

Letter to the Editor
BCR-ABL1 fusion tyrosine kinase transforms hematopoietic stem cells (HSCs) and cause chronic myeloid leukemia in chronic phase (CML-CP), which is a stem cell (leukemia stem cell=LSC) -derived but a progenitor (leukemia progenitor cell=LPC)-driven disease.1 BCR-ABL1 kinase is leukemogenic only when expressed in a HSC with self-renewal capacity thereby inducing LSC. LSCs are capable to generate large numbers of LPCs: leukemia common myeloid (LCMPs) and leukemia granulocyte/macrophage (LGMPs) progenitors, which cannot self-renew and eventually differentiate to mature elements. Thus, CML-CP is a stem cell-derived but a progenitor-driven disease.1 In CML-BC leukemia progenitors (LCMP, LGMP) also display the ability of self-renewal and to sustain leukemogenesis therefore they are LSC candidates.
CML cells display the signature of genomic instability.2 Resistance of CML cells to tyrosine kinase inhibitors (TKIs) such as imatinib is often caused by point mutations in BCR-ABL1 kinase domain, which may emerge under TKI therapy. Overall, point mutations in the kinase domain of BCR-ABL1 have been detected in 50–90% of patients with acquired resistance to imatinib; TKI-resistant (TKIR) clones have been detected in Lin−CD34+ cells including Lin−CD34+CD38− LSCs and Lin−CD34+CD38+ LPCs.3 Generation of second- and third-generation of TKIs (dasatinib, nilotinib, ponatinib) to overcome TKI resistance may result in appearance of novel mutations including compound mutations (polymutants).4
We reported that TKI-naïve and TKI-treated LSCs and LPCs accumulate high levels of ROS and oxidative DNA damage producing mutations BCR-ABL1 kinase causing TKI resistance.5,6 There are several possible explanations for persistent elevated levels of ROS and oxidative DNA damage in CML-CP cells surviving TKI treatment. For example, the effect of TKIs on BCR-ABL1 kinase-induced signaling pathways stimulating ROS production may be obscured by growth factors, usually resulting in incomplete inhibition or even stimulation of STAT5, AKT, RAC2, and MAPK.7,8 Therefore instead of developing novel inhibitors to target the elusive BCR-ABL1 kinase, drugs downregulating ROS production should be combined with existing TKIs to prevent mutations and to cause more radical elimination of CML-CP cells.
Phosphatidylinositol 3-kinase (PI3K) kinase – mTOR signaling has been implicated in production of ROS in BCR-ABL1 –positive cell lines.9 Recently we showed that RAC2, a putative downstream effector of PI3K can alter the electron flow through mitochondrial respiratory chain complex III (MRC-cIII) to elevate ROS in LSCs and LPCs.8 Here we evaluated the role of AKT serine/threonine kinase, another PI3K downstream effector, in generation of ROS-induced oxidative DNA damage and TKI resistance in LSCs and LPCs.
As described before AKT and RAC2 were inhibited in BCR-ABL1 –positive 32Dcl3 cells either by expression of specific dominant negative mutants AKT(K179M) and RAC(T17N) 8,10, respectively, and in Lin−CD34+ CML-CP cells by AKT activation inhibitor perifosine 11 and RAC inhibitor NSC23766, respectively (Figure 1a). Inhibition of AKT does not affect the activity of RAC and inhibition of RAC did not affect AKT activity clearly indicating that their activation status does not depend on each other.
Fig. 1. RAC-independent AKT-induced ROS caused oxidative DNA damage resulting in accumulation of imatinib-resistant clones.
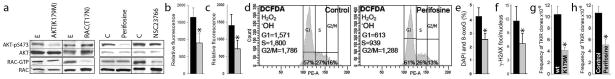
(a) BCR-ABL1-transformed 32Dcl3 cells transfected with AKT(K179M) and Rac(T17N) dominant-negative mutants or empty plasmids (E) 8,10, and Lin−CD34+ CML-CP cells treated with 10 μM AKT inhibitor perifosine, 25 μM NSC23766 or diluent (C) 8,11 were tested for activation of AKT and RAC. Western analyses detect AKT phosphorylated on serine 473 (AKT-pS473) and Rac bound to GTP as described before 8,15; total levels of AKT and RAC were also determined as loading controls. (b) ROS were measured with DCFDA in BCR-ABL1 -32Dcl3 cells transfected with empty plasmid (black bar) and AKT(K179M) mutant (grey bar). (c–f) Lin−CD34+ CML-CP cells were left untreated (black bars) or incubated with 10 μM perifosine (grey bars) in the presence of growth factors.8 (c) ROS were measured with DCFDA in annexin V-negative cells as described before 5,8. (d) ROS were detected by DCFDA in G1, S and G2/M phase determined by Vybrant DyeCycle Orange live cell staining (Invitrogen/Molecular Probes) as described before. 8 ROS measurements are at the left sides, and percentages of cells in cell cycle phases are indicated at the bottom. (e) 8-oxoG and (f) γ-H2AX detected by specific immunofluorescence as described before.8 (g, h) BCR-ABL1 –positive 32Dcl3 cells transfected with AKT(K179M) mutant or empty plasmid (g) and untreated (Control) or treated with 1 μM perifosine (h) were cultured for 10 weeks. The frequency of TKI resistant (TKIR) clones was determined as described before.8 *p<0.05.
In the presence of growth factors AKT(K179M) mutant and perifosine diminished ROS levels in annexin V-negative living BCR-ABL1 -32Dcl3 cells and Lin−CD34+ cells, respectively (Figure 1b, c). Perifosine effectively downregulated ROS in Lin−CD34+ CML-CP cells in the G0/G1, S and G2/M cell cycle phases (Figure 1d), which was associated with reduction of oxidative DNA lesions, 8-oxoG and DSBs (Figure 1e, f). Finally, inhibition of AKT either by AKT(K179M) mutant or perifosine resulted in reduction of accumulation of TKIR clones in BCR-ABL1 -32Dcl3 cells (Figure 1g, h).
To determine if AKT is responsible for overproduction of ROS in imatinib-treated LSCs and LPCs, Lin−CD34+ CML-CP cells were incubated with imatinib in the presence of growth factors and ABL1 and AKT activation and ROS levels were measured. Despite inhibition of ABL1 kinase activity, AKT activation was mostly preserved in imatinib-treated Lin−CD34+ cells (Figure 2a) in concordance with other report 7. As expected from previous studies with PI3K inhibitors LY294002 or wortmannin and BCR-ABL1 kinase inhibitor imatinib, 13 targeting AKT, a downstream effector of PI3K, with perifosine enhanced the inhibitory effect of imatinib on clonogenic growth of Lin−CD34+ CML-CP cells (Supplemental Figure 1). This effect may depend on perifosine-mediated elevation of ROS (including mitochondrial ROS) in a sub-population of annexin V-positive leukemia cells committed to apoptosis (Supplemental Figure 2) in concordance with other report 12.
Fig. 2. AKT elevated ROS in imatinib-treated LPCs but not in LSCs.
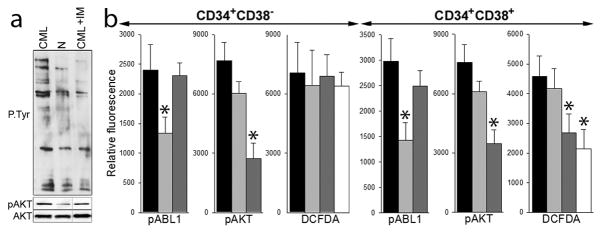
(a) Lin−CD34+cells from normal donor (N) and from CML-CP patient were untreated (CML) or treated with 1 μM imatinib (CML+IM) in the presence of growth factors. Total tyrosine phosphorylated proteins (P-Tyr), AKT phosphorylated on serine 473 (pAKT) and total AKT protein were detected by Western analysis. (b) Lin−CD34+ cells from 3–6 CML-CP patients were untreated (black bars) and treated with 1 μM imatinib (light grey bars), 10 μM perifosine (dark grey bars), or 1 μM imatinib + 10 μM perifosine (white bars) in the presence of growth factors. Phospho-ABL1 (pABL1; phospho-Y245), phospho-AKT (pAKT; phospho-T308) and ROS (DCFDA) were detected in annexin V-negative Lin−CD34+CD38− LSCs and Lin−CD34+CD38+ LPCs as described before 5,8. *p<0.05 in comparison to untreated cells.
Sustained AKT activation was also detected in imatinib-treated Lin−CD34+CD38− LSCs and Lin−CD34+CD38+ LPCs when compared to imatinib-naïve counterparts (Figure 2b). However, perifosine reduced ROS levels in imatinib-naïve and imatinib-treated Lin−CD34+CD38+ LPCs but not in Lin−CD34+CD38− LSCs (Figure 2b). Therefore it appears that AKT kinase plays an important role in generation of ROS in imatinib-naïve and imatinib-treated Lin−CD34+CD38+ LPCs, but it is expendable in Lin−CD34+CD38− LSCs. Since accumulation of DNA lesions such as 8-oxoG and DSBs directly depends on ROS levels in Lin−CD34+CD38− LSCs and Lin−CD34+CD38+ LPCs, 5,8 we postulate that AKT kinase regulates oxidative DNA damage in LPCs, but not in LSCs.
In conclusion, we postulate that in imatinib-treated CML-CP patients AKT serine/threonine kinase plays a prominent role in accumulation of TKIR clones emerging from Lin−CD34+CD38+ LPCs, but probably not form Lin−CD34+CD38− LSCs. The mechanisms responsible for this cell compartment specific AKT-mediated effect on genomic instability in CML-CP are unknown. Although AKT remained active in imatinib-treated Lin−CD34+CD38+ LPCs and Lin−CD34+CD38− LSCs, intrinsic differences between leukemic progenitor and stem cells may contribute to the selective AKT effect in LPCs.14 Moreover, it remains to be determined if AKT and RAC employ overlapping or different downstream signaling pathways.
Supplementary Material
Acknowledgments
This work was supported by NIH/NCI CA134458 to T.Skorski.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary information is available at Leukemia’s website.
References
- 1.Marley SB, Gordon MY. Chronic myeloid leukaemia: stem cell derived but progenitor cell driven. Clin Sci (Lond) 2005;109(1):13–25. doi: 10.1042/CS20040336. [DOI] [PubMed] [Google Scholar]
- 2.Perrotti D, Jamieson C, Goldman J, Skorski T. Chronic myeloid leukemia: mechanisms of blastic transformation. J Clin Invest. 2010;120(7):2254–2264. doi: 10.1172/JCI41246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sorel N, Bonnet ML, Guillier M, Guilhot F, Brizard A, Turhan AG. Evidence of ABL-kinase domain mutations in highly purified primitive stem cell populations of patients with chronic myelogenous leukemia. Biochem Biophys Res Commun. 2004;323(3):728–730. doi: 10.1016/j.bbrc.2004.08.169. [DOI] [PubMed] [Google Scholar]
- 4.Gibbons DL, Pricl S, Posocco P, et al. Molecular dynamics reveal BCR-ABL1 polymutants as a unique mechanism of resistance to PAN-BCR-ABL1 kinase inhibitor therapy. Proc Natl Acad Sci U S A. 2014;111(9):3550–3555. doi: 10.1073/pnas.1321173111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bolton-Gillespie E, Schemionek M, Klein HU, et al. Genomic instability may originate from imatinib-refractory chronic myeloid leukemia stem cells. Blood. 2013;121(20):4175–4183. doi: 10.1182/blood-2012-11-466938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nieborowska-Skorska M, Hoser G, Hochhaus A, Stoklosa T, Skorski T. Anti-oxidant vitamin E prevents accumulation of imatinib-resistant BCR-ABL1 kinase mutations in CML-CP xenografts in NSG mice. Leukemia. 2013;27(11):2253–2254. doi: 10.1038/leu.2013.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Konig H, Holtz M, Modi H, et al. Enhanced BCR-ABL kinase inhibition does not result in increased inhibition of downstream signaling pathways or increased growth suppression in CML progenitors. Leukemia. 2008;22(4):748–755. doi: 10.1038/sj.leu.2405086. [DOI] [PubMed] [Google Scholar]
- 8.Nieborowska-Skorska M, Kopinski PK, Ray R, et al. Rac2-MRC-cIII-generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors. Blood. 2012;119(18):4253–4263. doi: 10.1182/blood-2011-10-385658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim JH, Chu SC, Gramlich JL, et al. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood. 2005;105(4):1717–1723. doi: 10.1182/blood-2004-03-0849. Epub 2004 Oct 1714. [DOI] [PubMed] [Google Scholar]
- 10.Skorski T, Bellacosa A, Nieborowska-Skorska M, et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. Embo J. 1997;16(20):6151–6161. doi: 10.1093/emboj/16.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kondapaka SB, Singh SS, Dasmahapatra GP, Sausville EA, Roy KK. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol Cancer Ther. 2003;2(11):1093–1103. [PubMed] [Google Scholar]
- 12.Rahmani M, Reese E, Dai Y, et al. Coadministration of histone deacetylase inhibitors and perifosine synergistically induces apoptosis in human leukemia cells through Akt and ERK1/2 inactivation and the generation of ceramide and reactive oxygen species. Cancer Res. 2005;65(6):2422–2432. doi: 10.1158/0008-5472.CAN-04-2440. [DOI] [PubMed] [Google Scholar]
- 13.Klejman A, Rushen L, Morrione A, Slupianek A, Skorski T. Phosphatidylinositol-3 kinase inhibitors enhance the anti-leukemia effect of STI571. Oncogene. 2002;21(38):5868–5876. doi: 10.1038/sj.onc.1205724. [DOI] [PubMed] [Google Scholar]
- 14.Kornblau SM, Qutub A, Yao H, et al. Proteomic profiling identifies distinct protein patterns in acute myelogenous leukemia CD34+CD38− stem-like cells. PLoS One. 2013;8(10):e78453. doi: 10.1371/journal.pone.0078453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren SY, Bolton E, Mohi MG, Morrione A, Neel BG, Skorski T. Phosphatidylinositol 3-kinase p85{alpha} subunit-dependent interaction with BCR/ABL-related fusion tyrosine kinases: molecular mechanisms and biological consequences. Mol Cell Biol. 2005;25(18):8001–8008. doi: 10.1128/MCB.25.18.8001-8008.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.