

Abstract
Background
The phylogenetic history of genes underlying phenotypic diversity can offer insight into the evolutionary origin of adaptive traits. This is especially true where single genes have large phenotypic effects, for example in determining polymorphic mimicry in butterflies. Here, we characterise the evolutionary history of two candidate genes for the mimicry switch in the polymorphic Batesian mimic Papilio dardanus coding for the transcription factors engrailed and invected.
Results
We show that phased haplotypes associated with the dominant morphs f. poultoni and f. planemoides are phylogenetically highly divergent, in particular at non-synonymous sites. Some non-synonymous changes are shared between the divergent alleles suggesting either convergence or a shared ancestry. Gene trees for invected do not show this pattern. Despite their great divergence, all engrailed alleles of P. dardanus were monophyletic with respect to alleles of closely related species. Phylogenetic analyses therefore reveal no evidence for introgression from other species. A McDonald-Kreitman test conducted on a population sample from South Africa confirms a significant excess of intraspecific non-synonymous diversity in P. dardanus engrailed, suggesting long-term balanced polymorphism at this locus.
Conclusions
The divergence between engrailed haplotypes suggests an evolutionary history distorted by selection with multiple changes reflecting recurrent selective sweeps. The high level of intraspecific polymorphism observed is characteristic of balancing selection on this locus, as expected if the gene engrailed is under phenotypic selection for the maintenance of multiple mimetic morphs. Non-synonymous changes in key functional portions of a major transcription factor are likely to be deleterious but if maintained in a dominant allele at low frequency, heterozygosity would reduce the associated genetic load.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2148-14-140) contains supplementary material, which is available to authorized users.
Keywords: Mimicry, Balanced polymorphism, Supergene, Engrailed, Phylogenetics, Molecular evolution
Background
A key aim of evolutionary biology is to understand the processes that give rise to novel traits. What is the nature of the genetic changes underlying adaptation? How are new alleles introduced into a population, and how are they maintained in the face of varying types of selection? One way of solving these questions is to study polymorphic species – the genetic control of a polymorphism within species can be used to test hypotheses regarding modification of developmental processes at broader taxic scales [1]. To find the genes responsible, we can study nucleotide variation and search for characteristic signatures that selection will leave in the diversity of alleles in a species [2]. Such ‘signatures of selection’ can corroborate the role of the genes in determining the polymorphism. The identity of these genes will contribute to our understanding of the processes generating and maintaining evolutionary diversity.
Another key question underlying studies of phenotypic evolution is how complex phenotypes come under precise genetic control. Locally polymorphic species pose a particular challenge: multiple phenotypic optima are occupied by individuals from a single population requiring precise determination of the various discrete phenotypes despite mating between individuals of different morphs. Such situations can lead to the evolution of supergenes [3] where a single genetic locus comes to determine the polymorphism. Whatever the exact genomic architecture of the supergene, it seems likely that switching between alternate phenotypes is accomplished by differential regulation of effector genes. The ‘cis-regulatory’ hypothesis for morphological adaptation states that the regulatory regions of genes can evolve at a higher rate (and with fewer constraints) than the protein-coding regions of genes [4, 5]. This hypothesis predicts that morphological evolution will tend to arise through regulatory changes, often in cis-regulatory control of conserved genes involved in developmental processes [6].
Lepidopteran wing patterns are some of the best known examples of adaptive colouration and include some textbook examples of natural selection in action, including industrial melanism in Biston betularia and Müllerian mimicry in Heliconius butterflies [7–9]. These systems have revealed that wing pattern diversification is controlled by a small number of genes with alleles of large effect [10–12]. When the genomic regions containing these genes are analysed phylogenetically, they often display an evolutionary history that is discordant with that of the rest of the genome, producing topologies that group similar phenotypes together irrespective of species boundaries and geographic structure [13, 14]. In some cases these patterns have resulted from adaptive introgression, or collateral evolution by allele sharing [10, 15]. Reconstructing phylogenies of genomic regions that control phenotypic diversity can therefore be a powerful method for verifying their involvement in generating and maintaining phenotypic diversity, and in increasing our understanding of the processes giving rise to novel and adaptive phenotypes (e.g. [16]). Recent advances in unpicking the supergene underlying Batesian mimicry in Papilio polytes [12] offers insight into the nature and function of supergenes. In this case, a single coding region, doublesex, was found to determine female polymorphism, possibly through differential expression of isoforms of doublesex. High levels of synonymous and non-synonymous polymorphism were found in the doublesex coding region and alternative alleles were found to be highly divergent.
The African Mocker Swallowtail Papilio dardanus is a polymorphic female-limited Batesian mimic. At least 14 different female wing pattern morphs can be distinguished in P. dardanus ([17, 18], Figure 1). Wing pattern determination in females of P. dardanus maps to a single supergene locus, termed H [19]. Dominance of colour morphs is complete for most crosses between sympatric individuals, although there is partial breakdown of dominance when butterflies from different subspecies are crossed [19–23].
Figure 1.

Phenotypic variation in P. dardanus and the organisation of the H locus. (A) Female forms of P. dardanus (below the horizontal line) and examples of their model species in a number of African mimicry rings. The P. dardanus male pattern is shown boxed at the left along with male-like female forms. (B) Genomic organisation of the H locus showing the extent of exons (vertical blue bars) and introns (blue line) of the en and inv genes along the known sequence of the region. The bottom panels show the significance level for associations of SNP in the respective exons with three dominant morphs. The shading indicates the significance threshold for association. Note that significant association is found only in en (mostly exon 1). The figure is reproduced in an altered form from [25].
The H locus has been mapped to a 13.9 cM linkage group and shown to co-segregate with the gene coding for the transcription factor invected and possibly its paralogue engrailed that maps to the same genomic region [24]. More recent work has used a comparative genomic approach to physically map this candidate region and carry out further recombination and SNP-association analyses to test for differences among morphs in this region [25]. Around 24 genes co-segregate with H and are broadly syntenic with previously sequenced lepidopteran genomes. Despite the lack of resolution in positional cloning of H using pedigree information, this study furthermore demonstrated that the forms f. cenea, f. poultoni and f. planemoides showed significant association with SNP variants in engrailed [25] (see Figure 1). None of the other 23 genes showed these SNP associations with all three phenotypes, leading to the conclusion that engrailed alone is the prime candidate for the mimicry switch locus, H. Additionally, linkage disequilibrium in the region was found to be relatively low, but was high for SNPs within engrailed, as would be expected if this locus is a supergene harbouring multiple allelic sites which determine alternative phenotypes and which need to be maintained without recombination to avoid the formation of maladaptive intermediates. Finally, the comparative genomics analysis also revealed evidence for balancing selection based on the non-neutral distribution of SNP variation in the engrailed coding region [25].
These results indicate that engrailed and possibly invected are strong candidates for the P. dardanus mimicry switch. These genes are highly conserved developmental regulatory genes present in all hexapods [26]. If these genes do indeed determine the wing pattern, nucleotide variation will be affected by selection on the phenotype and therefore may have an evolutionary history discordant with that of unlinked markers, which were shown to follow a predominantly geographic structure [27]. Specifically, phylogenetic analysis of mitochondrial markers in P. dardanus has revealed deeply diverging lineages comprising Eastern and Western mainland African clades and an Indian Ocean clade, but no comparable separation has been detected in nuclear markers including the invected gene and two linked AFLP loci [27]. We additionally predict that similar phenotypes will share alleles at H regardless of their geographic origin; individuals of a given morph are therefore expected to be phylogenetically closely related at loci determining the phenotype (i.e. the H locus).
Here we use gene phylogenies for coding regions of engrailed and invected to study the phylogenetic history of these regions within P. dardanus and across the genus Papilio. Previous work has also demonstrated that P. dardanus forms hybrids in the wild with other Papilio species [28] and gene flow from other species may be a significant source of evolutionary novelty [10]. We therefore test the monophyly of P. dardanus alleles and the possibility that divergent alleles have undergone introgression, while also establishing a baseline of divergence in these loci beyond the P. dardanus clade. A phylogenetic approach will also be useful for the analysis of engrailed and invected alleles within the P. dardanus lineage to characterise the kind of nucleotide divergence distinguishing the various morphs. Given the known LD in the engrailed coding region the phase of SNP variation can be computationally inferred to obtain distinct alleles whose variation may be tree like and amenable to phylogenetic inference. Variation at these loci within P. dardanus is then further investigated, relative to population divergence in mitochondrial markers. Finally, tests for selection are applied to reveal associations of haplotypes in engrailed and invected with wing pattern and to give insight into the molecular evolution of this region.
Materials and methods
Papilio dardanus specimens were chosen to maximise overlap with previously published taxon sets [25, 27], whilst also adding samples from additional populations from either side of the documented mitochondrial lineage break (East African Rift Valley). Specimens were taken from existing collections of the Natural History Museum (NHM, London), the Afrotropical Butterfly Research Institute Kenya (ABRI, Nairobi) and Stratford Butterfly Farm (Stratford, UK). Additional sampling was carried out in Ghana and South Africa, with further specimens of P. dardanus dardanus from Western Kenya (Kakamega Forest) provided by S. C. Collins (ABRI). Sampling locations are plotted in Figure 2. In addition, P. phorcas, P. constantinus and other outgroups from the genus Papilio were purchased from Stratford Butterfly Farm. A full list of samples used is presented in Additional file 1: Tables S1.
Figure 2.
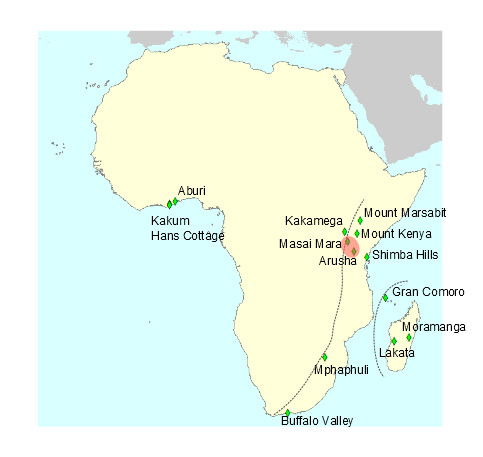
Sampling locations of P. dardanus used in this study. Dotted gray lines separate the Eastern, Western and Indian Ocean lineages. The shaded area is P. dardanus meseres, believed to be a contact zone between the Eastern and Western mainland lineages.
DNA was extracted from legs of fresh or frozen specimens using a Qiagen DNeasy kit. For dried museum collections material (either pinned or stored in envelopes) the protocols of Thomsen et al. [29] were followed. Briefly, this involves removal of a leg of the butterfly, incubation overnight in a buffer with protease and a purification step using a Qiagen QIAquick PCR cleanup kit. Primer sequences [25, 28, 30, 31] are provided in Additional file 2: Table S2. PCR products were bi-directionally Sanger sequenced using ABI technology. Sequences were checked and edited using Geneious (Version 6.1 [32] and aligned using the MAFFT [33] plugin for Geneious with default settings. Numbers of variable sites (including parsimony-informative, synonymous and non-synonymous sites) were calculated for each alignment in SITES [34].
To improve the accuracy of haplotype inference, allelic phase data was directly obtained for 2 individuals per morph through cloning of engrailed-derived PCR amplicons with Invitrogen TOPO TA vector and Invitrogen chemically competent E. coli grown on kanamycin/LB agar. PCR was performed on picked colonies directly prior to Sanger sequencing (sequences submitted to Genbank, accessions KJ507618-KJ507653). In addition, haplotypic phase data was obtained by sequencing H-linked PCR amplicons for 8 specimens (Additional file 3: Table S3) on a Illumina MiSeq (200 bp PE with v2 500 cycle kit, Nextera library prep at an NHM in-house sequencing facility). Resulting reads were trimmed in Geneious version 6.1 [32] using default settings and mapped to a BAC reference (Genbank accession FM995623.2) using Geneious read-mapper ([32], settings: 15% gaps per read, gap size 50, no words >20x, word length 14, index length 12. 30% mismatches per read, maximum ambiguity 4-fold, iterated 5 times). Paired-end information was used to improve the quality of the mapping, using only matches mapping nearby and ignoring multiple matches. The mapping resulted in coverage for each targeted region in excess of 100x, files of reads mapping to the BAC reference have been submitted to SRA (study PRJEB5625, ERP005044). The resulting pileups were used to call haplotypes using GATK read-backed phasing (GATK version 2.3-9, [36, 37]). Only those haplotypes obtained through cloning or Illumina amplicon sequencing were used to represent P. dardanus in the genus-level phylogenies (note that this sample includes all studied morphs). Individuals of P. dardanus f. lamborni were excluded from the present study due to the existence of a duplication of the engrailed/invected genome region in this morph [25], potentially confounding phylogenetic or haplotype-level analyses.
Tree searches
Nucleotide substitution models were obtained from jModeltest (version 2.1, [38, 39], testing 11 substitution schemes, each with 4 categories of rate variation among sites) and using the corrected AIC criterion (AICc) to find the best fitting model. Tree searches were performed under the selected model using PhyML (version 20120412, [39]) with support values assessed using 200 replicates and other settings set to default. Tree statistics (tree length, consistency index, retention index and homoplasy index) were calculated in PAUP* 4.0 [40].
Haplotype analysiswithin P. dardanus
Sequenced haplotypes from cloned PCR products and Illumina amplicon sequencing were added to P. dardanus genotypic data to assist in inferring phase in the remaining specimens using the program PHASE (version 2.1.1, [41, 42]), with settings 400 iterations, thinning interval = 4 and a burn-in of 150. Haplotypes that were not inferred with certainty (P = 1.000) were discarded. A list of samples for which engrailed and invected haplotypes were successfully inferred is presented in Additional file 4: Table S4. Unrooted phylogenetic trees of P. dardanus engrailed and invected inferred haplotypes were produced using PhyML as described above. The branch-lengths of these trees were rescaled to reflect the numbers of synonymous and non-synonymous substitutions in HyPhy (version 2.1.2, [43]). Monophyly of the morph-associated engrailed alleles was assessed using the Shimodaira-Hasegawa test for tree selection based on constrained trees (monophyly constraint) that keep target alleles as monophyletic against the unconstrained tree using PAUP* 4.0b10 [40]; heuristic search performed under likelihood criterion with 10 random stepwise addition replicates and tree bisection-reconnection branch swapping, using nucleotide substitution model given by jModeltest).
McDonald-Kreitman tests were performed in DnaSP (version 5.10.1, [44]) using 405 bp amplicon engrailed haplotypes inferred with PHASE from a population sample of 35 wild-caught specimens of P. dardanus subspecies cenea (Additional file 5: Table S5). The McDonald-Kreitman test requires a sufficiently divergent outgroup, such that there is little or no shared polymorphism, so we used a specimen of P. rex, rather than the more closely related P. phorcas or P. constantinus.
Results
Phylogeny ofengrailed andinvected exons across the genusPapilio
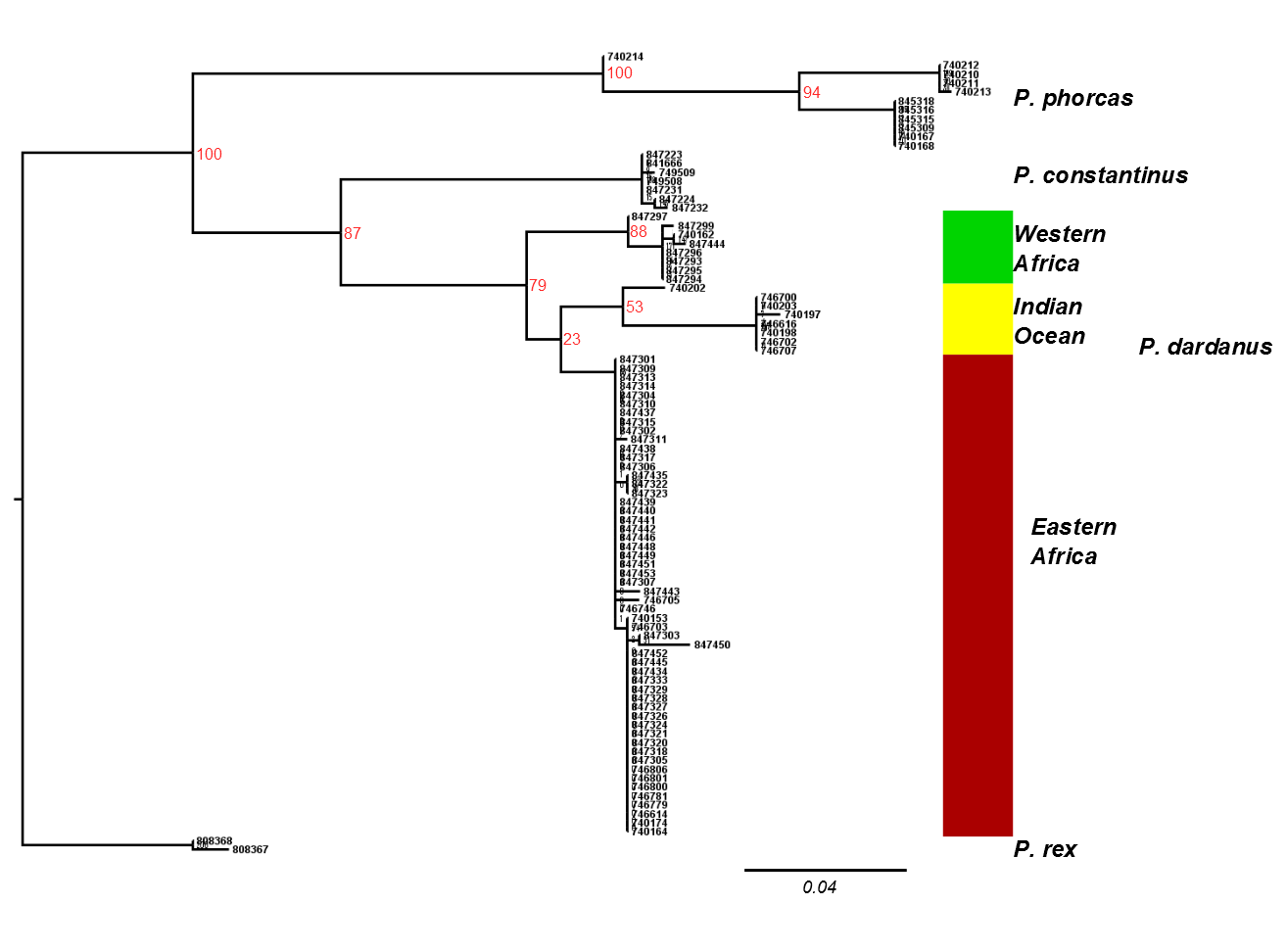
To study the evolution of the candidate wing-pattern switch genes, we inferred the phylogeny of engrailed and invected coding sequences across the genus Papilio (Figures 3 and 4 respectively). For engrailed, we used 93 terminal taxa (30 species) of exon-1 resulting in an alignment of 479 bp of coding region. The analysis places all haplotypes of P. dardanus in a clade as sister to its two closest relatives, P. constantinus and P. phorcas, and reveals a large diversity of alleles within P. dardanus. In particular, alleles from individuals of f. planemoides and f. poultoni represent highly-divergent sequences derived from within a cluster of alleles associated with the other morphs. Monophyly of P. dardanus alleles indicates that these alleles have evolved within P. dardanus, rather than through introgression from a related species.
Figure 3.
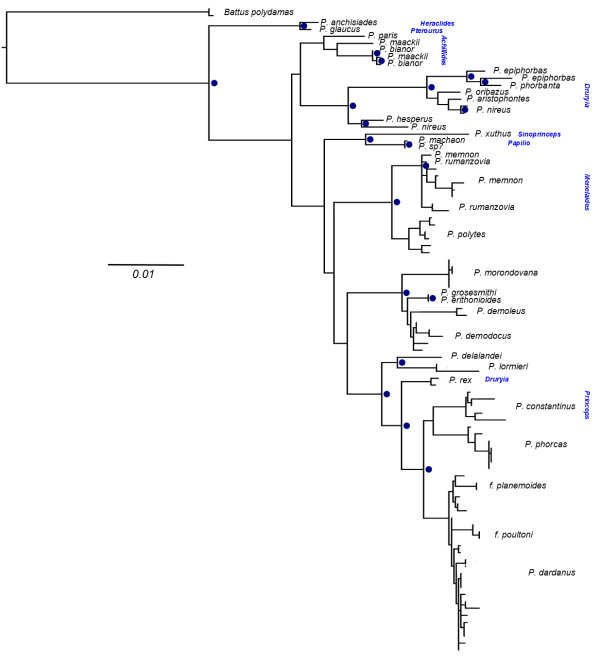
ML phylogeny of 5’ engrailed across the genus Papilio , with a Battus outgroup. Papilio dardanus individuals are represented by haplotypes from either cloning or MiSeq amplicon sequencing. Blue dots indicate nodes with bootstrap support >70%. Bootstraps at intraspecific nodes are ignored.
Figure 4.
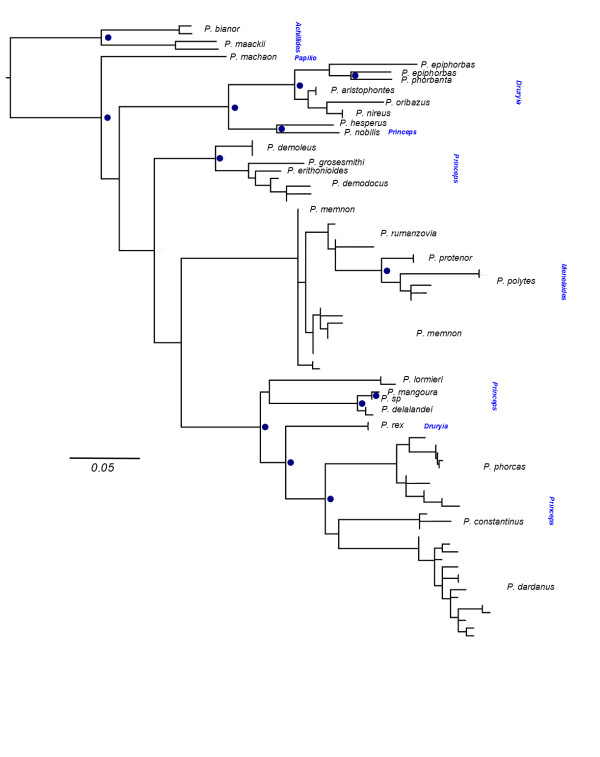
Maximum likelihood tree with 200 bootstrap replicates for Papilio 5’ invected . Papilio dardanus individuals are represented by haplotypes from MiSeq amplicon sequencing. Note that P. demodocus and P. memnon are paraphyletic. Blue dots indicate nodes with bootstrap support >70%. Bootstrap support at intraspecific nodes is not presented.
Outside of the P. dardanus clade, the overall topology is largely consistent with previous findings [45, 46] with the included subgenera, principally Princeps, Druryia and Menelaides recovered as monophyletic. Within the subgenus Menelaides, the species P. memnon and P. rumanzovia are not reciprocally monophyletic with one specimen of each species in an unresolved position at the base of this clade. Consistent with previous analyses [45] our tree places P. rex within subgenus Princeps; P. rex is therefore corroborated as sister to the clade (P. dardanus, P. constantinus, P. phorcas).
The dataset for invected consisted of 81 terminals (26 species) within Papilio and an alignment of 332 bps. The resulting tree also reveals a monophyletic P. dardanus group, with P. rex as sister, and is composed of highly divergent alleles that separate the monophyletic P. dardanus from its closest relatives P. phorcas and P. constantinus. The divergences within P. dardanus are more uniform than in the engrailed locus and there is no association of haplotypes with particular phenotypes.
Intraspecific variation inP. dardanus engrailed haplotypes
Haplotype inference for P. dardanus engrailed resulted in 51 unique haplotypes from 55 individuals inferred successfully for a 712 bp 5` region of engrailed from a sample of 174 individuals. The haplotype phylogram (Figure 5) shows highly divergent engrailed alleles that were associated with the dominant morphs f. planemoides and f. poultoni. Out of nine individuals of f. poultoni sequenced, eight share two very closely related haplotypes. All six f. planemoides sequenced have one allele in common, which is also shared with the single f. poultoni specimen lacking the f. poultoni-associated allele (BMNH746707). This individual may represent a ‘synthetic niobe’, i.e. the heterozygote between the f. planemoides and f. trophonius H alleles known to produce a phenotype similar to that of f. poultoni [20], or it may be f. salaami, a form with a similar, but distinguishable phenotype which may be specified by a different allele. Unfortunately the wings of this specimen are too damaged to be certain in its morph assignment. One individual (BMNH746604) of f. poultoni was found to be a heterozygote of the two divergent alleles in agreement with the documented dominance of the f. poultoni allele over f. planemoides [20, 22] and the pattern of SNP association observed for this specimen [25]. All individuals of these morphs possess another allele in addition to the divergent and morph-associated allele, indicating heterozygosity at H and consistent with the fact that f. planemoides and f. poultoni are dominant alleles.
Figure 5.
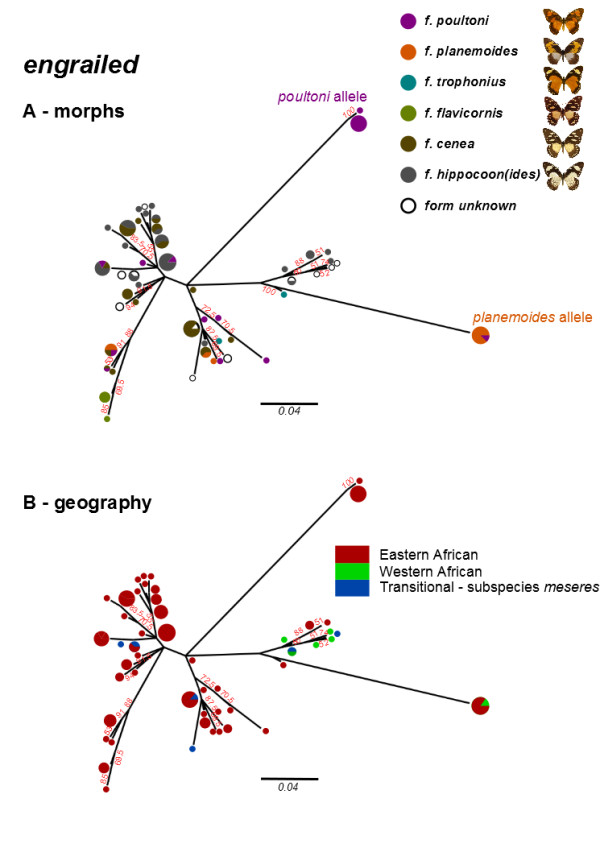
Maximum likelihood phylogeny of inferred 712 bp haplotypes of engrailed in P. dardanus , labelled by either the morph (A) or the population (B) the haplotypes were sampled from. Divergent alleles are associated with the dominant forms f. poultoni and f. planemoides, whilst there is no geographic structure. Percentage bootstrap support values above 50% are labelled in red.
The haplotypes can also be labelled according to the population from which they were sampled (Figure 5B). All of the haplotypes from Western Africa (subspecies P. d. dardanus) can be found within a single clade, however this lineage also contains a few Eastern African haplotypes from P. d. polytrophus. Subspecies P. d. meseres is treated separately as this has been suggested to be a ‘transitional’ or hybrid race where the Eastern and Western lineages meet around Lake Victoria [47–49]. Consistent with this hypothesis, individuals from this region are found to possess haplotypes of both Eastern and Western groups (Figure 5B). Finally, the f. planemoides-associated allele is found in individuals from both Eastern and Western African populations, possibly demonstrating a history of this allele independent of the population-level biogeography [27]. In summary, there is some geographic structure within our sampling of engrailed alleles, but allelic divergence within P. dardanus is deeper than any geographic structure.
P. dardanusinvected haplotypes
Intra-specific haplotype inference for the 5` region of invected (385 bp) was successful for 89 out of 114 individuals, yielding 52 unique haplotypes. The unrooted phylogram (Figure 6) is similar to that for engrailed (Figure 5), in that there is variation within the sampled population and there is no geographic structure. However, variation is overall lower than engrailed with only 27 variable sites in the nucleotide alignment of 333 positions.
Figure 6.
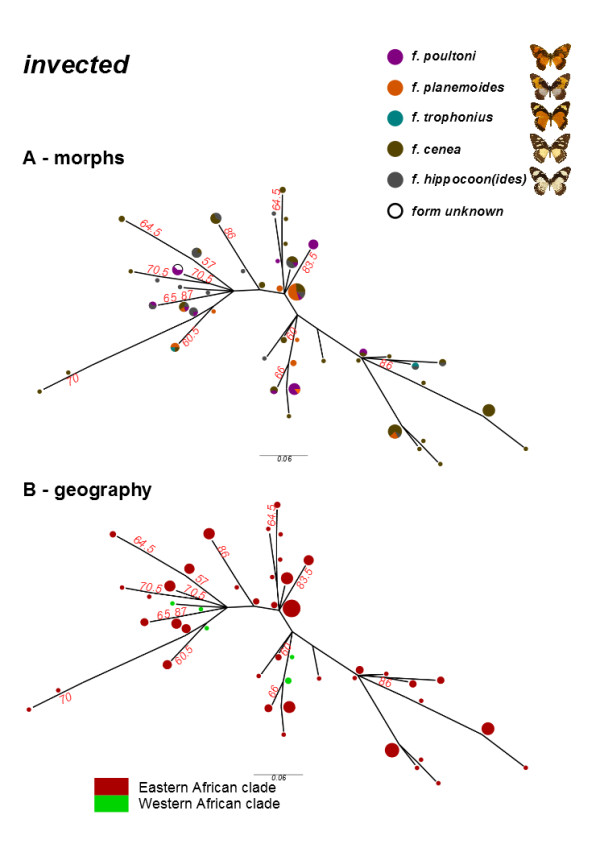
Phylogeny of inferred haplotypes for P. dardanus invected , labelled by either the morph (A) or the population (B) the haplotypes were sampled from. Percentage bootstrap support values >50% are labelled in red.
Nature of changes
Haplotype trees for engrailed exon 1 with synonymous and nonsynonymous changes mapped demonstrate that much of the divergence of the morph-associated alleles from the rest of the P. dardanus alleles is at non-synonymous (replacement) sites (Figure 7). The f. poultoni and f. planemoides alleles are characterised by multiple unique changes, primarily at replacement sites (Figure 7C). Additionally, the f. poultoni and f. planemoides alleles share 3 non-synonymous and 2 synonymous changes. To test for a shared origin of the two divergent alleles, we compared the best tree with an alternative topology in which f. poultoni and f. planemoides were constrained to monophyly. The likelihood of the constrained tree (lnL = -1846.602) was only 0.284 lnL units lower than in the unconstrained tree. This was not a significantly worse (P = 0.98) fit to the data in the SH test, indicating that we cannot rule out a shared origin for these alleles. Unlike the extensive non-synonymous changes in engrailed, all changes within the P. dardanus complex in invected were synonymous.
Figure 7.
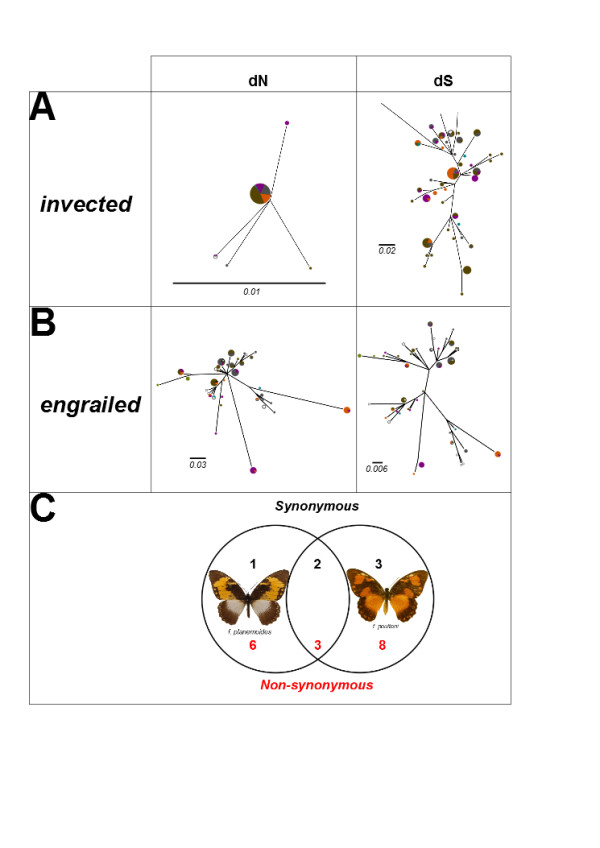
The structure of genetic variation in invected and engrailed. Gene trees were established by maximum likelihood for invected (A) and engrailed (B) haplotypes, separately from non-synonymous and synonymous nucleotide changes. Branch lengths correspond to the number of changes. The area of each pie is scaled in proportion to the frequency of that haplotype among the morphs sampled, and colours of pies reflect proportion of haplotypes recovered from each morph, labelled as in Figure 4. (C) Unique SNPs in the two morph-associated alleles relative to the consensus sequence for all other inferred P. dardanus alleles.
To test for non-neutral coding evolution within P. dardanus engrailed, the McDonald-Kreitman test [50] was applied to inferred engrailed haplotypes from a South African P. d. cenea population (specimens in Additional file 5: Table S5). We used this population as it was the largest wild-collected sample from a single locality and time point available to us. This population has neither of the dominant morphs f. poultoni and f. planemoides, but is polymorphic for f. hippocoonides, f. cenea, f. natalica and f. trophonius. Haplotypes were inferred for a 405 bp region (the same amplicon targeted for the genus phylogeny), yielding 26 unique haplotypes from 35 individuals. Haplotypes were also inferred for the same region in the outgroup species P. rex (2 haplotypes from 1 individual). The test values (Table 1) indicated a significant departure from neutrality, due to a large excess of non-synonymous polymorphisms within P. dardanus (P = 0.0128, two-tailed Fisher’s exact test), suggestive of diversifying selection acting on the engrailed coding sequence.
Table 1.
Segregating and fixed synonymous and non-synonymous differences in the comparison P . dardanus cenea - P. rex
| P. dardanus cenea – P. rex | Synonymous | Non-synonymous |
|---|---|---|
| Fixed | 21 | 4 |
| Polymorphic | 16 | 15 |
Discussion
We show here that phased haplotypes of engrailed associated with wing pattern forms are highly divergent phylogenetically. It has recently been shown that sets of SNPs across engrailed show strong association with the wing patterns of f. poultoni and f. planemoides and that this region shows elevated linkage disequilibrium (Figure 1; [25]). The presence of multiple linked SNPs is a prerequisite for the recognition of phylogenetically distinct lineages, and we show here that these morph-associated SNPs define a small number of morph-associated haplotypes. Each individual of these two morphs possesses at least one of these haplotypes, which occupy exceptionally long branches in the haplotype tree, even by comparison to the corresponding sequences across distantly related species of Papilio (Figure 3). The divergence of the morph-associated haplotypes is contributed disproportionately by non-synonymous changes. The allele diversity does not follow the strong geographic structure suggested by the mtDNA, which splits the populations in eastern and western branches ([27] and Additional file 6: Figure S1) in roughly the same way as genitalic characters [49], although other nuclear markers also show only weak subdivision ([27] and unpublished). Taken together, these findings strongly suggest that the genomic region around the first exon of engrailed diverges from patterns of neutral variation at phylogenetic and genetic levels, which further supports a role of this region for controlling wing pattern variation in P. dardanus.
The phylogenetic analysis revealed several details about the evolutionary history of engrailed and the adjacent invected loci. First, the higher-level analyses of the genus Papilio (Figures 3 and 4 respectively) did not yield many surprises; expected subclades were largely recovered for both loci, albeit with generally poor support due to the short (<800 bp) fragment length and high homoplasy (Additional file 7: Table S6). Hence it can be concluded that the engrailed and invected loci are useful to track the lineage history, and are not generally distorted by selection outside of P. dardanus. Another key result is that P. dardanus is recovered as monophyletic in the engrailed phylogeny, despite the inclusion of the divergent haplotypes, arguing against an origin for divergent alleles in other species. The intraspecific trees of P. dardanus alleles were characterised by a largely unresolved polytomy at the base in both loci, with very little internal structure related to geography, confirming the conclusions of [27] based on a more limited sample. Additionally, there is no obvious phylogenetic structure beyond the divergent alleles associated with f. poultoni and f. planemoides in the sequenced region of engrailed (Figure 5). Hence, aside from these divergent alleles, the phylogenies corroborate the findings from the SNP associations [25], that did not relate the recessive morphs to particular SNPs.
The existence of multiple fixed differences in the two divergent alleles is likely a result of recurrent fixation of novel mutations. The two groups of morph-associated alleles may share some changes due to a common ancestry or alternatively, if these non-synonymous changes have a direct functional role, this might represent convergence between the two alleles. Indeed, all of the changes that unite the f. poultoni and f. planemoides alleles are non-synonymous, consistent with the second hypothesis. However, the observation of an elevated level of non-synonymous intraspecific polymorphism in P. dardanus engrailed is not limited to the obvious cases of the morph-associated alleles. The results of the McDonald-Kreitman test provide strong evidence for non-neutral evolution in this gene. This has been observed with the SNP variation in the highly polymorphic Kenyan population used for the initial association studies [25], and is confirmed here for a second population from a different region (South Africa) that does not even include the most divergent alleles. The engrailed locus in P. dardanus therefore shows a pattern of evolution that differs from the remainder of the genus Papilio and also from the adjacent invected locus, in that non-synonymous changes have accumulated at a much increased evolutionary rate.
It is highly surprising to find such rapid coding sequence evolution in a gene that is so highly conserved across the arthropods. In particular, one of the changes unique to f. planemoides occurs in the conserved EH1 domain of the Engrailed protein, changing the core motif from FSISNIL in all other P. dardanus to YSISNIL which confers a change in the otherwise conserved core of FxIxxIL [51]. Given this conserved sequence it seems likely that this change may affect binding of Engrailed to the transcriptional repressor Groucho [52, 53]. Whether or not the coding changes in the two divergent alleles affect the function of the Engrailed protein remains an open question. Similarly, we cannot be certain whether these variant sites are themselves the focus of selection, or whether their fixation is the result of hitchhiking due to linkage with other changes. In the latter case, we hypothesise that favourable selection due to a novel mimetic resemblance outweighs the negative effects of a linked genetic load; selection coefficients for mimicry are likely to be very high [54].
Theory predicts that alleles with a high linked genetic load, as might be conferred by the amino acid changes in the EH1 domain, are likely to be under selection to increase in dominance: linked genetic load is ‘sheltered’ in rare dominant alleles as individuals will nearly always be heterozygous for these alleles [55]. In the Batesian mimicry system of P. dardanus, the equilibrium frequency is the abundance of the mimic relative to their model at which fitness is maximised, without predators associating a pattern with palatability rather than toxicity [56]. The equilibrium allele frequency will therefore be on average lower for a dominant allele as compared to alleles further down the dominance hierarchy. Hence, we speculate that whether the coding sequence changes at engrailed are in any way functional or not, their likely negative pleiotropic effects may be shielded from selection by the recessive-morph alleles at engrailed. This shielding of deleterious mutations through heterozygosity may therefore explain why it is especially the dominant morphs which show such a large excess of non-synonymous mutations.
Finally, we may ask what these results contribute to our understanding of the evolution of the supergene that was hypothesized to underlie the phenotypic variation in P. dardanus. The evolution of linkage disequilibrium to maintain co-adapted alleles is a core facet of classical supergene theory [57, 58] and other supergene systems have been shown to possess complex genomic architectures that may act to reduce recombination between supergene alleles [12, 59–61]. Here we show that the genomic signature of selection evident from the phylogenetic trees and increased rates of non-synonymous changes does not extend beyond engrailed; we find little evidence for morph-associated variants at linked genes such as invected. A similar pattern was seen in the SNP analysis [25], along with the low level of linkage disequilibrium observed elsewhere in the H region. The high level of diversity uncovered in a population sample of P. dardanus is indicative of negative frequency-dependent selection, as would be expected for a locus underlying polymorphic Batesian mimicry, as selection against over-abundant phenotypes results in a balanced polymorphism, which can be detected by a signature of increased diversity. The existence of multiple fixed changes in these alleles could be the result of recurrent selective sweeps fixing the variants, with either the coding region sites under selection themselves or hitchhiked to fixation as the result of selection on linked variants. The changes might also reflect the build-up of genetic load due to a reduction of recombination as predicted by classic supergene theories of polymorphic mimicry. The previous observation of low linkage disequilibrium beyond the engrailed locus [25] means that this predicted effect only affects a single gene which, however, spans a large genomic region of ~70 kb [25].
The findings of high levels of non-synonymous diversity, potentially affecting protein structure, are similar to patterns observed in the doublesex coding region in P. polytes [12]. Both the P. dardanus engrailed locus and P. polytes doublesex have alternative alleles associated with different female mimetic morphs and these alternative alleles differentiated by multiple changes with a high ratio of non-synonymous to synonymous SNPs in the coding regions. These two mimetic butterflies have evolved similar systems of Batesian mimetic polymorphism through a complex sequence of changes in single developmentally important genes, although the genes involved are very different.
Conclusions
The gene engrailed is a well-supported candidate for the mimicry switch locus in P. dardanus. Following the comparative genomics work of [25], we here investigated the evolutionary genetics of the engrailed and adjacent invected genes using phased alleles within the P. dardanus lineage and also provided the wider geographic and phylogenetic context by including additional populations of P. dardanus and other members of the genus Papilio. We also expanded the tests for non-neutral variation to a geographically distinct population, along with the use of a less divergent outgroup. These analyses revealed that dominant morphs are associated with highly divergent haplotypes of this gene in P. dardanus, with a large proportion of the divergence occurring at non-synonymous sites, but not in other species. In addition, there is no evidence for introgression from other species to explain the high level of divergence from other P. dardanus haplotypes. Furthermore, the high levels of non-synonymous polymorphism observed in P. dardanus engrailed are consistent with long-term balancing selection, mirroring similar findings in P. polytes [12] . The study provides new insights into the fascinating evolution of mimetic polymorphisms in the genus Papilio and shows that, while the polymorphism is generated by different genomic regions, the evolutionary processes that build up the phenotypic diversity at the genome level are similar between species of Papilio.
Availability of supporting data
DNA sequences are in Genbank under accession numbers given in a supplemental table.
Electronic supplementary material
Additional file 1: Table S1: Table of samples of Papilio used for phylogenetic analysis of engrailed and invected. (XLSX 41 KB)
Additional file 2: Table S2: Table of PCR primers. (XLSX 8 KB)
Additional file 3: Table S3: Table of samples used for Illumina MiSeq amplicon sequencing to infer haplotypes. (XLSX 9 KB)
Additional file 4: Table S4: Table of samples of P. dardanus used for phylogenetic and haplotype-level analysis. (XLSX 13 KB)
Additional file 5: Table S5: Table of samples of P. dardanus cenea from Mpaphuli Cycad Reserve population used in population genetic analyses. (XLSX 17 KB)
{kind=link}
Additional file 6: Figure S1: Maximum likelihood phylogeny of cytohrome b amplicon within the P. dardanus species group. P. dardanus is presented as sister to a clade of P. phorcas and P. constantinus, consistent with the engrailed phylogeny in Figure 3. This phylogeny recovers 3 deeply-coalescing lineages within P. dardanus, consistent with the mitochondrial phylogenies of Clark and Vogler [27]. (PNG 106 KB)
Additional file 7: Table S6: Table of summary statistics for alignments, details of nucleotide substitution models used in maximum likelihood tree searches and summary statistics for the resulting phylogenies. (XLSX 9 KB)
Acknowledgements
We are grateful to Steve Collins of the Afrotropical Butterfly Research Institute, Nairobi for the provision of samples and assistance with fieldwork, Butterfly Conservation Ghana, the Lepidopterists Society of Africa, Bennie and Andre Coetzer, Dr Oskar Brattstrom and Erik van Bergen for assistance with fieldwork and Andrew Knapp and Alessandra Pollara for assistance with preparation of samples for sequencing. Funded by NE/F006225/1 of the Natural Environment Research Council of the UK. MJTN was funded through a NERC Postdoctoral Fellowship (NE/I021578/1).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MJT conceived of the experiments, performed DNA extractions, amplification of target regions, cloning of haplotypes, performed the alignment, haplotype inference, population genetic and phylogenetic analyses. MJTNT conceived of the experiments, performed DNA extractions and amplification of target genes, cloning of haplotypes, contributed to analysis and interpretation of the results. CDJ assisted with choice of appropriate techniques, interpretation of results. APV conceived of the experiments and assisted with choice of appropriate techniques, interpretation of results. All authors participated in the writing of the manuscript.
Contributor Information
Martin J Thompson, Email: mjt68@cam.ac.uk.
Martijn JTN Timmermans, Email: martt@nhm.ac.uk.
Chris D Jiggins, Email: c.jiggins@zoo.cam.ac.uk.
Alfried P Vogler, Email: apv@nhm.ac.uk.
References
- 1.Joron M, Jiggins CD, Papanicolaou A, McMillan WO. Heliconius wing patterns: an evo-devo model for understanding phenotypic diversity. Heredity. 2006;97:157–167. doi: 10.1038/sj.hdy.6800873. [DOI] [PubMed] [Google Scholar]
- 2.Nielsen R. Molecular signatures of natural selection. Annu Rev Genet. 2005;39:197–218. doi: 10.1146/annurev.genet.39.073003.112420. [DOI] [PubMed] [Google Scholar]
- 3.Thompson MJ, Jiggins CD. Supergenes and their role in evolution. Heredity. 2014;113(1):1–8. doi: 10.1038/hdy.2014.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carroll SB. Evolution at two levels: on genes and form. PLoS Biol. 2005;3:e245. doi: 10.1371/journal.pbio.0030245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rebeiz M, Pool JE, Kassner VA, Aquadro CF, Carroll SB. Stepwise modification of a modular enhancer underlies adaptation in a Drosophila population. Science. 2009;326:1663–7. doi: 10.1126/science.1178357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prud’homme B, Gompel N, Carroll SB. Emerging principles of regulatory evolution. Proc Natl Acad Sci U S A. 2007;104(Suppl 1):8605–12. doi: 10.1073/pnas.0700488104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van’t Hof AE, Edmonds N, Dalíková M, Marec F, Saccheri IJ. Industrial melanism in British peppered moths has a singular and recent mutational origin. Science. 2011;332:958–960. doi: 10.1126/science.1203043. [DOI] [PubMed] [Google Scholar]
- 8.Counterman B, Araujo-Perez F, Hines HM, Baxter SW, Morrison CM, Lindstrom DP, Papa R, Ferguson LC, Joron M, Ffrench-Constant RH, Smith CP, Nielsen DM, Chen R, Jiggins CD, Reed RD, Halder G, Mallet J, McMillan WO. Genomic hotspots for adaptation: the population genetics of Müllerian mimicry in Heliconius erato. PLoS Genet. 2010;6:e1000796. doi: 10.1371/journal.pgen.1000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baxter SW, Nadeau NJ, Maroja LS, Wilkinson P, Counterman B, Dawson A, Beltrán M, Perez-Espona S, Chamberlain NL, Ferguson LC, Clark R, Davidson C, Glithero R, Mallet J, McMillan WO, Kronforst MR, Joron M, Ffrench-Constant RH, Jiggins CD. Genomic hotspots for adaptation: the population genetics of Müllerian mimicry in the Heliconius melpomene clade. PLoS Genet. 2010;6:e1000794. doi: 10.1371/journal.pgen.1000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.The Heliconius Genome Consortium: Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature. 2012;487:94–8. doi: 10.1038/nature11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naisbit RE, Jiggins CD, Mallet J. Mimicry: developmental genes that contribute to speciation. Evol Dev. 2007;5:269–80. doi: 10.1046/j.1525-142X.2003.03034.x. [DOI] [PubMed] [Google Scholar]
- 12.Kunte K, Zhang W, Tenger-Trolander A, Palmer DH, Martin A, Reed RD, Mullen SP, Kronforst MR. Doublesex is a mimicry supergene. Nature. 2014;507:229–32. doi: 10.1038/nature13112. [DOI] [PubMed] [Google Scholar]
- 13.Hines HM, Counterman BA, Papa R, Albuquerque de Moura P, Cardoso MZ, Linares M, Mallet J, Reed RD, Jiggins CD, Kronforst MR, McMillan WO. Wing patterning gene redefines the mimetic history of Heliconius butterflies. Proc Natl Acad Sci U S A. 2011;108:19666–71. doi: 10.1073/pnas.1110096108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pardo-Diaz C, Salazar C, Baxter SW, Merot C, Figueiredo-Ready W, Joron M, McMillan WO, Jiggins CD. Adaptive introgression across species boundaries in Heliconius butterflies. PLoS Genet. 2012;8:e1002752. doi: 10.1371/journal.pgen.1002752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stern DL. The genetic causes of convergent evolution. Nat Rev Genet. 2013;14:751–764. doi: 10.1038/nrg3483. [DOI] [PubMed] [Google Scholar]
- 16.Colosimo PF, Hosemann KE, Balabhadra S, Villarreal G, Dickson M, Grimwood J, Schmutz J, Myers RM, Schluter D, Kingsley DM. Widespread parallel evolution in sticklebacks by repeated fixation of Ectodysplasin alleles. Science. 2005;307:1928–33. doi: 10.1126/science.1107239. [DOI] [PubMed] [Google Scholar]
- 17.Nijhout HF. Polymorphic mimicry in Papilio dardanus: mosaic dominance, big effects, and origins. Evol Dev. 2003;5:579–92. doi: 10.1046/j.1525-142X.2003.03063.x. [DOI] [PubMed] [Google Scholar]
- 18.Thompson MJ, Timmermans MJTN. Characterising the phenotypic diversity of Papilio dardanus wing patterns using an extensive museum collection. Plos one. 2014;9(5):e96815. doi: 10.1371/journal.pone.0096815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clarke CA, Sheppard PM. The genetics of Papilio dardanus Brown. I. Race cenea from South Africa. Genetics. 1959;44:1347–1358. doi: 10.1093/genetics/44.6.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clarke CA, Sheppard PM. The genetics of Papilio dardanus Brown. II. Races dardanus, polytrophus, meseres, and tibullus. Genetics. 1960;45:439–457. doi: 10.1093/genetics/45.4.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clarke CA, Sheppard PM. The genetics of Papilio dardanus Brown. III. Race antinorii from Abyssinia and race meriones from Madagascar. Genetics. 1960;45:683–698. doi: 10.1093/genetics/45.6.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clarke CA, Sheppard PM. The genetics of Papilio dardanus Brown. IV. Data on race ochracea, race flavicornis, and further information on races polytrophus and dardanus. Genetics. 1962;47:909–920. doi: 10.1093/genetics/47.7.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clarke CA, Sheppard PM. The genetics of some mimetic forms of Papilio dardanus Brown, and Papilio glaucus Linn. J Genet. 1959;56:236–259. doi: 10.1007/BF02984747. [DOI] [Google Scholar]
- 24.Clark R, Brown SM, Collins SC, Jiggins CD, Heckel DG, Vogler AP. Colour pattern specification in the mocker swallowtail Papilio dardanus: the transcription factor invected is a candidate for the mimicry locus H. Proc R Soc B Biol Sci. 2008;275:1181–1188. doi: 10.1098/rspb.2007.1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Timmermans MJTN, Baxter SW, Clark R, Heckel DGG, Vogel H, Collins S, Papanicolaou A, Fukova I, Joron M, Thompson MJ, Jiggins CD, ffrench-Constant RH, Vogler AP. Comparative genomics of the mimicry switch in Papilio dardanus. Proc R Soc B Biol Sci. 2014;281(1787):20140465. doi: 10.1098/rspb.2014.0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peel AD, Telford MJ, Akam M. The evolution of hexapod engrailed-family genes: evidence for conservation and concerted evolution. Proc R Soc B Biol Sci. 2006;273:1733–42. doi: 10.1098/rspb.2006.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clark R, Vogler AP. A phylogenetic framework for wing pattern evolution in the mimetic mocker swallowtail Papilio dardanus. Mol Ecol. 2009;18:3872–84. doi: 10.1111/j.1365-294X.2009.04259.x. [DOI] [PubMed] [Google Scholar]
- 28.Thompson MJ, Vane-Wright RI, Timmermans MJTN. Hybrid origins: DNA techniques confirm that Papilio nandina is a species hybrid (Papilionidae) J Lepid Soc. 2011;65:199–201. [Google Scholar]
- 29.Thomsen PF, Elias S, Gilbert TM, Haile J, Munch K, Kuzmina S, Froese DG, Sher A, Holdaway RN, Willerslev E. Non-destructive sampling of ancient insect DNA. PLoS One. 2009;4:e5048. doi: 10.1371/journal.pone.0005048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kronforst MR. Primers for the amplification of nuclear introns in Heliconius butterflies. Mol Ecol Notes. 2005;5(1):158–162. doi: 10.1111/j.1471-8286.2004.00873.x. [DOI] [Google Scholar]
- 31.Barraclough TG, Hogan JE, Vogler AP. Testing whether ecological factors promote cladogenesis in a group of tiger beetles (Coleoptera: Cicindelidae) Proc R Soc B Biol Sci. 1999;266:1061–1067. doi: 10.1098/rspb.1999.0744. [DOI] [Google Scholar]
- 32.Drummond A, Ashton B, Buxton S, Cheung M, Cooper A, Duran C, Field M, Heled J, Kearse M, Markowitz S, Moir R, Stones-Havas S, Sturrock S, Thierer T, Wilson A. Geneious v5.3. 2010. [Google Scholar]
- 33.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–66. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hey J, Wakeley J. A coalescent estimator of the population recombination rate. Genetics. 1997;145:833–846. doi: 10.1093/genetics/145.3.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sturrock S, Meintjes P: The Geneious 6.0.3 Read Mapper.
- 36.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25:1253–6. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- 39.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 40.Swofford DL. Phylogenetic Analysis Using Parsimony (*and Other Methods) 2003. [Google Scholar]
- 41.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–89. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stephens M, Scheet P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am J Hum Genet. 2005;76:449–62. doi: 10.1086/428594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kosakovsky Pond SL, Frost SDW, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–9. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- 44.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 45.Zakharov EV, Caterino MS, Sperling FAH. Molecular phylogeny, historical biogeography, and divergence time estimates for swallowtail butterflies of the genus Papilio (Lepidoptera: Papilionidae) Syst Biol. 2004;53:193–215. doi: 10.1080/10635150490423403. [DOI] [PubMed] [Google Scholar]
- 46.Caterino MS, Sperling FAH. Papilio phylogeny based on mitochondrial cytochrome oxidase I and II genes. Mol Phylogenet Evol. 1999;11:122–37. doi: 10.1006/mpev.1998.0549. [DOI] [PubMed] [Google Scholar]
- 47.Poulton EB. The most interesting butterfly in the world. J East African Nat Hist Soc. 1924;20:4–22. [Google Scholar]
- 48.Ford EB. The genetics of Papilio dardanus Brown (Lep.) Trans R Entomol Soc London. 1936;85:435–466. doi: 10.1111/j.1365-2311.1936.tb00238.x. [DOI] [Google Scholar]
- 49.Turner JRG. Geographical variation and evolution in the males of the butterfly Papilio dardanus Brown (Lepidoptera: Papilionidae) Trans R Entomol Soc London. 1963;115:239–259. doi: 10.1111/j.1365-2311.1963.tb00821.x. [DOI] [Google Scholar]
- 50.McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- 51.Copley RR. The EH1 motif in metazoan transcription factors. BMC Genomics. 2005;6:169. doi: 10.1186/1471-2164-6-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith ST, Jaynes JB. A conserved region of engrailed, shared among all en-, gsc-, Nk1-, Nk2-and msh-class homeoproteins, mediates active transcriptional repression in vivo. Development. 1996;122:3141. doi: 10.1242/dev.122.10.3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jiménez G, Paroush Z, Ish-Horowicz D. Groucho acts as a corepressor for a subset of negative regulators, including Hairy and Engrailed. Genes Dev. 1997;11:3072–3082. doi: 10.1101/gad.11.22.3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kapan DD. Three-butterfly system provides a test of Müllerian mimicry. Nature. 2001;409:18–20. doi: 10.1038/35053066. [DOI] [PubMed] [Google Scholar]
- 55.Llaurens V, Billiard S, Castric V, Vekemans X. Evolution of dominance in sporophytic self-incompatibility systems: I. Genetic load and coevolution of levels of dominance in pollen and pistil. Evolution. 2009;63:2427–37. doi: 10.1111/j.1558-5646.2009.00709.x. [DOI] [PubMed] [Google Scholar]
- 56.Clarke B. Frequency-dependent selection for the dominance of rare polymorphic genes. Evolution. 1964;18:364–369. doi: 10.2307/2406348. [DOI] [Google Scholar]
- 57.Darlington CD, Mather K. Elements of Genetics. London: George Allen & Unwin Ltd; 1949. [Google Scholar]
- 58.Charlesworth D, Charlesworth B. Mimicry: the hunting of the supergene. Curr Biol. 2011;21:R846–8. doi: 10.1016/j.cub.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 59.Joron M, Frezal L, Jones RT, Chamberlain NL, Lee SF, Haag CR, Whibley AC, Becuwe M, Baxter SW, Ferguson LC, Wilkinson P, Salazar C, Davidson C, Clark R, Quail MA, Beasley H, Glithero R, Lloyd C, Sims S, Jones MC, Rogers J, Jiggins CD, Ffrench-Constant RH. Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature. 2011;477:203–6. doi: 10.1038/nature10341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huynh LY, Maney DL, Thomas JW. Chromosome-wide linkage disequilibrium caused by an inversion polymorphism in the white-throated sparrow (Zonotrichia albicollis) Heredity. 2010;106:537–546. doi: 10.1038/hdy.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang J, Wurm Y, Nipitwattanaphon M, Riba-Grognuz O, Huang Y-C, Shoemaker D, Keller L. A Y-like social chromosome causes alternative colony organization in fire ants. Nature. 2013;493:664–8. doi: 10.1038/nature11832. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1: Table of samples of Papilio used for phylogenetic analysis of engrailed and invected. (XLSX 41 KB)
Additional file 2: Table S2: Table of PCR primers. (XLSX 8 KB)
Additional file 3: Table S3: Table of samples used for Illumina MiSeq amplicon sequencing to infer haplotypes. (XLSX 9 KB)
Additional file 4: Table S4: Table of samples of P. dardanus used for phylogenetic and haplotype-level analysis. (XLSX 13 KB)
Additional file 5: Table S5: Table of samples of P. dardanus cenea from Mpaphuli Cycad Reserve population used in population genetic analyses. (XLSX 17 KB)
Additional file 6: Figure S1: Maximum likelihood phylogeny of cytohrome b amplicon within the P. dardanus species group. P. dardanus is presented as sister to a clade of P. phorcas and P. constantinus, consistent with the engrailed phylogeny in Figure 3. This phylogeny recovers 3 deeply-coalescing lineages within P. dardanus, consistent with the mitochondrial phylogenies of Clark and Vogler [27]. (PNG 106 KB)
Additional file 7: Table S6: Table of summary statistics for alignments, details of nucleotide substitution models used in maximum likelihood tree searches and summary statistics for the resulting phylogenies. (XLSX 9 KB)