

Abstract
The rewarding effects of nicotine are associated with activation of nicotine receptors. However, there is increasing evidence that the endogenous opioid system is involved in nicotine's rewarding effects. We employed PET imaging with [11C]carfentanil to test the hypotheses that acute cigarette smoking increases release of endogenous opioids in the human brain and that smokers have an upregulation of mu opioid receptors (MORs) when compared to nonsmokers. We found no significant changes in binding potential (BPND) of [11C]carfentanil between the placebo and the active cigarette sessions, nor did we observe differences in MOR binding between smokers and nonsmokers. Interestingly, we showed that in smokers MOR availability in bilateral superior temporal cortices during the placebo condition was negatively correlated with scores on the Fagerström Test for Nicotine Dependence (FTND). Also in smokers, smoking-induced decreases in [11C]carfentanil binding in frontal cortical regions were associated with self-reports of cigarette liking and wanting. Although we did not show differences between smokers and nonsmokers, the negative correlation with FTND corroborates the role of MORs in superior temporal cortices in nicotine addiction and provides preliminary evidence of a role of endogenous opioid signaling in frontal cortex in nicotine reward.
Introduction
Tobacco use is the largest preventable cause of death and disease in the United States. In 2011, 19% of adults (43.8 million) were current smokers [10]. The reinforcing effects of nicotine are mediated, in part, via its effects on α4β2 nicotinic acetylcholine receptors, which result in activation of dopamine (DA) neurons and increased release of DA in the nucleus accumbens (NAc) [11]. The ability of most drugs of abuse to increase DA in the NAc, is believed to be a common mechanism through which drugs of abuse exert their reinforcing effects [41]. Specifically, acute nicotine has been shown to change met-enkephalin in striatum in ways that are interpreted to indicate that nicotine enhances the release and synthesis of met-enkephalin in striatum [20]. However, preclinical studies have also shown that nicotine increases release of endogenous opioids [15]. These effects are likely to contribute to nicotine's reinforcing effects because nicotine is not reinforcing in knockout mice that do not express mu opioid receptors (MORs) [6]. Adaptations in endogenous opioids secondary to chronic smoking are also likely to contribute to the addictiveness of nicotine. Indeed, repeated nicotine administration results in increased expression of MORs [46]. Moreover, naloxone, a MOR antagonist drug, can trigger withdrawal in animals exposed chronically to nicotine [35] and in daily smokers [28]. Further, polymorphic variants in the mu receptor (Asn40Asp variant) predict response to nicotine replacement therapy [31]. Thus, understanding the acute and long-term effects of nicotine on the opioid system in humans might provide better strategies for the development of treatment medications for nicotine dependence.
In this study, we tested the hypothesis that nicotine at doses delivered through a cigarette increases the release of endogenous opioids in the human brain and that chronic smokers exhibit neuroadaptations in mu receptors. We assessed the effects of smoking a cigarette on the binding of the mu-opioid agonist receptor radioligand [11C]carfentanil using positron emission tomography (PET) and compared the responses in nonsmokers to those in smokers. [11C]carfentanil binding in the brain is sensitive to competition with endogenous opioids [49], and thus we hypothesized that its binding would be decreased after smoking a cigarette. We also hypothesized that chronic smokers would show decreased endogenous opioid release when not under the effects of nicotine and thus would have increases in MOR when tested at baseline.
Materials and Methods
Ethics Statement
The studies were approved by the Johns Hopkins Medicine (JHM) Office of Human Subjects Research - Institutional Review Boards and the NIH Combined Neuroscience Institutional Review Board. After explaining the procedure, written informed consent was obtained from each subject.
Subjects
Ten smokers and ten age-matched nonsmokers were recruited specifically for this study. The basic demographics and smoking-related measures are given in Table 1. Inclusion criteria for smokers were smoking 10–45 cigarettes per day for at least 2 years, urinary cotinine ≥200 ng/mL, and no desire to quit or reduce smoking. Inclusion criteria for nonsmokers were having smoked 1–20 cigarettes in their lifetime, no smoking in the past year, and urinary cotinine <30 ng/mL. Otherwise, inclusion criteria were the same for both groups: males and females 21–50 years old and estimated IQ ≥85. We followed the NIH policy for inclusion of men and women because we had no strong reasons to exclude by gender. Due to scan scheduling availability, female subjects were studied without controlling for menstrual cycle stage or use of birth control medication. Subjects were excluded if they had current or past psychiatric disorders (including drug abuse or dependence other than nicotine dependence), neurological diseases, significant medical illnesses, or those who were on psychoactive medications. Subjects were recruited using public advertisements, and were initially screened by phone, and subsequently evaluated for eligibility by a physician. As part of the screening procedure, subjects had a physical, psychiatric, and neurologic examination. They completed the Fagerström Test for Nicotine Dependence (FTND) [22], [3] and completed a neuropsychological battery to insure that they were not cognitively impaired. Routine laboratory tests were performed, including toxicology screening to rule out the use of common drugs of abuse. Subjects were instructed to abstain from alcohol and drugs (except caffeine, nicotine, and non-psychoactive prescription drugs) 24 hours before each session, and smokers abstained from smoking after 12:00 midnight the night before each session. Subjects were admitted overnight at the Johns Hopkins Hospital General Clinical Research Unit to insure that they did not smoke the night prior to the study. On the morning of the PET scans, subjects were provided with a low calorie breakfast and completed breathalyzer testing for alcohol and breath carbon monoxide (<10 parts per million) for smokers.
Table 1. Demographics of participants and radioligand information of placebo and active cigarette scans.
| Variables | Smokers | Nonsmokers |
| Demographics and smoking- and alcohol-related measures | ||
| Number of subjects (Sex) | 10 (8 M/2 F) | 10 (6 M/4 F) |
| Age (years) | 32.5±8.2 (range: 23–50) | 34.3±10.7 (range: 22–50) |
| Fagerström test for nicotine dependence | 6.8±1.8 (range: 5–10) | 0 |
| Number of cigarettes per day | 19.5±12.7 (range: 10–45) | 0 |
| Number of drinks per week | 1.2±1.7 | 1.3±1.6 |
| Drinking days per week | 0.7±1.0 | 0.8±1.1 |
Values are mean ± standard deviation. In demographics, M stands for males, and F, for females.
PET imaging
The subjects received two PET scans, each on a separate day with [11C]carfentanil. No statistical differences were noted in injected radioactivity, non-radioactive mass, and specific activity (Table 1) between placebo- and active-cigarette scans for smokers and non-smokers, or between smokers and non-smokers in the placebo- and active-cigarette scans. PET studies were performed on the High Resolution Research Tomograph (HRRT, CPS Innovations, Inc., Knoxville, TN). In preparation for the study, two intravenous catheters were placed, one for radiotracer injection and the other for blood withdrawal to measure nicotine concentrations in plasma (immediately after smoking, every 5 min for 15 min, then every 10 min until the PET scan was completed). Prior to each PET scan, subjects smoked either a placebo or a nicotine-containing cigarette (see below) and within 10 min after completion of smoking underwent dynamic PET scanning. Dynamic scans were obtained using three-dimensional list mode acquisition for 80 min following the intravenous bolus injection of [11C]carfentanil. A 6-min transmission scan was acquired prior to each dynamic scan using a rotating Cs-137 source for attenuation correction. A custom-made thermoplastic mask was employed to reduce head motion during the PET data acquisition times. [11C]carfentanil was synthesized via the reaction of [11C]methyliodide and a nor-methyl precursor as previously described [14] and was injected via a venous catheter.
Cigarette smoking procedure
Subjects smoked either a Quest 1 cigarette (active, 0.6 mg nicotine) or a Quest 3 cigarette (placebo, <0.05 mg nicotine) before each PET session in a portable smoking booth attached to the ventilation system of a room adjacent to the scanner. Subjects took 8 puffs over a 10-min period using a CReSS smoking topography system (Plowshares, Inc., Baltimore, MD) to approximate equivalent smoking topography. The order of placebo and active cigarette conditions was counterbalanced across subjects.
Self-report Measures
Subjects were administered the Minnesota Nicotine Withdrawal Scale (MNWS [24]) and the Tobacco Craving Questionnaire-Short Form (TCQ-SF) [23] before and after each PET scan session. The following Visual Analog Scale (VAS) items assessed the effects of the cigarette “right now”: feel the effect, good effect, bad effect, like the effect, and want a cigarette. Subjects verbally rated each item on a scale from 0 to 10. VAS items were completed at baseline and at 5, 10, 15, 20, 25, 35, 40, 50, 60, 70, 80, and 90 min post-smoking.
Measurement of Nicotine Concentrations in Plasma
Blood specimens (5 mL) were collected in 7-mL green-topped Vacutainer tubes containing lithium oxalate, and immediately placed on ice. Specimens were centrifuged within 1 h and 1.0 mL aliquots of plasma stored in cryotubes at −80°C until analysis. Nicotine, cotinine, trans-3′-hydroxycotinine (OH-cotinine) and norcotinine were measured concurrently in 0.5 mL plasma specimens by a previously validated liquid chromatography tandem mass spectrometry (LCMSMS) method [21]. Briefly, 2 mL 0.1% formic acid were added to plasma specimens and centrifuged at 4,000×g for 5 min at 4°C. Supernatants were submitted to solid phase extraction with Strata-XC cartridges (Phenomenex, San Jose, CA), with final elution in 3% NH4OH in methanol. LCMSMS analysis was performed with a Shimadzu liquid chromatography system (Shimadzu Corporation, Columbia, MD, USA), a Synergi Polar-RP 100A interfaced to a 3200 QTrap (AB Sciex, Foster City, CA, USA) with a Turbo V ESI source. Standard mobile phases were used with gradient elution and a total run time of 12 min. Mass spectrometric data were acquired in positive electrospray ionization mode and multiple reaction monitoring mode (MRM). The following transitions were monitored (quantification transition in bold): 163.2>132.2 and 163.2>84.2 for nicotine; 177.2>80.1 and 177.2>98.1 for cotinine; 193.2>80.2 and 193.2>134 for OH-Cotinine; 163.2>80.2 and 163.2>118.2 for norcotinine; 167.2>136.1 and 167.2>121 for Nicotine-d4; 180.2>80.2 and 180.2>101.2 for Cotinine-d3; 196.2>79.9 and 196.2>134.1 for OH-Cotinine-d3; and 167.2>84.2 and 167.2>139.2 for norcotinine-d4. Linearity ranges with 1/x weighting for nicotine and 1/x2 for metabolites were 1 to 500 ng/mL for cotinine, OH-cotinine and norcotinine, and from 2.5 to 500 ng/mL for nicotine. Assay accuracy at low, medium and high QCs was 90.1–103.5% (n = 20) and imprecision was 4–13.8% (n = 20).
Reconstruction of PET data
Emission PET scans were reconstructed using the iterative ordered-subset expectation-maximization algorithm correcting for attenuation, scatter, and dead-time [42]. The radioactivity was corrected for physical decay to the injection time and re-binned to 30 dynamic PET frames of 256 (left-to-right) by 256 (nasion-to-inion) by 207 (neck-to-cranium) voxels. The frame schedules were six 30 s, seven 60 s, five 120 s, and twelve 300 s frames. The final spatial resolution is expected to be less than 2.5 mm full-width at half-maximum in three directions [45].
MRI acquisition
On a separate occasion, a spoiled gradient (SPGR) sequence 1.5 or 3 T MRI was obtained on each subject for anatomical identification of the structures of interest using the following parameters: Repetition time, 35 ms; echo time, 6 ms; flip angle, 458; slice thickness, 1.5 mm with no gap; field of view, 24×18 cm2; image acquisition matrix, 256×192, reformatted to 256×256 for the 1.5 T. Repetition time, 2110 ms; echo time, 2.73 ms; flip angle, 8; slice thickness, 0.8 mm with no gap; field of view, 24×18 cm2; image acquisition matrix, 320×288, reformatted to 256×256 for the 3 T.
PET data analysis
Volumes of interest (VOIs)
VOIs were manually defined for putamen (Pu), caudate nucleus (CN), hippocampus (HP), and cerebellum (Cb) using locally developed VOI tool (VOILand). Striatal VOIs were divided into ventral striatum (vS) and anterior or posterior dorsal subdivisions by the anterior-commissure plane, as previously described [5], [37], [39]. Other subcortical VOIs, including globus pallidus (GP), thalamus (Th), and amygdala (Am) were defined with FIRST software [40] and manually adjusted on individual MRIs. Cortical VOIs were automatically defined using Freesurfer [19] software including subdivisions of frontal (Fr), temporal (Tp), parietal (Pa), and occipital (Oc) cortices, fusiform gyrus (Fs), cingulate (Cg), and insula (In). VOIs for In and Oc were manually adjusted on individual MRIs. The Oc VOI included cuneus, lingual, lateraloccipital, and pericalcarine lobules given by Freesurfer, and served as reference region, after confirming that these regions showed similarly low radioactivity. A total of 78 VOIs were transferred from MRI to PET spaces using MRI-to-PET coregistration parameters given by the SPM5 coregistration module [2], [33] to obtain time-activity curves (TACs) of regions.
Derivation of PET outcome variables
A set of reference tissue methods were employed to obtain the binding potentials (BPND) [27] of regions (reference region = Oc), including the reference region graphical analysis (RTGA) [32] with k2 R (the brain-to-blood efflux rate constant of Oc) set at 0.104 min−1 [17], multilinear reference region method with 2 parameters (MRTM2) [26], and the bolus-plus-infusion transformation of bolus-only scans [29]. Images of BPND were generated by the three methods and transferred to the Montreal Neurological Institute (MNI) standard space applying parameters of PET-to-MRI coregistration (See above) and spatial normalization given by SPM unified segmentation method [1] in one step, and smoothed by a Gaussian kernel (8 mm Full with at half maximum) to submit to SPM5 statistical methods.
Voxel-wise statistical analysis
SPM5 was used to examine group differences and correlations of [11C]carfentanil BPND to smoking status measures, and visual analogue scales on smoking, as described in the results section. A significance level of p<0.001, uncorrected was employed for SPM analyses with the cluster volume threshold set at 0.4 mL (k>50). Locally prepared gray matter probability, tissue classification, and Brodmann area maps were used for anatomical identification and visualization of SPM clusters. Briefly, Freesurfer-derived brain region VOIs of 399 subjects (Age range: 18–40 years) were transferred to the SPM standard space using the SPM unified segmentation method [1] to generate probability maps of individual structures. The gray matter probability map was generated by summing individual gray matter probability maps to visually confirm whether clusters fell within gray matter areas. The tissue classification map was generated by assigning voxels to brain structures, starting from the largest (white matter) to the smallest (the ventral striatum) structures with a probability threshold of 0.2 to identify anatomical locations of peaks, and calculate structure compositions of clusters. The Brodmann area map was prepared by spatially aligning a published atlas [34] to the local standard brain to report Brodmann areas that were closest to the peaks and within the clusters, if any.
Results
None of participants fulfilled criteria of alcohol abuse or dependence, and no statistical differences were noted regarding alcohol-related metrics between smokers and nonsmokers (Table 1). For smokers, plots of mean plasma nicotine concentration peaked at 5 min and reached a plateau around 30 min in the active cigarette scan, but remained around the baseline level throughout the placebo cigarette scan (Fig. 1). The active cigarette scan condition showed higher plasma nicotine levels than the placebo cigarette scan condition (Table 2), while no statistical differences were observed between the two conditions for its metabolites. For nonsmokers, concentrations increased initially and declined slowly thereafter (Fig. 1). Plasma nicotine concentrations were substantially greater for smokers than nonsmokers for both cigarette conditions (Table 2).
Figure 1. Line plots of mean concentrations across subjects of nicotine in plasma versus time of active and placebo cigarette scans.
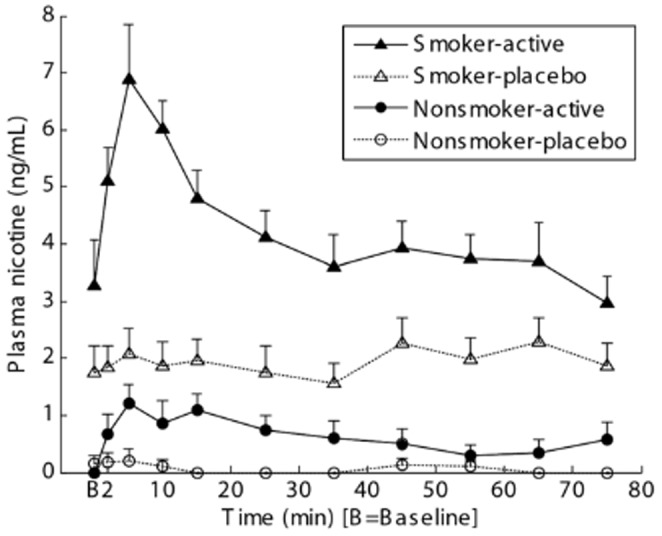
PET data acquisition began between 5 and 10 min on the time axis in individual subjects.
Table 2. Concentrations of nicotine and metabolites in plasma during active and placebo cigarette scans.
| Variables | Smokers | Nonsmokers | ||
| Cigarette type for PET Sessions | Placebo | Active | Placebo | Active |
| Nicotine (ng/mL) | 1.9±0.9# | 4.4±1.4* # | 0.1±0.1 | 0.7±0.7 |
| Nicotine (2–10 min; ng/mL) | 1.9±1.2# | 5.8±1.9* # | 0.2±0.4 | 0.9±0.9 |
| Cotinine (ng/mL) | 217±106 | 230±97 | <1 | <1 |
| Trans-3-hydroxy-cotinine (ng/mL) | 70.5±38.7 | 63.6±38.6 | 0 | 0 |
| Norcotinine (ng/mL) | 2.6±2.3 | 3.2±1.9 | 0 | 0 |
Mean ± standard deviation (ng/mL) of individual subjects' means across 2–75 min, except for the second nicotine row representing nicotine concentrations only from 2–10 min.
Limits of quantification were 1 ng/mL for cotinine, OH-cotinine and norcotinine, and 2.5 ng/mL for nicotine at individual time point.
* Active cigarette scan values > placebo cigarette scan value at p<0.01; paired t-test.
Smoker > nonsmoker at p<0.00001; t-test.
PET data were examined first for pharmacological effects (i.e., not including behavioral effects) of smoking active cigarettes on [11C]carfentanil binding. Of 78 VOIs, the right parahippocampus alone showed a significant decrease of [11C]carfentanil BPND in the active-cigarette scan (t = -5.96; p = 0.0002; df = 9; paired t-test) in smokers. However, this region showed relatively low [11C]carfentanil BPND (0.21±0.06 vs. 0.25±0.06 (mean ± SD; unitless) for placebo and active scans, respectively). Other regions did not show changes in [11C]carfentanil BPND between the two conditions separately for smokers and nonsmokers. The voxel-wise tests (SPM) showed no changes of [11C]carfentanil BPND between two conditions for smokers as well as for nonsmokers despite significant differences in plasma nicotine concentrations. Magnitudes of changes of [11C]carfentanil BPND between the two conditions remained undistinguishable between the two groups. No correlations of [11C]carfentanil BPND to plasma nicotine concentrations were observed including all plasma data points (2–75 min) or plasma samples around the peak (i.e., using 2, 5, and 10 min data) alone.
Correlations of changes of [11C]carfentanil BPND (Δ[11C]carfentanil BPND, defined as placebo-cigarette scan values minus active-cigarette scan values) with changes of self-reported VAS items (ΔVAS, defined as active minus placebo using means of data recorded between 20 and 80 min after the tracer injection) were examined. Smokers had single positive correlation clusters in left rostral frontal lobe for VAS items feel and like the effect, and good effect (Table 3 and Fig. 2). The three clusters spatially overlapped each other substantially. These correlations remained statistically significant (the coefficient of determination, R2>0.704) after removing one subject who showed larger ΔVAS values than other smokers in all three scores. The three clusters should be considered to represent one cluster of indistinguishable contributions from these three ΔVAS categories because the three ΔVAS categories were mutually correlated (R2>0.77; p<0.0001). Smokers also showed one cluster in right rostral frontal cortex for the VAS want a cigarette, although the cluster volume (0.24 mL) did not reach the set criterion (0.4 mL). SPM correlation analyses were not performed on nonsmokers because ΔVAS values were skewed heavily around 0 for this group. VOI-based analysis did not identify Δ[11C]carfentanil BPND, - ΔVAS correlations in any regions in smokers. No correlations were noted for nonsmokers in SPM and VOI-based analyses.
Table 3. Clusters of Δ[11C]carfentanil BPND (placebo - active) to ΔVAS (active - placebo) correlation.
| VAS | Peak coordinates | Peak t-values | Cluster volumes | Anatomical descriptions |
| Positive correlations | ||||
| Feel effect | −16 48 30 | 13.07 | 0.83 mL | Lt. rostral frontal lobe (53.9%) White matter (38.5%) |
| Good effect | −16 48 30 | 10.86 | 0.57 mL | Lt. rostral frontal lobe (43.7%) White matter (45.1%) |
| Like effect | −18 48 26 | 10.05 | 1.14 mL | Lt. rostral frontal lobe (71.8%) White matter (22.5%) |
Significance criteria: p<0.001, uncorrected and volume >0.4 mL.
Percentages in the last column indicate anatomical constituents of clusters.
Figure 2. Positive correlation clusters of Δ[11C]carfentanil binding potential (BPND) (placebo - active) versus ΔVAS of feel the effect category in smokers, displayed on trans-axial images of a gray-matter probability maps.
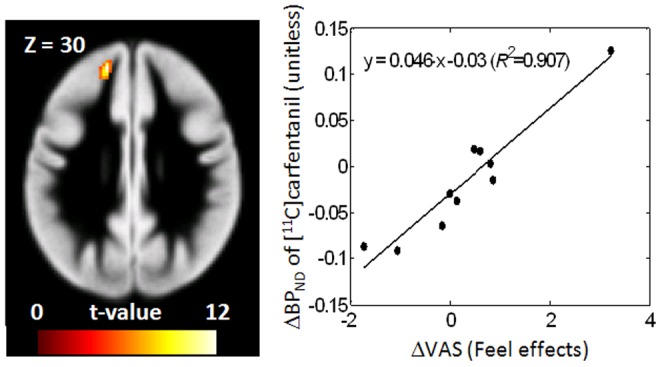
Scatter plots of cluster Δ[11C]carfentanil BPND values to ΔVAS are shown together with regression lines. VAS stands for the visual analog scale of smoking effects, and R2 stands for the coefficient of determination of linear regression.
We also compared [11C]carfentanil BPND for the placebo and for the differences between placebo and active cigarette scans between smokers and nonsmokers. No regions showed differences in VOI-based analyses, and no differences in clusters were identified in voxel-wise tests separately for active- and placebo-cigarette scans.
Finally, the following exploratory tests were performed on smokers alone. Correlation analysis between [11C]carfentanil BPND and FTND showed symmetrical negative correlation in superior temporal lobes (Table 4 and Fig. 3). A left-side negative correlation cluster also was observed for [11C]carfentanil BPND to current smoking status (cigarettes per day, CPD). The right side cluster did not reach the set significance criteria for this correlation. Spatial agreement of correlation clusters between the two smoking measures may be explained by the observation that these measures were highly correlated (CPD = 6.7•FTND - 26.2; R2 = 0.909). A positive correlation of lesser significance and volume was observed in left precentral gyrus with CPD.
Table 4. Clusters of placebo-cigarette [11C]carfentanil BPND to current nicotine-dependence and smoking statuses correlations in smokers.
| N/P | Peak coordinates | Peak t-values | Cluster volumes | Anatomical descriptions |
| Placebo-cigarette scan [11C]carfentanil BPND vs. FTDN | ||||
| N | −62 −2 −8 | 10.31 | 1.01 mL | Lt. superior temporal lobe (73.5%) Lt. precentral gyrus (22.1%) |
| N | 62 4 2 | 7.47 | 0.54 mL | Rt. superior temporal lobe (62.5%) Rt. precentral gyrus (29.2%) |
Significance criteria: p<0.001, uncorrected and volume>0.4 mL
P and N in the first column stand for clusters of positive and negative correlations, respectively.
Percentages in the last column indicate anatomical constituents of clusters.
Figure 3. Correlation clusters of [11C]carfentanil binding potential (BPND) of placebo-cigarette scans versus the Fagerström Test for Nicotine Dependence (FTND) in smokers, displayed on trans-axial images of a gray-matter probability maps.
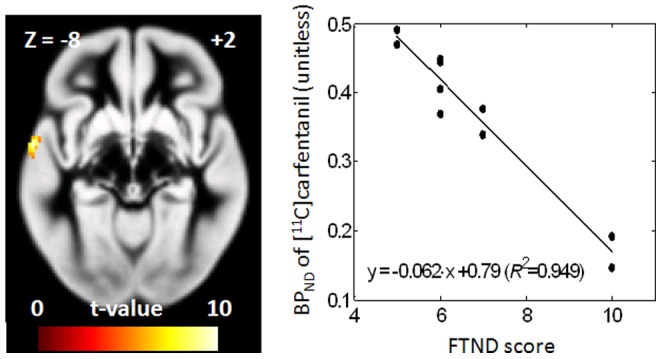
Right panels show scatter plots using cluster [11C]carfentanil BPND, together with regression line. In regression equations, R2 stands for the coefficient of determination.
Discussion
Active cigarettes in this study were expected to cause nearly 50% occupancy of nicotinic acetylcholine receptors (nAChRs) across brain regions (e.g., [18]), whereas Brody et al. [7] reported 26% and 79% occupancy of nACHRs by Quest 1 and 3 cigarettes, respectively. Although similar levels of occupancies of nAChRs were expected, no regions showed changes of [11C]carfentanil BPND in this study. This finding conflicts with results of a similar study [44] that reported clusters of decreased [11C]carfentanil BPND in the anterior cingulate and three increased clusters in the left amygdala, left ventral striatum, and right thalamus in the active-cigarette (1.01 mg nicotine/cigarette) condition compared to the placebo cigarette (0.08 mg nicotine/cigarette) condition. This discrepancy is likely to reflect the much lower nicotine concentrations achieved in our study (three fold lower plasma nicotine concentrations) and could be interpreted to suggest that higher nicotine doses than the ones achieved in our study (peak plasma level of 7 ng/mL versus average levels of 18 ng/mL in Scott et al. [44] might be necessary for endogenous opioid release. Differences in peak plasma nicotine concentration between smokers and nonsmokers were likely due to minimal inhalation on the part of nonsmokers. However, the differences in PET methodology (two 90-min scans on separate days in this study versus one 90-min scan for two cigarette sessions in one day) might also contribute to the differences in findings. Using the same Quest cigarettes as this study, Ray et al. [43] reported no changes in [11C]carfentanil BPND between the two cigarette conditions (see below), although they analyzed smokers of A/A genotype carriers of OPRM1 A118G genotype and G allele carriers separately. Thus, further studies are needed to investigate the pharmacological effects of nicotine in endogenous opioid release, including its potential modulation by OPRM1 A118G genotypes.
This and aforementioned studies [43], [44] identified different clusters of (Δ[11C]carfentanil BPND to ΔVAS correlations). Scott et al. [44] identified one cluster in the thalamus for the 'alert' score alone (i.e., the study did not identify correlations in VAS categories used in our study). Ray et al. [43] reported positive association of [11C]carfentanil BPND with changes of self-reported nicotine reward in right amygdala, caudate nucleus, anterior cingulate cortex, and thalamus using VOI-based analysis. Interestingly, the correlation was found in G allele carrier smokers (OPRM1 A118G genotype), but not in A/A genotype carriers. The closest equivalent to the reward measures in Ray et al. [43] would be the VAS items like the effect and good effect in our study for which the correlation was found in the left rostral frontal lobe (corresponding to Brodmann area 10), in the mixed genotype population. Moreover, positive correlations of cerebral blood flow changes to the amount of monetary rewards (suggestive of positive effects) were reported in a vicinity of current Δ[11C]carfentanil BPND to ΔVAS correlations ([x, y, z] = −20 12 42 [36]) in smokers, but not in nonsmokers. It is intriguing to speculate that the left rostral frontal lobe cluster (Fig. 2) might be indicative of the dopamine-opioid interaction because the hedonic effects of cigarettes, such as euphoria and craving, are associated with dopamine discharge in the striatum (e.g., [4]). Regarding the dopamine-opioid interaction, Colasanti et al. [12] demonstrated decreases of [11C]carfentanil BPND in multiple brain regions in the high dose scan (0.5 mg/kg) compared to a low dose (0.017 mg/kg) of oral amphetamine in healthy male nonsmokers. Because of no known direct actions of amphetamine on opioid neurotransmission and established amphetamine-induced dopamine release, the study suggested that dopamine-opioid interaction underpin the change of [11C]carfentanil BPND. Visual inspection (Fig. 3) of SPM clusters and tabulated results (Table S3) of Colasanti et al. [12] suggested the left middle frontal gyrus cluster (volume: 816 voxels or 65.3 mL) span the positive correlation cluster in the left rostral frontal lobe we observed for the feel/like/good effect ΔVAS categories. Therefore, this cluster might be related to the potential dopamine-opioid interaction. Interestingly, the location in the rostral frontal region corresponds to a region where functional connectivity was positively correlated with decreases in withdrawal symptoms with nicotine replacement therapy in abstinent smokers [13]. Since dysphoria is a central symptom in nicotine withdrawal [25], this also implicates endogenous opioid signaling in medial prefrontal regions in the reversal of negative symptoms by nicotine replacement.
Smoker versus nonsmoker differences in [11C]carfentanil BPND should be discussed within the limitation that subjects received either active or placebo cigarettes in each scan. Although the current study identified no differences in VOI-based (except in parahippocampal gyrus but not corroborated by SPM) and in voxel-wise statistical tests, Scott et al. [44] demonstrated robust (>13%) differences (smokers < nonsmokers) in the rostral anterior cingulate, thalamus, nucleus accumbens, and amygdala. Ray et al. [43] did not examine smoker versus nonsmoker differences. Correlation analysis of [11C]carfentanil BPND with nicotine dependence and current smoking status measures in our study should also be interpreted with caution because those data were obtained under the placebo cigarette condition. Numerous studies have documented the expectancy and/or sensorimotor effects of denicotinized cigarettes in reducing tobacco deprivation-induced withdrawal symptoms [8], [9], [16]. Thus, expectancy effects might have contributed to the release of endogenous opioids in smokers and nonsmokers.
Here, we report correlations of [11C]carfentanil BPND with smoking two smoking metrics that though related represent different smoking-related properties. A recent paper [47] reported negative correlations of baseline (i.e., no-specific tasks) [11C]carfentanil BPND to FTND scores in the cingulate cortex, thalamus, amygdala, and insula cortex, in alcohol-dependent subjects (n = 21) after partial correlations accounting for gender and recent drinking status. Although these correlations were observed in alcohol-dependent subjects [48], our study and the Weerts et al. [48] study indicated negative associations of [11C]carfentanil BPND to nicotine dependence/smoking status in selected brain regions, including superior temporal lobes. Involvement of these lobes in nicotine dependence was implicated in a number of studies. Interestingly, cue-induced changes in fMRI BOLD signal negatively correlated to FTND in the right superior temporal gyrus in a study involving 30 smokers with similar FTND scores to our study [38], suggesting that a decrease in MOR might underpin decreased BOLD response at least in this region. In a separate study, the left superior temporal gyrus alone showed cue-induced changes of BOLD signals [30] in abstaining smokers (7 hours), although correlations to FTND or CPD were not examined.
These findings need to be confirmed with a larger sample size. One limitation of our study was the possible psychological cues provided by smoking the placebo cigarette. The behaviors, the paraphernalia, and the environment associated with cigarette smoking, even in the absence of nicotine, likely contribute to the craving and reward of cigarette smoking in some smokers. Future studies without the cues of the smoking environment could control for this confounding influence.
In summary, smokers demonstrated correlations in [11C]carfentanil BPND with nicotine dependence and smoking status. This suggests the need for further investigation of the role of MOR in nicotine dependence.
Acknowledgments
Authors thank members of the JHU PET Center for synthesis of the radioligand and technical support that made performance of PET studies possible. Authors also thank Emily Gean for assistance with manuscript preparation.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are provided within the paper.
Funding Statement
Financial support was provided by the Intramural Research Program of NIH, National Institute on Drug Abuse (SH, CC, KM, RT), and an award from NIDA, K24 DA000412 (DFW), as well as Radiology Internal Funds (DFW). All authors declare no conflicts of interest. Additionally, resources were provided by the Johns Hopkins Institute for Clinical and Translational Research (ICTR), which is funded in part by grant number UL1 TR 001079 from the National Center for Advancing Translational Sciences (NCATS) a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the Johns Hopkins ICTR, NCATS or NIH. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ashburner J, Friston KJ (2005) Unified segmentation. Neuroimage 26:839–51 10.1016/j.neuroimage.2005.02.018 [DOI] [PubMed] [Google Scholar]
- 2.Ashburner J, Friston KJ (2003) Rigid body registration. In: Frackowiak RSJ, Friston KJ, Frith C, Dolan R, Friston KJet al. (ed)Human Brain Function. Academic Press, pp. 635–654. [Google Scholar]
- 3. Balfour DJ, Fagerström KO (1996) Pharmacology of nicotine and its therapeutic use in smoking cessation and neurodegenerative disorders. Pharmacol Ther 72:51–81. [DOI] [PubMed] [Google Scholar]
- 4. Barrett SP, Boileau I, Okker J, Pihl RO, Dagher A (2004) The hedonic response to cigarette smoking is proportional to dopamine release in the human striatum as measured by positron emission tomography and [11C]raclopride. Synapse 54:65–71 10.1002/syn.20066 [DOI] [PubMed] [Google Scholar]
- 5. Baumann B, Danos P, Krell D, Diekmann S, Leschinger A, et al. (1999) Reduced volume of limbic system-affiliated basal ganglia in mood disorders: preliminary data from a postmortem study. J Neuropsychiatry Clin Neurosci 11:71–78. [DOI] [PubMed] [Google Scholar]
- 6. Berrendero F, Kieffer BL, Maldonado R (2002) Attenuation of nicotine-induced antinociception, rewarding effects, and dependence in mu-opioid receptor knock-out mice. J. Neurosci 22:10935–10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brody AL, Mandelkern MA, Costello MR, Abrams AL, Scheibal D, et al. (2009) Brain nicotinic acetylcholine receptor occupancy: effect of smoking a denicotinized cigarette. Int J Neuropsychopharmacol. 2009 12:305–16 10.1017/S146114570800922X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brody AL, Mandelkern MA, Olmstead RE, Allen-Martinez Z, Scheibal D, et al. (2009) Ventral striatal dopamine release in response to smoking a regular vs a denicotinized vigarette. Neuropsychopharmacology. 34:282–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Buchhalter AR, Acosta MC, Evans SE, Breland AB, Eissenberg T (2005) Tobacco abstinence symptom suppression: the role played by the smoking-related stimuli that are delivered by denicotinized cigarettes. Addiction 100:550–559. [DOI] [PubMed] [Google Scholar]
- 10. CDC (2005) State-specific prevalence of cigarette smoking and quitting among adults—United States, 2004. MMWR Morb Mortal Wkly Rep 54:1124–1127. [PubMed] [Google Scholar]
- 11. Champtiaux N, Gotti C, Cordero-Erausquin M, David DJ, Przybylski C, et al. (2003) Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knock-out mice. J Neurosci 23:7820–7829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Colasanti A, Searle GE, Long CJ, Hill SP, Reiley RR, et al. (2012) Endogenous opioid release in the human brain reward system induced by acute amphetamine administration. Biol Psychiatry 72:371–377 10.1002/syn.20611 [DOI] [PubMed] [Google Scholar]
- 13. Cole DM, Beckmann CF, Long CJ, Matthews PM, Durcan MJ, et al. (2010) Nicotine replacement in abstinent smokers improves cognitive withdrawal symptoms with modulation of resting brain network dynamics. Neuroimage 52:590–599 10.1016/j.neuroimage.2010.04.251 [DOI] [PubMed] [Google Scholar]
- 14. Dannals RF, Ravert HT, Frost JJ, Wilson AA, Burns HD, et al. (1985) Radiosynthesis of an opiate receptor binding radiotracer: [11C]carfentanil. Int J Appl Radiat Isot 36:303–306. [DOI] [PubMed] [Google Scholar]
- 15. Dhatt RK, Gudehithlu KP, Wemlinger TA, Tejwani GA, Neff NH, et al. (1995) Preproenkephalin mRNA and methionine-enkephalin content are increased in mouse striatum after treatment with nicotine. J Neurochem 64:1878–1883. [DOI] [PubMed] [Google Scholar]
- 16. Donny EC, Houtsmuller E, Stitzer ML (2006) Smoking in the absence of nicotine: behavioral, subjective and physiological effects over 11 days. Addiction 102:324–334. [DOI] [PubMed] [Google Scholar]
- 17. Endres CJ, Bencherif B, Hilton J, Madar I, Frost JJ (2003) Quantification of brain mu-opioid receptors with [11C]carfentanil: reference-tissue methods. Nucl Med Biol 30:177–186. [DOI] [PubMed] [Google Scholar]
- 18. Esterlis I, Cosgrove KP, Batis JC, Bois F, Stiklus SM, et al. (2010) Quantification of smoking-induced occupancy of beta2-nicotinic acetylcholine receptors: estimation of nondisplaceable binding. J Nucl Med 51:1226–1233 10.2967/jnumed.109.072447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fischl B, van der Kouwe A, Destrieux C, Halgren E, Ségonne F, et al. (2004) Automatically parcellating the human cerebral cortex. Cereb Cortex 14:11–22. [DOI] [PubMed] [Google Scholar]
- 20. Hadjiconstantinou M, Neff NH (2011) Nicotine and endogenous opioids: neurochemical and pharmacological evidence. Neuropharmacology. 60:1209–20. [DOI] [PubMed] [Google Scholar]
- 21. Hahn B, Harvey AN, Concheiro-Guisan M, Huestis MA, Holcomb HH, et al. (2013) A Test of the Cognitive Self-Medication Hypothesis of Tobacco Smoking in Schizophrenia. Biol Psychiat. 2013 74:436–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heatherton TF, Kozlowski LT, Frecker RC, Fagerström KO (1991) The Fagerström Test for Nicotine Dependence: a revision of the Fagerström Tolerance Questionnaire. Br J Addict 86:1119–1127. [DOI] [PubMed] [Google Scholar]
- 23. Heishman SJ, Singleton EG, Pickworth WB (2008) Reliability and validity of a short form of the Tobacco Craving Questionnaire. Nicotine and Tobacco Research 10:643–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hughes J, Hatsukami DK (1998) Errors in using tobacco withdrawal scale. Tob Control 7:92–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hughes JR (2006) Clinical significance of tobacco withdrawal. Nicotine Tob Res 8:153–156 10.1080/14622200500494856 [DOI] [PubMed] [Google Scholar]
- 26. Ichise M, Liow JS, Lu JQ, Takano A, Model K, et al. (2003) Linearized reference tissue parametric imaging methods: application to [11C]DASB positron emission tomography studies of the serotonin transporter in human brain. J Cereb Blood Flow Metab 23:1096–1112. [DOI] [PubMed] [Google Scholar]
- 27. Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, et al. (2007) Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab 27:1533–1539 10.1038/sj.jcbfm.9600493 [DOI] [PubMed] [Google Scholar]
- 28. Krishnan-Sarin S, Rosen MI, O'Malley SS (1999) Naloxone challenge in smokers. Preliminary evidence of an opioid component in nicotine dependence. Arch Gen Psychiatry 56:663–668 10.1038/sj.jcbfm.9600493 [DOI] [PubMed] [Google Scholar]
- 29. Kuwabara H, McCaul ME, Wand GS, Earley CJ, Allen RP, et al. (2012) Dissociative changes in the Bmax and KD of dopamine D2/D3 receptors with aging observed in functional subdivisions of the striatum: a revisit with an improved data analysis method. J Nucl Med 53:805–812 10.2967/jnumed.111.098186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee JH, Lim Y, Wiederhold BK, Graham SJ (2005) A functional magnetic resonance imaging (FMRI) study of cue-induced smoking craving in virtual environments. Appl Psychophysiol Biofeedback 30:195–204 10.1007/s10484-005-6377-z [DOI] [PubMed] [Google Scholar]
- 31. Lerman C, Wileyto EP, Patterson F, Rukstalis M, Audrain-McGovern J, et al. (2004) The functional mu opioid receptor (OPRM1) Asn40Asp variant predicts short-term response to nicotine replacement therapy in a clinical trial. Pharmacogenomics J 4:184–192 10.1038/sj.tpj.6500238 [DOI] [PubMed] [Google Scholar]
- 32. Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, et al. (1996) Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab 16:834–840 10.1097/00004647-199609000-00008 [DOI] [PubMed] [Google Scholar]
- 33. Maes F, Collignon A, Vandermeulen D, Marchal G, Suetens P (1997) Multimodality image registration by maximization of mutual information. IEEE Trans Med Imaging 16:187–198 10.1109/42.563664 [DOI] [PubMed] [Google Scholar]
- 34. Maldjian JA, Laurienti PJ, Kraft RA, Burdette JH (2003) An automated method for neuroanatomic and cytoarchitectonic atlas-based interrogation of fMRI data sets. Neuroimage 19:1233–1239. [DOI] [PubMed] [Google Scholar]
- 35. Malin DH, Lake JR, Carter VA, Cunningham JS, Wilson OB (1993) Naloxone precipitates nicotine abstinence syndrome in the rat. Psychopharmacology (Berl.) 112:339–342. [DOI] [PubMed] [Google Scholar]
- 36. Martin-Soelch C, Missimer J, Leenders KL, Schultz W (2003) Neural activity related to the processing of increasing monetary reward in smokers and nonsmokers. Eur J Neurosci 18:680–688. [DOI] [PubMed] [Google Scholar]
- 37. Mawlawi O, Martinez D, Slifstein M, Broft A, Chatterjee R, et al. (2001) Imaging human mesolimbic dopamine transmission with positron emission tomography: I. Accuracy and precision of D(2) receptor parameter measurements in ventral striatum. J Cereb Blood Flow Metab 21:1034–57 10.1097/00004647-200109000-00002 [DOI] [PubMed] [Google Scholar]
- 38. McClernon FJ, Kozink RV, Rose JE (2008) Individual differences in nicotine dependence, withdrawal symptoms, and sex predict transient fMRI-BOLD responses to smoking cues. Neuropsychopharmacology 33:2148–2157 10.1038/sj.npp.1301618 [DOI] [PubMed] [Google Scholar]
- 39. Oswald LM, Wong DF, McCaul M, Zhou Y, Kuwabara H, et al. (2005) Relationships among ventral striatal dopamine release, cortisol secretion, and subjective responses to amphetamine. Neuropsychopharmacology 30:821–832 10.1038/sj.npp.1300667 [DOI] [PubMed] [Google Scholar]
- 40. Patenaude B, Smith SM, Kennedy DN, Jenkinson M (2011) A Bayesian model of shape and appearance for subcortical brain segmentation. Neuroimage 56:907–922 10.1016/j.neuroimage.2011.02.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pontieri FE, Tanda G, Orzi F, Di Chiara G (1996) Effects of nicotine on the nucleus accumbens and similarity to those of addictive drugs. Nature 382:255–257 10.1038/382255a0 [DOI] [PubMed] [Google Scholar]
- 42. Rahmin A, Cheng JC, Blinder S, Camborde ML, Sossi V (2005) Statistical dynamic image reconstruction in state-of-the-art high-resolution PET. Phys Med Biol 50:4887–4912. [DOI] [PubMed] [Google Scholar]
- 43. Ray R, Ruparel K, Newberg A, Wileyto EP, Loughead JW, et al. (2011) Human Mu Opioid Receptor (OPRM1 A118G) polymorphism is associated with brain mu-opioid receptor binding potential in smokers. Proc Natl Acad Sci USA 108:9268–9273 10.1073/pnas.1018699108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Scott DJ, Domino EF, Heitzeg MM, Koeppe RA, Ni L, et al. (2007) Smoking modulation of mu-opioid and dopamine D2 receptor-mediated neurotransmission in humans. Neuropsychopharmacology 32:450–457 10.1038/sj.npp.1301238 [DOI] [PubMed] [Google Scholar]
- 45.Sossi V, De Jong M, Barker W, Bloomfield P, Burbar Z, et al. (2005) The second generation HRRT: a multi-centre scanner performance investigation. IEEE Nucl. Sci. Symp. Conf, pp. 2195–2199.
- 46. Walters CL, Cleck JN, Kuo YC, Blendy JA (2005) Mu-opioid receptor and CREB activation are required for nicotine reward. Neuron 46:933–943 10.1016/j.neuron.2005.05.005 [DOI] [PubMed] [Google Scholar]
- 47. Weerts EM, McCaul ME, Kuwabara H, Yang X, Xu X, et al. (2013) Influence of OPRM1 Asn40Asp variant (A118G) on [11C]carfentanil binding potential: preliminary findings in human subjects. Int J Neuropsychopharmacol 16:47–53 10.1017/S146114571200017X [DOI] [PubMed] [Google Scholar]
- 48. Weerts EM, Wand GS, Kuwabara H, Munro CA, Dannals RF, et al. (2011) Positron emission tomography imaging of mu- and delta-opioid receptor binding in alcohol-dependent and healthy control subjects. Alcohol Clin Exp Res 35:2162–2173 10.1111/j.1530-0277.2011.01565.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zubieta JK, Smith YR, Bueller JA, Xu Y, Kilbourn MR, et al. (2001) Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science 293:311–315 10.1126/science.1060952 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are provided within the paper.