

Abstract
Muscular dystrophies (MD) are a group of heterogeneous genetic disorders characterized by progressive striated muscle wasting and degeneration. Although the genetic basis for many of these disorders has been identified, the exact mechanism for disease pathogenesis remains unclear. The presence of oxidative stress (OS) is known to contribute to the pathophysiology and severity of the MD. Mitochondrial dysfunction is observed in MD and likely represents an important determinant of increased OS. Experimental antioxidant therapies have been implemented with the aim of protecting against disease progression, but results from clinical trials have been disappointing. In this study, we explored the capacity of the cacao flavonoid (−)-epicatechin (Epi) to mitigate OS by acting as a positive regulator of mitochondrial structure/function endpoints and redox balance control systems in skeletal and cardiac muscles of dystrophic, δ-sarcoglycan (δ-SG) null mice. Wild type or δ-SG null 2.5 month old male mice were treated via oral gavage with either water (control animals) or Epi (1 mg/kg, twice/day) for 2 weeks. Results evidence a significant normalization of total protein carbonylation, recovery of reduced/oxidized glutathione (GSH/GSSG ratio) and enhanced superoxide dismutase 2, catalase and citrate synthase activities with Epi treatment. These effects were accompanied by increases in protein levels for thiolredoxin, glutathione peroxidase, superoxide dismutase 2, catalase and mitochondrial endpoints. Furthermore, we evidence decreases in heart and skeletal muscle fibrosis, accompanied with an improvement in skeletal muscle function with treatment. These results warrant the further investigation of Epi as a potential therapeutic agent to mitigate MD associated muscle degeneration.
Keywors: Epicatechin, mitocondria, oxidative stress, muscular dystrophy
Introduction
Muscular dystrophies (MD) comprise a heterogeneous group of inherited diseases that mainly affect striated muscle. MD are characterized by progressive muscle weakness, atrophy and in many instances, premature death [1]. MD share common histopathological features, such as muscle necrosis followed by replacement with connective (scar) tissue and fat [2]. These disorders are due to selective mutations in a number of molecules, including extracellular matrix, cytoskeletal, cytosolic, nuclear membrane and sarcolemmal proteins (such as members of the dystrophin-associated protein complex; DAPC) [3]. Many of these mutations cause cell membrane disruption [4] which may trigger cytoplasmic and/or mitochondrial calcium overload [5]. Calcium overload can induce mitochondrial swelling and organelle dysfunction, which can lead to high levels of reactive oxygen species (ROS) and thus, tissue oxidative stress (OS) [6]. The redox balance control system is comprised of physiological buffers and multiple enzymes and is responsible for the physiological management of ROS. It is thought that the progressive muscle degeneration process seen with MD is exacerbated by alterations in redox balance control systems leading to dramatically increased OS. OS may then, further accelerate the damage process and as such, be an early aggravating event in the initiation and progression of MD leading to tissue necrosis [7]. Multiple MD clinical trials using antioxidants were implemented but have failed to attenuate disease progression [8]. Failure may be related to the unidimensional approach used of trying to eliminate or scavenge free radicals, while not also targeting the redox balance control systems.
Flavonoids are a class of natural compounds recognized for their health benefits [9]. Health benefits are attributed to their “direct” antioxidant potential. However, for flavonoids with similar antioxidant activity end-biological effects vary widely, suggesting other mechanistic venues for attenuating OS [9]. We recently reported that (−)-epicatechin (Epi), the most abundant flavonoid found in cacao substantially ameliorates OS in skeletal muscle (SkM) in a mouse model of insulin resistance [9]. Similar effects were observed in the SkM of heart failure and type 2 diabetes mellitus patients treated with Epi-rich cocoa [9]. Effects were accompanied by the recovery of key elements of the redox balance control systems including physiological antioxidants (e.g., glutathione) and enzymes such as mitochondrial superoxide dismutase (SOD2) and catalase, as well as multiple mitochondrial structure/function proteins [9].
In the present study, we examined the effects of two weeks of Epi treatment on SkM and myocardial redox balance control systems including mitochondrial related proteins as well as mitochondrial function and tissue fibrosis in δ-sarcoglycan (SG) knock out (δ-KO) mice. Furthermore, we evaluated SkM function in the δ-KO mice. This mouse model recapitulates several characteristics of the human form of limb-girdle muscular dystrophy 2F in which DAPC defects are reflected in SkM as well as in myocardium, leading to the development of a severe cardiomyopathy.
Results
Protein carbonylation
Protein carbonylation significantly increased (~60%) in SkM (Fig. 1A) and approximately doubled in heart (Fig. 1B) of Ctrl δ-KO mice group vs. Ctrl WT group. In the δ-KO group treated with Epi, carbonylation significantly decreased vs. Ctrl δ-KO. No significant differences were observed between the Epi δ-KO vs. Ctrl WT groups.
Figure 1. Effect of Epi on protein carbonylation.

Total protein carbonylation was determined by dot blots in quadriceps (A) and hearts (B) of water (vehicle) and Epi treated WT and δ-KO mice. Bargraphs report on the densytometric analysis of dot blots. Protein carbonylation was increased in the quadriceps and heart of Ctrl δ-KO as compared vs. Ctrl WT mice (*P<0.05). Epi treatment significantly reduced carbonylation levels (#P<0.05 Epi δ-KO vs Ctrl δ-KO). n=5 animals/group.
Reduced/oxidized glutathione (GSH/GSSG) ratio
The Ctrl δ-KO group evidenced a significant decrease in SkM and heart GSH/GSSG ratios vs. Ctrl WT (Fig. 2A and B). In the Epi δ-KO group, treatment significantly increased GSH/GSSG ratio in SkM (Fig. 2A) and heart (Fig. 2B) vs. Ctrl δ-KO. The Epi WT group also yielded a significant increase in the ratio vs. Ctrl WT in both tissues (Fig. 2A and B).
Figure 2. Effect of Epi on GSH/GSSG ratio.

GSH/GSSG ratio was measured in quadriceps (A) and heart (B) of WT and δ-KO mice. GSH/GSSG ratio significantly decreased in Ctrl δ-KO as compared vs. Ctrl WT mice (*P<0.05). Epi treatment of δ-KO significantly increased GSH/GSSG ratio (#P<0.05 Epi δ-KO vs Ctrl δ-KO). Epi WT mice GSH/GSSG ratio significantly increased vs. Ctrl WT (‡P<0.05). n=5 animals/group.
OS Regulatory Protein System
SIRT3, TRX, GPx, SOD2 and catalase protein levels were measured as relevant redox control system endpoints. In SkM and hearts, all proteins evidenced significant decreases in Ctrl δ-KO vs. Ctrl WT mice (Fig 3A and B). In the Epi δ-KO group, the flavonoid was able to induce a significant recovery of all proteins measured in SkM and heart as compared vs. Ctrl δ-KO mice (Fig 3A and 3B). The recovery of protein levels in SkM fluctuated from ~50% for GPx and SIRT3 to ~80% for SOD2 (Fig. 3A). In the heart, recovery fluctuated from ~60% for GPx to ~80% for SOD2 (Fig. 3B). Interestingly, there was a significant increase in the levels of TRX and SOD2 in the Epi WT group vs. Ctrl WT in SkM (Fig. 3A), while in the heart increases were noted in SIRT3, TRX and SOD2 (Fig. 3B).
Figure 3. Effect of Epi on oxidative stress regulatory systems.

Protein levels of sirtuin-3 (SIRT3), thioredoxine (TRX), glutathione peroxidase (GPx), mitochondrial superoxide dismutase (SOD2) and catalase were analyzed by Western blot. Significant decreases were observed in Ctrl δ-KO mice as compared vs. Ctrl WT mice (*P<0.05). In the Epi δ-KO, treatment either partially restored or fully recovered their levels. (#P<0.05 Epi δ-KO vs Ctrl δ-KO). In Epi WT animals there was a significant increase in the levels of TRX and SOD2 in SkM, while in heart there was an increase in SIRT3, TRX, and SOD2 vs. Ctrl WT (‡P<0.05). The bargraphs report on densitometric analysis as: protein/GAPDH for each group. n=5 animals/group.
SOD and catalase activities
SkM and heart SOD2 activity decreased significantly by ~50% in the Ctrl δ-KO group vs. the Ctrl WT group (Fig. 4A and B). Catalase activity also decreased by ~60–65% in both organs (Fig. 4C and D). Treatment with Epi, fully restored the activity of both enzymes in SkM and heart. The Epi WT group evidenced a significant increase in the activities of the enzymes vs. Ctrl WT in both organs (Fig. 4A–D).
Figure 4. Effect of Epi on SOD2 and catalase activities.

SOD2 (A and B) and catalase (C and D) activities were evaluated by colorimetric assays in quadriceps (A and C) and heart (B and D). A loss of enzymatic activities was noted in Ctrl δ-KO mice as compared vs. Ctrl WT mice (*P<0.05). In the Epi δ-KO group, treatment fully recovered enzyme activities (#P<0.05 Epi δ-KO vs Ctrl δ-KO). Epi WT mice significantly increased enzyme activities vs. Ctrl WT (‡P<0.05). n=5 animals/group.
Mitochondrial related proteins
SIRT1, PGC1α, TFAM, porin, mitofilin and complex V were evaluated as mitochondrial relevant endpoints. In both SkM and heart, all proteins demonstrated a significant decrease (of up to ~60%) in the Ctrl δ-KO vs. the Ctrl WT group (Fig. 5A and B). Treatment with Epi either partially restored or fully recovered SkM protein levels (Fig. 5A) a pattern similarly observed for heart samples (Fig. 5B). PGC1α, TFAM, porin, mitofilin and complex V protein levels evidenced significant increases in the Epi WT group vs. Ctrl WT in both organs (Fig. 5A and B).
Figure 5. Effects of Epi on mitochondrial related proteins.

Protein levels for the mitochondrial related proteins SIRT1, PGC1α, TFAM, porin, mitofilin and CV were analyzed by Westerns. Ctrl δ-KO mice evidenced decreased levels of the proteins as compared vs. Ctrl WT mice (*P<0.05). In the Epi δ-KO, treatment either partially restored or fully recovered their levels (#P<0.05 Epi δ-KO vs Ctrl δ-KO). In Epi WT group there was a significant increase in the levels of PGC1α, TFAM, porin, mitofilin and CV in SkM and heart vs. Ctrl WT (‡P<0.05). The bargraphs report on densitometric analysis as: protein/GAPDH for each group. n=5 animals/group.
Citrate synthase (CS) activity
SkM and heart CS activity decreased significantly by ~60% in the Ctrl δ-KO group vs. the Ctrl WT group (Fig. 6A and B). Treatment with Epi fully restored the activity of the enzyme in the SkM and heart of the δ-KO mice (Epi δ-KO vs. Ctrl δ-KO). The Epi WT group also evidenced a significant increase in the activity of the enzyme vs. Ctrl WT in both organs (Fig. 6A and B).
Figure 6. Effect of Epi on citrate synthase activity.
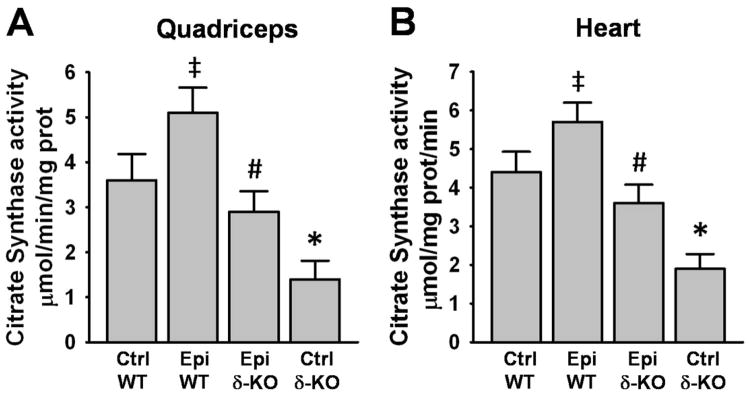
Citrate synthase activity was quantified in quadriceps (A) and heart (B). A decrease in enzymatic activity was observed in Ctrl δ-KO mice as compared vs. Ctrl WT mice (*P<0.05). In the Epi δ-KO group, treatment fully restored enzyme activity (#P<0.05 Epi δ-KO vs Ctrl δ-KO). Epi WT mice significantly increased enzyme activities vs. Ctrl WT (‡P<0.05). n=5 animals/group.
Cell death signaling pathway related proteins
Proteins isolated from mitochondria of SkM and heart were analyzed for changes in cytoplasmic and mitochondrial levels of Bax and Cyt C (Fig. 7). Bargraph results are expressed as ratio of mitochondrial over cytoplasmic Cyt C and ratio of cytoplasmic over mitochondrial for Bax. SkM and heart Cyt C levels increased significantly in the cytoplasm of Ctrl δ-KO vs. Ctrl WT group. However, with Epi treatment Cyt C decreased to almost normal levels in both organs (Fig. 7A and B). In contrast, SkM and heart Bax levels were higher in SkM and heart mitochondria of Ctrl δ-KO vs. Ctrl WT group. With treatment, Epi δ-KO group, Bax levels decreased to normal in mitochondria (Fig. 7A and B).
Figure 7. Effect of Epi on Bax and Cyt C protein levels.

Quadriceps femoris (A) and heart (B) enriched mitochondrial (M) and cytoplasmic (C) fractions of water and Epi treated WT and δ-KO mice were analyzed for Cyt C and Bax protein levels by Western blot. In Ctrl δ-KO mice, high levels of cytoplasmic Cyt C were observed as the ratio mitochondrial/cytoplasmic levels show. Mitochondrial Bax was highly present in Ctrl δ-KO tissues and Epi treatment significantly reversed it into the cytoplasm as the ratio cytoplasmic/mitochondrial show. (*P<0.05 when compares δ-KO vs. Ctrl WT mice or #P<0.05 Epi δ-KO vs Ctrl δ-KO). n=5 animals/group.
Tissue fibrosis
As evidenced by Masson’s trichrome staining and the quantification of collagen area fraction, SkM (Fig. 8A–E) and heart (Fig. 8G–K) tissue fibrosis increased significantly in Ctrl δ-KO (Fig. 8B and H) vs. the Ctrl WT (Fig. 8A and G). Epi treatment essentially normalized SkM (Fig. 8D and E) and heart (Fig. 8J and K) fibrosis to levels comparable to those of Ctrl WT animals. These results are consistent with the collagen III protein levels noted by Westerns in SkM (Fig. 8F) and heart (Fig. 8L).
Figure 8. Effects of Epi on tissue fibrosis.

Masson’s trichrome staining was used in tissue sections of quadriceps (A–D) and heart (G–J). δ-KO mice evidenced high levels of fibrosis in SkM (B) and heart (H) which were normalized with Epi treatment (D and J respectively). Fibrous tissue quantification was performed as a fraction of the total area (blue area/total area) for SkM (E) and heart (K). As a surrogate of tissue fibrosis levels of collagen III were analyzed by Western blot. Ctrl δ-KO mice evidenced increased levels of the protein vs. Ctrl WT mice (*P<0.05). Epi treatment fully normalized their levels (#P<0.05 Epi δ-KO vs Ctrl δ-KO). The bargraphs report on densitometric analysis as: protein/GAPDH for each group. n=5 animals/group.
Skeletal muscle function
Five Ctrl δ-KO and 5 Epi δ-KO mice were evaluated using a hanging wire time test to determine whether Epi treatment had an impact on SkM function. Under the conditions used, Epi treatment of δ-KO mice demonstrated a statisticaly significant incfrease in normalized hanging wire time as compared vs. Ctrl δ-KO (Fig. 9)
Figure 9. Effects of Epi on hanging wire time.
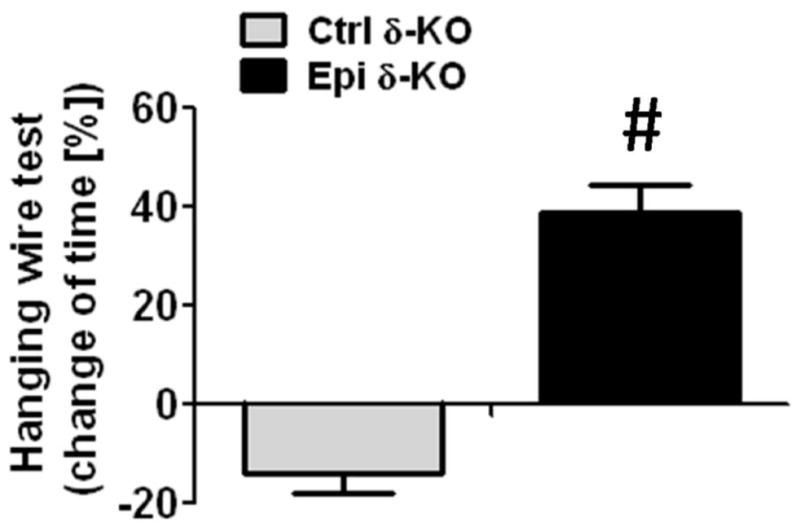
Skeletal muscle function was evaluated in Epi δ-KO and Ctrl δ-KO mice using a wire test and calculating the percent changes observed in hanging time (time normalized vs. pre-treatment). After 15 days of treatment, there was an improvement muscle function in Epi δ-KO mice vs. the moderate loss observed in Ctrl δ-KO animals.
Discussion
Unique results from this study indicate that 2 weeks of Epi treatment of δ-KO mice yields the normalization of total protein carbonylation, increases reduced/oxidized glutathione ratio and enhances SOD2, catalase and citrate synthase activities. These effects are accompanied by increases in protein levels for TRX, GPx, SOD2, catalase and multiple mitochondrial endpoints. Furthermore, a normalization of SkM and heart fibrosis, as well as an improvement in SkM function is observed with treatment.
ROS are generated during normal cell metabolism mainly in mitochondria and although they are essential for cell signaling, when present in excess they can adversely modify cell structure/function. Excess ROS can be attained when endogenous cell defense mechanisms involving buffering systems, as well as redox control enzymes are outweighed by its generation thus, resulting in OS [10]. Dysfunctional mitochondria are an important source of excess ROS. OS causes cellular damage by directly and irreversibly altering macromolecules such as DNA, lipids and proteins [10]. OS can also activate proteolytic enzymes and pro-inflammatory/fibrotic cytokines. OS has been also been implicated in the pathophysiology of SkM conditions and diseases such as those seen with aging (sarcopenia) or metabolic disorders (type 2 diabetes) as well as in multiple cardiac pathologies [9, 11–14]. Although most of the MDs have a well identified genetic basis, the exact mechanism for their pathogenesis remains unclear. Emerging evidence indicates that interactions between the primary genetic defect and the ensuing OS contributes notably to the progression of the disease leading to tissue necrosis and fibrosis [15]. Human mutations in the δ-SG gene causes limb-girdle MD type 2F, an autosomal recessive MD that triggers progressive weakness and wasting of proximal limb muscles which is often accompanied with cardiac involvement [16]. Patient survival depends on the ability to sustain proper cardiac and respiratory function consequently, many succumb to dilated cardiomyopathy and/or respiratory failure [17, 18]. There are a number of animal models of δ-sarcoglycanopathy that have aided in the understanding of its pathogenesis [19, 20]. δ-KO mice from young age, develop typical histological features (including fibrosis and SkM waste) of MD in most of their SkM [16, 21]. δ-KO mice also develop a severe cardiomyopathy with focal areas of fibrosis that is thought to be related to structural and functional abnormalities in the coronary vasculature yielding local vasospasms [16, 21]. Therefore δ-KO mice recapitulate many of the important features of the human disease, making it a useful preclinical research tool.
It has been demonstrated that in the δ-KO mouse, the absence of δ-SG is accompanied by the concomitant loss of α-, β-, γ, and ε-SGs and sarcospan in SkM and cardiac muscle, which has been also observed in human forms of δ-sarcoglycanopathy [16], [22]. Similar observations have been made for β-SG [23]. The altered disposition of these molecules is likely due to the central role of β- and δ-SG as part of an assembly core for the rest of the SGs and to their association with the C-terminus of dystrophin [24]. Although several types of MD animal models develop OS, there is limited knowledge regarding δ-KO mice. In this study, we report the presence of elevated SkM and heart OS in δ-KO animals as evidenced by high levels of markers (i.e. total protein carbonylation) as well as important decreases in the levels or activities of key regulators of the redox control systems (enzymatic and non-enzymatic). Treatment with Epi was able to largely normalize SkM and heart OS related abnormalities. There is precedent for catechins to positively impact OS related endpoints. The use of green tea extracts (tipically rich in epicatechins) in a mouse model of Duchenne’s MD (mdx), reduced in a dose-dependent manner extensor digitorium longus muscle OS and necrosis, as well as OS in cultured C2C12 cells [7]. Epi effects on OS related endpoints observed in this study also closely mirror those previously reported by us in the SkM of a high fat diet mouse model of insulin resistance and obesity where the redox control systems are adversely impacted. In the clinical part of that study, we also reported on the beneficial effects that Epi-rich cocoa has on heart failure and type 2 diabetes patient SkM OS regulatory systems such as glutathione, SOD2 and catalase, where we identified the nuclear regulatory mechanisms implicated in mediating the effects of treatment which involved the activation of the transcriptional factors PGC1α and FOXO1 [9].
Damage and/or loss of mitochondria in the setting of MD has been well documented and as discussed bellow, has critical implications in the development of cell death. A strategy that can be used to counteract mitochondrial loss is to stimulate its biogenesis. Multiple studies have reported on the key importance of the sirtuin-PGC-1α-TFAM pathway in the process of mitochondrial biogenesis. SIRT1 activates PGC-1α through deacetylation leading to its translocation into the nucleus [25]. PGC-1α then activates several transcription factors leading to the expression of a broad set of mitochondrial genes including oxidative phosphorylation complexes (OXPHOS) and TFAM [26, 27]. Reduced levels of gene programs regulated by PGC-1α have been reported in MD [28] and it has been suggested as an important contributor to mitochondrial dysfunction in multiple muscle-associated diseases [29]. Results from this study evidence the restorative effects of Epi treatment on protein markers of mitochondrial biogenesis and organelle structure/function (i.e. citrate synthase activity) in the SkM and heart of δ-KO mice. There is precedent for other polyphenols (e.g. resveratrol) yielding beneficial effects via increasing SIRT1 activity in δ-SG deficient hamster and mdx mice [30]. Previously, we reported that 2 weeks of treatment with Epi resulted in increases in OXPHOS complexes I, II, III and V, mitofilin, porin, and TFAM protein levels as well as mitochondrial volume and cristae abundance in hindlimb and cardiac muscles of normal middle aged mice (12 months old) [31]. We also previously reported on the abnormal microstructural features of SkM in heart failure/diabetic patients. Noteworthy, there was loss of mitochondrial cristae [32] and sarcomere organization [33]. Protein levels for multiple members of the DAPC including dystrophin, dystroglycans and sarcoglycans were very low as compared to normal muscle [33]. Treatment with Epi rich cocoa for 3 months, increased cristae abundance and was associated with higher protein levels of multiple regulators of mitochondrial biogenesis and of DAPC members [32, 33]. Therefore, Epi appears uniquely capable of restoring mitochondrial related endpoints which has the potential to translate into improved muscle structure/function likely by favorably impacting cellular bioenergetics.
OS in δ-KO mice has been attributed at least partially, to abnormalities in the structure/function of mitochondria. Mitochondrial swelling which compromises organelle function is commonly observed by electron microscopy in MD tissues [1, 34, 35]. It has been reported that treatment with a cyclophilin inhibitor (Debio-025) which reduces mitochondrial swelling mitigates necrotic disease manifestations in mdx and in δ-KO mice [1]. Mitochondrial swelling can also lead to the release of mitochondrial proteins such as Cyt C which in concert with the abnormally high levels of Bax in the organelle, can activate cell death (apoptotic) pathways [5]. In this study, we document in δ-KO mice, the abnormal presence of Cyt C in SkM and heart sample cytoplasm while evidencing increases of Bax in mitochondria. With Epi treatment, we observe a resegregation of Cyt C into mitochondria and Bax to the cytoplasm, an effect that can be interpreted as reflecting the preservation of mitochondrial structure and thus, avoidance from cell death commitment.
OS is also tightly related to the development of tissue fibrosis, a process activated in part by ROS-triggered profibrotic signaling pathways (i.e. transforming growth factor-β pathway) [36, 37]. Fibrosis development/progression can also depend on unmitigated wound-healing process in response to chronic tissue inflammation and necrosis [38]. Muscle fibrosis is recognized as a prominent pathological feature of dystrophic patients. Fibrosis contributes to muscle dysfunction and importantly to the lethal phenotype of MD. Although there are no effective therapies for MD, recent studies have demonstrated that ameliorating muscle fibrosis may represent a viable therapeutic approach [38]. In this study, we report the normalization of SkM and heart tissue fibrosis in δ-KO mice with Epi treatment. The normalization of tissue fibrosis with Epi is likely to arise from the multiple benefits attained such as reduced OS, inflammation, necrosis and improved cell viability. Reductions in collagen production may also be impacted by the inhibition of its synthesis or by enhancing degradation [39] issues which await further research. Together with decreases in tissue fibrosis, we observed with Epi treatment an improvement in SkM function as evidenced by the increased hanging wire time recorded in δ-KO animals.
In the present study, we postulate that the beneficial effects of Epi in δ-KO mice are related to its capacity to favorably impact multiple molecular regulatory mechanisms rather than exerting direct antioxidant actions. As Epi is known to be safe and non-toxic, we believe it offers an opportunity to be rigorously tested in the clinical setting in particular, as the consumption of dark chocolate has been associated with multiple beneficial effects on cardiometabolic endpoints.
Materials and Methods
Animal Model
δ-KO (B6.129-Sgcdtm1Mcn/J) mice were purchased from Jackson Laboratories (Bar Harbor, ME). The genotype of δ-SG locus was verified using a protocol provided by Jackson Laboratory to differentiate wild type (B6.129-Sgcd wild type [WT]) vs. the δ-KO mice (http://jaxmice.jax.org/strain/004582.html). All procedures were performed in accordance to The Code of Ethics of the World Medical Association (Declaration of Helsinki) and the U.S. Guidelines for the Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources (http://www.nal.usda.gov/awic/pubs/noawicpubs/careuse.htm) and approved in Mexico by the National Academy of Medicine following the Mexican Official Standard NOM-062-ZOO-1999.
Experimental Design and Approach
At the age of 2.5 months, δ-KO mice develop most of the disease features which resemble human limb girdle muscle dystrophy [16]. We examined the effects of Epi on skeletal muscle (quadriceps femoris) and heart (left ventricle) muscles of 2.5 month old male WT and δ-KO mice. A total of 20 animals (10 WT and 10 δ-KO mice) were allocated into four groups of 5 animals each: 1) water WT (Ctrl WT); 2) Epi WT; 3) Epi δ-KO and 4) Ctrl δ-KO groups. Mice were provided either vehicle (water) or 1 mg/kg body mass of Epi (Sigma-Aldrich, St Louis, MO, USA) by oral gavage twice a day (morning and evening) for 2 weeks. After 14 days of treatment, mice were euthanized by using isoflurane sedation followed by cervical dislocation. The quadriceps femoris (SkM) and heart were harvested and used for histological, biochemical and molecular analysis.
Protein Carbonylation
Protein carbonylation was used as an indicator of tissue OS levels [9]. Approximately 100 mg of SkM and heart were rinsed with PBS pH 7.4. Tissues were homogenized in 1 ml of cold buffer (50 mM MES pH 6.7, containing 1 mM EDTA). Homogenates were centrifuged at 10,000 g for 15 min at 4 °C. Supernatant was recovered and incubated at room temperature for 15 min with streptomycin sulfate at a final concentration of 1%. Samples were centrifuged at 6000 g for 10 min at 4 °C. Supernatant was recovered and an aliquot was used to measure protein content using a Bradford assay (Bio-Rad). A total of 50 μg of protein was used for dot blot assays. Dot blot assays relied on protein carbonyl functional group derivatization with 2,4-dinitrophenyldrazone (DNP) from the protein carbonyl assay kit (Cayman Chemicals) to render stable DNP-proteins. Protein samples were then applied to polivydinil fluoride membranes and used for carbonylation detection via a specific anti-DNP antibody (Millipore) following the method outlined bellow for Western blot detection.
Measurement of reduced (GSH) and oxidized (GSSG) glutathione
Reduced glutathione (GSH) is an important intracellular antioxidant and their levels can be impacted (reduced) in the setting of OS [40]. Tissue samples (25 mg) were homogenized with a polytron in 250 μL of cold buffer (50 mM potassium phosphate, pH 7, containing 1 mM EDTA), centrifuged at 10, 000 × g for 15 min at 4°C. The supernatants were deproteinated as per manufacturer instructions. Briefly, samples were mixed with an equal volume of 1.25M metaphosphoric acid (MPA) and centrifugated at 2000 × g for 2 min. Supernatants were recovered and incubated with triethanolamine (TEAM) 4M, then samples were used to measure GSH and GSSG using a colorimetric detection assay kit according to the manufacturer’s instructions (Cayman Chemicals, intra-assay coefficient of variation of 1.6%). All samples were tested in duplicates and measured at room temperature.
SOD2 Activity
SOD2 transforms superoxide, a byproduct of the mitochondrial electron transport chain, into hydrogen peroxide and diatomic oxygen and thus, constitutes an important enzyme member of the redox control system [9]. To measure its activity, ~25 mg of SkM and heart were rinsed with PBS pH 7.4, homogenized using teflon homogenizer with 300 μL sucrose buffer (0.25 M sucrose, 10 mM Tris, 1 mM EDTA, pH 7.4). The homogenate was sonicated on an ice bath for 5 min and centrifuged at 10,000 g for 60 min at 4 °C. The supernatant was used to measure SOD2 activity using a colorimetric kit (Dojindo Molecular Technologies, intra-assay coefficient of variation of < 5%) in which KCN (1 mM) was added to the sample so as to inactivate non-SOD2 activities.
Catalase Activity
Catalase is another important member of the redox control system and it catalyzes the decomposition of hydrogen peroxide to water and oxygen [9]. To measure its activity, ~25 mg of the SkM and heart were rinsed with PBS (pH 7.4). Tissues were homogenized with polytron in 250 ml cold 50 mM potassium phosphate, pH 7.0, containing 1 mM EDTA. Homogenates were centrifuged at 10,000 × g for 15 minutes at 4°C. The supernatant was used to measure catalase activity using a colorimetrically-based detection assay (Cayman Chemical).
Western blotting
Approximately 50 mg of SkM and heart were homogenized with a polytron in 500 μl lysis buffer (1% Triton X-100, 20 mM Tris, 140 mM NaCl, 2 mM EDTA, and 0.1% SDS) with protease and phosphatase inhibitor cocktails (P2714 and P2850, Sigma-Aldrich), supplemented with 0.15 mM PMSF, 5 mM Na3VO4 and 3 mM NaF. Homogenates were sonicated 20 min at 4°C and centrifuged 12,000 g for 10min. Total protein content was measured in the supernatant using the Bradford method. A total of 40 μg of protein was loaded onto a 4–15% precast polyacrylamide gel (Bio-Rad), electrotransferred to a polyvinylidene difluoride (PVDF) membrane using a semidry system. Membranes were incubated 1 h in blocking solution (5% non-fat dry milk in TBS plus 0.1% Tween 20 [TBS-T]), followed by a 3h incubation at room temperature with primary antibodies: glutathione peroxidase (GPx; Abcam), thiolredoxin (TRX), SOD2, catalase (Cell Signaling) were used to examine redox control related proteins [9]. To examine mitochondrial biogenesis/cell metabolism related proteins we evaluated, sirtuin-1 (SIRT1), SIRT3, peroxisome proliferator-activated receptor gamma, coactivator-1α (PGC1α) (Cell Signaling) and TFAM (Sigma-Aldrich) [32]. To examine mitochondria structure/function related proteins we examined, porin (a membrane protein), mitofilin (a cristae protein) and oxidative phosphorylation complex V (MitoSciences) [31]. To examine cell death pathway related proteins Bax (Abcam) and Cytochome C (Cyt C) (Santa Cruz Biotechnologies) levels were evaluated in cytoplasmic and mitochondrial samples. To examine tissue fibrosis, collagen III (Col3; GeneTex) levels were evaluated. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Cell Signaling) was used as a loading control. All primary antibodies were diluted 1:1000–2000 in blocking solution. Membranes were washed (3 × for 5 min) in TBS-T and incubated 1 h at room temperature with specific HRP-conjugated secondary antibodies. Membranes were again washed 3 times in TBS-T, and the immunoblots developed using an ECL Plus detection kit (Amersham-GE). The band intensities were digitally quantified using ImageJ software (http://www.nih.gov).
Citrate synthase activity
As an indicator of mitochondrial function we evaluated CS activity in heart and SkM samples. Tissue samples (25 mg) were homogenized with a polytron in 250 μL of cold extraction buffer (20 mM Tris-HCl, 140 mM NaCl, 2 mM EDTA, and 0.1 % sodium dodecyl sulfate) with protease inhibitors (P2714, Sigma-Aldrich), 5 mM Na3VO4, and 3 mM NaF. Homogenates were centrifuged at 10,000 × g for 15 min at 4°C. Supernatants were recovered and used to measure CS as the rate of production of the mercaptide ion based on conversion of acetyl-CoA and oxaloacetate into citrate synthase and CoA-SH. CoA-SH in the presence of 5,5-disthiobis-2-nitrobenzoic acid (DTNB) produces mercaptide ion. Samples were analysed in a Beckman DU 730 spectrophotomer (Beckman, Fullerton, CA, USA) at 412 nm [31]. All samples were tested in duplicates and measured at room temperature.
Mitochondrial protein isolation
Bax plays a key role in apoptotic signaling in cells by allowing the release of mitochondrial Cyt C into the cytoplasm [41, 42]. In healthy muscle Bax is found mainly in the cytoplasm and Cyt C in mitochondria. However, when cell death processes are activated, Bax moves into mitochondria and Cyt C is released to the cytoplasm [41, 42]. Thus, the isolation of mitochondria was used to evaluate the presence of such proteins in SkM and heart. Mitochondria were isolated using a Mitochondria Isolation kit (Thermo Sientific). Once mitochondria were isolated, proteins were extracted, quantified by Bradford and analyzed by Western blot as above.
Histology
SkM and heart were embedded in paraffin blocks and 4 μm serial sections were obtained in order to quantify SkM and heart fibrosis by using Masson’s trichome staining as published [36]. Collagen area fraction was measured by 2 observers blinded to genotypes, by visualizing blue-stained collagenous tissue vs. non-stained muscle from each section (n=3–5 animals/group, SkM and heart 12 sections/animal, 10 microscopic fields/section) observed at 20X magnification under light microscope. Quantification of collagen area fraction was performed using an image analyzer software (ImageJ) with color-based thresholding. The percentage of fibrosis was determined as blue-stained area divided by total tissue area [36].
Hanging wire test
A total of ten δ-KO mice (n=5 animals/group) were randomly assigned to water (Ctrl δ-KO) or Epi treatment (Epi δ-KO). Before the start and at the end of treatment, mice were subjected to a hanging wire test. The apparatus was lab-built using a polyethylene plastic pail of approximately 8 cm in diameter, with an affixed handle. The bottom surface of the pail was removed to yield an open cylinder. Subsequently, a circular piece of wire mesh (1 mm diameter wires and 1 cm × 1 cm spacing) was securely fastened to the entire surface top of the pail using standard cable-ties. The pail handle was mounted onto a ring stand such that the pail could be rotated freely about the handle over a cotton padded cage. To perform the wire hanging test, the pail was inverted and an animal was placed inside the pail on the wire mesh. The pail was then flipped around, thereby inverting the animal over the open bottom such that it had to hold on to the cage wire in order to avoid falling [43]. The hanging time was then recorded. Values are reported as percent normalized vs. the pre-treatment time value recorded for each animal.
Statistical Analysis
All data are presented as mean ± SD. ANOVA was performed to compare the group means for each variable. When appropriate, post hoc Tukey’s analysis was used to determine which means were significantly different from each other. Statistical significance was defined when P<0.05.
Acknowledgments
De los Santos S. and Gonzalez-Basurto S. are Ph.D. candidates supported by Consejo Nacional de Ciencia y Tecnología (CONACyT, México) grant 383805/255784 and 336212/229339 respectively. This work was partially supported by the Instituto Politecnico Nacional grant 20130528. This work was submitted in partial fulfillment of the requirements for the D.Sc. degree for D.S.S. and G.B.S. at Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México. This work was supported by NIH HL43617, AT4277, MD000220 and DK92154 to Dr. Villarreal, and CONACyT Mexico 129889 and an unrestricted grant by Cardero Therapeutics Inc. to Dr. Ceballos. IRS and RCV designed research; SDS, SGB, PML, PC, AZD and CPF conducted research; GC and FV analyzed data and performed statistical analysis; IRS, CVR, GC and FV wrote the paper.
Footnotes
Conflict of interest. F. Villarreal and G. Ceballos-Reyes are stockholders of Cardero Therapeutics. All other autors declare no conflict of interest
References
- 1.Millay DP, Sargent MA, Osinska H, Baines CP, Barton ER, Vuagniaux G, Sweeney HL, Robbins J, Molkentin JD. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat Med. 2008;14:442–447. doi: 10.1038/nm1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manzur AY, Muntoni F. Diagnosis and new treatments in muscular dystrophies. J Neurol Neurosurg Psychiatry. 2009;80:706–714. doi: 10.1136/jnnp.2008.158329. [DOI] [PubMed] [Google Scholar]
- 3.Mercuri E, Muntoni F. The ever-expanding spectrum of congenital muscular dystrophies. Ann Neurol. 2012;72:9–17. doi: 10.1002/ana.23548. [DOI] [PubMed] [Google Scholar]
- 4.Durbeej M, Campbell KP. Muscular dystrophies involving the dystrophin-glycoprotein complex: an overview of current mouse models. Curr Opin Genet Dev. 2002;12:349–361. doi: 10.1016/s0959-437x(02)00309-x. [DOI] [PubMed] [Google Scholar]
- 5.Fraysse B, Nagi SM, Boher B, Ragot H, Laine J, Salmon A, Fiszman MY, Toussaint M, Fromes Y. Ca2+ overload and mitochondrial permeability transition pore activation in living delta-sarcoglycan-deficient cardiomyocytes. Am J Physiol Cell Physiol. 2010;299:C706–713. doi: 10.1152/ajpcell.00545.2009. [DOI] [PubMed] [Google Scholar]
- 6.Pellegrini C, Zulian A, Gualandi F, Manzati E, Merlini L, Michelini ME, Benassi L, Marmiroli S, Ferlini A, Sabatelli P, et al. Melanocytes--a novel tool to study mitochondrial dysfunction in Duchenne muscular dystrophy. J Cell Physiol. 2013;228:1323–1331. doi: 10.1002/jcp.24290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buetler TM, Renard M, Offord EA, Schneider H, Ruegg UT. Green tea extract decreases muscle necrosis in mdx mice and protects against reactive oxygen species. Am J Clin Nutr. 2002;75:749–753. doi: 10.1093/ajcn/75.4.749. [DOI] [PubMed] [Google Scholar]
- 8.Fenichel GM, Brooke MH, Griggs RC, Mendell JR, Miller JP, Moxley RT, 3rd, Park JH, Provine MA, Florence J, Kaiser KK, et al. Clinical investigation in Duchenne muscular dystrophy: penicillamine and vitamin E. Muscle Nerve. 1988;11:1164–1168. doi: 10.1002/mus.880111110. [DOI] [PubMed] [Google Scholar]
- 9.Ramirez-Sanchez I, Taub PR, Ciaraldi TP, Nogueira L, Coe T, Perkins G, Hogan M, Maisel AS, Henry RR, Ceballos G, et al. (−)-Epicatechin rich cocoa mediated modulation of oxidative stress regulators in skeletal muscle of heart failure and type 2 diabetes patients. Int J Cardiol. 2013;168:3982–3990. doi: 10.1016/j.ijcard.2013.06.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Terrill JR, Radley-Crabb HG, Iwasaki T, Lemckert FA, Arthur PG, Grounds MD. Oxidative stress and pathology in muscular dystrophies: focus on protein thiol oxidation and dysferlinopathies. Febs J. 2013;280:4149–4164. doi: 10.1111/febs.12142. [DOI] [PubMed] [Google Scholar]
- 11.Droge W. Oxidative stress and aging. Adv Exp Med Biol. 2003;543:191–200. doi: 10.1007/978-1-4419-8997-0_14. [DOI] [PubMed] [Google Scholar]
- 12.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 13.Rando TA. Oxidative stress and the pathogenesis of muscular dystrophies. Am J Phys Med Rehabil. 2002;81:S175–186. doi: 10.1097/01.PHM.0000029774.56528.A6. [DOI] [PubMed] [Google Scholar]
- 14.Arthur PG, Grounds MD, Shavlakadze T. Oxidative stress as a therapeutic target during muscle wasting: considering the complex interactions. Curr Opin Clin Nutr Metab Care. 2008;11:408–416. doi: 10.1097/MCO.0b013e328302f3fe. [DOI] [PubMed] [Google Scholar]
- 15.Tidball JG, Wehling-Henricks M. The role of free radicals in the pathophysiology of muscular dystrophy. J Appl Physiol (1985) 2007;102:1677–1686. doi: 10.1152/japplphysiol.01145.2006. [DOI] [PubMed] [Google Scholar]
- 16.Coral-Vazquez R, Cohn RD, Moore SA, Hill JA, Weiss RM, Davisson RL, Straub V, Barresi R, Bansal D, Hrstka RF, et al. Disruption of the sarcoglycan-sarcospan complex in vascular smooth muscle: a novel mechanism for cardiomyopathy and muscular dystrophy. Cell. 1999;98:465–474. doi: 10.1016/s0092-8674(00)81975-3. [DOI] [PubMed] [Google Scholar]
- 17.Politano L, Nigro V, Passamano L, Petretta V, Comi LI, Papparella S, Nigro G, Rambaldi PF, Raia P, Pini A, et al. Evaluation of cardiac and respiratory involvement in sarcoglycanopathies. Neuromuscul Disord. 2001;11:178–185. doi: 10.1016/s0960-8966(00)00174-7. [DOI] [PubMed] [Google Scholar]
- 18.Fayssoil A. Cardiac diseases in sarcoglycanopathies. Int J Cardiol. 2010;144:67–68. doi: 10.1016/j.ijcard.2008.12.048. [DOI] [PubMed] [Google Scholar]
- 19.Blain AM, Straub VW. delta-Sarcoglycan-deficient muscular dystrophy: from discovery to therapeutic approaches. Skelet Muscle. 2011;1:13. doi: 10.1186/2044-5040-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allamand V, Campbell KP. Animal models for muscular dystrophy: valuable tools for the development of therapies. Hum Mol Genet. 2000;9:2459–2467. doi: 10.1093/hmg/9.16.2459. [DOI] [PubMed] [Google Scholar]
- 21.Hack AA, Lam MY, Cordier L, Shoturma DI, Ly CT, Hadhazy MA, Hadhazy MR, Sweeney HL, McNally EM. Differential requirement for individual sarcoglycans and dystrophin in the assembly and function of the dystrophin-glycoprotein complex. J Cell Sci. 2000;113 (Pt 14):2535–2544. doi: 10.1242/jcs.113.14.2535. [DOI] [PubMed] [Google Scholar]
- 22.Cohn RD, Durbeej M, Moore SA, Coral-Vazquez R, Prouty S, Campbell KP. Prevention of cardiomyopathy in mouse models lacking the smooth muscle sarcoglycan-sarcospan complex. J Clin Invest. 2001;107:R1–7. doi: 10.1172/JCI11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sandona D, Betto R. Sarcoglycanopathies: molecular pathogenesis and therapeutic prospects. Expert Rev Mol Med. 2009;11:e28. doi: 10.1017/S1462399409001203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J, Shi W, Zhang Y, Sokol R, Cai H, Lun M, Moore BF, Farber MJ, Stepanchick JS, Bonnemann CG, et al. Identification of functional domains in sarcoglycans essential for their interaction and plasma membrane targeting. Exp Cell Res. 2006;312:1610–1625. doi: 10.1016/j.yexcr.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 25.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J Biol Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 26.Aquilano K, Vigilanza P, Baldelli S, Pagliei B, Rotilio G, Ciriolo MR. Peroxisome proliferator-activated receptor gamma co-activator 1alpha (PGC-1alpha) and sirtuin 1 (SIRT1) reside in mitochondria: possible direct function in mitochondrial biogenesis. J Biol Chem. 2010;285:21590–21599. doi: 10.1074/jbc.M109.070169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hock MB, Kralli A. Transcriptional control of mitochondrial biogenesis and function. Annu Rev Physiol. 2009;71:177–203. doi: 10.1146/annurev.physiol.010908.163119. [DOI] [PubMed] [Google Scholar]
- 28.Handschin C, Kobayashi YM, Chin S, Seale P, Campbell KP, Spiegelman BM. PGC-1alpha regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev. 2007;21:770–783. doi: 10.1101/gad.1525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Austin S, St-Pierre J. PGC1alpha and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J Cell Sci. 2012;125:4963–4971. doi: 10.1242/jcs.113662. [DOI] [PubMed] [Google Scholar]
- 30.Hori YS, Kuno A, Hosoda R, Tanno M, Miura T, Shimamoto K, Horio Y. Resveratrol ameliorates muscular pathology in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. J Pharmacol Exp Ther. 2011;338:784–794. doi: 10.1124/jpet.111.183210. [DOI] [PubMed] [Google Scholar]
- 31.Nogueira L, Ramirez-Sanchez I, Perkins GA, Murphy A, Taub PR, Ceballos G, Villarreal FJ, Hogan MC, Malek MH. (−)-Epicatechin enhances fatigue resistance and oxidative capacity in mouse muscle. J Physiol. 2011;589:4615–4631. doi: 10.1113/jphysiol.2011.209924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taub PR, Ramirez-Sanchez I, Ciaraldi TP, Perkins G, Murphy AN, Naviaux R, Hogan M, Maisel AS, Henry RR, Ceballos G, et al. Alterations in skeletal muscle indicators of mitochondrial structure and biogenesis in patients with type 2 diabetes and heart failure: effects of epicatechin rich cocoa. Clin Transl Sci. 2012;5:43–47. doi: 10.1111/j.1752-8062.2011.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taub PR, Ramirez-Sanchez I, Ciaraldi TP, Gonzalez-Basurto S, Coral-Vazquez R, Perkins G, Hogan M, Maisel AS, Henry RR, Ceballos G, et al. Perturbations in skeletal muscle sarcomere structure in patients with heart failure and type 2 diabetes: restorative effects of (−)-epicatechin-rich cocoa. Clin Sci (Lond) 2013;125:383–389. doi: 10.1042/CS20130023. [DOI] [PubMed] [Google Scholar]
- 34.Thakar JH, Thede A, Strickland KP. On the ultrastructure of primary cultures of normal and dystrophic hamster tongue muscle. Muscle Nerve. 1980;3:340–344. doi: 10.1002/mus.880030411. [DOI] [PubMed] [Google Scholar]
- 35.Makita T, Sasaki K. A possible mode of formation of mitochondrial dense bodies in cardiac muscle of a dystrophic hamster. Cytobios. 1979;25:183–192. [PubMed] [Google Scholar]
- 36.Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM, et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J Clin Invest. 2010;120:3520–3529. doi: 10.1172/JCI42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao W, Zhao T, Chen Y, Ahokas RA, Sun Y. Oxidative stress mediates cardiac fibrosis by enhancing transforming growth factor-beta1 in hypertensive rats. Mol Cell Biochem. 2008;317:43–50. doi: 10.1007/s11010-008-9803-8. [DOI] [PubMed] [Google Scholar]
- 38.Zhou L, Lu H. Targeting fibrosis in Duchenne muscular dystrophy. J Neuropathol Exp Neurol. 2010;69:771–776. doi: 10.1097/NEN.0b013e3181e9a34b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bishop JE, Laurent GJ. Collagen turnover and its regulation in the normal and hypertrophying heart. Eur Heart J. 1995;16(Suppl C):38–44. doi: 10.1093/eurheartj/16.suppl_c.38. [DOI] [PubMed] [Google Scholar]
- 40.Sies H. Glutathione and its role in cellular functions. Free Radic Biol Med. 1999;27:916–921. doi: 10.1016/s0891-5849(99)00177-x. [DOI] [PubMed] [Google Scholar]
- 41.Tews DS. Role of Apoptosis in Myopathies. Basic Applied Myology. 2003;13:181–190. [Google Scholar]
- 42.Hand SC, Menze MA. Mitochondria in energy-limited states: mechanisms that blunt the signaling of cell death. J Exp Biol. 2008;211:1829–1840. doi: 10.1242/jeb.000299. [DOI] [PubMed] [Google Scholar]
- 43.Klein SM, Vykoukal J, Lechler P, Zeitler K, Gehmert S, Schreml S, Alt E, Bogdahn U, Prantl L. Noninvasive in vivo assessment of muscle impairment in the mdx mouse model--a comparison of two common wire hanging methods with two different results. J Neurosci Methods. 2012;203:292–297. doi: 10.1016/j.jneumeth.2011.10.001. [DOI] [PubMed] [Google Scholar]