Scholar]
- Lann D. LeRoith D. Insulin resistance as the underlying cause for the metabolic syndrome. Med Clin North Am. 2007;91:1063–1077. doi: 10.1016/j.mcna.2007.06.012. [DOI] [PubMed] [Google Scholar]
- Lee JG, Lee S, Kim YJ, Jin HK, Cho BM, Kim YJ, Jeong DW, Park HJ. Kim JE. Multiple biomarkers and their relative contributions to identifying metabolic syndrome. Clin Chim Acta. 2009;408:50–55. doi: 10.1016/j.cca.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Lepper P, Schumann C, Triantafilou K, Rasche F, Schuster T, Frank H, Schneider E, Triantafilou M. von Eynatten M. Association of lipopolysaccharide-binding protein and coronary artery disease in men. J Am Coll Cardiol. 2007;50:25–31. doi: 10.1016/j.jacc.2007.02.070. [DOI] [PubMed] [Google Scholar]
- Ley RE. Obesity and the human microbiome. Curr Opin Gastroenterol. 2010;26:5–11. doi: 10.1097/MOG.0b013e328333d751. [DOI] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S. Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- Lindberg AA, Weintraub A, Zähringer U. Rietschel ET. Structure-activity relationships in lipopolysaccharides of Bacteroides fragilis. Rev Infect Dis. 1990;12:S133–S141. doi: 10.1093/clinids/12.supplement_2.s133. [DOI] [PubMed] [Google Scholar]
- Liou AP, Paziuk M, Luevano J-M, Jr, Machineni S, Turnbaugh PJ. Kaplan LM. Conserved shifts in the gut microbiota due to gastric bypass reduce host weight and adiposity. Sci Transl Med. 2013;5:178ra141. doi: 10.1126/scitranslmed.3005687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manco M, Putignani L. Bottazzo GF. Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr Rev. 2010;31:817–844. doi: 10.1210/er.2009-0030. [DOI] [PubMed] [Google Scholar]
- Mansfield MW, Heywood DM. Grant PJ. Circulating levels of factor VII, fibrinogen, and von Willebrand factor and features of insulin resistance in first-degree relatives of patients with NIDDM. Circulation. 1996;94:2171–2176. doi: 10.1161/01.cir.94.9.2171. [DOI] [PubMed] [Google Scholar]
- Matsuura M. Structural modifications of bacterial lipopolysaccharide that facilitate Gram-negative bacteria evasion of host innate immunity. Front Immunol. 2013;4:109. doi: 10.3389/fimmu.2013.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews D, Hosker J, Rudenski A, Naylor B, Treacher D. Turner R. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- Meigs JB, Muller DC, Nathan DM, Blake DR. Andres R. The natural history of progression from normal glucose tolerance to type 2 diabetes in the Baltimore Longitudinal Study of Aging. Diabetes. 2003;52:1475–1484. doi: 10.2337/diabetes.52.6.1475. [DOI] [PubMed] [Google Scholar]
- Moller D. Berger J. Role of PPARs in the regulation of obesity-related insulin sensitivity and inflammation. Int J Obes (Lond) 2003;27:S17–S21. doi: 10.1038/sj.ijo.0802494. [DOI] [PubMed] [Google Scholar]
- Monteiro R. Azevedo I. Chronic inflammation in obesity and the metabolic syndrome. Mediators Inflamm. 2010;2010:289645. doi: 10.1155/2010/289645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monzillo LU. Hamdy O. Evaluation of insulin sensitivity in clinical practice and in research settings. Nutr Rev. 2003;61:397–412. doi: 10.1301/nr.2003.dec.397-412. [DOI] [PubMed] [Google Scholar]
- Moreira APB, Texeira TFS, Ferreira AB, do Carmo Gouveia Peluzio M. de Cássia Gonçalves Alfenas R. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br J Nutr. 2012;108:801–809. doi: 10.1017/S0007114512001213. [DOI] [PubMed] [Google Scholar]
- Moreno-Navarrete JM, Ortega F, Serino M, et al. Circulating lipopolysaccharide-binding protein (LBP) as a marker of obesity-related insulin resistance. Int J Obes (Lond) 2012;36:1442–1449. doi: 10.1038/ijo.2011.256. [DOI] [PubMed] [Google Scholar]
- Morrison DC. Ryan JL. Endotoxins and disease mechanisms. Annu Rev Med. 1987;38:417–432. doi: 10.1146/annurev.me.38.020187.002221. [DOI] [PubMed] [Google Scholar]
- Musso G, Gambino R. Cassader M. Obesity, diabetes, and gut microbiota: the hygiene hypothesis expanded? Diabetes Care. 2010;33:2277–2284. doi: 10.2337/dc10-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso G, Gambino R. Cassader M. Interactions between gut microbiota and host metabolism predisposing to obesity and diabetes. Annu Rev Med. 2011;62:361–380. doi: 10.1146/annurev-med-012510-175505. [DOI] [PubMed] [Google Scholar]
- Neuschwander-Tetri BA. Caldwell SH. Nonalcoholic steatohepatitis: summary of an AASLD Single Topic Conference. Hepatology. 2003;37:1202–1219. doi: 10.1053/jhep.2003.50193. [DOI] [PubMed] [Google Scholar]
- Neyrinck A, Van Hée V, Piront N, De Backer F, Toussaint O, Cani P. Delzenne N. Wheat-derived arabinoxylan oligosaccharides with prebiotic effect increase satietogenic gut peptides and reduce metabolic endotoxemia in diet-induced obese mice. Nutr Diabetes. 2012;2:e28. doi: 10.1038/nutd.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklas JM, Sacks FM, Smith SR, LeBoff MS, Rood JC, Bray GA. Ridker PM. Effect of dietary composition of weight loss diets on high-sensitivity C-reactive protein: the Randomized POUNDS LOST trial. Obesity. 2013;21:681–689. doi: 10.1002/oby.20072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novitsky TJ. Limitations of the Limulus amebocyte lysate test in demonstrating circulating lipopolysaccharides. Ann NY Acad Sci. 1998;851:416–421. doi: 10.1111/j.1749-6632.1998.tb09018.x. [DOI] [PubMed] [Google Scholar]
- Olsen MH, Hansen TW, Christensen MK, Gustafsson F, Rasmussen S, Wachtell K, Borch-Johnsen K, Ibsen H, Jorgensen T. Hildebrandt P. N-terminal pro brain natriuretic peptide is inversely related to metabolic cardiovascular risk factors and the metabolic syndrome. Hypertension. 2005;46:660–666. doi: 10.1161/01.HYP.0000179575.13739.72. [DOI] [PubMed] [Google Scholar]
- Onat A, Uyarel H, Hergenc G, Karabulut A, Albayrak S, Sari I, Yazici M. Keleş I. Serum uric acid is a determinant of metabolic syndrome in a population-based study. Am J Hypertens. 2006;19:1055–1062. doi: 10.1016/j.amjhyper.2006.02.014. [DOI] [PubMed] [Google Scholar]
- Parnell JA. Reimer RA. Weight loss during oligofructose supplementation is associated with decreased ghrelin and increased peptide YY in overweight and obese adults. Am J Clin Nutr. 2009;89:1751–1759. doi: 10.3945/ajcn.2009.27465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepys MB. Baltz ML. Acute phase proteins with special reference to C-reactive protein and related proteins (pentaxins) and serum amyloid A protein. Adv Immunol. 1983;34:141–212. doi: 10.1016/s0065-2776(08)60379-x. [DOI] [PubMed] [Google Scholar]
- Piéroni L, Bastard JP, Piton A, Khalil L, Hainque B. Jardel C. Interpretation of circulating C-reactive protein levels in adults: body mass index and gender are a must. Diabetes Metab. 2003;29:133–138. doi: 10.1016/s1262-3636(07)70019-8. [DOI] [PubMed] [Google Scholar]
- Pradhan AD, Manson JE, Rifai N, Buring JE. Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. J Am Med Assoc. 2001;286:327–334. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- Prasad A. Quyyumi AA. Renin-angiotensin system and angiotensin receptor blockers in the metabolic syndrome. Circulation. 2004;110:1507–1512. doi: 10.1161/01.CIR.0000141736.76561.78. [DOI] [PubMed] [Google Scholar]
- Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y. Shen D. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- Rydén L, Standl E, Małgorzata B, et al. Guidelines on diabetes, pre-diabetes, and cardiovascular diseases: executive summary. Eur Heart J. 2007;28:88–136. doi: 10.1093/eurheartj/ehl260. [DOI] [PubMed] [Google Scholar]
- Santacruz A, Marcos A, Wärnberg J, et al. Interplay between weight loss and gut microbiota composition in overweight adolescents. Obesity. 2009;17:1906–1915. doi: 10.1038/oby.2009.112. [DOI] [PubMed] [Google Scholar]
- Santacruz A, Collado M, García-Valdés L, Segura M, Martín-Lagos J, Anjos T, Marti-Romero M, Lopez R, Florido J. Campoy C. Gut microbiota composition is associated with body weight, weight gain and biochemical parameters in pregnant women. Br J Nutr. 2010;104:83–92. doi: 10.1017/S0007114510000176. [DOI] [PubMed] [Google Scholar]
- Sanz Y, Santacruz A. Gauffin P. Gut microbiota in obesity and metabolic disorders. Proc Nutr Soc. 2010;69:434–441. doi: 10.1017/S0029665110001813. [DOI] [PubMed] [Google Scholar]
- Scholze J, Alegria E, Ferri C, Langham S, Stevens W, Jeffries D. Uhl-Hochgraeber K. Epidemiological and economic burden of metabolic syndrome and its consequences in patients with hypertension in Germany, Spain and Italy; a prevalence-based model. BMC Public Health. 2010;10:529. doi: 10.1186/1471-2458-10-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder NWJ. Schumann RR. Non-LPS targets and actions of LPS binding protein (LBP) J Endotoxin Res. 2005;11:237–242. doi: 10.1179/096805105X37420. [DOI] [PubMed] [Google Scholar]
- Schumann RR, Leong SR, Flaggs GW, Gray PW, Wright SD, Mathison JC, Tobias PS. Ulevitch RJ. Structure and function of lipopolysaccharide binding protein. Science. 1990;249:1429–1433. doi: 10.1126/science.2402637. [DOI] [PubMed] [Google Scholar]
- Scott KP, Gratz SW, Sheridan PO, Flint HJ. Duncan SH. The influence of diet on the gut microbiota. Pharmacol Res. 2012;69:52–60. doi: 10.1016/j.phrs.2012.10.020. [DOI] [PubMed] [Google Scholar]
- Seino S, Shibasaki T. Minami K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J Clin Invest. 2011;121:2118–2125. doi: 10.1172/JCI45680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servais A, Giral P, Bernard M, Bruckert E, Deray G. Isnard Bagnis C. Is serum cystatin-C a reliable marker for metabolic syndrome? Am J Med. 2008;121:426–432. doi: 10.1016/j.amjmed.2008.01.040. [DOI] [PubMed] [Google Scholar]
- Shen J, Obin MS. Zhao L. The gut microbiota, obesity and insulin resistance. Mol Aspects Med. 2012;34:39–58. doi: 10.1016/j.mam.2012.11.001. [DOI] [PubMed] [Google Scholar]
- Sonnenburg JL, Xu J, Leip DD, Chen CH, Westover BP, Weatherford J, Buhler JD. Gordon JI. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science. 2005;307:1955–1959. doi: 10.1126/science.1109051. [DOI] [PubMed] [Google Scholar]
- Staten MA, Stern MP, Miller WG, Steffes MW. Campbell SE. Insulin assay standardization leading to measures of insulin sensitivity and secretion for practical clinical care. Diabetes Care. 2010;33:205–206. doi: 10.2337/dc09-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern MP, Williams K. Haffner SM. Identification of persons at high risk for type 2 diabetes mellitus: do we need the oral glucose tolerance test? Ann Intern Med. 2002;136:575–581. doi: 10.7326/0003-4819-136-8-200204160-00006. [DOI] [PubMed] [Google Scholar]
- Stern MP, Williams K, González-Villalpando C, Hunt KJ. Haffner SM. Does the metabolic-syndrome improve identification of individuals at risk of type 2 diabetes and/or cardiovascular disease? Diabetes Care. 2004;27:2676–2681. doi: 10.2337/diacare.27.11.2676. [DOI] [PubMed] [Google Scholar]
- Sun L, Yu Z, Ye X, et al. A marker of endotoxemia is associated with obesity and related metabolic disorders in apparently healthy Chinese. Diabetes Care. 2010;33:1925–1932. doi: 10.2337/dc10-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabák AG, Jokela M, Akbaraly TN, Brunner EJ, Kivimäki M. Witte DR. Trajectories of glycaemia, insulin sensitivity, and insulin secretion before diagnosis of type 2 diabetes: an analysis from the Whitehall II study. Lancet. 2009;373:2215–2221. doi: 10.1016/S0140-6736(09)60619-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Bäckhed F, Fulton L. Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3:213–223. doi: 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R. Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009;1:6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzounis X, Rodriguez-Mateos A, Vulevic J, Gibson GR, Kwik-Uribe C. Spencer JP. Prebiotic evaluation of cocoa-derived flavanols in healthy humans by using a randomized, controlled, double-blind, crossover intervention study. Am J Clin Nutr. 2011;93:62–72. doi: 10.3945/ajcn.110.000075. [DOI] [PubMed] [Google Scholar]
- Verdam FJ, Fuentes S, de Jonge C, Zoetendal EG, Erbil R, Greve JW, Buurman WA, de Vos WM. Rensen SS. Human intestinal microbiota composition is associated with local and systemic inflammation in obesity. Obesity. 2013 doi: 10.1002/oby.20466. DOI: 10.1002/oby.20466. [DOI] [PubMed] [Google Scholar]
- Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE. Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–231. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrieze A, Nood EV, Holleman F, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143:913–916. doi: 10.1053/j.gastro.2012.06.031. [DOI] [PubMed] [Google Scholar]
- Vulevic J, Juric A, Tzortzis G. Gibson GR. A mixture of trans-galactooligosaccharides reduces markers of metabolic syndrome and modulates the fecal microbiota and immune function of overweight adults. J Nutr. 2013;143:324–331. doi: 10.3945/jn.112.166132. [DOI] [PubMed] [Google Scholar]
- Wellen KE. Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. Preventing Chronic Diseases: A Vital Investment. Geneva: World Health Organization; 2005. WHO technical report series: 894, [Google Scholar]
- Wild SH. Byrne CD. The global burden of the metabolic syndrome and its consequences for diabetes and cardiovascular disease. In: Byrne CD, editor; Wild SH, editor. The Metabolic Syndrome. Chichester, UK: John Wiley & Sons, Inc; 2006. pp. 1–41. [Google Scholar]
- Xiao S, Fei N, Pang X, et al. A gut microbiota-targeted dietary intervention for amelioration of chronic inflammation underlying metabolic syndrome. FEMS Microbiol Ecol. 2013 doi: 10.1111/1574-6941.12228. DOI: 10.1111/1574-6941.12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS. Tartaglia LA. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeni-Komshian H, Carantoni M, Abbasi F. Reaven GM. Relationship between several surrogate estimates of insulin resistance and quantification of insulin-mediated glucose disposal in 490 healthy nondiabetic volunteers. Diabetes Care. 2000;23:171–175. doi: 10.2337/diacare.23.2.171. [DOI] [PubMed] [Google Scholar]
- Zeyda M. Stulnig TM. Adipose tissue macrophages. Immunol Lett. 2007;112:61–67. doi: 10.1016/j.imlet.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Zhang C, Zhang M, Wang S, et al. Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. ISME J. 2010;4:232–241. doi: 10.1038/ismej.2009.112. [DOI] [PubMed] [Google Scholar]
- Zhang C, Zhang M, Pang X, Zhao Y, Wang L. Zhao L. Structural resilience of the gut microbiota in adult mice under high-fat dietary perturbations. ISME J. 2012a;6:1848–1857. doi: 10.1038/ismej.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhao Y, Zhang M, Pang X, Xu J, Kang C, Li M, Zhang C, Zhang Z. Zhang Y. Structural changes of gut microbiota during berberine-mediated prevention of obesity and insulin resistance in high-fat diet-fed rats. PLoS One. 2012b;7:e42529. doi: 10.1371/journal.pone.0042529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Li S, Yang L, et al. Structural modulation of gut microbiota in life-long calorie-restricted mice. Nat Commun. 2013;4:2163. doi: 10.1038/ncomms3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L. The gut microbiota and obesity: from correlation to causality. Nat Rev Microbiol. 2013;11:639–647. doi: 10.1038/nrmicro3089. [DOI] [PubMed] [Google Scholar]
- Zhao L. Shen J. Whole-body systems approaches for gut microbiota-targeted, preventive healthcare. J Biotechnol. 2010;149:183–190. doi: 10.1016/j.jbiotec.2010.02.008. [DOI] [PubMed] [Google Scholar]
- Zuo H, Shi Z, Yuan B, Dai Y, Wu G. Hussain A. Association between serum leptin concentrations and insulin resistance: a population-based study from China. PLoS One. 2013;8:e54615. doi: 10.1371/journal.pone.0054615. [DOI] [PMC free article] [PubMed] [Google Scholar]