

Abstract
Neurofibromatosis type 2 (NF2) is an autosomal dominant genetic disorder resulting from germline mutations in the NF2 gene. Bilateral vestibular schwannomas, tumors on cranial nerve VIII, are pathognomonic for NF2 disease. Furthermore, schwannomas also commonly develop in other cranial nerves, dorsal root ganglia and peripheral nerves. These tumors are a major cause of morbidity and mortality, and medical therapies to treat them are limited. Animal models that accurately recapitulate the full anatomical spectrum of human NF2-related schwannomas, including the characteristic functional deficits in hearing and balance associated with cranial nerve VIII tumors, would allow systematic evaluation of experimental therapeutics prior to clinical use. Here, we present a genetically engineered NF2 mouse model generated through excision of the Nf2 gene driven by Cre expression under control of a tissue-restricted 3.9kbPeriostin promoter element. By 10 months of age, 100% of Postn-Cre; Nf2flox/flox mice develop spinal, peripheral and cranial nerve tumors histologically identical to human schwannomas. In addition, the development of cranial nerve VIII tumors correlates with functional impairments in hearing and balance, as measured by auditory brainstem response and vestibular testing. Overall, the Postn-Cre; Nf2flox/flox tumor model provides a novel tool for future mechanistic and therapeutic studies of NF2-associated schwannomas.
INTRODUCTION
Neurofibromatosis type 2 (NF2) is an autosomal dominant disorder that arises from germline heterozygosity of the NF2 tumor suppressor gene, and affected individuals have a high propensity for the acquisition of multiple schwannomas. Vestibular schwannomas that develop on cranial nerve VIII (CN VIII) are pathognomonic for NF2, arise with nearly complete penetrance and cause progressive hearing loss and balance abnormalities (1). In addition, vestibular schwannomas can also contribute to early mortality owing to brainstem compression and surgical complications (1).
Although vestibular schwannomas are pathognomonic for NF2, schwannomas can occur on any cranial, spinal or distal peripheral nerve. In addition, sporadic schwannomas observed in patients without familial NF2 disease are almost universally NF2 deficient, underscoring the critical function of the NF2 tumor suppressor gene in Schwann cells (1). Sporadic schwannomas are often treated with surgery and/or radiotherapy, but the latter option is controversial for NF2 patients already harboring a germline mutation in a tumor suppressor gene (2). Thus, there exists a large unmet clinical need for medical alternatives to treatment of schwannomas, especially in NF2 patients.
A useful preclinical animal model of human disease requires broad physiological relevance at both genotypic and phenotypic levels (3). Therefore, genetically engineered murine (GEM) models of human disease should harbor mutations in gene(s) homologous to gene(s) mutated in human cancer. Moreover, this genetic perturbation should occur in cell types and gene doses similar to the hypothetical human tumor cell of origin. These tissue-specific genetic mutations should induce tumors that share histological and biological characteristics with the corresponding human tumors. Additionally, the GEM tumors should be expected to develop in a similar anatomical distribution and with similar functional consequence as human tumors. Importantly, the first-generation murine NF2 model demonstrated that Nf2 disruption in Schwann cell lineages during embryogenesis promotes schwannoma formation (4,5). In existing models, however, schwannomas are incompletely penetrant (only occurring in about one-third of mice) and, generally, arise only late in life. Furthermore, vestibular schwannomas have not been observed in existing murine models of NF2. These temporal, anatomical and functional divergences from the human phenotype are critical barriers to in vivo genetic and pharmacologic studies that could facilitate better understanding of the molecular pathogenesis of NF2-associated schwannomas. Additionally, lack of an optimal mouse model NF2-associated schwannomas prevents the development and preclinical testing of novel therapeutics for these tumors.
The prior published murine Nf2 conditional knockout model depends on Cre-mediated Nf2 allele excision driven by the P0 promoter element, which is expressed in neural crest cells at embryonic day 9.5 (E9.5) and in Schwann cell progenitors at E12.5 (5). We hypothesized that an alternate promoter element with distinct developmental timing and anatomical distribution may provide an additional opportunity to recapitulate the phenotypic consequences of human NF2 mutations and disease manifestations in mice. Therefore, we investigated Cre expression driven by the 3.9-kb upstream promoter region of the Periostin gene (Postn-Cre), which prior studies have shown to drive robust reporter gene expression in Schwann cell progenitors beginning at E10 (6). We intercrossed mice carrying loxp sequences flanking exon 2 of the Nf2 gene (Nf2flox/flox) (5) with Postn-Cre mice, generating Postn-Cre; Nf2flox/flox mice and their Cre-negative control littermates. Here, we present this intercross, which results in the consistent development of schwannomas that closely recapitulate features of the human disease, including hearing loss and vestibular impairment associated with vestibular schwannoma development.
RESULTS
Initial studies demonstrated that all Postn-Cre; Nf2flox/flox mice sacrificed at 15 months of age possessed significant enlargements of the dorsal root ganglion (DRG) and proximal spinal nerve roots (Fig. 1A and B). Subsequent histological analyses indicated that these enlargements are present at every vertebral level in Postn-Cre; Nf2flox/flox mice. To better understand the time course of these enlargements, we sacrificed and dissected mice at multiple time points. By 5 months of age, all Postn-Cre; Nf2flox/flox mice acquired enlargements of their DRG and proximal spinal nerve roots, with microscopic evidence of Schwann cell hyperplasia and/or schwannoma, featuring dense, intersecting fascicles of Schwann cells (Supplementary Material, Fig. S1). By 8 months, all Postn-Cre; Nf2flox/flox mice demonstrated DRG and proximal spinal nerve tumors with histological characteristics of frank schwannoma (Fig. 1C and D). Further analyses demonstrated diffuse S100-positivity of tumors from Postn-Cre; Nf2flox/flox mice, consistent with the established diagnostic criteria for genetically engineered mouse (GEM) Grade I schwannoma (Fig. 1E) (9). The histological pattern of growth observed in these tumors largely resembled the Antoni A pathologic subtype observed in human tumors, which has been noted in previous studies of murine schwannomas (9). At 15 months, dissected spinal cords from Postn-Cre; Nf2flox/flox mice demonstrated more than a five-fold increase in DRG volume as compared with Cre-negative control littermates (Fig. 1F). Thus, Postn-Cre; Nf2flox/flox mice develop slow-growing, low-grade schwannomas highly reminiscent of schwannomas observed in individuals with NF2.
Figure 1.

Postn-Cre; Nf2flox/flox mice develop schwannomas of the DRG and spinal nerves. (A and B) Dissected spinal cord of 15-month-old Cre-negative control (A) and Postn-Cre; Nf2flox/flox (B) mice, with Postn-Cre; Nf2flox/flox mice displaying diffusely enlarged DRG and spinal nerves (red arrowheads). (C and D) Hematoxylin and eosin (H&E) stain of the DRG from 7-month Cre-negative control and Postn-Cre; Nf2flox/flox mice, respectively. The whorls of Schwann cell proliferation observed in D are characteristic of schwannoma histology. Original magnification ×200. (E) Immunohistochemical S100 stain indicating tumors are comprised of mature Schwann cells. Original magnification ×400. (F) DRG volume quantitation of 15-month-old Cre-negative control mice (4) and Postn-Cre; Nf2flox/flox mice (6). P < 0.0001, unpaired Student's t-test with Welch's correction. (G) Kaplan–Meier survival analysis of Cre-negative control mice (7) and Postn-Cre;Nf2flox/flox mice (8). P < 0.001, log-rank (Mantel–Cox) test.
Detailed temporal analysis of cohorts revealed that Postn-Cre; Nf2flox/flox mice had a reduced lifespan as compared with Cre-negative Nf2flox/flox control mice (Fig. 1G). In the majority of mice, only multiple large schwannomas were observed. However, 5 of 16 Postn-Cre; Nf2flox/flox mice analyzed required sacrifice owing to the development of tumors that, upon histological examination, were found to be GEM Grade III lesions consistent with malignant peripheral nerve sheath tumors (MPNSTs) (Supplementary Material, Fig. S2). MPNSTs, malignant sarcomas and malignant carcinomas have also been observed with disruption of Nf2 in other genetically engineered models (5).
Detailed examination of schwannomas excised from Postn-Cre; Nf2flox/flox mice indicated that these tumors possessed an architecture highly reminiscent of schwannomas found in human NF2 disease. Some nerves demonstrated discrete nodules of schwannoma, similar to the Schwann cell tumorlets commonly observed in the cauda equina of NF2 patients (Fig. 2A) (7). In multiple cases, proliferating schwann cells caused diffuse expansion of the nerve and extended into its ramifications, simulating plexiform schwannomas frequently encountered in human NF2 patients (Fig. 2B). Other focal lesions demonstrated pseudo-onion bulb formations comprised of proliferating Schwann cells tightly whorled around a central axon, a histological feature also found in some human NF2-associated schwannomas (Fig. 2C). Finally, in some nerves, proliferating Schwann cells did not form a discrete mass but rather exhibited diffuse regions of hypercellularity, possibly representing an early neoplastic change prior to schwannoma development (Fig. 2D).
Figure 2.

Schwannomas observed in Postn-Cre; Nf2flox/flox mice histologically resemble those observed in human patients. (A) H&E stain showing a multifocal pattern of schwannoma growth along a nerve (red arrowheads). Original magnification ×100. (B) Example of a plexiform schwannoma involving a nerve twig with peripheral axons. Original magnification ×400. (C) Pseudo-onion bulb formation of proliferating Schwann cells forming whorls around centrally located axons within a nerve. Original magnification ×200. (D) Diffuse spinal nerve hyperplasia observed in the nerve of a Postn-Cre; Nf2flox/flox mice. Original magnification ×400.
NF2 patients develop schwannomas in multiple anatomic regions, including peripheral, cranial and spinal nerves (10). Interestingly, by 10 months of age, thorough dissection and histological analysis of each Postn-Cre; Nf2flox/flox mouse revealed schwannomas in nearly all histological specimens of peripheral, cranial and spinal nerves. For example, all Postn-Cre; Nf2flox/flox mice examined for cranial nerve pathology featured schwannomas of the trigeminal (CN V) and facial (CN VII) nerves (Supplementary Material, Fig. S3). Given the high correlation between human NF2 disease and the presence of bilateral vestibular schwannomas, we focused on CN VIII and its associated functions. Histological analysis of Postn-Cre; Nf2flox/flox mice revealed aberrant Schwann cell growth in CN VIII proximal to its entry into the inner ear, including tumors observed in Scarpa's ganglion, the anatomic site of the afferent nerve cell bodies prior to their synapse with vestibular hair cells (Fig. 3A and 3B). Postn-Cre; Nf2flox/flox mice also demonstrated Schwann cell hyperplasia in the spiral ganglion (Fig. 3C), a structure containing nerve cell bodies that transmit sound-derived nerve impulses from the cochlea to the brain. One-hundred percent of Postn-Cre; Nf2flox/flox mice analyzed for CN VIII pathology had vestibular schwannomas, whereas no remarkable pathology was observed in CN VIII of Cre-negative littermates. Importantly, the cochlea did not display any other obvious structural differences between the two genotypes (Supplementary Material, Fig. S4).
Figure 3.

Postn-Cre; Nf2flox/flox mice develop schwannomas on CN VIII. (A1) H&E stain of a vestibular schwannoma located in Scarpa's ganglion (ScG). Original magnification ×100. In the 200× magnified inset image (A2), notice the Schwann cell proliferation displacing nerve cell bodies (black arrows) and pushing axons to the periphery of the nerve (red arrowheads). (B) Diffuse hypercellularity observed in CN VIII. Original magnification ×100. (C1) Schwann cell proliferation in the spiral ganglion (SpG), expanding and disrupting the architecture of the ganglion. Original magnification ×100. In the magnified (400×) inset C2, notice the dysplastic, irregularly spaced Schwann cells. Cochlea (Co), scala media (SM), scala vestibuli (SV) and scala tympani (ST).
To correlate these CN VIII lesions with potential functional hearing deficits, we used click-evoked auditory brainstem response (ABR) testing, a standard method to assess hearing loss in humans and small animals (11). ABRs are electrical potentials recorded from electrodes placed on the scalp and near the ear. In response to sound stimuli, a characteristic ABR waveform with a series of peaks and troughs is observed, where peaks correspond to different components of the ascending auditory pathway (12). We tested cohorts of mice at 3, 6, 8, and 10 months of age, finding a progressively increasing ABR threshold in Postn-Cre; Nf2flox/flox mice, whereas control mice maintained a relatively consistent hearing threshold throughout the study (Fig. 4A and B). Given that this click-ABR screening was limited to a stimulus range from 30 to 90 decibel sound pressure level (dB SPL) and that the majority of Cre-negative mice exhibited robust waveforms at 30 dB SPL, it is likely that the mean differences between the two groups are even larger than those depicted in Figure 4B. Thus, the present data indicate a hearing loss that is temporally correlated with tumorigenesis in this mouse model. Functionally, a sensorineural hearing loss of this type would negatively impact a patient's quality of life by limiting communication and social interaction in everyday listening environments with background noise.
Figure 4.
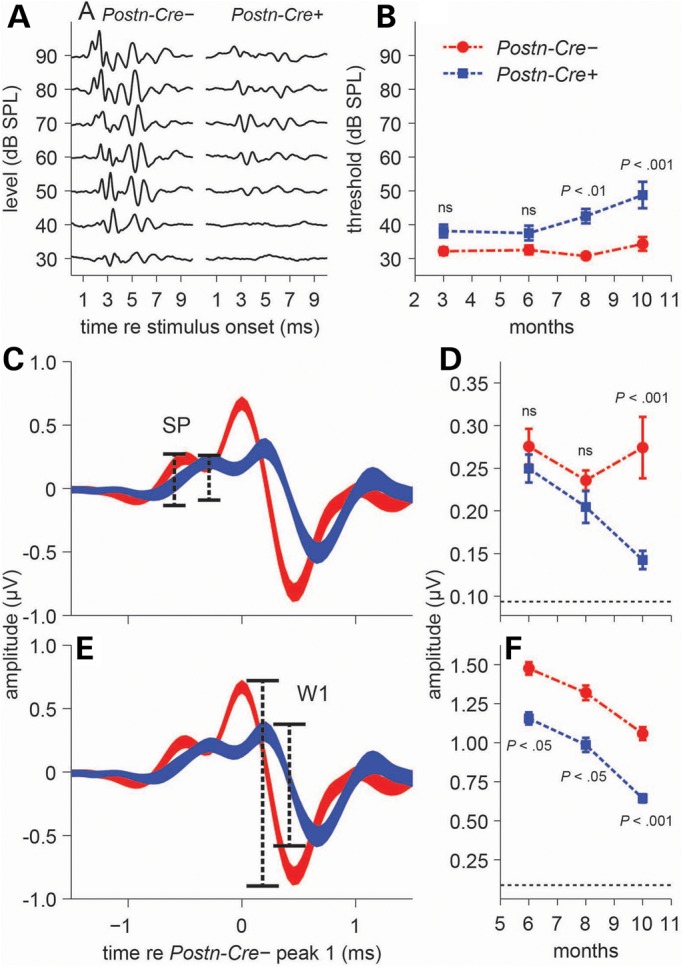
Postn-Cre; Nf2flox/flox mice show deficits in both threshold and supra-threshold ABRs. (A) Representative ABR waveforms from individual Cre-negative (left) and Postn-Cre; Nf2flox/flox mice (right) at 10 months of age. (B) Time-course click-ABR study quantitating differences between Cre-negative and Postn-Cre; Nf2flox/flox mice. Significant threshold elevations were seen in Postn-Cre; Nf2flox/flox mice at 8 (P < 0.01) and 10 (P < 0.001) months of age (n = 15, 12, 15 and 19 mice for 3, 6, 8 and 10 months, respectively). Thresholds were averaged across animals (mean ± SEM). (C and D) The SP (C), dominated by inner-hair-cell receptor potentials and evident in the ABR waveform immediately preceding peak 1, is comparable in amplitude across groups at 6 and 8 months of age but is reduced in Postn-Cre; Nf2flox/flox mice at 10 months (P < 0.001) (D). (E and F) ABR wave 1 (W1) (E), representing the summed response of the cochlear nerve, is reduced in Postn-Cre; Nf2flox/flox mice compared with Cre-negative controls at all ages (P < 0.05, P < 0.05, P < 0.001) (F), indicating deficits in pre- and/or post-synaptic cochlear-nerve function. All statistical tests: two-way ANOVA with Bonferonni post hoc analysis. Thickness of waveforms (C and E) represents mean ± SEM. Amplitudes (D and F) represent high-level responses (across 60–90 dB SPL) averaged across animals (mean ± SEM). Horizontal dotted lines (D and F) at the base of the y-axis indicate the noise floor measurement.
To expand our analysis of the hearing impairment in Postn-Cre; Nf2flox/flox mice, we studied the individual peaks present in click-ABR waveforms (Fig. 4C and E). The first ABR wave (W1) represents potentials generated from the cochlear nerve (13). The summating potential (SP), which appears as a small shoulder preceding W1, is largely comprised of potentials originating from the inner hair cells of the cochlea, with some contributions from outer hair cells (14,15). No significant differences were observed between Postn-Cre; Nf2flox/flox mice and Cre-negative control SPs at both the 6- and 8-month time points, but a significant reduction in SP amplitude was observed in the Postn-Cre; Nf2flox/flox mice at 10 months (Fig. 4D). W1 amplitude was significantly attenuated in the Postn-Cre; Nf2flox/flox mice compared with Cre-negative controls at all of the tested time points (Fig. 4F). The reduction in W1 amplitude at the 6- and 8-month time points, when contrasted with the non-significant differences in SP amplitude, specifically points to a functional disruption in the cochlear synapse and/or cochlear nerve in Postn-Cre; Nf2flox/flox mice. These data also indicate a more extensive disruption of auditory function at the 10-month time point, with decreased SPs suggesting diminished hair-cell function in addition to deficits in the cochlear synapse and/or cochlear nerve. Altogether, these studies reveal that Postn-Cre; Nf2flox/flox mice acquire sensorineural hearing loss that, in consideration of the schwannoma histopathology described in Figure 3, likely originates in the cochlear nerve.
Vestibular schwannomas can also severely impair a patient's sense of normal balance and orientation, leading to vertigo, nausea, accidents related to falls, and, thus, substantially decreased quality of life. In our studies with Postn-Cre; Nf2flox/flox mice, we observed that some mice acquired a phenotype suggestive of vestibular dysfunction, as they exhibited head tilting, head tossing, head bobbing, and locomotive circling behavior. This behavior is collectively referred to as shaker/waltzer behavior and has been observed in mice possessing defects in their vestibular organs (8,16). To test for vestibular dysfunction, we conducted three different behavioral tests to examine the integrity of the vestibular system (17). Each mouse underwent a trunk curl, contact righting and swim test on three occasions over the course of 3 consecutive days, with the results scored by two independent observers experimentally blinded to the genotype of the mice. Postn-Cre; Nf2flox/flox mice demonstrated profound impairment in their ability to successfully complete each of the three tests when compared with their Cre-negative littermates (Table 1).
Table 1.
Postn-Cre; Nf2flox/flox mice develop vestibular impairment
| Test | Genotype | Number of mice | Successes | Failures | P |
|---|---|---|---|---|---|
| Trunk curl | Postn-Cre− | 7 | 19 | 2 | <0.0001 |
| Postn-cre+ | 7 | 4 | 17 | ||
| Contact righting | Postn-Cre− | 6 | 18 | 0 | <0.0001 |
| Postn-Cre+ | 7 | 4 | 17 | ||
| Swimming | Postn-Cre− | 7 | 21 | 0 | <0.0001 |
| Postn-Cre+ | 7 | 5 | 16 |
Results of examination of the vestibular system in Postn-Cre; Nf2flox/flox mice and Cre-negative controls. Tests were conducted on 3 consecutive days and scored by two independent scorers experimentally blinded to the animal's genotype. Mice were scored on a two-point scale, either succeeding or failing a particular test. A detailed description of the three tests can be found in the section Materials and methods. All mice were 13 months old at the time of testing. Fischer's exact test was used to test for statistically significant differences.
To gain a more detailed understanding of the temporal, anatomical, and cellular origins of Nf2-associated schwannomas, we further characterized gene expression driven by the 3.9kbPeriostin promoter element. First, recombination of Nf2 was confirmed in the cranial and spinal nerves (Supplementary Material, Fig. S5). Next, we performed embryologic studies with an intercross between Postn-Cre and Rosa26 lox-stop-lox LacZ reporter mice. By E12.5, robust β-galactosidase expression was observed in the DRG, facial ganglia and the developing cranial, spinal and peripheral nerves (Supplementary Material, Fig. S6A, B, and C). Approximately half of Schwann cells derived from the sciatic nerve of adult mice demonstrated positive X-gal staining with universal S100-positivity (Supplementary Material, Fig. S6D and E). In the DRG, nerve roots and peripheral nerves of E14 and E14.5 tissue, β-galactosidase expression coincided with Brain Lipid-Binding Protein (Blbp) expression, an established immunological marker of Schwann cell precursors and immature Schwann cells (Supplementary Material, Fig. S6F and G) (18). Schwann cell precursors and immature Schwann cells are multipotential embryonic Schwann cell lineages that descend from neural crest cells and represent the progenitor cell populations that give rise to mature Schwann cells. Importantly, the time points of the X-gal-stained embryos in Supplementary Material, Figure S6 represent the transition from Schwann cell precursors (E12–E13) to immature Schwann cells (E13–E15) (18). These data indicate that Postn-Cre is expressed in early multipotent glial cell populations, likely representing the tumor cell of origin for this model.
DISCUSSION
In this study, we describe a novel NF2 mouse model recapitulating schwannoma phenotypes found in human patients. NF2-associated schwannomas are most frequently located on CN VIII and/or the spinal roots, and these tumors are seen in nearly all NF2 patients (1). The NF2 mouse model presented in this study develops tumors on both structures with complete penetrance, and it also demonstrates functional impairments in hearing and balance that are commonly associated with human vestibular schwannomas.
The characteristics of this model permit three broad areas of translational study. First, the genesis of genetically engineered models and the use of genetic intercrosses to provide in vivo proofs of concept have recently resulted in advances in the understanding of tumor suppressor genes and their roles in regulating specific signaling pathways, including other neurocutaneous disorders such as NF1 and tuberous sclerosis (19–24). While the NF2 gene product was identified as mutated in familial NF2 disease over 25 years ago, it remains unclear which pathways are critical to tumor development downstream of NF2 (25). This genetic model may provide the means for elucidating which signaling pathways downstream of NF2 are essential to schwannoma development.
Second, the utility of accurate preclinical models that allow physiologic testing of clinical phenotypes is particularly important for orphan genetic diseases such as NF2, where patient numbers limit the ability to conduct multiple meaningful trials. In the entire USA, it is estimated that there are 12 000 individuals that have NF2. Though nearly all patients acquire vestibular schwannomas, at any given time, only a small number of patients are available for clinical trials. Thus, preclinical models that can effectively screen for pharmaceutical agents that have meaningful therapeutic benefit for schwannomas are essential for the advancement of NF2 therapies. Functional ABR assessment of hearing during studies utilizing this model provides a unique, non-invasive and physiologically relevant measure for determining therapeutic efficacy of compounds.
Finally, a preclinical model can assist in understanding the basic biology of the hearing loss associated with vestibular schwannomas, which is incompletely understood. Tumor size resulting in local compression of CN VIII does not appear to account completely for the onset and progression of hearing loss in NF2 patients (26). Interestingly, the recent short-term improvements in hearing with Bevacizumab treatment of vestibular schwannomas in NF2 patients are not consistently associated with a reduction in tumor size (27). It has also been suggested that biochemical disturbance of the inner-ear fluids could be the underlying cause of hearing loss related to these tumors (28). Otoacoustic-emission studies in patients with vestibular schwannomas support the hypothesis that some degree of hearing loss is cochlear in origin (29,30). Altogether, the degree to which hearing loss in these patients is retrocochlear (thus likely originating in the cochlear nerve) or cochlear in origin remains an open question.
Our studies suggest functional impairment of cochlear-nerve activity in Postn-Cre; Nf2flox/flox mice at both early (6 and 8 months) and later (10 months) stages of disease progression. In contrast, Postn-Cre; Nf2flox/flox mice have significantly reduced SPs only at 10 months, suggesting that the structures required for mechanoelectric transduction of sound prior to the inner-hair cell-cochlear-nerve synapse are not significantly impaired earlier in the disease process. These findings, in association with the neoplastic Schwann cell accumulation and extensive morphologic changes seen in the spiral ganglion, strongly suggest that a significant component of the hearing loss in this model originates in the cochlear nerve. Evidence for an additional, later-onset dysfunction of cochlear origin comes from the combination of increased thresholds at 10 months with further decreased wave 1 amplitude and decreased SP. In summary, these data suggest an early retrocochlear form of hearing loss beginning in the cochlear nerve, and, at later time points, a more extensive form of hearing loss incorporating both the cochlear nerve and cochlea. Further studies utilizing the Postn-Cre-based NF2 mouse model may allow additional insights into this clinical question regarding the mechanisms of hearing loss present in vestibular schwannoma patients.
MATERIALS AND METHODS
Study approval
All studies were carried out in accordance with, and approval of, the Institutional Animal Care and Use Committee of Indiana University Medical School, the U.S. Department of Agriculture's Animal Welfare Act (9 CFR Parts 1, 2 and 3) and the Guide for the Care and Use of Laboratory Animals.
Statistical methods
Statistical analyses were performed with GraphPad Prism 5.0. The Kaplan–Meier method was used to analyze the survival outcomes of mice, and the log-rank (Mantel–Cox) test was used to compare survival curves. As noted in the text, Analysis of Variance (ANOVA) with Bonferonni post hoc analysis or Student's t-test was used to test for differences in tumor volume and hearing threshold. Specific tests and significance levels can be found in the figures and figure legends. Fischer's exact test for independence was performed on the vestibular phenotypic data using the R Statistical Programming Environment. P-values of <0.05 were considered significant.
Hearing screening
Auditory brainstem responses were recorded from left and right ears of anesthetized mice (100 mg/kg ketamine and 10 mg/kg xylazine by intraperitoneal injection) using subdermal needle electrodes. Stimuli were produced, and responses were recorded with a Tucker-Davis Technologies (TDT) BioSigRZ system, using an RZ6 digital/analog converter (TDT). Responses to clicks (512 repetitions, 30–90 dB SPL in 10 dB steps, presented at 21/s with a closed-field system) were amplified, filtered (3 Hz–3 kHz), averaged and stored for offline analysis in Matlab (Mathworks, Natick, MA, USA). ABR waveforms were additionally high-pass-filtered (cut-off 200 Hz) to remove slow oscillations and emphasize characteristic ABR peaks. ABR threshold was defined as the lowest measured SPL at which a reproducible peak or trough was identified. Amplitude of the SP (Fig. 4C) was calculated as baseline-to-peak, and amplitude of wave 1 (W1, Fig. 4E) was calculated as peak-to-trough.
Mice and genotyping
Postn-Cre and Nf2flox/flox transgenic lines were maintained on Teklad Lab Animal Diet (TD 2014, Harlan Laboratories USA) using a 12:12 (light/dark) photoperiod at 22–24°C. Nf2flox2 and Nf2Δ2 bands were detected by PCR analysis as described in Giovannini et al. (4). The Postn-Cre transgene was detected by PCR analysis with the following primers: P1 (CAT-TTG-GGC-CAG-CTA-AAC-AT) and P2 (CCC-GGC-AAA-ACA-GGT-AGT-TA). Band sizes are reported in Supplementary Material, Figure S5. Postn-Cre; Nf2flox/flox mice and Cre-negative controls possessed the same mixed genetic background, with SNP analysis indicating close relation to the FVB/NTac strain (87.37% identity).
Histology and immunohistochemistry
Tumors were excised from recently sacrificed animals, fixed with 10% formalin, embedded in paraffin, sectioned and stained with H&E. Embryos were fixed in 4% paraformaldehyde (PFA), stained with X-gal, embedded in paraffin and then sectioned. Vector labs ABC (PK-6100), DAB (SK-4100) and Avidin/Biotin blocking (SP-2001) kits were used for immunoperoxidase staining. BLBP (Abcam, ab32423) primary antibody was detected with biotinylated goat anti-rabbit secondary antibody from Sigma (cat. B8896). For sectioning and staining of inner-ear structures, mice were fixed with 4% PFA injected via intracardiac perfusion into the right atrium. Samples were then sent to Dr Brian Faddis at the Otolaryngology Histology Core at Washington University in St. Louis for processing and staining.
Tumor volume quantitation
After fixation and decalcification in 5% formic acid, the DRG and spinal nerves were then dissected out under a microscope. Tumor volume was calculated using the length and width values for a particular tumor in the formula volume = length × width2 × 0.52, the approximate volume of a spheroid.
Vestibular studies
All of the functional vestibular studies (trunk curl, contact righting and swim) were performed on 3 consecutive days, with each test being performed once per day. The tests were scored by two independent observers experimentally blinded to the genotype of each animal. Fischer's exact test was used to test for differences in the two genotypes.
For the trunk curl test, mice were individually suspended by their tails and presented with a horizontal platform roughly 2 cm outside their reach. Control mice will reach toward this platform, but mice with vestibular impairment will tend to curl in toward their abdomen with their head and upper body. This inward curling was scored as a failure for the test. Mice were scored on a two-point system, either succeeding or failing the test.
In the contact righting test, mice were placed in a 45-cm Plexiglas tube with a diameter of 6 cm, and the tube was re-oriented until the mouse was at the approximate center of the tube, facing toward the closed end. The tube was then rotated, and the ability of the mice to correct their posture was assessed. Mice that had significant difficulty adjusting to the rotation fell on their sides or on their backs. This was scored as a failure of the test. Mice were scored on a two-point system, either succeeding or failing the test.
For the swim test, a large sink (50 × 40 × 20 cm) was filled with tepid water to a depth of 18 cm and given several hours to equilibrate to room temperature. Mice were placed in the sink individually, and their ability to swim was assessed for 1 min. Normal swimming behavior was noted when the animal kept its body aligned horizontally in the water with its nose clearly above the surface. Abnormal swimming was noted when the animal made vertical deviations, had substantial head tilt, assumed an immobile floating position or had significant lower limb thrashing. Any abnormal swimming was scored as a failure. Mice were scored on a two-point system, either succeeding or failing the test.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the Department of Defense (NF110107 to D.W.C.); National Institute of Health (P30 CA082709-14 and P30 DC004665 to R.A.C.); and the Children's Tumor Foundation (2012-01-013 to J.R.G.).
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge the thoughtful review of this manuscript by Dr Marco Giovannini. We also recognize the essential contribution of Dr Brian Faddis at the Research Core Center for Auditory and Vestibular Studies at Washington University in St. Louis.
Conflict of Interest statement: none declared.
REFERENCES
- 1.Asthagiri A.R., Parry D.M., Butman J.A., Kim H.J., Tsilou E.T., Zhuang Z., Lonser R.R. Neurofibromatosis type 2. Lancet. 2009;373:1974–1986. doi: 10.1016/S0140-6736(09)60259-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di Maio S., Akagami R. Prospective comparison of quality of life before and after observation, radiation, or surgery for vestibular schwannomas. J. Neurosurg. 2009;111:855–862. doi: 10.3171/2008.10.JNS081014. [DOI] [PubMed] [Google Scholar]
- 3.Sharpless N.E., Depinho R.A. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat. Rev. Drug. Discov. 2006;5:741–754. doi: 10.1038/nrd2110. [DOI] [PubMed] [Google Scholar]
- 4.Giovannini M., Robanus-Maandag E., Niwa-Kawakita M., van der Valk M., Woodruff J.M., Goutebroze L., Merel P., Berns A., Thomas G. Schwann cell hyperplasia and tumors in transgenic mice expressing a naturally occurring mutant NF2 protein. Genes Dev. 1999;13:978–986. doi: 10.1101/gad.13.8.978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giovannini M., Robanus-Maandag E., van der Valk M., Niwa-Kawakita M., Abramowski V., Goutebroze L., Woodruff J.M., Berns A., Thomas G. Conditional biallelic Nf2 mutation in the mouse promotes manifestations of human neurofibromatosis type 2. Genes Dev. 2000;14:1617–1630. [PMC free article] [PubMed] [Google Scholar]
- 6.Lindsley A., Snider P., Zhou H., Rogers R., Wang J., Olaopa M., Kruzynska-Frejtag A., Koushik S.V., Lilly B., Burch J.B., et al. Identification and characterization of a novel Schwann and outflow tract endocardial cushion lineage-restricted periostin enhancer. Dev. Biol. 2007;307:340–355. doi: 10.1016/j.ydbio.2007.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stemmer-Rachamimov A.O., Ino Y., Lim Z.Y., Jacoby L.B., MacCollin M., Gusella J.F., Ramesh V., Louis D.N. Loss of the NF2 gene and merlin occur by the tumorlet stage of schwannoma development in neurofibromatosis 2. J. Neuropathol. Experim. Neurol. 1998;57:1164–1167. doi: 10.1097/00005072-199812000-00008. [DOI] [PubMed] [Google Scholar]
- 8.Tsai H., Hardisty R.E., Rhodes C., Kiernan A.E., Roby P., Tymowska-Lalanne Z., Mburu P., Rastan S., Hunter A.J., Brown S.D., et al. The mouse slalom mutant demonstrates a role for Jagged1 in neuroepithelial patterning in the organ of Corti. Hum. Mol. Genet. 2001;10:507–512. doi: 10.1093/hmg/10.5.507. [DOI] [PubMed] [Google Scholar]
- 9.Stemmer-Rachamimov A.O., Louis D.N., Nielsen G.P., Antonescu C.R., Borowsky A.D., Bronson R.T., Burns D.K., Cervera P., McLaughlin M.E., Reifenberger G., et al. Comparative pathology of nerve sheath tumors in mouse models and humans. Cancer Res. 2004;64:3718–3724. doi: 10.1158/0008-5472.CAN-03-4079. [DOI] [PubMed] [Google Scholar]
- 10.Sperfeld A.D., Hein C., Schroder J.M., Ludolph A.C., Hanemann C.O. Occurrence and characterization of peripheral nerve involvement in neurofibromatosis type 2. Brain. 2002;125:996–1004. doi: 10.1093/brain/awf115. [DOI] [PubMed] [Google Scholar]
- 11.Zheng Q.Y., Johnson K.R., Erway L.C. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hearing Res. 1999;130:94–107. doi: 10.1016/s0378-5955(99)00003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henry K.R. Auditory brainstem volume-conducted responses: origins in the laboratory mouse. J. Am. Audit. Soc. 1979;4:173–178. [PubMed] [Google Scholar]
- 13.Melcher J.R., Kiang N.Y. Generators of the brainstem auditory evoked potential in cat. III: identified cell populations. Hearing Res. 1996;93:52–71. doi: 10.1016/0378-5955(95)00200-6. [DOI] [PubMed] [Google Scholar]
- 14.Santarelli R., Del Castillo I., Rodriguez-Ballesteros M., Scimemi P., Cama E., Arslan E., Starr A. Abnormal cochlear potentials from deaf patients with mutations in the otoferlin gene. J. Assoc. Res. Otolaryngol. 2009;10:545–556. doi: 10.1007/s10162-009-0181-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Durrant J.D., Wang J., Ding D.L., Salvi R.J. Are inner or outer hair cells the source of summating potentials recorded from the round window? J. Acoust. Soc. Am. 1998;104:370–377. doi: 10.1121/1.423293. [DOI] [PubMed] [Google Scholar]
- 16.Vidal P.P., Degallaix L., Josset P., Gasc J.P., Cullen K.E. Postural and locomotor control in normal and vestibularly deficient mice. J. Physiol. 2004;559:625–638. doi: 10.1113/jphysiol.2004.063883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hardisty-Hughes R.E., Parker A., Brown S.D. A hearing and vestibular phenotyping pipeline to identify mouse mutants with hearing impairment. Nat. Protoc. 2010;5:177–190. doi: 10.1038/nprot.2009.204. [DOI] [PubMed] [Google Scholar]
- 18.Jessen K.R., Mirsky R. The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci. 2005;6:671–682. doi: 10.1038/nrn1746. [DOI] [PubMed] [Google Scholar]
- 19.Yang F.C., Ingram D.A., Chen S., Zhu Y., Yuan J., Li X., Yang X., Knowles S., Horn W., Li Y., et al. Nf1-dependent tumors require a microenvironment containing Nf1+/− and c-kit-dependent bone marrow. Cell. 2008;135:437–448. doi: 10.1016/j.cell.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pollizzi K., Malinowska-Kolodziej I., Doughty C., Betz C., Ma J., Goto J., Kwiatkowski D.J. A hypomorphic allele of Tsc2 highlights the role of TSC1/TSC2 in signaling to AKT and models mild human TSC2 alleles. Hum. Mol. Genet. 2009;18:2378–2387. doi: 10.1093/hmg/ddp176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mo W., Chen J., Patel A., Zhang L., Chau V., Li Y., Cho W., Lim K., Xu J., Lazar A.J., et al. CXCR4/CXCL12 mediate autocrine cell-cycle progression in NF1-associated malignant peripheral nerve sheath tumors. Cell. 2013;152:1077–1090. doi: 10.1016/j.cell.2013.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee da Y., Gianino S.M., Gutmann D.H. Innate neural stem cell heterogeneity determines the patterning of glioma formation in children. Cancer Cell. 2012;22:131–138. doi: 10.1016/j.ccr.2012.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu J., Williams J.P., Rizvi T.A., Kordich J.J., Witte D., Meijer D., Stemmer-Rachamimov A.O., Cancelas J.A., Ratner N. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell. 2008;13:105–116. doi: 10.1016/j.ccr.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Birnbaum R.A., O'Marcaigh A., Wardak Z., Zhang Y.Y., Dranoff G., Jacks T., Clapp D.W., Shannon K.M. Nf1 and Gmcsf interact in myeloid leukemogenesis. Mol. Cell. 2000;5:189–195. doi: 10.1016/s1097-2765(00)80415-3. [DOI] [PubMed] [Google Scholar]
- 25.Seizinger B.R., Rouleau G., Ozelius L.J., Lane A.H., St George-Hyslop P., Huson S., Gusella J.F., Martuza R.L. Common pathogenetic mechanism for three tumor types in bilateral acoustic neurofibromatosis. Science. 1987;236:317–319. doi: 10.1126/science.3105060. [DOI] [PubMed] [Google Scholar]
- 26.Fisher L.M., Doherty J.K., Lev M.H., Slattery W.H. Concordance of bilateral vestibular schwannoma growth and hearing changes in neurofibromatosis 2: neurofibromatosis 2 natural history consortium. Otol. Neurotol. 2009;30:835–841. doi: 10.1097/MAO.0b013e3181b2364c. [DOI] [PubMed] [Google Scholar]
- 27.Plotkin S.R., Stemmer-Rachamimov A.O., Barker F.G., 2nd, Halpin C., Padera T.P., Tyrrell A., Sorensen A.G., Jain R.K., di Tomaso E. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N. Engl. J. Med. 2009;361:358–367. doi: 10.1056/NEJMoa0902579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Asthagiri A.R., Vasquez R.A., Butman J.A., Wu T., Morgan K., Brewer C.C., King K., Zalewski C., Kim H.J., Lonser R.R. Mechanisms of hearing loss in neurofibromatosis type 2. PloS one. 2012;7:e46132. doi: 10.1371/journal.pone.0046132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gouveris H.T., Victor A., Mann W.J. Cochlear origin of early hearing loss in vestibular schwannoma. Laryngoscope. 2007;117:680–683. doi: 10.1097/MLG.0b013e31803146c5. [DOI] [PubMed] [Google Scholar]
- 30.Prasher D.K., Tun T., Brookes G.B., Luxon L.M. Mechanisms of hearing loss in acoustic neuroma: an otoacoustic emission study. Acta Otolaryngol. 1995;115:375–381. doi: 10.3109/00016489509139332. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.