

Abstract
Introduction
The armamentarium of antileishmanials is small. It is further being threatened by development of resistance and decreasing sensitivity to the available drugs. Development of newer drugs are sorely needed.
Areas covered
Literature search on investigational drugs for visceral leishmaniasis (VL) was done on PubMed. Those candidates with at least in vitro and in vivo activity against leishmania species causing VL were reviewed. Among the investigational drugs the nitroimidazole compound fexinidazole is the one of the few drugs which has reached phase II trials. Although the (S)-PA-824 is in phase II trials for the treatment of tuberculosis its R enantiomer has shown good antileishmanial activity. Development of sitamaquin, which has completed phase II studies has been stopped for VL due to its low efficacy. Many novel delivery system and oral formulations of Amphotericin B which are cheap and less toxic are in investigational stages, and will go a long way in improving the treatment of VL.
Expert opinion
Very few new drugs have reached the clinical stage in the treatment of this neglected tropical disease. Thus, there is an urgent need for support from public private partnerships to ensure that drug candidates are promptly taken forward into development.
Keywords: Visceral leishmaniasis, investigational drugs, therapy
1. Introduction
1.1 Organism
Leishmaniasis, a vector-borne disease, is caused by an obligate intracellular protozoan of the genus Leishmania. It broadly manifests as visceral leishmaniasis (VL; also known as kalaazar), cutaneous leishmaniasis (CL) and mucocutaneous leishmaniasis (MCL). VL is caused by the Leishmania donovani complex: L. donovani, the causative organism of VL in the Indian subcontinent and Africa; L. infantum (L. chagasi) which causes VL in the Mediterranean basin, Central and South America. CL is caused by various Leishmania species1.
In South Asia and the Horn of Africa, the predominant mode of transmission of VL is anthroponotic, and humans with kala-azar or post--kala-azar dermal leishmaniasis (PKDL) provide the major reservoir for transmission2, 3. In the Mediterranean, the Middle East and Brazil, VL is zoonotic, with the domestic dog as the most important reservoir host sustaining transmission3. Most CL have zoonotic transmission except those caused by L. tropica, which is predominantly anthroponotic. The only proven vectors of human disease are sand fly of species Phlebotomus in the Old World and Lutzomyia in the New World1.
1.2 Disease
VL is the most severe form of leishmaniasis, characterized by prolonged fever, splenomegaly, hepatomegaly, pancytopenia, progressive anemia and weight loss. If untreated, VL is uniformly fatal. After recovery, about 50% of patients in Sudan and 1 to 3% in India develop dermal leishmaniasis characterized by indurated nodules or depigmented macules called PKDL4, 5. The clinical features of CL tend to vary between and within regions, reflecting different species of parasite or the type of zoonotic cycle concerned, immunological status and also perhaps genetically determined responses of patients6. In CL, Old World species mostly cause benign and often self limiting cutaneous disease, while New World species cause a wide spectrum of manifestation, from benign to severe disease including mucosal involvement.
1.3 Epidemiology
Approximately 0.2 to 0.4million VL cases and 0.7 to 1.2million CL cases occur each year. More than 90% of global VL cases occur in just six countries: India, Bangladesh, Sudan, South Sudan, Brazil and Ethiopia. CL is more widely distributed, with about one-third of cases occurring in each of three regions, the Americas, the Mediterranean basin and western Asia from the Middle East to Central Asia. The ten countries with the highest estimated case counts, Afghanistan, Algeria, Colombia, Brazil, Iran, Syria, Ethiopia, North Sudan, Costa Rica and Peru, together account for 70 to 75% of globally estimated incidence of CL7.
HIV—VL co-infection is reported from more than 35 countries. Initially, most of these cases were from south-western Europe, but the number of cases is increasing in sub-Saharan Africa, especially Ethiopia, Brazil and South Asia8-10.
1.4 Present treatment guidelines
Visceral leishmaniasis
At present, single dose of 10 mg/kg of Liposomal Amphotericin B (L-AmB) or combination therapy consisting of either (single injection of 5 mg/kg L AmB and 7-day 50 mg oral miltefosine or single injection of 5 mg/kg L AmB and 10-day 11 mg/kg intramuscular paromomycin (PM); or 10 days each of miltefosine and PM) are the preferred treatment options in the Indian subcontinent11, 12. The combination of Sodium stibogluconate (SSG) with paromomycin (PM) for 17 days is the treatment of choice in East Africa and Yemen, whereas L-AmB up to a total dose of 18 -- 21 mg/kg is the treatment of choice in Mediterranean Basin, Middle East, Central Asia and South America1, 13.
Post-kala-azar dermal leishmaniasis
In India, Amphotericin B 60 -- 80 doses of 1mg/kg over 4 months or miltefosine for 12 weeks are the recommended regimens but the compliance is poor. In East Africa, PKDL is not routinely treated, as the majority of cases (85%) heal spontaneously within 1 year. Only patients with severe or disfiguring disease, those with lesions that have remained for > 6 months, those with concomitant anterior uveitis and young children with oral lesions that interfere with feeding are treated, with either SSG (20 mg/kg/day per day) for up to 2 months or a 20-day course of L-AmB at 2.5 mg/kg/day1.
HIV-VL co-infection
Lipid formulations infused at a dose of 3 -- 5 mg/kg daily or intermittently for 10 doses (days 1-5, 10, 17, 24, 31 and 38) up to a total dose of 40 mg/kg are recommended. Antiretroviral therapy should be initiated and secondary prophylaxis should be given till the CD4 counts are > 200/μL1.
The armamentarium of antileishmanials is small, consisting of pentavalent antimonials, amphotericin B (AmB) and its lipid formulations, miltefosine and paromomycin. For several decades, pentavalent antimonials (Sbv) have been the standard first- line medicines for visceral leishmaniasis. However, widespread resistance to the drug has developed in North Bihar and neighbouring areas of Nepal14, 15. The efficacy of the only oral drug miltefosine has also declined dramatically in the Indian subcontinent16. With the dwindling efficacy of drugs, development of newer antileishmanials is the need of the hour. Here we have reviewed the various investigational drugs for the treatment of VL. Literature search was done on Pubmed. As our search revealed a large number of compounds with antileishmanial activity, we reviewed those with at least in vitro and in vivo activity against leishmania species causing visceral disease. Preference to drugs which can be administered orally, already in use for other indications, cheap and stable at different temperatures have been given in this review .
2. Investigational drugs
2.1 Nitroimidazole compound
2.1.1 Fexinidazole
Fexinidazole (1-methyl-2-((p-(methylthio)- phenoxy)methyl)-5-nitroimidazole (Fig: 1), CAS registry number 59729- 37-2) had been in preclinical development in the 1970s and early 1980s as a broad-spectrum antimicrobial agent by Hoechst AG (now sanofi-aventis)17, 18. Although the in vivo activity of fexinidazole against African trypanosomes was observed in early 80's further development of the drug was halted19. It was rediscovered as a promising drug candidate for the treatment of Human African trypanosomiasis (HAT) by the Drugs for Neglected Diseases initiative (DNDi) after extensive compound mining efforts of more than 700 new and existing nitroheterocycles mostly nitroimidazoles20.
Figure 1.
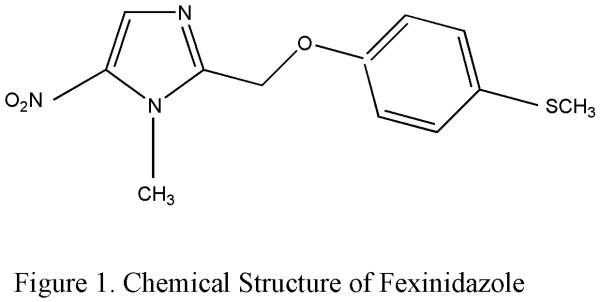
Chemical structure of fexinidazole
A bacteria-like nitroreductase has been implicated in both the mode of action and the mechanism of resistance to nitro-drugs in the related trypanosomatids, Trypanosoma brucei and T. cruzi21-23. Fexinidazole, is believed to act as prodrugs that require enzyme-mediated reduction by nitroreductases to generate cytotoxic species that cause DNA, lipid and protein damage 24. As the genomes of leishmania parasites contain a homologous nitroreductase gene a study to assess the leishmanicidal activity and preclinical profile of fexinidazole was done25.
2.1.1.2 In vitro and In vivo sensitivity of L. donovani to fexinidazole and its metabolites
The leishmanicidal activity of fexinidazole and its two predominant metabolites (fexinidazole sulfoxide and sulfone) was observed in vitro against promastigotes and axenic amastigotes of L. donovani (strain LdBOB).The EC50 values of fexinidazole against promastigotes and amastigotes was 5.6 ± 0.2 and 2.8 ± 0.1 μM while that of miltefosine was 6.1 ± 0.3 and 4.4± 0.2 μM respectively. The sulfoxide and sulfone metabolites of fexinidazole was as sensitive as the unmetabolized form of the drug. However, in intracellular L. donovani (LV9) amastigotes in peritoneal mouse macrophages fexinidazole itself had little effect while fexinidazole sulfoxide and sulfone had EC50s of 5.3 ± 0.1 and 5.3 ± 0.2 μM, respectively which was comparable to miltefosine (EC50 - 3.3 ± 0.3 μM).This suggests that the sulfoxide and sulfone and not the parent compound, were likely to be therapeutically important. In vivo sensitivity of L. donovani to the drug was excellent with five single daily doses of 200 mg/ kg in BALB/c mice suppressing infection by 98.4%.Lower doses of drug were also effective in treating the murine model of infection, with the ED50 and ED90 estimated at 12 and 57 mg/ kg, respectively25. In similar in vivo studies miltefosine had ED50, 4 mg/ kg and ED90, 27 mg/ kg and pentostam had ED50, 20 mg/ kg and ED90, 57 mg/ kg which shows that the results of fexinidazole were comparable to currently used antileishmanial26. To determine whether the drug was cytocidal or cytostatic axenic amastigotes were incubated with fexinidazole sulfone at a concentration equivalent to 10 times its EC50 value. Growth of drug-treated cultures ceased almost immediately with cell numbers declining after 10h with no intact or viable cells visible by 30h. Fexinidazole sulfoxide also had a similar cytotoxic profile suggesting that both fexinidazole sulfoxide and sulfone were leishmanicidal25.
2.1.1.3 In vivo pharmacokinetic properties
The oral absorption potential of fexinidazole was assessed in Caco-2 cell model for intestinal epithelial permeability which showed high absorption potential. The absolute bioavailability of oral fexinidazole was 41% in mice, 30% in rats and 10% in dogs. In all species tested, fexinidazole was rapidly and extensively metabolised to the sulfoxide and subsequently sulfone derivatives. Whole-body autoradiography in rats using [14C]-radiolabelled fexinidazole showed that the parent drug and/or its metabolites were broadly distributed to all organs and tissues, with peak concentrations in most tissues 2 h after oral dosing. After 48 h, most radioactivity was eliminated from the body and no tissue specific accumulation was noted20. In an in vivo sensitivity study of L.donovani to the drug in BALB/c mice the total blood concentrations of both the sulfoxide and sulfone comfortably exceeded their respective EC99 levels shortly after oral dosing, The sulfoxide accumulated rapidly in the blood then its concentration dropped below the EC99 after 8h.While blood concentrations of the sulfone were slower to accumulate, it remained above therapeutic levels for more than 24h.Thus their cumulative blood concentrations exceeded the EC99 for ~30h, underlining the potential of fexinidazole as a once daily25. It was metabolised extensively by multiple CYP450 isoforms but none of the enzymes tested metabolised either the sulfoxide or the sulfone to any significant degree20.
Overexpression of the leishmanial homologue of this nitroreductase in L. donovani increased sensitivity to fexinidazole sulfone by 19-fold indicating that nitroreductase played a crucial role in activation of fexinidazole and its metabolites in L. donovani 25.
2.1.1.4 Safety
Safety pharmacology and 4-weeks repeated-dose toxicokinetics in rat and dog showed that fexinidazole was well tolerated. The No Observed Adverse Event Levels in the 4-weeks repeated dose toxicity studies in rats and dogs was 200 mg/kg/day in both species, with no issues of concern identified for doses up to 800 mg/kg/day20. While fexinidazole and its metabolites was mutagenic in the Ames test due to bacterial specific metabolism however, mutagenicity was either attenuated or lost in Ames Salmonella strains that lacked one or more nitroreductase(s).It was not genotoxic to mammalian cells in an in vitro micronucleus test on human lymphocytes, an in vivo mouse bone marrow micronucleus test, and an ex vivo unscheduled DNA synthesis test in rats20, 27.
Redox measurements showed that fexinidazole and its sulfoxide and sulfone metabolites possessed highly negative single-electron redox potentials of - 511, - 493 and - 488 mV, respectively. This was consistent with the absence of mutagenic activity in mammalian cells as mammalian nitroreductases are expected to be incapable of nitro-reducing compounds with such negative single-electron redox potentials27.
2.1.1.5 Human Studies
Phase 1study of fexinidazole in healthy male volunteers has been completed28. Another phase 1study in which 3 or 4 tablets of fexinidazole 600mg per day were administered during 4 days (loading dose) then 2 tablets of fexinidazole 600mg for 6 days to 36 healthy male subsaharan volunteers was terminated due to poor tolerability at the higher dose.29 A phase 1 study to assess the bioavailability of fexinidazole tablets after single oral dose of 1200mg under different food intake conditions has been completed.30
In the ongoing phase II/III study the efficacy and safety of fexinidazole 1800 mg once a day for 4 days, followed by 1200 mg once a day for 6 days is being compared to Nifurtimox-Eflornithine combination therapy in patients with late-stage Human African Trypanosomiasis (HAT) due to T.b. Gambiense31. Curently, a phase II proof of concept trial to determine efficacy of fexinidazole at the daily dose of 1800 mg (3 tablets) once a day for 4 days continued by 1200mg (2 tablets)once a day for 6 days in VL patients in Sudan is recruiting patients.32
2.1.2 PA-824
Another nitroimidazole compound PA-824, (S)-2-nitro-6-[4-(trifluoromethoxy)benzyloxy]-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine), is a 4-nitroimidazo-oxazine. It exhibits potent bactericidal activity against both replicating and nonreplicating Mycobacterium tuberculosis, the causative agent of tuberculosis (TB), and is currently being tested in phase II clinical trials33-35.
2.1.2.1 In vitro sensitivity of L. donovani to (S)- and (R)-PA-824 ( Fig:2)
Figure 2.

Chemical structure of (S)- and (R)-PA-824
The potency of (S)-PA-824 was determined in vitro against L. donovani (LdBOB) promastigotes and against intracellular amastigotes in peritoneal mouse macrophages. (S)-PA- 824 showed antileishmanial activity against both developmental stages of the parasite, with EC50 of 0.9± 0.1 and 4.9± 0.3μM against promastigotes and amastigotes, respectively36. On testing the antileishmanial activity of R enantiomer of PA-284 which had minimal activity against M. tuberculosis (MIC, >100μM)37 it was observed to be 5-fold-more potent inhibitor of L. donovani growth in vitro than the S-enantiomer candidate, with EC50 of 0.16±0.03 and 0.9± 0.1 μM against promastigotes and intracellular amastigotes, respectively36.
The efficacy of both (R)- and (S)-PA-824 was assessed in a mouse model of VL. Seven days following infection with L. donovani LV9 ex vivo amastigotes, groups of BALB/c mice were dosed orally with (R)- or (S)-PA-824 (30 or 100 mg/ kg) twice daily for five days. Fourteen days following inoculation, parasite burdens in the livers of infected mice were determined. Mice dosed twice daily at 30 mg /kg with (R)- or (S)-PA-824 suppressed infection in the murine model by approximately 35% compared to untreated controls. Increased dose of 100 mg/ kg did not improve the efficacy of (S)-PA-824 in vivo, while treatment with (R)-PA-824 at 100 mg/ kg effectively cured the murine model of infection, suppressing infection by 99.9%. Dosing with (R)-PA-824 at this level proved to be superior to treatment with sodium stibogluconate (41.9% suppression) and miltefosine (68.7% suppression)36.
2.1.2.2 In vivo pharmacokinetic properties of (S)- and (R)-PA-824
Mice dosed with (S)- or (R)-PA-824 at 50 mg/ kg showed a maximum concentration in blood after 4 h of 11,600 or 10,500 ng/ ml, respectively Thereafter, blood concentrations decreased [elimination half-life (t1/2) ~5 h and 2 h for (S)- or (R)-PA-824, respectively], reaching undetectable levels at a time point between 8 and 24 h for (R)-PA-824. At 24 h, (S)-PA-824 still had a blood concentration of 900 ng/ ml.(R)-PA-824 had an EC50 of 0.93μM for L. donovani cultured in macrophages and EC90 value of 3.4μM(equivalent to 1,200 ng/ ml). (R)-PA-824 blood levels exceeded the EC90 1 h after dosing and remained above the EC90 for at least 7 h. Thus, it was predicted that 100 mg/ kg twice daily of (R)-PA-824 would provide adequate exposure. Although (S)- and (R)-PA-824 reached comparable blood levels, the lower potency of (S)-PA-824 (EC90= 22μM) resulted in a maximal free concentration below EC90 (37,000 ng ml−1) after oral dosing at 50 mg/ kg36.
(S)-PA-824 is believed to function as a prodrug which requires bioreductive activation prior to exhibiting antitubercular activity38, 39. In M. tuberculosis, this reduction is catalyzed by an unusual deazaflavin (F420)-dependent nitroreductase39, 40. However, overexpression of nitroreductase in promastigotes did not significantly alter the sensitivity to either (S)-PA-824 or (R)-PA-824 suggesting that this nitroreductase did not play a role in the activation of PA-824 in L. donovani. Drug combination studies in vitro indicated that fexinidazole and (R)-PA-824 are additive whereas (S)-PA-824 and (R)-PA-824 showed mild antagonistic behaviour36.
2.1.2.3 Human studies
Phase 1 study was done in 58 healthy male volunteers using single oral doses (50, 250, 500, 750, 1,000, 1,250, or 1,500 mg) or multiple doses of 200, 600 and 1,000 mg of (S)- PA-824 each day for 7 days. PA-824 reached maximal plasma levels in 4 to 5 h independently of the dose. Maximal blood levels averaged 3 μg/ml (1,500-mg dose) in the single-dose study and 3.8 μg/ml (600-mg dose) in the multiple-dose study. Steady state was achieved after 5 to 6 days of daily dosing, with an accumulation ratio of approximately 2. The elimination half-life averaged 16 to 20 h34. There was an increase in exposure to (S)- PA-824 when administered with high-calorie, high-fat meal compared to the fasted state41. It did not inhibit or induce CYP3A4 and is not likely to markedly affect the pharmacokinetics of CYP3A4 metabolized drugs42.
2.1.2.4 Safety
(S)-PA-824 was well tolerated at all doses in the phase 1 study, with no serious adverse events. Headache was the most common adverse event, followed by elevated serum creatinine levels, stomach discomfort (nausea, vomiting, flatulence, and/or diarrhea), and back pain. In the multiple-dose study, maximum elevation of serum creatinine was 1.3 (200-mg-dose group) and 1.4 mg/dl (600-mg-dose group). In the 1,000-mg-dose group by day 5 of dosing serum creatinine levels had risen in five of six subjects by an average of 0.28 mg/dl relative to the baseline level; the highest recorded absolute value was 1.6 mg/dl. Consequently, dosing was stopped on day 5, however, serum creatinine levels returned to clinically normal levels during the ensuing 7-day34. Another study was conducted to assess the effect of the drug in renal functions of healthy volunteers. In this study other than creatinine elevations, multiple doses of 800- and 1,000-mg (S)-PA-824 were not associated with clinically meaningful alterations in any of the other renal function measured by glomerular filtration rate (GFR), change in effective renal plasma flow (ERPF), filtration fraction (FF), blood urea nitrogen (BUN), and uric acid (UA)43. Currently (S)- PA-824 has completed a phase II trial in which the combination of (S)-PA-824-moxifloxacinpyrazinamide was found suitable for treating drug-sensitive and multidrug-resistant tuberculosis 44.
2.2 New Amphotericin B formulations
AmphotericinB( AmB)(Fig:3) is one of the most potent antileishmanial agent however its major drawbacks are need for prolonged injections, toxicity and cost. Recently, novel drug delivery systems including liposomes, niosomes, microspheres, nanoparticles, carbon nanotubes have been used to provide targeted delivery of the drug to macrophages 45. The lipid formulations have excellent efficacy and are less toxic but their cost are prohibitive. Thus efforts are ongoing to make cheaper and less toxic formulations of this drug and to make it orally bioavailable.
Figure 3.

Chemical structure of amphotericin B
2.2.1 Nano-amphotericin B
The rationale behind testing nanoparticles of amphotericin B deoxycholate in leishmaniasis is that nanoparticles are recognized as foreign bodies and phagocytosed by the macrophages which also harbours the parasite leading to target specific delivery of the drug 46. In vitro efficacy of nano-amphotericin B was done in intracellular amastigotes of L. donovani parasite (MZP 301strain). The ED50 of nano-amphotericin B was 3.69-fold lower than that of amphotericin B. Cytotoxicity study of this formulation and amphotericin B in the J774A cell line revealed that CC50 values were far higher (12.67 and 10.61 mg/L) than the ED50 doses (0.004 and 0.012 mg/L, respectively) for intracellular amastigotes. In vivo studies in hamster with nano-amphotericin B and conventional amphotericin B injected intraperitoneally at 5 mg/kg per day for 5 days revealed inhibition of amastigotes in the splenic tissue with nanoamphotericin B was significantly more than with conventional amphotericin B (92.18% versus 74.57%, P= 0.005). Similarly, the suppression of parasite replication in the spleen was also found to be significant (99.18% versus 97.17%, P= 0.05)47. Amphotericin B (AmB)-encapsulated chitosan nanocapsules (CNC-AmB) showed 86% parasite inhibition in Leishmania donovani-infected hamsters along with the upregulation of tumor necrosis factor alpha (TNF-α), interleukin-12 (IL-12), and inducible nitric oxide synthase and with the downregulation of transforming growth factor β (TGF-β), IL-10, and IL-448.
2.2.2 Carbon Nanotubes
In the family of nanomaterials, carbon nanotubes (CNTs) have emerged as a efficient tool for transporting drugs 49. Functionalized carbon nanotubes (f-CNTs) prepared by carboxylation and amidation with amphotericin B attached to it was tested for antileishmanial activity. In intracellular amastigotes the efficacy of f-CNT–AmB was significantly higher than that of AmB (IC50 0.00234+0.00075 mg/mL versus 0.03263+0.00123 mg/mL; P≤0.0001). In vivo toxicity assessment in BALB/c mice revealed no hepatic or renal toxicity. The percentage inhibition of amastigote replication in hamsters treated with f-CNT–AmB injected intraperitoneally (i.p.) at 5 mg/kg body weight per day for 5 days, was significantly more than that with AmB at the same dose (89.85%+2.93% versus 68.97%+1.84%; P=0.0004)50. As the chemical synthesis of f-CNT–AmB involves covalent coupling rather than biological molecules,its production could be cheaper than the existing liposomal AmB. Covalent modification by the organic functionalization of end groups and side walls of f-CNTs increases the solubility of functionalized carbon nanotubes in a range of solvents, including water51.With this idea the leishmanicidal efficacy of oral f-CNT-AmB was tested in hamsters. This experiment showed this formulation could be administered orally and resulted in 99% inhibition of parasite growth following a 5-day course at 15 mg/kg body weight52.
2.2.3 Oral lipid-based formulation of amphotericin B
An oral lipid-based formulation of amphotericin B (iCo-009) composed of monoglycerides, diglycerides and distearoylphosphatidylethanolamine polyethylene glycol 2000 significantly reduced liver parasitemia in a murine model of VL at 10 and 20 mg/kg twice daily for 5 days and was also effective in systemic fungal infections 53, 54 . However, instability of the product at tropical temperature limited the potential of iCo-009 and a new oral lipid-based formulation of AmB (iCo-010) composed of monoglycerides, diglycerides, polyethylene glycol glycerides and D-alpha-tocopheryl polyethylene glycol succinate was developed and tested against VL in a murine model55. The formulation showed good antileishmanial activity at both 10 mg/kg po bid for 5 days (<99% reduction in parasitic infection) and 20 mg/kg po qd for 5 days (95% inhibition when compared to control) and very good stability at tropical temperatures. Histopathological analysis detected no gastrointestinal, liver or kidney toxicity following multiple dose oral administration of iCo-010 in BALB/c mice 56. Multiple dosing of oral iCo-010 led to higher steady state concentrations in the tissues of rats , which could lead to enhanced eradication of tissue borne fungal and parasitic infections57.
2.2.4 Other Amphotericin B preparations
AmB has been formulated in lipo-polymerosome (L-Psome) by spontaneous self-assembly of synthesized glycol chitosan stearic acid copolymer as a low cost, stable and safe alternative. In vitro and in vivo toxicity studies revealed high plasma stability and less toxicity of AmBL-Psome compared to commercialized Fungizone (amphotericin deoxycholate) and Ambisome (L-AmB). The rank order of antileishmanial efficacy was AmB-L-Psome > Ambisome > Fungizone. AmB-L-Psome also upregulated Th-1 cytokines (TNF-α, IL-12 and IFN-γ) and inducible nitric oxide synthase, and downregulated of Th-2 cytokines (TGF-β, IL-10 and IL-4) in in vitro (macrophage amastigote system) and in vivo (hamsters) study58.
It was hypothesized that direct non-covalent association of AmB with poly(methacrylic acid) (PMAA) would replicate many of the properties of liposomal AmB (L-AmB) thus water-soluble AmB–PMAA complexes with AmB loadings ranging from 20 to 45% were reproducibly prepared. The AmB–PMAA complex had in vitro activity against Leishmania donovani amastigotes with no macrophage toxicity observed at an IC50 of 0.043 (±0.003) μM. It was well tolerated in vivo and a total dose of 6 mg/ kg achieved greater than 90% parasite inhibition in vivo after a single dose against L. donovani in HU3 infected BALB/c mice 59.
N-(2-hydroxypropyl)-methacrylamide-GFLG-amphotericin B copolymer conjugates decreased parasites by up to 94% in the liver of L. donovani infected BALB/c mice after intravenous administration of 1mg/kg amphotericin B equivalent on 3 alternate days and by up to 99.6% at a dose of 3mg/kg amphotericin B equivalent 60.
Besides these a number of novel drug delivery system have been used to carry AmB to the site of infection 61-66.
2.3 Quinoline derivatives
Quinoline scaffold-based derivatives, such as indolyl quinoline analogues , 4-substituted quinoline, 2-substituted quinoline, 8-amino quinoline derivatives (sitamaquine) , and 2 propylene quinoline derivatives , quinoline derivative (S-4), 2-(2-methylquinolin-4-ylamino)-N-phenylacetamide display antiparasitic activities 67-72.
2.3.1 2-substituted quinoline
2-substituted quinoline alkaloids were originally isolated from a Bolivian medicinal plant (Galipea longiflora ) and shown to have an effect in the treatment of experimental New World CL73. Activity of 2-substituted quinoline alkaloids was subsequently reported in the L.donovani – BALB/c mouse model with 2-n-propylquinoline showing significant activity after oral administration and chimanine D after subcutaneous administration74. Among more than one hundred 2-substituted quinolones, 2-n-propylquinoline was found to be the most stable and safe compound in various conditions75. As this compound was in oily state, a 2-npropylquinoline formulation as camphorsulfonic salt ( Fig:4) was prepared and characterised. In vivo studies in Balb/c mice model, oral treatment at 60 mmoles/kg/day for 10 days with this formulation was compared to 2-n-propylquinoline alone and to miltefosine. The salt formulation did not alter the activity of the 2-n-propylquinoline and reduced the parasite burden 76% compared to 89% for miltefosine 76.
Figure 4.
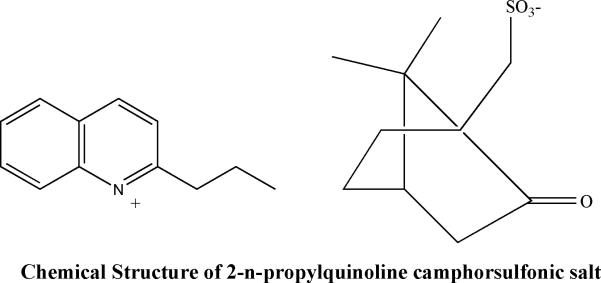
Chemical structure of 2-npropylquinoline camphorsulfonic salt
Recently 26g a new 2-substituted quinoline compound with morpholine at C4 position, chloro at C6 position and fluoro at C7 position (Fig:5) , was found to have in vitro and in vivo antileishmanial activity; it exhibited an IC50 value of 0.2 mM and >180 fold selectivity. The hydrochloride salt of 26g showed 84.26± 4.44 percent inhibition at 50 mg/kg for 5 days (twice daily, oral route) dose in L. donovani/hamster model77. Studies have shown the efficacy of 2-substituted quinolines against both HIV and Leishmania, suggesting that oral therapy with these compounds could be an effective therapy for Leishmania-HIV co infection 69, 78.
Figure 5.

Chemical structure of Compound 26G
2.3.2 4-Aminoquinaldine compounds
PP-9 and PP-10 are analogues of 4-aminoquinaldine with chlorine substituents( Fig:6). They showed antiparasitic activity against both SAG-sensitive and -resistant strains of Leishmania in in vitro and as well as in vivo studies. There was 95 to 98% reductions in the parasite burden of spleen and liver with only 4 doses, administered orally to BALB/c mice and they were more active than miltefosine at similar doses. Both the compounds did not cause any changes in haematological or cardiac functions in BALB/c mice and were not hepato or nephrotoxic. Investigation of their mode of action revealed that killing by PP-10 involved moderate inhibition of dihydrofolate reductase and elicitation of the apoptotic cascade79.
Figure 6.

Chemical structures of PP-9 and PP-10
2.3.3 8 aminoquinoline
Sitamaquine (WR- 6026) is a 8-aminoquinoline analogue (Fig:7) discovered by the Walter Reed Army Institute of Research (WRAIR, USA) being developed as a oral treatment for VL.
Figure 7.

Chemical structure of sitamaquine
2.3.3.1 In vivo efficacy
In 1950's 8-aminoquinoline had shown to have antileishmanial activity and oral availability against L. donovani in the hamster model80. Later, 8-[[6-(diethylamino) hexyl]amino]-6-methoxy-4-methylquinoline or WR6026, now called sitamaquine was shown to be 708 times more active than meglumine antimoniate (Glucantime®) against L. donovani in hamsters81.
Pharmacokinetics data in humans showed that sitamaquine has a short elimination half-life (about 26 hr) in contrast to miltefosine half-life (150-200 hr)82. The metabolism of sitamaquine was studied in a rat hepatic microsomal system83. Two metabolites were found: 8(-6-ethylaminohexylamino)-6-methoxy-lepidine and 8(-6-iethylaminohexylamino)-6-methoxy-4-hydroxy-methyl-quinoline84. The formation of both metabolites was catalyzed by different cytochrome P450 isozymes. Multiple-dose pharmacokinetic profile of sitamaquine and its metabolite desethyl-sitamaquine after administration of 2 mg/kg/day of sitamaquine with or without food for 21 days are as follows. Area under curve (AUC)(0−τ) = 6,627−8,903 ng.hr/mL, AUC(0−16) = 4,859−6,633 ng.hr/mL, maximum plasma concentration (Cmax) = 401−570 ng/mL, apparent terminal half-life (t1/2) = 18.3−22.8 hr, time to reach Cmax (tmax) = 3.5−6 hr; and desethyl-sitamaquine, AUC(0−τ) = 2,307−3,163 ng.hr/mL, Cmax = 109−154 ng/mL, t1/2 = 23.0−27.9 hr, tmax = 2−10 hr, with no significant food effect 85.
2.3.3.2 Human studies
The first phase 2 study was done in Kenya in 16 patients at a dose of 0.75-1mg/kg for 2-4 weeks with 50% cure rate for 28day treatment with 1mg/kg70. This was followed by a study in Brazil. Cure rates for Brazilian patients treated for 28 days were as follows: 1 mg/kg/day:0 of 4 (0%); 1.5 mg/kg/day: 1 of 6 (17%); 2.0 mg/kg/day: 4 of 6 (67%); 2.5 mg/kg/day: 1 of 5 (20%); and 3.25 mg/kg/day: 0 of 1 (0%)86. A randomized, open label, multicenter study was conducted in India to study the dose response and safety profile one of four sitamaquine doses (1.5, 1.75, 2.0, or 2.5 mg/ kg/ day) daily for 28 days. Final cure (primary efficacy outcome) was achieved in 92 of 106 (87%) patients overall and 25 of 31 (81%), 24 of 27 (89%), 23 of 23 (100%), and 20 of 25 (80%) patients at doses of 1.5, 1.75, 2.0, or 2.5 mg/ kg/day sitamaquine, respectively 87. In Africa, cure was achieved in 79 (83%) of 95 patients overall, and in 11 (92%) of 12, 49 (80%) of 61, 9 (82%) of 11, and 10 (91%) of 11 patients at sitamaquine doses of 1.75, 2.0, 2.5, or 3.0 mg/kg/day, respectively88. In another recent study with sitamaquin at 2mg/kg for 28 days in India the final clinical cure (day 180) was 85% (95% confidence interval = 70.8–94.4%)85.
2.3.3.3 Safety
Methhemoglobinemia and nephrotoxicity are the two main toxicity of the drug. Increased methemoglobin levels are a known class effect of 8-aminoquinolines; all patients are at risk regardless of glucose-6-phosphate dehydrogenase status. In the first phase2 study in India laboratory reports showed 40 (33%) of 120 patients had methemoglobin level increases ≥ 10% while six patients were symptomatic87. In another study only 5 patients had methemoglobin levels > 5%, the increase was reversible after the end of therapy and patients were asymptomatic85. Methemoglobinemia was not reported in the Kenyan study88.
The study from India reported nephritic syndrome in 3% and glomerulonephritis in 2% patients with doses ≥ 2.5 mg /mg/day87. Nephropathy was also reported in the study from Brazil in 3 patients86. In the recent study from India, proteinuria was the most significant renal safety finding observed in 7 of 41 patients, 6 with a protein:creatinine ratio increase of > 3.5 from baseline. These changes were reversible after drug discontinuation. Two patients had a transient decrease in creatinine clearance which reversed within two weeks of completion of therapy. Two patients had asymptomatic hematuria (normalized by day 90 and day 180)85.
2.3.3.4. Drug resistance
To study experimental resistance to the drug a L. donovani promastigote line resistant to 160 μM sitamaquine was selected by in vitro drug pressure. The resistant line was infective for murine peritoneal macrophages in vitro as its parent wild-type line but less infective for Balb/c mice, suggesting that a low transmission of resistant parasites could occur in the field. The sitamaquine IC50 of the resistant line was about five and three times higher than those of the wild-type line on promastigote and intramacrophage amastigote forms, respectively. No cross-resistance with other antileishmanial agents was observed. However, this resistance was stable when parasites were subcultured in drug-free medium for a long time or after in vivo passage89, 90.
2.4. Naphthoquinones
Plumbagin, a naphthoquinones derived from the stem barks of Pera benensis a medicinal plant was used by the Chimane Indians in the Bolivian Amazonia as treatment of cutaneous leishmaniasis91.
2.4.1. Buparvaquone and Derivatives
Buparvaquone a hydroxynaphtoquinone, was shown to be highly active in vitro against intracellular L. donovani amastigotes in macrophages, but less active in vivo in the BALB/c mouse 92. Buparvaquone-oxime demonstrated moderate in vitro activity against amastigotes of L. Donovani93. In vivo study of naphthoquinone buparvaquone and two phosphate prodrugs showed that hydrous gel and water-in-oil emulsion of buparvaquone significantly reduced cutaneous parasite burden. In the visceral model, both prodrugs were more effective at reducing liver parasite burden than the parent drug, buparvaquone,however the liver parasite burden was reduced only by 34%94. In vivo effectiveness of buparvaquone entrapped in phosphatidylserine liposomes (BPQ-PS-LP) was studied in L.infantum chagasi-infected hamsters. BPQ-PS-LP at 0.33 mg/kg/day for eight consecutive days reduced the number of amastigotes by 89.4% (P<0.05) in the spleen and by 67.2% (P>0.05) in the liver, compared to 84.3% (P<0.05) and 99.7% (P<0.05), respectively, following Glucantime® treatment at 50 mg/kg/day95. In vitro antileishmanial activity of 2-methyl-5-(3'-methyl-but-2'-enyloxy)- [1,4]naphthoquinone , a prenyloxy-naphthoquinone isolated and characterised from roots of the plant Plumbago zeylanica (family-Plumbaginaceae) was evaluated. The observed EC50 of this compound against promastigote and amastigote forms of L. donovani was significantly (p<0.001) lower than miltefosine96. In another in vitro study both naphthoquinones 2,3-dichloro-5,8-dihydroxy-1,4-naphthoquinone (TR 001) and 2,3-dibromo-1,4-naphthoquinone (TR 002) also showed antileismanial activity97. Treatment of infected BALB/c mice with D17 a di-epoxide derivative of diospyrin (bis- naphthoquinone ) at 2mg/kg/day reduced the hepatic parasite load only by 38% 98.
2.5 Doxorubicin
Doxorubicin, a well characterized anticancer drug, showed strong antileishmanial activity. It achieved up to 95% reduction of parasite in spleen of infected mouse model at a dose of 625 μg doxorubicin/kg body weight/day in 4 consecutive doses, which is far less than the toxic dose 99.
To provide improved delivery of the drug into macrophage, doxorubicin conjugated to mannose-human serum albumin (man-HSA) was tested in experimental VL to give excellent results100. However, the limitation was that the drug was being directed not only to the infected macrophages but also to the normal macrophages, thereby causing some toxicity. A 51-kDa Leishmania species-specific protein on the infected macrophage surface was characterized101 and active targeting of doxorubicin to infected macrophages was studied by incorporating it in immunoliposomes prepared by grafting F(ab)′2 of anti-51- kDa antibody onto the liposomal surface. In a 45-day mouse model of VL, complete elimination of splenic parasite burden was achieved by doxorubicin incorporated in immunoliposome (immunodoxosome) at a dose of 250 μg/kg/day for 4 consecutive days102. To improve the delivery of this toxic drug into the macrophages a number of drug delivery systems have been tried like gel-assisted layer-by-layer nanomatrix with high payload of doxorubicin, chitosan microparticles as a carrier of doxorubicin, phosphatidylserine specific ligand-anchored nanocapsules bearing doxorubicin and doxorubicin loaded nanocapsules 103-106.
3. Other investigational agents
Strategies to improve the antileishmanial activity and to decrease the side effects of currently available drugs by the use of different drug delivery system have also been undertaken45. Liposome encapsulated antimonials and pentamidine, niosomal formulation of SSG, pentamidine with polymethacrylate nanoparticles have shown superior activity as compared to the free drug107-110. Micellar systems composed of AMB molecules incorporated into miltefosine to provide both the drugs orally were evaluated. However, permeability studies showed that transport of both drugs across the intestinal barrier was reduced when they were present together111-113. With the large number of compounds being evaluated daily Nwaka et al114 proposed a set of criteria based on biological activity, physico-chemical characteristics and pharmacokinetics to facilitate the identification of effective antileishmanial drug candidates. According to the authors, a leishmanicidal hit should fit the following criteria: In in vitro studies, IC50 for the amastigote (in macrophages) ~1–2μg/mL, selectivity over parasites >20 (selectivity index, SI), in vivo activity in Leishmania infantum hamster model or L donovani mouse model with > 80% reduction in amastigote burden. Besides this the compound should have confirmed and elucidate structure, established synthetic route, good drug-likeness scores (DLS), no violation of Lipinski's Rule of Five and chemically exploitable 114. Many natural products have shown antileishmanial effect however, most of them are preliminary in vitro studies. To develop these natural products as antileishmanial drug candidates in vivo, toxicity and other studies to fulfil Nwaka's criteria needs to be done115-117. Some natural and synthetic products that have shown in vitro and in vivo antileishmanial activity have been enumerated in table 1.
Table 1.
Investigational agents in visceral leishmaniasis
| Reference | Active compound | In vitro efficacy | In vitro or Ex vivo toxicity | In vivo efficacy | Other characteristics |
|---|---|---|---|---|---|
| Mukherjee et al118 | Imipramine, N-(c-dimethylaminopropyl)-iminodibenzyl HCl, a tricyclic antidepressant | IC50 and EC50 same in SSG(S) or SSG (R) L. donovani strains | 60μM cleared 100% of intracellular parasites while 100% MO̵s remained viable upto 90 μM of imipramine | 5 mg/kg/day for 4 weeks, cleared 99.5% parasites in SSG-R or SSG-S hamster | FDA approved oral drug ↓ IL 10 and TGFβ level ↑ TNF α, IFN γ and iNOS in imipramine treated infected hamsters |
| Palit et al119 | Sertraline a selective serotonin reuptake inhibitor | IC50 against L. donovani promastigotes & amastigotes of 2.2 and 2.3 mg/L | 10 mg/kg body weight, two times per week for 4 weeks orally ↓ed parasite load in spleen by 72%& in liver by 70% in BALB/c mice | Induces ↓ in cytoplasmic ATP levels & oxygen consumption rate in promastigotes suggesting the involvement of an apoptosis mode of cell death. | |
| Suryawanshi et al120 | Triazole integrated phenyl heteroterpenoids compound 3a | IC50-6.4 ± 1.2 μM in amastigotes of L. donovani 100% Inhibition at 40 μM | CC 50 - 112.4 ± 10.9 μM SI - 18 | 79 ± 11% inhibition of parasite multiplication at 50 mg/ kg × 5 days I.P. in hamster model. | Inhibits growth of Leishmania amastigotes by preventing the 14 a-demethylation of lanosterol and effectively blocking synthesis of ergosterol. |
| Sharma et al121 | Quinazolinone-Triazine. 8a 8g |
IC50–against promastigote ± amastigotes - 7.05 ± 2.3 μM ± 3.95 ± 2.3 μM IC50 against promastigotes ± amastigotes - ±40 μM ± 4.39 ±1.4 μM |
CC50 > 400 SI >101.26 CC50 > 400 SI >91.1 |
73.15 ± 12.69% inhibition of parasite 80.93 ± 10.50%. against L.donovani in hamster model |
|
| Kyriazis et al122 | Olive tree extracts Oleuropein Hydroxytyrosol |
Oleuropein – IC50-110±32 in amastigotes Hydroxytyrosol- IC50-38.7±3 in amastigotes |
Oleuropein CC50 -356±23 SI 3.24 Hydroxytyrosol - CC50 180±16 SI 4.65 |
14 alternate day IP oleuropein reduced spleen parasitic burden >80% at 3 days & >95% in liver and spleen at 6weeks post treatment in BALB/c mice | |
| Bhattacharya et al123 | Bungarus caeruleus snake venom (BCV) | IC50 - L. donovani promastigote - 14.5 μg/ml Amastigote - 11.2 μg/ml. | 20μg/kg and 40μg/kg body ↓parasite count by 54.9% andj 74.2% in spleen and 41.4% and 60.4% in liver of infected BALB/c mice. | Increased production of TNF-α, IFN- γ, ROS, NO in infected mice. | |
| Khaliq et al124 | peganine hydrochloride. Peganum harmala seeds |
L. donovani promastigotes IC50 38 (±1.23) μg/ ml. In intracellular Amastigotes IC50 μg/ml |
Devoid of cytotoxic effect in J774A.1 macrophages | In-vivo activity, 79.6 (+/−8.07)% ± 87.5 ±9.10)% f inhibition of parasites in hamsters at a oral dose of 100mg/kg & 200mg/kg for 5 days respectively. | Causes apoptosis-like cell death due to L. donovani's topoisomerase I inhibition |
| Maes et al125 Germonprez et al126 | Maesabalide III triterpenoid saponins, from plant Maesa balansae | IC50 value 7 ng/ml in L. infantum amastigotes | CC50 > 32μg/mL in human fibroblast (MRC-5) cell line | Single dose of MB-III at 0.8 mg/kg causes 94.2% reduction of the liver amastigote burden in hamsters | Efficacy comparable to that of a single dose of liposomal amphotericin B at 5 mg/kg |
| Mittra et al127 | luteolin flavonoids | IC50 value 12.5μM in L.donovani promastigotes 12.5 μM reduced the intracellular parasite load by 70% in L. Donovani-infected BALB/c mice peritoneal macrophages | No effect on normal human T-cell blasts. | A dose of 3.5-mg/kg twice a week for 1month reduced the splenic parasite load by over 80% in hamsters | Inhibited DNA synthesis in promastigotes, and promoted topoisomerase-II mediated linearization of kDNA minicircles. |
| Zhai et al128 | oxygenated chalcones 35m4ac 24m4ac |
IC50 value −0.81 ± 0.3 IC50 value −0.65 ± 0.35μM in L.donovani amastigotes |
20mg/kg for 6 days ↓ in parasite load in liver 97% 88% |
Interferes with the function of the parasite mitochondria | |
| Chen et al129 | licochalcone A | 20 mg/kg per day i.p. for 6 days resulted in a > 96% ↓ in parasite load in the liver and the spleen 5to 150 mg/kg oral for 6days resulted in >65 and 85% ↓ in the parasite load in liver and spleen |
|||
| Medda et al130 | Amarogentin, a secoiridoid glycoside from Indian medicinal plant Swertia chirata | blood pathology, histological staining of tissues and liver enzyme levels showed no toxicity. | 2.5 mg/kg of amarogentin, in 0.5 mL niosomal suspension s.c. every 3 days for a total of 6 doses ↓ 9o% parasite load in spleen in hamsters | Inhibit the activity of DNA-topoisomerase I of L. donovani. |
SSG-S -antimony sensitive SSG-R- antimony resistant
MO̵ - peritoneal exudate cells of BALB/c mice
IC 50- half maximum inhibitory concentration
CC 50, half maximum cytotoxic con- centration
SI -Selectivity index -The selectivity index (SI) is defined as the ratio of CC 50 on Vero cells to IC 50 on L. donovani intramacrophagic amastigotes.
I.P.- intraperitoneal
ROS, reactive oxygen species; NO, nitric oxide;
4. Expert opinion
At present only a small number of antileishmanial drugs are available for clinical use, three injectables and the sole orally adminstrable miltefosine. There is a pressing need to develop more antileishmanial drugs as there are several handicaps with each of the available drug. Among all the investigational drugs fexinidazole, a nitroimidazole, has reached the stage of phase 2 clinical trial for VL. Fexinidazole has excellent in vitro and in vivo antileishmanial activity. The phase II trial in Sudanese VL patients which will be completed in 2015, will give us important efficacy and safety data. The oral advantage,comparable leishmanicidal activity to miltefosine, and safety reiterates the potential of fexinidazole as a much needed additional oral therapy for VL. As fexinidazole acts as a prodrug that must be activated by nitroreduction, this reliance on a single enzyme for activation makes it vulnerable to the emergence of drug resistance. Presently, monotherapy is not the preferred way of treating VL, and combination therapy with another antileishmanial agent would prevent the emergence of drug resistance. Work to identify an appropriate partner drug for fexinidazole is currently underway.
(R)-PA-824, another nitroimidazole compound with its in-vitro and in vivo leishmanicidal activity, oral bioavailability, safety, has the potential to be used as an antileishmanial drug. The fact that it shows additive effect in combination with fexinidazole is an added advantage. At present the (S) enantiomer has reached phase II trials for the treatment of tuberculosis therefore studies of the efficacy of the (R)-enantiomer in the treatment of VL are needed.
A number of novel delivery system of amphotericin B and oral formulations of Amphotericin B are in investigational stages for several years. Unfortunately, their development has been slow with none of them reaching the stage of a clinical trial.
Sitamaquine is the second orally active antileishmanial drug after miltefosine which has reached phase 2 trials. Unfortunately due to its low efficacy, development of this drug has been stopped for VL.
Naphthoquinone, buparvaquone except for the liposomal formulation is more active for CL than VL, but its other derivatives could be explored for activity against VL.
Doxorubicin an anticancer drug has shown strong antileishmanial activity at low doses. Active targeting of this drug to infected macrophages are being experimented, however at present its toxicity is its major drawback.
Leishmaniasis is a neglected tropical disease and pharmaceutical industries have very little interest in the development of drugs for such diseases due to lack of financial incentives. Thus, there is a huge gap between screening for antileishmanial hits and identification and optimization of leads, preclinical and clinical studies. As majority of these screening is being done by the academia they are not being further evaluated due to lack of resources. Recently, support from public-private partnerships (PPP) such as the Drugs for Neglected Diseases Initiative (DNDi) has helped in identifying new drug targets for neglected tropical diseases . Such PPPs could play an important role in ensuring that these drug candidates are promptly taken forward into development.
Article Highlight.
Nitroimidazole compound fexinidazole has reached phase II trials as oral therapy for VL.
Sitamaquin has completed phase II trials but has shown inadequate efficacy for monotherapy.
The R enantiomer of PA-824 has shown excellent antileishmanial activity and an additive effect with fexinidazole.
Novel delivery system and oral formulations of Amphotericin B are still in investigational stages.
Active targeting of doxorubicin to infected macrophages are being done to increase efficacy and decrease toxicity of this anticancer drug.
References
- 1**.Control of the Leishmaniasis. Report of a meeting of the WHO Expert Committee on the Control of Leishmaniases. 2010 Mar 22-26; [Available from: http://whqlibdoc.who.int/trs/WHO_TRS_949_eng.pdf. [Most recent comprehensive expert panel review of the treatment by the WHO expert.]
- 2.Magill AJ. Epidemiology of the leishmaniases. Dermatol Clin. 1995 Jul;13(3):505–23. [PubMed] [Google Scholar]
- 3.Pearson RDJS, deQueiroz Sousa A. Leishmaniasis. In: Guerrant RLWD, Weller PF, editors. Tropical infectious diseases: principles, pathogens and practice. Churchill Livingstone; Philadelphia: 1999. pp. 797–813. [Google Scholar]
- 4*.Zijlstra EE, Musa AM, Khalil EA, el-Hassan IM, el-Hassan AM. Post-kala-azar dermal leishmaniasis. Lancet Infect Dis. 2003;3(2):87–98. doi: 10.1016/s1473-3099(03)00517-6. [An exhaustive description of PKDL from different geographical region.] [DOI] [PubMed] [Google Scholar]
- 5.Thakur CP, Kumar K. Post kala-azar dermal leishmaniasis: a neglected aspect of kalaazar control programmes. Ann Trop Med Parasitol. 1992 Aug;86(4):355–9. doi: 10.1080/00034983.1992.11812678. [DOI] [PubMed] [Google Scholar]
- 6**.Reithinger R, Dujardin JC, Louzir H, Pirmez C, Alexander B, Brooker S. Cutaneous leishmaniasis. Lancet Infect Dis. 2007 Sep;7(9):581–96. doi: 10.1016/S1473-3099(07)70209-8. [Very good reference paper on CL; the authors have discussed various aspects including choice of drugs and regimens.] [DOI] [PubMed] [Google Scholar]
- 7.Alvar J, Velez ID, Bern C, Herrero M, Desjeux P, Cano J, et al. Leishmaniasis worldwide and global estimates of its incidence. PLoS One. 7(5):e35671. doi: 10.1371/journal.pone.0035671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alvar J, Canavate C, Gutierrez-Solar B, Jimenez M, Laguna F, Lopez-Velez R, et al. Leishmania and human immunodeficiency virus coinfection: the first 10 years. Clin Microbiol Rev. 1997 Apr;10(2):298–319. doi: 10.1128/cmr.10.2.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Desjeux P, Alvar J. Leishmania/HIV co-infections: epidemiology in Europe. Ann Trop Med Parasitol. 2003 Oct;97(Suppl 1):3–15. doi: 10.1179/000349803225002499. [DOI] [PubMed] [Google Scholar]
- 10**.Alvar J, Aparicio P, Aseffa A, Den Boer M, Canavate C, Dedet JP, et al. The relationship between leishmaniasis and AIDS: the second 10 years. Clin Microbiol Rev. 2008 Apr;21(2):334–59. doi: 10.1128/CMR.00061-07. [table of contents. An excellent description of various aspects of HIV--VL coinfection.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11**.Sundar S, Chakravarty J, Agarwal D, Rai M, Murray HW. Single-dose liposomal amphotericin B for visceral leishmaniasis in India. N Engl J Med. 2010 Feb 11;362(6):504–12. doi: 10.1056/NEJMoa0903627. [This is a landmark paper in which efficacy of the single dose of L-AmB in India was described.] [DOI] [PubMed] [Google Scholar]
- 12**.Sundar S, Sinha PK, Rai M, Verma DK, Nawin K, Alam S, et al. Comparison of short-course multidrug treatment with standard therapy for visceral leishmaniasis in India: an open-label, non-inferiority, randomised controlled trial. Lancet. 2011 Feb 5;377(9764):477–86. doi: 10.1016/S0140-6736(10)62050-8. [This was the Phase III trial for multidrug therapy for the treatment of VL.] [DOI] [PubMed] [Google Scholar]
- 13.Sundar S, Chakravarty J. Leishmaniasis: an update of current pharmacotherapy. Expert Opin Pharmacother. 2013 Jan;14(1):53–63. doi: 10.1517/14656566.2013.755515. [DOI] [PubMed] [Google Scholar]
- 14*.Sundar S, More DK, Singh MK, Singh VP, Sharma S, Makharia A, et al. Failure of pentavalent antimony in visceral leishmaniasis in India: report from the center of the Indian epidemic. Clin Infect Dis. 2000 Oct;31(4):1104–7. doi: 10.1086/318121. [First description of the large-scale failure of pentavalent antimony in India.] [DOI] [PubMed] [Google Scholar]
- 15.Rijal S, Chappuis F, Singh R, Bovier PA, Acharya P, Karki BM, et al. Treatment of visceral leishmaniasis in south-eastern Nepal: decreasing efficacy of sodium stibogluconate and need for a policy to limit further decline. Trans R Soc Trop Med Hyg. 2003 May-Jun;97(3):350–4. doi: 10.1016/s0035-9203(03)90167-2. [DOI] [PubMed] [Google Scholar]
- 16*.Sundar S, Singh A, Rai M, Prajapati VK, Singh AK, Ostyn B, et al. Efficacy of miltefosine in the treatment of visceral leishmaniasis in India after a decade of use. Clin Infect Dis. 2012 Aug;55(4):543–50. doi: 10.1093/cid/cis474. [First study to show the decreasing eficacy of miltefosine in India.] [DOI] [PubMed] [Google Scholar]
- 17.Winkelmann ERW. New chemotherapeutically active nitroimidazoles. Curr Chemother Infect Dis, Proc Int Congr Chemother. 1980;2:969–70. 11th. [Google Scholar]
- 18.Raether W, Seidenath H. The activity of fexinidazole (HOE 239) against experimental infections with Trypanosoma cruzi, trichomonads and Entamoeba histolytica. Ann Trop Med Parasitol. 1983 Feb;77(1):13–26. doi: 10.1080/00034983.1983.11811668. [DOI] [PubMed] [Google Scholar]
- 19.Jennings FW, Urquhart GM. The use of the 2 substituted 5-nitroimidazole, Fexinidazole (Hoe 239) in the treatment of chronic T. brucei infections in mice. Z Parasitenkd. 1983;69(5):577–81. doi: 10.1007/BF00926669. [DOI] [PubMed] [Google Scholar]
- 20.Torreele E, Bourdin Trunz B, Tweats D, Kaiser M, Brun R, Mazue G, et al. Fexinidazole--a new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness. PLoS Negl Trop Dis. 2010;4(12):e923. doi: 10.1371/journal.pntd.0000923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilkinson SR, Taylor MC, Horn D, Kelly JM, Cheeseman I. A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc Natl Acad Sci U S A. 2008 Apr;Jan;105(13):5022–7. doi: 10.1073/pnas.0711014105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sokolova AY, Wyllie S, Patterson S, Oza SL, Read KD, Fairlamb AH. Cross-resistance to nitro drugs and implications for treatment of human African trypanosomiasis. Antimicrob Agents Chemother. 2010 Jul;54(7):2893–900. doi: 10.1128/AAC.00332-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hall BS, Bot C, Wilkinson SR. Nifurtimox activation by trypanosomal type I nitroreductases generates cytotoxic nitrile metabolites. J Biol Chem. 2011 Apr 15;286(15):13088–95. doi: 10.1074/jbc.M111.230847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raether W, Hanel H. Nitroheterocyclic drugs with broad spectrum activity. Parasitol Res. 2003 Jun;90(Supp 1):S19–39. doi: 10.1007/s00436-002-0754-9. [DOI] [PubMed] [Google Scholar]
- 25**.Wyllie S, Patterson S, Stojanovski L, Simeons FR, Norval S, Kime R, et al. The anti-trypanosome drug fexinidazole shows potential for treating visceral leishmaniasis. Sci Transl Med. 2012 Feb 1;4(119):119re1. doi: 10.1126/scitranslmed.3003326. [First study to show the antileishmanial activity of fexinidazole.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Escobar P, Yardley V, Croft SL. Activities of hexadecylphosphocholine (miltefosine), AmBisome, and sodium stibogluconate (Pentostam) against Leishmania donovani in immunodeficient scid mice. Antimicrob Agents Chemother. 2001 Jun;45(6):1872–5. doi: 10.1128/AAC.45.6.1872-1875.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27*.Tweats D, Bourdin Trunz B, Torreele E. Genotoxicity profile of fexinidazole--a drug candidate in clinical development for human African trypanomiasis (sleeping sickness). Mutagenesis. 2012 Sep;27(5):523–32. doi: 10.1093/mutage/ges015. [Study showing the genotoxicity profile of fexinidazole.] [DOI] [PubMed] [Google Scholar]
- 28. [21st March, 2014];Human African Trypanosomiasis: First in Man Clinical Trial of a New Medicinal Product, the Fexinidazole. 2011 Mar 9th; Available from: http://clinicaltrials.gov/show/NCT00982904.
- 29.Multiple Dose Study to Evaluate Security, Tolerance and Pharmacokinetic of Fexinidazole (Drug Candidate for Human African Trypanosomiasis) Administered With a Loading Dose and With Food. 2012 Feb 10; [Available from: http://clinicaltrials.gov/show/NCT01483170.
- 30. [April 27, 2011];Fexinidazole (1200mg) Bioavailability Under Different Food Intake Conditions. Available from: http://clinicaltrials.gov/ct2/show/NCT01340157.
- 31. [October 22, 2012];Pivotal Study of Fexinidazole for Human African Trypanosomiasis in Stage 2. Available from: http://clinicaltrials.gov/show/NCT01685827.
- 32. [November 12, 2013];Trial to Determine Efficacy of Fexinidazole in Visceral Leihmaniasis Patients in Sudan. Available from: http://clinicaltrials.gov/show/NCT01980199.
- 33.Barry CE, 3rd, Boshoff HI, Dowd CS. Prospects for clinical introduction of nitroimidazole antibiotics for the treatment of tuberculosis. Curr Pharm Des. 2004;10(26):3239–62. doi: 10.2174/1381612043383214. [DOI] [PubMed] [Google Scholar]
- 34.Ginsberg AM, Laurenzi MW, Rouse DJ, Whitney KD, Spigelman MK. Safety, tolerability, and pharmacokinetics of PA-824 in healthy subjects. Antimicrob Agents Chemother. 2009 Sep;53(9):3720–5. doi: 10.1128/AAC.00106-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nuermberger E, Tyagi S, Tasneen R, Williams KN, Almeida D, Rosenthal I, et al. Powerful bactericidal and sterilizing activity of a regimen containing PA-824, moxifloxacin, and pyrazinamide in a murine model of tuberculosis. Antimicrob Agents Chemother. 2008 Apr;52(4):1522–4. doi: 10.1128/AAC.00074-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36**.Patterson S, Wyllie S, Stojanovski L, Perry MR, Simeons FR, Norval S, et al. The R enantiomer of the antitubercular drug PA-824 as a potential oral treatment for visceral Leishmaniasis. Antimicrob Agents Chemother. 2013 Oct;57(10):4699–706. doi: 10.1128/AAC.00722-13. [First study to show the activity of R enantiomer of PA-824 against VL.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gurumurthy M, Mukherjee T, Dowd CS, Singh R, Niyomrattanakit P, Tay JA, et al. Substrate specificity of the deazaflavin-dependent nitroreductase from Mycobacterium tuberculosis responsible for the bioreductive activation of bicyclic nitroimidazoles. FEBS J. 2012 Jan;279(1):113–25. doi: 10.1111/j.1742-4658.2011.08404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh R, Manjunatha U, Boshoff HI, Ha YH, Niyomrattanakit P, Ledwidge R, et al. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science. 2008 Nov 28;322(5906):1392–5. doi: 10.1126/science.1164571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manjunatha UH, Boshoff H, Dowd CS, Zhang L, Albert TJ, Norton JE, et al. Identification of a nitroimidazo-oxazine-specific protein involved in PA-824 resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2006 Jan 10;103(2):431–6. doi: 10.1073/pnas.0508392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stover CK, Warrener P, VanDevanter DR, Sherman DR, Arain TM, Langhorne MH, et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature. 2000 Jun 22;405(6789):962–6. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- 41.Winter H, Ginsberg A, Egizi E, Erondu N, Whitney K, Pauli E, et al. Effect of a high-calorie, high-fat meal on the bioavailability and pharmacokinetics of PA-824 in healthy adult subjects. Antimicrob Agents Chemother. 2013 Nov;57(11):5516–20. doi: 10.1128/AAC.00798-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Winter H, Egizi E, Erondu N, Ginsberg A, Rouse DJ, Severynse-Stevens D, et al. Evaluation of pharmacokinetic interaction between PA-824 and midazolam in healthy adult subjects. Antimicrob Agents Chemother. 2013 Aug;57(8):3699–703. doi: 10.1128/AAC.02632-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ginsberg AM, Laurenzi MW, Rouse DJ, Whitney KD, Spigelman MK. Assessment of the effects of the nitroimidazo-oxazine PA-824 on renal function in healthy subjects. Antimicrob Agents Chemother. 2009 Sep;53(9):3726–33. doi: 10.1128/AAC.00112-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Diacon AH, Dawson R, von Groote-Bidlingmaier F, Symons G, Venter A, Donald PR, et al. 14-day bactericidal activity of PA-824, bedaquiline, pyrazinamide, and moxifloxacin combinations: a randomised trial. Lancet. 2012 Sep 15;380(9846):986–93. doi: 10.1016/S0140-6736(12)61080-0. [DOI] [PubMed] [Google Scholar]
- 45**.Pham TT, Loiseau PM, Barratt G. Strategies for the design of orally bioavailable antileishmanial treatments. Int J Pharm. 2013 Sep 15;454(1):539–52. doi: 10.1016/j.ijpharm.2013.07.035. [An excellent review of different strategies for designing oral therapy for leishmaniasis.] [DOI] [PubMed] [Google Scholar]
- 46.Muller RH, Jacobs C, Kayser O. Nanosuspensions as particulate drug formulations in therapy. Rationale for development and what we can expect for the future. Adv Drug Deliv Rev. 2001 Mar 23;47(1):3–19. doi: 10.1016/s0169-409x(00)00118-6. [DOI] [PubMed] [Google Scholar]
- 47.Manandhar KD, Yadav TP, Prajapati VK, Kumar S, Rai M, Dube A, et al. Antileishmanial activity of nano-amphotericin B deoxycholate. J Antimicrob Chemother. 2008 Aug;62(2):376–80. doi: 10.1093/jac/dkn189. [DOI] [PubMed] [Google Scholar]
- 48.Asthana S, Jaiswal AK, Gupta PK, Pawar VK, Dube A, Chourasia MK. Immunoadjuvant chemotherapy of visceral leishmaniasis in hamsters using amphotericin B-encapsulated nanoemulsion template-based chitosan nanocapsules. Antimicrob Agents Chemother. 2013 Apr;57(4):1714–22. doi: 10.1128/AAC.01984-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Foldvari M, Bagonluri M. Carbon nanotubes as functional excipients for nanomedicines: II. Drug delivery and biocompatibility issues. Nanomedicine. 2008 Sep;4(3):183–200. doi: 10.1016/j.nano.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 50.Prajapati VK, Awasthi K, Gautam S, Yadav TP, Rai M, Srivastava ON, et al. Targeted killing of Leishmania donovani in vivo and in vitro with amphotericin B attached to functionalized carbon nanotubes. J Antimicrob Chemother. 2011 Apr;66(4):874–9. doi: 10.1093/jac/dkr002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Georgakilas V, Kordatos K, Prato M, Guldi DM, Holzinger M, Hirsch A. Organic functionalization of carbon nanotubes. J Am Chem Soc. 2002 Feb 6;124(5):760–1. doi: 10.1021/ja016954m. [DOI] [PubMed] [Google Scholar]
- 52.Prajapati VK AK, Yadav TP, Rai M, Srivastava ON, Sundar S. An oral formulation of amphotericin B attached to functionalized carbon nanotubes is an effective treatment for experimental visceral leishmaniasis. J Infect Dis. 2012;205(2):333–6. doi: 10.1093/infdis/jir735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wasan EK, Bartlett K, Gershkovich P, Sivak O, Banno B, Wong Z, et al. Development and characterization of oral lipid-based amphotericin B formulations with enhanced drug solubility, stability and antifungal activity in rats infected with Aspergillus fumigatus or Candida albicans. Int J Pharm. 2009 May 8;372(1-2):76–84. doi: 10.1016/j.ijpharm.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 54.Wasan KM, Wasan EK, Gershkovich P, Zhu X, Tidwell RR, Werbovetz KA, et al. Highly effective oral amphotericin B formulation against murine visceral leishmaniasis. J Infect Dis. 2009 Aug 1;200(3):357–60. doi: 10.1086/600105. [DOI] [PubMed] [Google Scholar]
- 55.Wasan EK, Gershkovich P, Zhao J, Zhu X, Werbovetz K, Tidwell RR, et al. A novel tropically stable oral amphotericin B formulation (iCo-010) exhibits efficacy against visceral Leishmaniasis in a murine model. PLoS Negl Trop Dis. 2010;4(12):e913. doi: 10.1371/journal.pntd.0000913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sivak O, Gershkovich P, Lin M, Wasan EK, Zhao J, Owen D, et al. Tropically stable novel oral lipid formulation of amphotericin B (iCo-010): biodistribution and toxicity in a mouse model. Lipids Health Dis. 2011;10:135. doi: 10.1186/1476-511X-10-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ibrahim F, Gershkovich P, Sivak O, Wasan EK, Wasan KM. Pharmacokinetics and tissue distribution of amphotericin B following oral administration of three lipid-based formulations to rats. Drug Dev Ind Pharm. 2013 Sep;39(9):1277–83. doi: 10.3109/03639045.2012.719908. [DOI] [PubMed] [Google Scholar]
- 58.Gupta PK, Jaiswal AK, Kumar V, Verma A, Dwivedi P, Dube A, et al. Covalent Functionalized Self-Assembled Lipo-Polymerosome Bearing Amphotericin B for Better Management of Leishmaniasis and Its Toxicity Evaluation. Mol Pharm. 2014 Feb 19; doi: 10.1021/mp400603t. [DOI] [PubMed] [Google Scholar]
- 59.Les KAM-AA, Balan S, Choi J, Martin D, Yardley V, Powell K, Godwin A, Brocchini S. Poly(methacrylic acid) complexation of amphotericin B to treat neglected diseases. Polym Chem. 2014;5:1037–48. [Google Scholar]
- 60.Nicoletti S, Seifert K, Gilbert IH. N-(2-hydroxypropyl)methacrylamide-amphotericin B (HPMA-AmB) copolymer conjugates as antileishmanial agents. Int J Antimicrob Agents. 2009 May;33(5):441–8. doi: 10.1016/j.ijantimicag.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Singodia D, Verma A, Verma RK, Mishra PR. Investigations into an alternate approach to target mannose receptors on macrophages using 4-sulfated N-acetyl galactosamine more efficiently in comparison with mannose-decorated liposomes: an application in drug delivery. Nanomedicine. 2012 May;8(4):468–77. doi: 10.1016/j.nano.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 62.Alsaadi M, Italia JL, Mullen AB, Ravi Kumar MN, Candlish AA, Williams RA, et al. The efficacy of aerosol treatment with non-ionic surfactant vesicles containing amphotericin B in rodent models of leishmaniasis and pulmonary aspergillosis infection. J Control Release. 2012 Jun 28;160(3):685–91. doi: 10.1016/j.jconrel.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 63.Sanchez-Brunete JA, Dea MA, Rama S, Bolas F, Alunda JM, Raposo R, et al. Treatment of experimental visceral leishmaniasis with amphotericin B in stable albumin microspheres. Antimicrob Agents Chemother. 2004 Sep;48(9):3246–52. doi: 10.1128/AAC.48.9.3246-3252.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nahar M, Jain NK. Preparation, characterization and evaluation of targeting potential of amphotericin B-loaded engineered PLGA nanoparticles. Pharm Res. 2009 Dec;26(12):2588–98. doi: 10.1007/s11095-009-9973-4. [DOI] [PubMed] [Google Scholar]
- 65.Nahar M, Dubey V, Mishra D, Mishra PK, Dube A, Jain NK. In vitro evaluation of surface functionalized gelatin nanoparticles for macrophage targeting in the therapy of visceral leishmaniasis. J Drug Target. 2010 Feb;18(2):93–105. doi: 10.3109/10611860903115290. [DOI] [PubMed] [Google Scholar]
- 66.Pruthi J, Mehra NK, Jain NK. Macrophages targeting of amphotericin B through mannosylated multiwalled carbon nanotubes. J Drug Target. 2012 Aug;20(7):593–604. doi: 10.3109/1061186X.2012.697168. [DOI] [PubMed] [Google Scholar]
- 67.Chakrabarti G, Basu A, Manna PP, Mahato SB, Mandal NB, Bandyopadhyay S. Indolylquinoline derivatives are cytotoxic to Leishmania donovani promastigotes and amastigotes in vitro and are effective in treating murine visceral leishmaniasis. J Antimicrob Chemother. 1999 Mar;43(3):359–66. doi: 10.1093/jac/43.3.359. [DOI] [PubMed] [Google Scholar]
- 68.Galvao LO, Moreira Junior S, Medeiros Junior P, Lemos GJ, Cunha NF, Antonino RM, et al. Therapeutic trial in experimental tegumentary leishmaniasis caused by Leishmania (Leishmania) amazonensis. A comparative study between mefloquine and aminosidine. Rev Soc Bras Med Trop. 2000 Jul-Aug;33(4):377–82. doi: 10.1590/s0037-86822000000400008. [DOI] [PubMed] [Google Scholar]
- 69.Nakayama H, Loiseau PM, Bories C, Torres de Ortiz S, Schinini A, Serna E, et al. Efficacy of orally administered 2-substituted quinolines in experimental murine cutaneous and visceral leishmaniases. Antimicrob Agents Chemother. 2005 Dec;49(12):4950–6. doi: 10.1128/AAC.49.12.4950-4956.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sherwood JA, Gachihi GS, Muigai RK, Skillman DR, Mugo M, Rashid JR, et al. Phase 2 efficacy trial of an oral 8-aminoquinoline (WR6026) for treatment of visceral leishmaniasis. Clin Infect Dis. 1994 Dec;19(6):1034–9. doi: 10.1093/clinids/19.6.1034. [DOI] [PubMed] [Google Scholar]
- 71.Mishra BB, Singh RK, Srivastava A, Tripathi VJ, Tiwari VK. Fighting against Leishmaniasis: search of alkaloids as future true potential anti-Leishmanial agents. Mini Rev Med Chem. 2009 Jan;9(1):107–23. doi: 10.2174/138955709787001758. [DOI] [PubMed] [Google Scholar]
- 72.Sahu NP, Pal C, Mandal NB, Banerjee S, Raha M, Kundu AP, et al. Synthesis of a novel quinoline derivative, 2-(2-methylquinolin-4-ylamino)-N-phenylacetamide--a potential antileishmanial agent. Bioorg Med Chem. 2002 Jun;10(6):1687–93. doi: 10.1016/s0968-0896(02)00046-9. [DOI] [PubMed] [Google Scholar]
- 73.Fournet A, Barrios AA, Munoz V, Hocquemiller R, Cave A, Bruneton J. 2-substituted quinoline alkaloids as potential antileishmanial drugs. Antimicrob Agents Chemother. 1993 Apr;37(4):859–63. doi: 10.1128/aac.37.4.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fournet A, Gantier JC, Gautheret A, Leysalles L, Munos MH, Mayrargue J, et al. The activity of 2-substituted quinoline alkaloids in BALB/c mice infected with Leishmania donovani. J Antimicrob Chemother. 1994 Mar;33(3):537–44. doi: 10.1093/jac/33.3.537. [DOI] [PubMed] [Google Scholar]
- 75.Vieira NC, Herrenknecht C, Vacus J, Fournet A, Bories C, Figadere B, et al. Selection of the most promising 2-substituted quinoline as antileishmanial candidate for clinical trials. Biomed Pharmacother. 2008 Dec;62(10):684–9. doi: 10.1016/j.biopha.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 76.Campos Vieira N, Vacus J, Fournet A, Baudouin R, Bories C, Seon-Meniel B, et al. Antileishmanial activity of a formulation of 2-n-propylquinoline by oral route in mice model. Parasite. 2011 Nov;18(4):333–6. doi: 10.1051/parasite/2011184333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gopinath VS, Pinjari J, Dere RT, Verma A, Vishwakarma P, Shivahare R, et al. Design, synthesis and biological evaluation of 2-substituted quinolines as potential antileishmanial agents. Eur J Med Chem. 2013 Nov;69:527–36. doi: 10.1016/j.ejmech.2013.08.028. [DOI] [PubMed] [Google Scholar]
- 78.Fakhfakh MA, Fournet A, Prina E, Mouscadet JF, Franck X, Hocquemiller R, et al. Synthesis and biological evaluation of substituted quinolines: potential treatment of protozoal and retroviral co-infections. Bioorg Med Chem. 2003 Nov 17;11(23):5013–23. doi: 10.1016/j.bmc.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 79.Palit P, Hazra A, Maity A, Vijayan RS, Manoharan P, Banerjee S, et al. Discovery of safe and orally effective 4-aminoquinaldine analogues as apoptotic inducers with activity against experimental visceral leishmaniasis. Antimicrob Agents Chemother. 2012 Jan;56(1):432–45. doi: 10.1128/AAC.00700-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Beveridge E, Goodwin LG, Walls LP. A new series of leishmanicides. Nature. 1958 Aug 2;182(4631):316–7. doi: 10.1038/182316b0. [DOI] [PubMed] [Google Scholar]
- 81.Kinnamon KE, Steck EA, Loizeaux PS, Hanson WL, Chapman WL, Jr., Waits VB. The antileishmanial activity of lepidines. Am J Trop Med Hyg. 1978 Jul;27(4):751–7. doi: 10.4269/ajtmh.1978.27.751. [DOI] [PubMed] [Google Scholar]
- 82.Theoharides AD KM, Ashmore RW, Shipley LA. Identification and quantification of human urinary metabolites of a candidate 8-aminoquinoline antileishmanial drug WR-6026. Proceedings of American Society of Experimental Biology. 1987;46:865. [Google Scholar]
- 83.Theoharides AD, Chung H, Velazquez H. Metabolism of a potential 8-aminoquinoline antileishmanial drug in rat liver microsomes. Biochem Pharmacol. 1985 Jan 15;34(2):181–8. doi: 10.1016/0006-2952(85)90122-4. [DOI] [PubMed] [Google Scholar]
- 84.Yeates C. Sitamaquine (GlaxoSmithKline/Walter Reed Army Institute). Curr Opin Investig Drugs. 2002 Oct;3(10):1446–52. [PubMed] [Google Scholar]
- 85*.Sundar S, Sinha PK, Dixon SA, Buckley R, Miller AK, Mohamed K, et al. Pharmacokinetics of oral sitamaquine taken with or without food and safety and efficacy for treatment of visceral leishmaniais: a randomized study in Bihar, India. Am J Trop Med Hyg. 2011 Jun;84(6):892–900. doi: 10.4269/ajtmh.2011.10-0409. [Most recent study of sitamaquin in India.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dietze R, Carvalho SF, Valli LC, Berman J, Brewer T, Milhous W, et al. Phase 2 trial of WR6026, an orally administered 8-aminoquinoline, in the treatment of visceral leishmaniasis caused by Leishmania chagasi. Am J Trop Med Hyg. 2001 Dec;65(6):685–9. doi: 10.4269/ajtmh.2001.65.685. [DOI] [PubMed] [Google Scholar]
- 87.Jha TK, Sundar S, Thakur CP, Felton JM, Sabin AJ, Horton J. A phase II dose-ranging study of sitamaquine for the treatment of visceral leishmaniasis in India. Am J Trop Med Hyg. 2005 Dec;73(6):1005–11. [PubMed] [Google Scholar]
- 88.Wasunna MK, Rashid JR, Mbui J, Kirigi G, Kinoti D, Lodenyo H, et al. A phase II dose-increasing study of sitamaquine for the treatment of visceral leishmaniasis in Kenya. Am J Trop Med Hyg. 2005 Nov;73(5):871–6. [PubMed] [Google Scholar]
- 89.Bories C, Cojean S, Huteau F, Loiseau PM. Selection and phenotype characterisation of sitamaquine-resistant promastigotes of Leishmania donovani. Biomed Pharmacother. 2008 Mar;62(3):164–7. doi: 10.1016/j.biopha.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 90.Loiseau PM, Cojean S, Schrevel J. Sitamaquine as a putative antileishmanial drug candidate: from the mechanism of action to the risk of drug resistance. Parasite. 2011 May;18(2):115–9. doi: 10.1051/parasite/2011182115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fournet A, Angelo A, Munoz V, Roblot F, Hocquemiller R, Cave A. Biological and chemical studies of Pera benensis, a Bolivian plant used in folk medicine as a treatment of cutaneous leishmaniasis. J Ethnopharmacol. 1992 Sep;37(2):159–64. doi: 10.1016/0378-8741(92)90074-2. [DOI] [PubMed] [Google Scholar]
- 92.Croft SL, Hogg J, Gutteridge WE, Hudson AT, Randall AW. The activity of hydroxynaphthoquinones against Leishmania donovani. J Antimicrob Chemother. 1992 Dec;30(6):827–32. doi: 10.1093/jac/30.6.827. [DOI] [PubMed] [Google Scholar]
- 93.Mantyla A, Rautio J, Nevalainen T, Vepsalainen J, Juvonen R, Kendrick H, et al. Synthesis and antileishmanial activity of novel buparvaquone oxime derivatives. Bioorg Med Chem. 2004 Jul 1;12(13):3497–502. doi: 10.1016/j.bmc.2004.04.032. [DOI] [PubMed] [Google Scholar]
- 94.Garnier T, Mantyla A, Jarvinen T, Lawrence J, Brown M, Croft S. In vivo studies on the antileishmanial activity of buparvaquone and its prodrugs. J Antimicrob Chemother. 2007 Oct;60(4):802–10. doi: 10.1093/jac/dkm303. [DOI] [PubMed] [Google Scholar]
- 95.Reimao JQ, Colombo FA, Pereira-Chioccola VL, Tempone AG. Effectiveness of liposomal buparvaquone in an experimental hamster model of Leishmania (L.) infantum chagasi. Exp Parasitol. 2012 Mar;130(3):195–9. doi: 10.1016/j.exppara.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 96.Mishra BB, Gour JK, Kishore N, Singh RK, Tripathi V, Tiwari VK. An antileishmanial prenyloxy-naphthoquinone from roots of Plumbago zeylanica. Nat Prod Res. 2012 Mar;27(4-5):480–5. doi: 10.1080/14786419.2012.696254. [DOI] [PubMed] [Google Scholar]
- 97.Lezama-Davila CM, Isaac-Marquez AP, Kapadia G, Owens K, Oghumu S, Beverley S, et al. Leishmanicidal activity of two naphthoquinones against Leishmania donovani. Biol Pharm Bull. 2012;35(10):1761–4. doi: 10.1248/bpb.b12-00419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hazra S, Ghosh S, Das Sarma M, Sharma S, Das M, Saudagar P, et al. Evaluation of a diospyrin derivative as antileishmanial agent and potential modulator of ornithine decarboxylase of Leishmania donovani. Exp Parasitol. 2013 Oct;135(2):407–13. doi: 10.1016/j.exppara.2013.07.021. [DOI] [PubMed] [Google Scholar]
- 99.Sett R, Basu N, Ghosh AK, Das PK. Potential of doxorubicin as an antileishmanial agent. J Parasitol. 1992 Apr;78(2):350–4. [PubMed] [Google Scholar]
- 100.Sett R, Sarkar K, Das PK. Macrophage-directed delivery of doxorubicin conjugated to neoglycoprotein using leishmaniasis as the model disease. J Infect Dis. 1993 Oct;168(4):994–9. doi: 10.1093/infdis/168.4.994. [DOI] [PubMed] [Google Scholar]
- 101.Basu N, Kole L, Ghosh A, Das PK. Expression and characterization of a parasite-specific antigen on macrophages after infection with Leishmania donovani. Mol Cell Biochem. 1994 Mar 16;132(1):1–6. doi: 10.1007/BF00925668. [DOI] [PubMed] [Google Scholar]
- 102.Mukherjee S, Das L, Kole L, Karmakar S, Datta N, Das PK. Targeting of parasite-specific immunoliposome-encapsulated doxorubicin in the treatment of experimental visceral leishmaniasis. J Infect Dis. 2004 Mar 15;189(6):1024–34. doi: 10.1086/382048. [DOI] [PubMed] [Google Scholar]
- 103.Gupta GK, Kansal S, Misra P, Dube A, Mishra PR. Uptake of biodegradable gel-assisted LBL nanomatrix by Leishmania donovani-infected macrophages. AAPS PharmSciTech. 2009;10(4):1343–7. doi: 10.1208/s12249-009-9334-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kunjachan S, Gupta S, Dwivedi AK, Dube A, Chourasia MK. Chitosan-based macrophage-mediated drug targeting for the treatment of experimental visceral leishmaniasis. J Microencapsul. 2011;28(4):301–10. doi: 10.3109/02652048.2011.559281. [DOI] [PubMed] [Google Scholar]
- 105.Kansal S, Tandon R, Dwivedi P, Misra P, Verma PR, Dube A, et al. Development of nanocapsules bearing doxorubicin for macrophage targeting through the phosphatidylserine ligand: a system for intervention in visceral leishmaniasis. J Antimicrob Chemother. 2012 Nov;67(11):2650–60. doi: 10.1093/jac/dks286. [DOI] [PubMed] [Google Scholar]
- 106.Kansal S, Tandon R, Verma PR, Dube A, Mishra PR. Development of doxorubicin loaded novel core shell structured nanocapsules for the intervention of visceral leishmaniasis. J Microencapsul. 2013;30(5):441–50. doi: 10.3109/02652048.2012.752532. [DOI] [PubMed] [Google Scholar]
- 107.Chapman WL, Jr., Hanson WL, Alving CR, Hendricks LD. Antileishmanial activity of liposome-encapsulated meglumine antimonate in the dog. Am J Vet Res. 1984 May;45(5):1028–30. [PubMed] [Google Scholar]
- 108.Banerjee G, Nandi G, Mahato SB, Pakrashi A, Basu MK. Drug delivery system: targeting of pentamidines to specific sites using sugar grafted liposomes. J Antimicrob Chemother. 1996 Jul;38(1):145–50. doi: 10.1093/jac/38.1.145. [DOI] [PubMed] [Google Scholar]
- 109.Nieto J, Alvar J, Mullen AB, Carter KC, Rodriguez C, San Andres MI, et al. Pharmacokinetics, toxicities, and efficacies of sodium stibogluconate formulations after intravenous administration in animals. Antimicrob Agents Chemother. 2003 Sep;47(9):2781–7. doi: 10.1128/AAC.47.9.2781-2787.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Paul M, Durand R, Boulard Y, Fusai T, Fernandez C, Rivollet D, et al. Physicochemical characteristics of pentamidine-loaded polymethacrylate nanoparticles: implication in the intracellular drug release in Leishmania major infected mice. J Drug Target. 1998;5(6):481–90. doi: 10.3109/10611869808997874. [DOI] [PubMed] [Google Scholar]
- 111.Menez C, Legrand P, Rosilio V, Lesieur S, Barratt G. Physicochemical characterization of molecular assemblies of miltefosine and amphotericin B. Mol Pharm. 2007 Mar-Apr;4(2):281–8. doi: 10.1021/mp0601143. [DOI] [PubMed] [Google Scholar]
- 112.Menez C, Buyse M, Besnard M, Farinotti R, Loiseau PM, Barratt G. Interaction between miltefosine and amphotericin B: consequences for their activities towards intestinal epithelial cells and Leishmania donovani promastigotes in vitro. Antimicrob Agents Chemother. 2006 Nov;50(11):3793–800. doi: 10.1128/AAC.00837-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Menez C, Buyse M, Chacun H, Farinotti R, Barratt G. Modulation of intestinal barrier properties by miltefosine. Biochem Pharmacol. 2006 Feb 14;71(4):486–96. doi: 10.1016/j.bcp.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 114**.Nwaka S, Ramirez B, Brun R, Maes L, Douglas F, Ridley R. Advancing drug innovation for neglected diseases-criteria for lead progression. PLoS Negl Trop Dis. 2009;3(8):e440. doi: 10.1371/journal.pntd.0000440. [Excellent article which clearly elucidates the criteria for drug development.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115**.Singh N, Mishra BB, Bajpai S, Singh RK, Tiwari VK. Natural product based leads to fight against leishmaniasis. Bioorg Med Chem. 2014 Jan 1;22(1):18–45. doi: 10.1016/j.bmc.2013.11.048. [Recent review of natural products showing antileishmanial activity.] [DOI] [PubMed] [Google Scholar]
- 116.Mishra BB, Kale RR, Singh RK, Tiwari VK. Alkaloids: future prospective to combat leishmaniasis. Fitoterapia. 2009 Mar;80(2):81–90. doi: 10.1016/j.fitote.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 117**.Salem MM, Werbovetz KA. Natural products from plants as drug candidates and lead compounds against leishmaniasis and trypanosomiasis. Curr Med Chem. 2006;13(21):2571–98. doi: 10.2174/092986706778201611. [Excellent review of natural products showing antileishmanial activity.] [DOI] [PubMed] [Google Scholar]
- 118.Mukherjee S, Mukherjee B, Mukhopadhyay R, Naskar K, Sundar S, Dujardin JC, et al. Imipramine is an orally active drug against both antimony sensitive and resistant Leishmania donovani clinical isolates in experimental infection. PLoS Negl Trop Dis. 2012;6(12):e1987. doi: 10.1371/journal.pntd.0001987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Palit P, Ali N. Oral therapy with sertraline, a selective serotonin reuptake inhibitor, shows activity against Leishmania donovani. J Antimicrob Chemother. 2008 May;61(5):1120–4. doi: 10.1093/jac/dkn046. [DOI] [PubMed] [Google Scholar]
- 120.Suryawanshi SN, Tiwari A, Kumar S, Shivahare R, Mittal M, Kant P, et al. Chemotherapy of leishmaniasis. Part XII: design, synthesis and bioevaluation of novel triazole integrated phenyl heteroterpenoids as antileishmanial agents. Bioorg Med Chem Lett. 2013 May 15;23(10):2925–8. doi: 10.1016/j.bmcl.2013.03.055. [DOI] [PubMed] [Google Scholar]
- 121.Sharma M, Chauhan K, Shivahare R, Vishwakarma P, Suthar MK, Sharma A, et al. Discovery of a new class of natural product-inspired quinazolinone hybrid as potent antileishmanial agents. J Med Chem. 2013 Jun 13;56(11):4374–92. doi: 10.1021/jm400053v. [DOI] [PubMed] [Google Scholar]
- 122.Kyriazis JD, Aligiannis N, Polychronopoulos P, Skaltsounis AL, Dotsika E. Leishmanicidal activity assessment of olive tree extracts. Phytomedicine. 2012 Feb 15;20(3-4):275–81. doi: 10.1016/j.phymed.2012.11.013. [DOI] [PubMed] [Google Scholar]
- 123.Bhattacharya S, Ghosh P, De T, Gomes A, Dungdung SR. In vivo and in vitro antileishmanial activity of Bungarus caeruleus snake venom through alteration of immunomodulatory activity. Exp Parasitol. 2013 Sep;135(1):126–33. doi: 10.1016/j.exppara.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 124.Khaliq T, Misra P, Gupta S, Reddy KP, Kant R, Maulik PR, et al. Peganine hydrochloride dihydrate an orally active antileishmanial agent. Bioorg Med Chem Lett. 2009 May 1;19(9):2585–6. doi: 10.1016/j.bmcl.2009.03.039. [DOI] [PubMed] [Google Scholar]
- 125.Maes L, Germonprez N, Quirijnen L, Van Puyvelde L, Cos P, Vanden Berghe D. Comparative activities of the triterpene saponin maesabalide III and liposomal amphotericin B (AmBisome) against Leishmania donovani in hamsters. Antimicrob Agents Chemother. 2004 Jun;48(6):2056–60. doi: 10.1128/AAC.48.6.2056-2060.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Germonprez N, Maes L, Van Puyvelde L, Van Tri M, Tuan DA, De Kimpe N. In vitro and in vivo anti-leishmanial activity of triterpenoid saponins isolated from Maesa balansae and some chemical derivatives. J Med Chem. 2005 Jan 13;48(1):32–7. doi: 10.1021/jm031150y. [DOI] [PubMed] [Google Scholar]
- 127.Mittra B, Saha A, Chowdhury AR, Pal C, Mandal S, Mukhopadhyay S, et al. Luteolin, an abundant dietary component is a potent anti-leishmanial agent that acts by inducing topoisomerase II-mediated kinetoplast DNA cleavage leading to apoptosis. Mol Med. 2000 Jun;6(6):527–41. [PMC free article] [PubMed] [Google Scholar]
- 128.Zhai L, Chen M, Blom J, Theander TG, Christensen SB, Kharazmi A. The antileishmanial activity of novel oxygenated chalcones and their mechanism of action. J Antimicrob Chemother. 1999 Jun;43(6):793–803. doi: 10.1093/jac/43.6.793. [DOI] [PubMed] [Google Scholar]
- 129.Chen M, Christensen SB, Theander TG, Kharazmi A. Antileishmanial activity of licochalcone A in mice infected with Leishmania major and in hamsters infected with Leishmania donovani. Antimicrob Agents Chemother. 1994 Jun;38(6):1339–44. doi: 10.1128/aac.38.6.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Medda S, Mukhopadhyay S, Basu MK. Evaluation of the in-vivo activity and toxicity of amarogentin, an antileishmanial agent, in both liposomal and niosomal forms. J Antimicrob Chemother. 1999 Dec;44(6):791–4. doi: 10.1093/jac/44.6.791. [DOI] [PubMed] [Google Scholar]