

Abstract
Tunable control of protein degradation in bacteria would provide a powerful research tool. We use components of the Mesoplasma florum tmRNA system to create a synthetic degradation system that provides both independent control of the steady-state protein level and inducible degradation of targeted proteins in Escherichia coli. We demonstrate application of this system in synthetic circuit development and control of core bacterial processes and antibacterial targets, and transfer the system to Lactococcus lactis to establish its broad functionality in bacteria. We create a 238-member library of tagged essential proteins in E. coli that can serve as both a research tool to study essential gene function and an applied system for antibiotic discovery. Our synthetic protein degradation system is modular, does not require disruption of host systems, and can be transferred to diverse bacteria with minimal modification.
Exogenous control of protein biosynthesis through transcriptional and translational regulation is well-established1–4, but robust and tunable control of protein degradation in bacteria remains elusive. Controlled protein degradation would provide biologists with the ability to probe protein function without disrupting the transcriptional and translational regulation that control the expression of its cognate gene, and biological engineers with a tool to develop more complex synthetic gene circuits.
Protein degradation in bacteria occurs in part through the tmRNA system, which uses C-terminal fusion of the ssrA peptide to direct proteins to the endogenous ClpXP and ClpAP proteases for rapid degradation in E. coli5. Variants of the E. coli ssrA tag (ec-ssrA) are commonly used to modify the degradation rate of attached proteins both in bacteria and eukaryotes, but these tags do not provide inducible control of degradation. Recently developed inducible eukaryotic systems rely on degradation machinery not present in bacteria6, 7, and bacterial systems such as the one developed by Davis et al.8 require disruption of the endogenous tmRNA system and are therefore not easily transferred to other organisms.
Here we present a synthetic degradation system based on the Gram-positive M. florum tmRNA system that does not rely on host degradation systems and can function in a wide range of bacteria. Gur and Sauer9 showed that the M. florum ssrA tag (mf-ssrA) is degraded by its endogenous Lon protease (mf-Lon) but not by E. coli Lon or ClpXP, and mf-Lon does not recognize or degrade ec-ssrA, providing a protease and cognate degradation tag with orthogonal functionality in E. coli.
We renamed the mf-ssrA tag “pdt” (protein degradation tag) to minimize confusion with the E. coli ssrA tag, and incorporated it into a GFP-based test platform for inducible protein degradation in E. coli (Fig. 1a). To first engineer pdt variants that modify steady-state GFP levels in the absence of mf-Lon expression, we chose to target pdt residues 24–27 for mutagenesis due to the region’s partial homology with the ec-ssrA ClpA binding site5 and altered GFP-pdt stability in clpA, clpX, and clpP deletion strains (Supplementary Figs. 1 and 2). As seen in Figure 1b, we identified several pdt variants, denoted with numbers, that alter GFP steady-state levels and maintained near wild-type GFP degradation rates following mf-Lon expression. Importantly, untagged GFP remained largely unaffected by mf-Lon expression while the wild-type GFP-pdt fusion was reduced to 3% of its initial levels, confirming the specificity of pdt-mediated mf-Lon degradation seen by Gur and Sauer for LacZ degradation9. Sequence analysis of the identified pdt number variants showed that a majority contained multiple arginine and glutamine residues in the mutagenized region and none of them contained negatively charged residues known to disrupt mf-Lon recognition9 (Supplementary Table 1).
Figure 1. Protein degradation tag characterization.
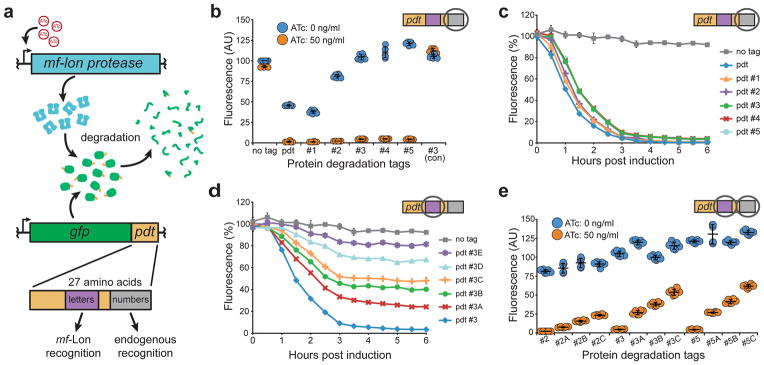
(a) Schematic of the tunable protein degradation system where anhydrotetracycline (ATc) induced mf-Lon expression allows the protease to degrade constitutively expressed GFP in a pdt-dependent manner. Mutations in two pdt regions produced tag variants with altered recognition by mf-Lon (letters) or endogenous E. coli proteases (numbers). (b) Dot plot of pdt number variants that show altered steady-state levels. Fluorescence was measured by flow cytometry 6 hours after ATc induction of cells in exponential growth. As an experimental control, the pdt#3 variant was tested in a strain that did not contain the mf-Lon expression cassette (#3 con). Fluorescence units are arbitrary, with untagged GFP set to 100, and show the mean of six biological replicates. P < 0.001 for ATc induction of each pdt variant except #3 (con). (c–d) Flow cytometry measurements of GFP degradation following mf-Lon induction with 50 ng/ml ATc. Data show the geometric mean fluorescence of at least 5,000 cells as a percentage of the non-induced control for each pdt variant. (c) Pdt number variants maintain similar mf-Lon-mediated degradation dynamics. (d) Pdt letter variants display altered mf-Lon-mediated degradation rates. (e) Dot plot of hybrid pdt variants. Strains expressing the indicated GFP-pdt fusion were measured by flow cytometry 6 hours after ATc induction. Fluorescence units are arbitrary with untagged GFP set to 100, and show the mean of six biological replicates. P < 0.001 for ATc induction of each pdt variant. The data in all panels show the mean of at least three biological replicates, and the error bars show the standard deviation.
We used flow cytometry to further characterize mf-Lon-mediated GFP-pdt degradation and found that the pdt number variants displayed temporal degradation dynamics similar to wild-type pdt, reducing GFP levels to 1–5% of initial levels within 4 hours (Fig. 1c). GFP degradation did not occur in the absence of either mf-Lon or the pdt tag, and the tight monomodal shift in the fluorescent population distribution showed that degradation occurred across all cells in the experimental population (Supplementary Fig. 3).
We next sought to identify pdt variants (denoted with letters) that alter mf-Lon dependent degradation but not recognition by endogenous E. coli proteases. We used GFP-pdt#3 as the parental tag and targeted pdt residues 13–15 for mutagenesis because the region is essential for Lon-mediated degradation in Mycoplasma pneumoniae10 and does not show homology to known ClpA, ClpX or SspB binding sites5. Pdt variants that maintained steady-state GFP levels and displayed a range of mf-Lon dependent degradation rates are shown in Figure 1d.
To determine if these letter variants could be combined with other number variants to produce hybrid tags with predictable control over both the steady-state protein level and induced degradation rate, we created a panel of hybrid pdt variants and measured GFP fluorescence in the presence and absence of mf-Lon induction. When combined with the number variants pdt#2 and pdt#5, the letter variants displayed the same rank order of degradation rates that were initially identified using pdt#3 (Fig. 1e). In the absence of mf-Lon induction, the hybrid pdt variants also showed steady-state levels that largely conformed to the level dictated by the number variant used, although there was significant variation in some hybrid tag combinations suggesting partial recognition of the letter variant region by E. coli proteases.
To determine if this GFP-pdt characterization can be used to predict pdt-mediated degradation of other protein targets, we placed pdt variants on the fluorescent protein mCherry and measured degradation following mf-Lon induction. As seen in Figure 2a, the letter variants produced mCherry degradation dynamics that correlated strongly with GFP degradation, displaying linear regression with an R2 value of 0.98. The slope of the regression line (1.09) and its y-intercept (−0.01) suggest that mf-Lon-mediated degradation of GFP and mCherry occurred at similar relative rates for all pdt letter variants tested. Pdt number variants also showed strong correlation for mCherry and GFP, with a linear regression R2 value of 0.97 (Supplementary Fig. 4a).
Figure 2. Pdt system characterization.

(a) Comparative analysis of pdt-mediated degradation of mCherry and GFP. Pdt letter variants were fused to GFP and mCherry, and the percent fluorescence remaining after mf-Lon induction is shown (50 ng/ml ATc for 6 h). Fluorescent data were collected by flow cytometry, and the pdt variants shown are pdt#3, #3A, #3B, #3C, #3D, #3E, listed in order of increasing percent fluorescence. The best-fit line by linear regression is y=1.09× – 0.01 with an R2 value of 0.98 and standard errors of 0.03 and 0.01 for the slope and y-intercept, respectively. (b) Pdt-dependent degradation of mCherry in L. lactis. Nisin induced mf-Lon expression in L. lactis causes pdt-dependent mCherry degradation. Data show the geometric mean fluorescence as a percent of the fluorescence of uninduced cells. Nisin induction was 3 ng/ml. (c) Comparative analysis of pdt letter variants in E. coli and L. lactis. Pdt letter variants were fused to mCherry, and the percent fluorescence remaining after mf-Lon induction is shown (6 hour induction, E. coli: 50 ng/ml ATc and L. lactis: 3 ng/ml nisin). Fluorescent data were collected by flow cytometry, and the pdt variants shown are pdt#3, #3A, #3B, #3C, #3D, #3E, listed in order of increasing percent fluorescence. The best-fit line by linear regression is y=0.51× – 0.04 with an R2 value of 0.91 and standard errors of 0.03 and 0.02 for the slope and y-intercept, respectively. (d) Transcription and post-translation-based control of mf-Lon-mediated pdt degradation. Inducible transcription provides control of mf-Lon-mediated degradation of GFP-pdt#3 across a range of ATc induction levels. Fusion of the E. coli ssrA tag variants ec-AAV and ec-ASV to mf-Lon shift the GFP degradation profile, and inactivation of mf-Lon protease activity (S692A) blocks GFP degradation. Data were collected 6 hours after ATc induction using GFP-pdt#3 as the degradation target. For all panels, the data show the mean of at least three biological replicates and the error bars show the standard deviation.
To determine if this targeted degradation system can function in other bacteria, we transferred the inducible protease and pdt variants to Lactococcus lactis, an industrially important Gram-positive bacterium that is phylogenetically distant from E. coli (a Gram-negative bacterium). As seen in Figure 2b, mf-Lon expression from the inducible nisA promoter11 in L. lactis resulted in efficient pdt-mediated degradation of mCherry, and the relative degradation strength of the pdt letter variants in L. lactis correlated well with their corresponding strength in E. coli (R2 = 0.91) (Fig. 2c). As expected for pdt number variants that were chosen for their altered recognition by E. coli specific proteases, their effect on mCherry steady-state levels in L. lactis showed only weak correlation to E. coli (Supplementary Fig. 4b).
Targeted protein degradation is dependent not only on the target protein and the pdt variant but also on mf-Lon expression levels, providing an additional mechanism to control target protein levels. As shown in Figure 2d, transcriptional control of mf-Lon, based on anhydrotetracycline (ATc) induction of the PLtetO promoter, provided a well-defined range mf-Lon expression levels, as defined by targeted GFP-pdt#3 degradation. To enable post-translational control of mf-Lon, we fused variants of the ec-ssrA tag to mf-Lon and measured their effect on GFP-pdt#3 degradation. The ec-AAV variant caused a significant shift in mf-Lon dependent GFP-pdt#3 degradation throughout the range of ATc levels tested, while the weaker ec-ASV variant had only a small effect. An inactivating mutation in the conserved active site of the mf-Lon proteolytic domain (S692A) fully blocked mf-Lon mediated GFP-pdt#3 degradation.
To demonstrate the use of this system to control engineered genetic circuits, we used pdt fusions to provide post-translational control of a transcription-based toggle switch12. As shown in Figure 3a, LacI and TetR were previously engineered to form a bistable circuit based on reciprocal repression, and concomitant regulation of GFP and mCherry allows facile fluorescence-based identification of the toggle switch state13. We fused pdt#3 to the C-terminus of LacI in the toggle circuit and used the arabinose-inducible PBAD promoter1 to drive mf-Lon expression from a second plasmid. Upon mf-Lon induction, the circuit containing LacI-pdt#3 switched from the LacI+/GFP+ state to the TetR+/mCherry+ state within 8 hours of mf-Lon induction, while the untagged circuit remained unchanged (Fig. 3b). Moreover, substitution of LacI-pdt#3 with the hybrid tags pdt#3A and pdt#3B provided temporal control over the circuit switch rate (Fig. 3c), and pdt fusions to TetR enabled mf-Lon to switch the toggle in the opposite direction (Supplementary Fig. 5). Importantly, the LacI-pdt circuits maintained transcription-based bistability in the absence of mf-Lon induction, demonstrating the ability of the system to leave existing regulatory networks intact (Supplementary Fig. 6).
Figure 3. Protease-driven control of a synthetic toggle switch.

(a) Schematic of the synthetic toggle switch in which reciprocal transcriptional repression by TetR and LacI form a bistable circuit. GFP and mCherry serve as fluorescent reporters for the LacI+ and TetR+ toggle states, respectively. Addition of a pdt tag to LacI enables a protease-driven switch from the GFP+ to the mCherry+ state. (b) Flow cytometry scatter plots show GFP and mCherry fluorescence 0, 4 and 8 hours after mf-Lon expression from the inducible promoter PBAD. Degradation of LacI-pdt#3 causes the toggle to switch from the GFP+ state to the mCherry+ state by 8 hours, while the untagged toggle remains in the GFP+ state. Magenta lines indicate the gate parameters used to define the GFP+ and mCherry+ states: cells bounded in the lower left quadrant are considered negative for both GFP and mCherry. (c) The percentage of cells in the mCherry+ state following mf-Lon induction with 1 mM arabinose. Data collected by flow cytometry were measured using the parameters shown in (b) and represent the mean of three biological replicates. For all LacI-pdt variants, P < 0.001 when compared to untagged LacI at 24 hours after induction. Error bars show the standard deviation. See Supplementary Figure 6 for data showing that non-induced strains did not shift to mCherry+.
The relatively slow switch rate for this degradation-dependent toggle switch reflects the time needed for arabinose-induced mf-Lon expression and LacI-pdt#3 degradation, and the additional three hours it takes compared to ATc or IPTG induction13 correlates well with the time needed for inducible degradation of GFP-pdt#3 by mf-Lon (see Fig. 1d). As shown below, induced mf-Lon degradation of endogenous proteins takes as little as 45 minutes, likely reflecting the relatively low abundance of most endogenous proteins compared to LacI and GFP, which are strongly expressed from multi-copy plasmids as is typical for synthetic gene circuits.
A major goal in microbial biotechnology is to develop tools to control and manipulate endogenous bacterial systems, so next we sought to target native E. coli pathways for control by our system. We developed a modified recombineering method to insert pdt tags into the E. coli genome (Fig. 4a) and began by targeting MurA, an essential enzyme involved in peptidoglycan biosynthesis14 whose depletion causes cell lysis measurable by a drop in optical density. The murA-pdt#1 genomic fusion caused observable cell lysis 45 minutes after mf-Lon induction (Fig. 4b), and the delayed phenotypic response of the hybrid variants pdt#1A and pdt#1B correlates well with the temporal delay seen for letter variants in the toggle switch and GFP degradation assays (see Fig. 2d and Fig. 3c). Importantly, cells containing murA-pdt fusions display the same growth rate as wild-type cells in the absence of mf-Lon induction, demonstrating that the pdt variants do not interfere with MurA function or regulation (Supplementary Fig. 7a). Sequence analysis of cells with murA-pdt#1 that escape ATc-induced cell lysis showed mutations in mf-lon or its promoter that block its expression.
Figure 4. Tunable control of endogenous bacterial systems and antibacterial targets.

(a) Schematic of our recombineering method for genomic insertion of pdt variants, adapted from Datsenko and Wanner30. Red recombinase-assisted insertion of the desired pdt variant is followed by Flp recombinase-mediated excision of the accompanying kanR cassette. The resulting insertion contains the C-terminal pdt variant fusion and a 106 bp scar including the remaining FRT site. (b) Growth of strains following protease-driven depletion of MurA. Protease induction during early exponential phase growth (arrow) causes cells containing murA-pdt#1 to lyse within 1 hour, as measured by optical density (OD600). Cells containing the weakened pdt letter variants show a delayed response. Error bars show the standard deviation from the mean of six biological replicates. See Supplementary Figure 7 for data showing wild-type growth of non-induced cells. (c) DIC-fluorescence overlay images of cells after ATc induction for 3 hours. Cells containing ftsZ-pdt#5 form filaments while untagged wild-type bacteria maintain normal length. The fluorescence micrograph overlay showing constitutive GFP expression serves as a visual aid. Scale bars are 10 μm. (d) Disk diffusion assay on a chemotactic motility plate shows loss of chemotactic motility due to pdt-dependent CheZ degradation. Cells were stabbed into the chemotaxis plate following addition of 250 ng ATc to the center disk. Scale bar is 6 mm. (e) Cells containing murA-pdt#1D show increased sensitivity to fosfomycin upon simultaneous induction with 4 ng/ml ATc (induced). OD600 measurements were taken 4 hours after ATc and fosfomycin treatment and are presented as a percent of the OD600 of cells not exposed to fosfomycin (untreated). In the murA-pdt#1D strain, P < 0.001 when comparing uninduced and induced cells for fosfomycin concentrations between 0.05 and 2 μg/ml. See Supplementary Figure 8 for additional data. (f) Pdt-dependent degradation of RecA causes hypersensitivity to the quinolone norfloxacin that matches the known hypersensitivity of a recA deletion strain (ΔrecA). Where indicated, cells were induced with 50 ng/ml ATc for 2 hours before treatment with norfloxacin (25 ng/ml) for 2 hours. Survival was measured by colony forming units (CFU) and is presented as a percent of CFUs measured immediately before norfloxacin treatment. P < 0.001 for ATc induction of the recA-pdt#3 strain. (g) Scatter plot displaying the relative growth and CFU count of EPD library members after targeted mf-Lon degradation. Growth and CFU measurements are displayed as a ratio of induced/uninduced cells at 4 hours after ATc induction (50 ng/ml). CFU data points were placed at 1.0 × 10−6, the limit of detection, when colonies were not recovered from the induced well. Error bars show the standard deviation from the mean of three biological replicates. CFU ratios represent a single experiment. See Supplementary Table 2 for details. (h) Histogram of propidium iodide (PI) staining for EPD member plsB-pdt#1. PI was measured by flow cytometry 2 hours after induction with 50 ng/ml ATc. The percentage of cells that are PI+ is displayed. The data are normalized to mode and are representative of three biological replicates.
We next targeted FtsZ, a tubulin homologue that forms the ring structure necessary for cell septation following genome replication15. As seen in Figure 4c, mf-Lon induction caused distinct filamentation in ftsZ-pdt#5 cells but not wild-type cells within 3 hours of ATc induction. The pdt#5 fusion had no discernible effect on FtsZ function under non-inducing conditions (0 hour images), and its growth rate was identical to wild-type cells (Supplementary Fig. 7b). We then targeted CheZ, a member of the chemotaxis signaling system whose disruption prevents directed flagellar motility16. In a disk diffusion assay on motility agar, mf-Lon induction caused bacteria containing cheZ-pdt#5 to lose chemotactic motility (Fig. 4d), but bacteria that did not contain the cheZ-pdt#5 fusion or did not express mf-Lon maintained normal chemotactic motility, confirming the specificity of ATc induced mf-Lon degradation of CheZ-pdt#5.
We sought to determine if this system could serve as a tool for antibiotic discovery and characterization. Large-scale chemical and natural product libraries can be efficiently screened for antimicrobial activity, but the next step of target identification is difficult and represents a major bottleneck in antibiotic development17. To identify compounds that target a specific protein for inhibition, one method is to reduce expression of the desired protein target to levels that induce a hypersensitive phenotype, in which antimicrobial compounds that inhibit the targeted protein show increased potency18. To demonstrate target-specific antibiotic hypersensitivity using our system, we returned our focus to MurA, the known target of fosfomycin19. We used the weak hybrid tag pdt#1D to inducibly degrade MurA and tested a range of ATc concentrations to identify mf-Lon induction conditions that produce a small murA-pdt#1D dependent growth defect (4 ng/ml ATc, see Supplementary Fig. 8). Under this low induction condition, cells that contained murA-pdt#1D displayed increased sensitivity to fosfomycin, while untagged or uninduced cells remained equally sensitive to the MurA-specific antibiotic (Fig. 4e).
The slow pace of antibiotic discovery in recent years has led to alternative approaches to drug development, including an increased focus on chemical adjuvants that boost the potency of antibiotics currently in clinical use. Inducible control of adjuvant targets with our system could provide a method to both identify novel cellular targets for adjuvant development and screen for chemical adjuvants that inhibit a desired protein. As a proof of principle demonstration, we used our platform to control endogenous levels of RecA, a DNA-damage repair protein that helps to protect cells against norfloxacin and is actively being pursued as an antibiotic adjuvant target20. Upon mf-Lon induction, cells containing a recA-pdt#3 fusion become hypersensitive to norfloxacin, and the induced phenotype is nearly identical to a recA deletion strain (Fig. 4f). As shown above for MurA-dependent fosfomycin sensitivity, intermediate-level RecA degradation could be used to screen for molecules that inhibit RecA.
Current classes of antibiotics target only a small subset of essential cellular proteins for inhibition, and emerging resistance to these antibiotics has led to an urgent search for additional targets for antibiotic development. To aid in this search, we targeted every essential gene in E. coli for pdt fusion to create a strain library that could be screened for antibiotic hypersensitivity and degradation-dependent cell death. Using high-throughput electroporation, we recovered pdt#1 insertions in 238 of the 305 essential genes targeted, and we arrayed the strains in multi-well format to form the Essential Protein Degradation (EPD) library (see Supplementary Table 2). Despite repeated attempts, we could not recover pdt insertions in the remaining 67 genes, including 28 ribosomal genes, possibly due to pdt interference with protein function, altered target protein stability caused by the pdt#1 variant, or a polar effect of the insertion cassette on downstream genes.
To establish the functionality of the EPD library and search for potential antibiotic targets, we screened for growth inhibition and cell death following ATc induced mf-Lon expression. As shown in Figure 4g and Supplementary Table 2, 112 strains (46%) showed at least a 10-fold reduction in colony forming units (CFUs), including 54 that showed at least a 6-log reduction, and this correlated well with their observed growth defect as measured by optical density. As similarly proposed by Wei et al. in Mycobacterium smegmatis 21, these degradation-sensitive strains represent a list of potential drug targets, and as documented for MurA above they also provide a means to screen for target-specific antibiotics. For EPD members with no inducible growth defect, possible explanations include pdt inaccessibility due to protein folding, multi-protein complex formation, export from the cytoplasm, or post-translational C-terminal processing. High expression or proteolytic stability of the target protein may also prevent mf-Lon from degrading it to levels low enough to affect cell viability.
Antibiotics that disrupt bacterial membrane integrity are particularly desirable, as they not only kill cells but may also enhance the potency of other antibiotics that are excluded from their cytoplasmic targets by intrinsic or acquired resistance mechanisms. To identify protein targets that affect membrane integrity, we screened the EPD library using propidium iodide (PI), a hydrophilic nucleic acid intercalating agent that cannot normally permeate the E. coli inner membrane. Strains were assayed by high-throughput flow cytometry two hours after mf-Lon induction, and the percentage of cells that show cytoplasmic PI staining was quantified. As shown in Figure 4h and Supplementary Table 2, targeted degradation of phospholipid and peptidoglycan biogenesis enzymes such PlsB and MurC caused a distinct membrane permeability phenotype, and in particular, isoprenoid biosynthesis enzymes including Dxr, Dxs, IspD, IspG, IspH and IspU showed high membrane permeability following ATc induction. These data correlate well with the antimicrobial activity of the Dxr inhibitor fosmidomycin and several IspU inhibitors22, and it suggests that additional isoprenoid biosynthesis enzymes may serve as effective antibiotic targets, especially because the pathway is not found in humans. Other interesting PI+ strains include the carbonic anhydrase Can and the NAD kinase NadK (Fig. 4h), suggesting that they play an essential role in maintaining membrane integrity and could also be effective antibiotic targets.
The synthetic degradation system presented here is facile and modular, comprising a single protease gene and a small peptide tag that provides control over both the steady-state level and inducible degradation rate of attached proteins. This system represents an important advance in synthetic biology, where protein level control will provide an additional regulatory mechanism to aid in complex circuit design23, 24. As demonstrated here for a transcription-based toggle switch, existing synthetic circuitry can be readily modified with our system to enable post-translational control while leaving the original regulatory framework intact, and use of our system to integrate multiple synthetic circuits can easily be envisioned25, 26. Recent work by Huang et al.27 also uses mf-Lon to create a toggle switch, and Prindle et al.28 uses competition for the endogenous ClpXP protease to couple synthetic oscillatory circuits in E. coli, further demonstrating the utility of protease-driven control in engineered systems.
This platform provides tunable control of endogenous bacterial systems and can serve as both a basic research tool and as an applied system to aid in antibiotic discovery. Single-step genomic insertion provides a simple and efficient method to target pdt fusions to almost any E. coli gene, and the ability to control endogenous systems without disrupting the existing regulatory networks should prove particularly useful in metabolic engineering29. For the EPD library presented here, the system’s inducible protease effectively converts the inducer into an ‘antibiotic’ that can specifically target nearly 80% of the essential proteins in E. coli. By this perspective, the EPD library may facilitate screens for synergistic and antagonistic relationships among existing and potential antibiotic targets and may also serve as a basic research tool to study essential gene function. As seen for murA-pdt#1 in Figure 4b, the selective pressure caused by essential protein degradation may result in the outgrowth of cells containing protease mutations, potentially limiting the experimental window following mf-Lon induction.
Mf-Lon and pdt are based on a unique tmRNA system found only in Mycoplasma5, so as shown here for L. lactis, the system should be transferable to many other organisms, including microbial pathogens such as Staphylococcus aureus and Listeria monocytogenes, where protocols for genomic integration and inducible protein expression are well established. As in E. coli, the degradation system may best be used to study essential genes, but the inducible nature of the system may also provide a means to study the temporal function of virulence genes during infection. Additional pdt number variants may be needed to control degradation by endogenous proteases in these organisms, but the pdt letter variants that determine mf-Lon degradation rates should function as predicted in this study.
METHODS
Strains and reagents
The E. coli K-12 derivative strain MG1655Pro (F-, λ-, Spr, lacI, tetR) published previously2, 31 was used as the wild-type strain in all cases except for the synthetic toggle experiments where we used MG1655ΔlacIΔaraBAD. Unless otherwise noted, E. coli were grown in Luria broth (LB) at 30° with shaking and mf-Lon expression was induced with 50 ng/ml ATc. L. lactis strain NZ900011 was used for all L. lactis experiments and grown in M17 broth containing 0.5% glucose. Antibiotics carbenicillin (100 μg/ml), kanamycin (30 μg/ml) and erythromycin (10 μg/ml) were added to the media when appropriate.
Strain construction
The parental strains for all experiments are E. coli MG1655 (ATCC no. 47076) and L. lactis NZ900011. MG1655ΔlacIΔaraBAD was created through P1 phage transduction of lacI::kanR from the Keio collection32 into MG1655, and Red-recombinase-mediated homologous recombination was used to create the in-frame deletion of araBAD according to published methods30. Flp recombinase, expressed on pECA102 (10mM arabinose for 4 hours) was used to remove the kanR cassette in each case. DH5αλpir33 was used for cloning. Endogenous E. coli protease deletions were constructed by P1 phage transduction of the corresponding mutations from the Keio collection into MG1655pro31 followed by kanR cassette removal as detailed above. To construct the mf-Lon expression cassette, we codon optimized mf-lon for expression in E. coli, forward engineered a strong RBS to enable high expression34, and cloned the cassette into pZE11. We used overlapping PCR to add a transcriptional terminator 5′ of the PLtetO promoter in this plasmid, then cloned this expression cassette, which includes the 5′ terminator, PLtetO promoter, mf-lon gene, and 3′ terminator, into pWM91-lacZ, a derivative of pWM9135 that includes a 1kb lacZ targeting region. The resulting plasmid, pECL275 (GenBank accession no. KM521209), was introduced into MG1655pro by conjugation from Sm10λpir, single integrants were selected on carbenicillin plates, grown in rich media for 8 hours, and then selected on plates containing 1% tryptone, 0.5% yeast extract, 8% sucrose and 1.5% agar to select for plasmid excision. The resulting colonies were screened by PCR for the mf-Lon expression cassette. Mf-Lon variants that contain ec-AAV and ec-ASV fusions and the S692A point mutation were constructed in the same manner.
E. coli-based degradation platform
The mf-lon gene was codon optimized for E. coli expression, placed under control of the PLtetO promoter, and integrated into the lacZ locus along with 5′ and 3′ transcriptional terminators to block unwanted mf-Lon expression from any proximal genomic promoters. The GFP variant GFPmut3b was used for all GFP expression. To express GFPmut3b36 and mCherry-pdt#3 fusions, the constitutive PlacIq promoter37 was fused to GFP and mCherry and inserted into pZE21-MCS2 to form pZE27GFP3 and pZE27MC3, respectively. For these plasmids, the last number and letter indicate the pdt variant used (e.g. pZE27GFP5B contains pdt#5B).
PDT mutagenesis screens
Pdt mutant libraries were created by polymerase chain reaction (PCR) using primers containing randomized nucleotides at the indicated pdt codons. Strains containing the GFP-pdt mutants were individually picked into 96-well plates and measured by plate fluorimetry during exponential phase growth. Strains that exhibited the desired GFP degradation dynamics following induction with ATc were further characterized by flow cytometry.
Synthetic toggle switch
The plasmid pKDL071R8, based on pKDL07113, was altered to contain a weakened tetR ribosome binding site to enhance toggle bistability in the minimal media conditions used. This plasmid served as the parental strain for all LacI-pdt toggle switch experiments. Pdt#3 was fused to lacI by overlapping PCR and cloned into pKDL071R8 to make pECJ3 (GenBank accession no. KM521210). To express mf-Lon, the arabinose inducible PBAD promoter was fused to mf-Lon and cloned into pZA11 to form pZA16mflon. Cells containing the toggle switch and mf-Lon expression plasmids were grown in 200 μl in 96-well round bottom plates at 37° in M9 minimal media containing 0.2% glycerol and 0.05% casamino acids, and care was taken to maintain exponential growth throughout the experiment. Cells were grown for 6 hours with either 30 ng/ml ATc or 500 μM Isopropyl β-D-1-thiogalactopyranoside (IPTG) to induce cells into the GFP+ or mCherry+ states, respectively. Cells were diluted 1:1000 into non-inducing media and allowed to grow for an additional 12 h. To induce mf-Lon, cells were grown at 37° shaking with 1 mM arabinose and passaged every 4 hours (~1:10 dilution) into media containing the same inducing conditions. At each time point, cells were fixed with 1% paraformaldehyde in PBS and stored at 4° for up to 5 days. Cells that did not contain the toggle switch plasmid were used to define the GFP- / mCherry-state shown in Figure 3b.
L. lactis-based degradation platform
Plasmid pECGMC was created to enable expression of mf-Lon and mCherry in L. lactis. Based on the plasmid pZE11-MCS2, it contains the ColE1 origin of replication and ampR ampicillin resistance cassette to enable cloning in E. coli, and it contains the AMβ1 origin of replication and ermR erythromycin resistance cassette from pVE552338 to enable replication and selection in L. lactis. In pECGMC, mCherry is expressed from the constitutive P32 promoter39 and mf-Lon is expressed from the inducible promoter PnisA11 with 3 ng/ml nisin. Plasmids containing pdt fusions to mCherry are labeled according to the pdt variant used (e.g. pECGMC3 contains pdt#3). Plasmids were transformed into L. lactis according to established protocols40, and the sequence of pECGMC3 was deposited in GenBank under accession number KM521211.
Genomic insertion of pdt variants
The pECT plasmids were created to serve as a template for PCR amplification of the pdt variant cassettes shown in Figure 4a. The kanR cassette and surrounding FRT sites from pKD1332 were cloned into pWM9135 to generate pECT, which was further named according to the cloned pdt variant (e.g., pECT3A contains pdt#3A). To generate pECA102, Flp recombinase was cloned into pBAD241 using KpnI, and the constitutively expressed sacB cassette was subsequently amplified from pWM9135 and cloned into the plasmid using partial MluI and SalI digestion. Plasmids pECT3 and pECA102 were deposited in GenBank under accession numbers KM521207 and KM521212, respectively. To generate PCR products for genomic integration, pdt variants were amplified from their pECT plasmid using primers P1 and P2 that contained additional 42 base 5′ extensions with homology to the C-terminus and immediate 3′ UTR of the targeted gene, respectively. Note that the endogenous gene’s stop codon should not be included in the P1 5′ extension. The base P1 and P2 primer sequences and the full length primers used to target murA, ftsZ, cheZ and recA are listed below. PCR products were transformed into E. coli containing pKD46 using published methods30, and successful genomic pdt insertions were verified by PCR. Plasmid pECA102 was used to remove the kanR cassette and was subsequently cured by selection on LB plates containing 8% sucrose.
P1: GCGGCGAACAAAAACGAA
P2: TTATGTAGGCTGGAGCTGC
P1-murA: CTGCGCGCTTTAGGTGCAAATATTGAGCGTGTGAAAGGCGAAGCGGCGAACAAAAACGAA
P2-murA: CTGGCGGTAGCCCCGCGAACGGGGCTGCCAGCTCTCAGACGATTATGTAGGCTGGAGCTGC
P1-ftsZ: GATTATCTGGATATCCCAGCATTCCTGCGTAAGCAAGCTGATGCGGCGAACAAAAACGAA
P2-ftsZ: GTTTAGCACAAAGAGCCTCGAAACCCAAATTCCAGTCAATTCTTATGTAGGCTGGAGCTGC
P1-cheZ: AGTCAGGATCAGGTGGACGATTTGTTGGATAGTCTTGGATTTGCGGCGAACAAAAACGAA
P2-cheZ: CCGCCTGATATGACGTGGTCACGCCACATCAGGCAATACAAATTATGTAGGCTGGAGCTGC
P1-recA: GTAGATGATAGCGAAGGCGTAGCAGAAACTAACGAAGATTTTGCGGCGAACAAAAACGAA
P2-recA: AAAAGGGCCGCAGATGCGACCCTTGTGTATCAAACAAGACGATTATGTAGGCTGGAGCTGC
Essential Protein Degradation library
The EPD library was created using pECM1, a derivative of pECT1 that includes MmeI sites on both sides of the insertion cassette, resulting in an extended pdt tag that includes the additional N-terminal amino acids VG. The full pdt#1 tag is VGAANKNEENTNEVPTFMLNAGQANRLQL. Genomic integration was performed according the protocol described above for individual pdt insertions, except that electroporation was performed using the BTX Gemini X2 System with an HT100 plate handler, and the kanamycin cassette was not removed. The list of targeted essential genes was manually collated from Ecogene41 and Baba et al.32, and a list of the primers used can be found in Supplementary Table 3. Plamid pECM1 was deposited in GenBank under accession number KM521208
Flow cytometry
Data for GFP and mCherry degradation dynamics and propidium iodide staining were collected using an LSRFortessa cell analyzer equipped with a High Throughput Sampler (BD Biosciences), and data for the synthetic toggle switch were collected using a FACSAriaII flow cytometer (BD Biosciences). For each GFP and mCherry measurement, cells were fixed in 1% paraformaldehyde (PFA), held at 4° for up to 5 days, and then diluted 1:10 in PBS for analysis. At least 5,000 cells were collected for each measurement and FloJo (Treestar) was used for data analysis.
Plate fluorimetry and optical density
Fluorescence and optical density measurements were made with a SpectraMax M5 microplate reader (Molecular Devices) using excitation and emission wavelengths of 488 nm and 520 nm, respectively, with an emission filter cutoff at 515 nm. Optical density was measured at 600 nm (OD600). All measurements were made in 200 μl in 96-well flat bottom plates.
MurA-induced lysis growth conditions
Strains were grown in 200 μl LB in 96-well flat bottom plates with lids at 30° shaking in a SpectraMax M5 plate reader. OD600 measurements were taken every 15 minutes and normalized using media-only wells. Wells on the perimeter of the plate were filled with water and not used for bacterial growth.
FtsZ microscopy
Differential interference contrast (DIC) and fluorescence microscopy images were taken with a Nikon Eclipse Ti microscope using a 100× objective and a Coldsnap HQ2 CCD camera (Photometrics) operated with NIS-Elements Advanced Research 3.2 software. Cells in exponential growth in liquid cultures were induced with ATc, grown for 3 hours at 30°, placed on a 300 μl pad containing PBS and 0.75% low-melt agarose (Boston Bioproducts) and immediately imaged.
Chemotactic motility plates
Cells in exponential growth were stabbed into soft agar plates containing 1% tryptone, 0.5% NaCl and 0.3% agar. ATc dissolved in 10 μl water was added to sterile 6mm disks in the center of the plates immediately prior to bacterial inoculation. Plates were incubated for 18 hours at 30° before imaging with a Gel Logic 6000 Pro (Carestream).
Hypersensitivity assay
Norfloxacin and ATc were simultaneously added at the indicated concentrations to cells in exponential growth in 96-well flat bottom plates, and OD600 was measured after 6 hours, with media only wells serving as absorbance controls. For targeted RecA degradation, cells in exponential growth were induced with 50 ng/ml ATc for 2 hours, if indicated, before treatment with 25 ng/ml norfloxacin for 2 hours. Cells were then serially diluted in PBS and spotted on LB plates without selection, and visible colonies forming units (CFU) were counted after incubation overnight at 30°.
Colony Forming Unit Assay
Cells with and without ATc induction were grown shaking in 96-well round bottom plates for 4 hours and then serially diluted in PBS before plating on LB agar without selection for growth overnight at 30 degrees.
Propidium Iodide (PI) assay
Cells in exponential growth were induced with ATc for 4 hours in 96-well round bottom plates, diluted 1:100 in PBS containing 5 μM propidium iodide and analyzed by flow cytometry within 20 minutes. The threshold for PI+ cells was defined using untagged wild-type cells and is presented as a percentage of the total whole cell population for each strain.
Statistical analysis
Linear regression models used the ordinary least-squares approach to determine the best-fit line, coefficient of determination (R2) and standard deviation. All observed y-axis data was used to calculate the best-fit lines. p-values were calculated using a paired sample, one-tailed t-test.
Supplementary Material
Acknowledgments
We are grateful to Caleb Bashor, Michael Lobritz, Mo Khalil and Dan Dwyer for helpful discussions and critical reviews of the manuscript. This work was supported by funding from the Office of Naval Research (ONR) MURI Program, Defense Threat Reduction Agency grant HDTRA1-14-1-0006, and the Howard Hughes Medical Institute.
Footnotes
AUTHOR CONTRIBUTIONS
D.E.C. and J.J.C. conceived the study, analyzed data and wrote the paper. D.E.C. designed and performed the experiments.
COMPETING FINANCIAL INTERESTS STATEMENT
The authors have submitted a patent application related to the work described here.
References
- 1.Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. Journal of bacteriology. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lutz R, Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997;25:1203–1210. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Isaacs FJ, et al. Engineered riboregulators enable post-transcriptional control of gene expression. Nat Biotechnol. 2004;22:841–847. doi: 10.1038/nbt986. [DOI] [PubMed] [Google Scholar]
- 4.Lou C, Stanton B, Chen YJ, Munsky B, Voigt CA. Ribozyme-based insulator parts buffer synthetic circuits from genetic context. Nat Biotechnol. 2012;30:1137–1142. doi: 10.1038/nbt.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janssen BD, Hayes CS. The tmRNA ribosome-rescue system. Adv Protein Chem Struct Biol. 2012;86:151–191. doi: 10.1016/B978-0-12-386497-0.00005-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neklesa TK, et al. Small-molecule hydrophobic tagging-induced degradation of HaloTag fusion proteins. Nat Chem Biol. 2011;7:538–543. doi: 10.1038/nchembio.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonger KM, Chen LC, Liu CW, Wandless TJ. Small-molecule displacement of a cryptic degron causes conditional protein degradation. Nat Chem Biol. 2011;7:531–537. doi: 10.1038/nchembio.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis JH, Baker TA, Sauer RT. Small-molecule control of protein degradation using split adaptors. ACS Chem Biol. 2011;6:1205–1213. doi: 10.1021/cb2001389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gur E, Sauer RT. Evolution of the ssrA degradation tag in Mycoplasma: specificity switch to a different protease. Proc Natl Acad Sci U S A. 2008;105:16113–16118. doi: 10.1073/pnas.0808802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ge Z, Karzai AW. Co-evolution of multipartite interactions between an extended tmRNA tag and a robust Lon protease in Mycoplasma. Mol Microbiol. 2009;74:1083–1099. doi: 10.1111/j.1365-2958.2009.06923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mierau I, Kleerebezem M. 10 years of the nisin-controlled gene expression system (NICE) in Lactococcus lactis. Applied microbiology and biotechnology. 2005;68:705–717. doi: 10.1007/s00253-005-0107-6. [DOI] [PubMed] [Google Scholar]
- 12.Gardner TS, Cantor CR, Collins JJ. Construction of a genetic toggle switch in Escherichia coli. Nature. 2000;403:339–342. doi: 10.1038/35002131. [DOI] [PubMed] [Google Scholar]
- 13.Litcofsky KD, Afeyan RB, Krom RJ, Khalil AS, Collins JJ. Iterative plug-and-play methodology for constructing and modifying synthetic gene networks. Nat Methods. 2012;9:1077–1080. doi: 10.1038/nmeth.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown ED, Vivas EI, Walsh CT, Kolter R. MurA (MurZ), the enzyme that catalyzes the first committed step in peptidoglycan biosynthesis, is essential in Escherichia coli. Journal of bacteriology. 1995;177:4194–4197. doi: 10.1128/jb.177.14.4194-4197.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adams DW, Errington J. Bacterial cell division: assembly, maintenance and disassembly of the Z ring. Nat Rev Microbiol. 2009;7:642–653. doi: 10.1038/nrmicro2198. [DOI] [PubMed] [Google Scholar]
- 16.Silversmith RE. Auxiliary phosphatases in two-component signal transduction. Curr Opin Microbiol. 2010;13:177–183. doi: 10.1016/j.mib.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roemer T, Boone C. Systems-level antimicrobial drug and drug synergy discovery. Nat Chem Biol. 2013;9:222–231. doi: 10.1038/nchembio.1205. [DOI] [PubMed] [Google Scholar]
- 18.DeVito JA, et al. An array of target-specific screening strains for antibacterial discovery. Nat Biotechnol. 2002;20:478–483. doi: 10.1038/nbt0502-478. [DOI] [PubMed] [Google Scholar]
- 19.Kim DH, et al. Characterization of a Cys115 to Asp substitution in the Escherichia coli cell wall biosynthetic enzyme UDP-GlcNAc enolpyruvyl transferase (MurA) that confers resistance to inactivation by the antibiotic fosfomycin. Biochemistry. 1996;35:4923–4928. doi: 10.1021/bi952937w. [DOI] [PubMed] [Google Scholar]
- 20.Peterson EJ, Janzen WP, Kireev D, Singleton SF. High-throughput screening for RecA inhibitors using a transcreener adenosine 5′-O-diphosphate assay. Assay and drug development technologies. 2012;10:260–268. doi: 10.1089/adt.2011.0409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei JR, et al. Depletion of antibiotic targets has widely varying effects on growth. Proc Natl Acad Sci U S A. 2011;108:4176–4181. doi: 10.1073/pnas.1018301108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu W, et al. Antibacterial drug leads targeting isoprenoid biosynthesis. Proc Natl Acad Sci U S A. 2013;110:123–128. doi: 10.1073/pnas.1219899110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cameron DE, Bashor CJ, Collins JJ. A brief history of synthetic biology. Nat Rev Microbiol. 2014;12:381–390. doi: 10.1038/nrmicro3239. [DOI] [PubMed] [Google Scholar]
- 24.Weber W, Fussenegger M. Emerging biomedical applications of synthetic biology. Nat Rev Genet. 2012;13:21–35. doi: 10.1038/nrg3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slusarczyk AL, Lin A, Weiss R. Foundations for the design and implementation of synthetic genetic circuits. Nat Rev Genet. 2012;13:406–420. doi: 10.1038/nrg3227. [DOI] [PubMed] [Google Scholar]
- 26.Callura JM, Cantor CR, Collins JJ. Genetic switchboard for synthetic biology applications. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:5850–5855. doi: 10.1073/pnas.1203808109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang DC, Holtz WJ, Maharbiz MM. A genetic bistable switch utilizing nonlinear protein degradation. J Biol Eng. 2012;6:9. doi: 10.1186/1754-1611-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prindle A, et al. Rapid and tunable post-translational coupling of genetic circuits. Nature. 2014;508:387–391. doi: 10.1038/nature13238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holtz WJ, Keasling JD. Engineering static and dynamic control of synthetic pathways. Cell. 2010;140:19–23. doi: 10.1016/j.cell.2009.12.029. [DOI] [PubMed] [Google Scholar]
- 30.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Callura JM, Dwyer DJ, Isaacs FJ, Cantor CR, Collins JJ. Tracking, tuning, and terminating microbial physiology using synthetic riboregulators. Proc Natl Acad Sci U S A. 2010;107:15898–15903. doi: 10.1073/pnas.1009747107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Molecular systems biology. 2006;2:0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Metcalf WW, Jiang W, Wanner BL. Use of the rep technique for allele replacement to construct new Escherichia coli hosts for maintenance of R6K gamma origin plasmids at different copy numbers. Gene. 1994;138:1–7. doi: 10.1016/0378-1119(94)90776-5. [DOI] [PubMed] [Google Scholar]
- 34.Salis HM, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol. 2009;27:946–950. doi: 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Metcalf WW, et al. Conditionally replicative and conjugative plasmids carrying lacZ alpha for cloning, mutagenesis, and allele replacement in bacteria. Plasmid. 1996;35:1–13. doi: 10.1006/plas.1996.0001. [DOI] [PubMed] [Google Scholar]
- 36.Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 37.Muller-Hill B, Crapo L, Gilbert W. Mutants that make more lac repressor. Proc Natl Acad Sci U S A. 1968;59:1259–1264. doi: 10.1073/pnas.59.4.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dieye Y, Usai S, Clier F, Gruss A, Piard JC. Design of a protein-targeting system for lactic acid bacteria. J Bacteriol. 2001;183:4157–4166. doi: 10.1128/JB.183.14.4157-4166.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van de Guchte M, Kok J, Venema G. Gene expression in Lactococcus lactis. FEMS microbiology reviews. 1992;8:73–92. doi: 10.1111/j.1574-6968.1992.tb04958.x. [DOI] [PubMed] [Google Scholar]
- 40.Holo H, Nes IF. Transformation of Lactococcus by electroporation. Methods in molecular biology. 1995;47:195–199. doi: 10.1385/0-89603-310-4:195. [DOI] [PubMed] [Google Scholar]
- 41.Zhou J, Rudd KE. EcoGene 3.0. Nucleic Acids Res. 2013;41:D613–624. doi: 10.1093/nar/gks1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.