

Abstract
A sequence polymorphism (rs738409, I148M) in patatin-like phospholipid domain containing protein 3 (PNPLA3) is strongly associated with nonalcoholic fatty liver disease (NAFLD), but the mechanistic basis for this association remains enigmatic. Neither ablation nor overexpression of wild-type PNPLA3 affects liver fat content in mice, whereas hepatic overexpression of the human 148M transgene causes steatosis. To determine whether the 148M allele causes fat accumulation in the liver when expressed at physiological levels, we introduced a methionine codon at position 148 of the mouse Pnpla3 gene. Knockin mice had normal levels of hepatic fat on a chow diet, but when challenged with a high-sucrose diet their liver fat levels increased 2 to 3-fold compared to wild-type littermates without any associated changes in glucose homeostasis. The increased liver fat in the knockin mice was accompanied by a 40-fold increase in PNPLA3 on hepatic lipid droplets, with no increase in hepatic PNPLA3 messenger RNA (mRNA). Similar results were obtained when the catalytic dyad of PNPLA3 was inactivated by substituting the catalytic serine with alanine (S47A). Conclusion: These data provide the first direct evidence that physiological expression of PNPLA3 148M variant causes NAFLD, and that the accumulation of catalytically inactive PNPLA3 on the surfaces of lipid droplets is associated with the accumulation of TG in the liver. (Hepatology 2015;61:108–118)
See Editorial on Page 18
Cirrhosis caused by fatty liver disease is emerging as a major indication for liver transplantation.1,2 Recent population-based surveys have revealed that one out of every three adults in the United States has hepatic steatosis,3,4 the first stage of fatty liver disease.5 The accumulation of hepatic triglyceride (TG) in hepatocytes per se is not associated with any apparent adverse clinical sequelae, but a subset of individuals with steatosis develop an inflammatory response and fibrosis; these individuals are at increased risk of developing cirrhosis.6 This continuum of liver disease is referred to as nonalcoholic fatty liver disease (NAFLD) and individuals with this disorder are at increased risk of hepatocellular carcinomas.7,8
Despite the clinical importance of NAFLD, major questions remain unanswered regarding its pathogenesis.9 The first molecular handle on the genetic underpinnings of NAFLD was the identification of a missense sequence variation (I148M) in patatin-like phospholipase domain protein family member 3 (PNPLA3), which is associated with increased hepatic TG content and liver enzymes.10 More than 50 studies have confirmed the association between this variant and the full spectrum of NAFLD, including simple steatosis, nonalcoholic steatohepatitis, cirrhosis, and hepatocellular carcinoma.11
The mechanism by which the I148M substitution promotes liver fat accumulation remains enigmatic. Recombinant PNPLA3 has TG hydrolase activity that is markedly attenuated by the I148M substitution.12–14 Based on this observation, it was hypothesized that the accumulation of hepatic TG in the liver was due to the loss of PNPLA3-mediated TG hydrolysis in hepatic lipid droplets. This hypothesis was quickly challenged when two groups reported that Pnpla3 knockout mice do not develop fatty liver.15,16 Moreover, mice expressing high levels of the human mutant protein in the liver (PNPLA3148MTg mice) had hepatic steatosis, whereas mice expressing the wild-type protein (PNPLA3WTTg mice) had hepatic TG levels that were similar to nontransgenic littermates. Thus, loss of PNPLA3 activity is not sufficient to cause hepatic steatosis and expressing high levels of the enzyme in the liver does not decrease hepatic TG content, as would be expected if it functions as a lipase.17
Zechner and colleagues showed that PNPLA3 catalyzes the acylation of lysophosphatidic acid to phosphatidic acid, a step in the TG biosynthetic pathway, when expressed in bacteria as a fusion protein with a trigger factor.18 In this expression system, the 148M variant increased lysophosphatidic acid acyltransferase (LPAAT) activity. LPAAT activity was not detected in recombinant PNPLA3 purified from Sf9 cells,13 and was not increased in livers of mice expressing high levels of wild-type PNPLA3 or PNPLA3-148M.17
Metabolic studies in the PNPLA3148MTg mice provided evidence consistent with PNPLA3-148M conferring both a loss, and a gain, of function. Glycerol release from TG was reduced in primary hepatocytes from PNPLA3148MTg mice when compared to the PNPLA3WTTg mice. Endogenous fatty acid synthesis was significantly increased in both transgenic lines of mice when compared to nontransgenic animals, with the levels of TG synthesis being higher in PNPLA3148MTg animals.
Although the PNPLA3148MTg mice developed hepatic steatosis, this model has two important limitations: 1) human PNPLA3 is only 68% identical to the mouse protein and may have nonphysiological effects in mice, and 2) the PNPLA3 transgene is expressed constitutively at very high levels and does not exhibit the ∼80-fold changes in response to food intake that characterize the endogenous gene.19 To address these limitations, we developed mice in which isoleucine at residue 148 of the endogenous Pnpla3 gene was replaced with methionine. To determine the effect of an obligate loss-of-function mutation in PNPLA3 on hepatic fat content, we generated mice in which the catalytic serine in the patatin domain was replaced by alanine.
Materials and Methods
Animals
The I148M and S47A mutations were introduced into the mouse Pnpla3 gene by homologous recombination as described in the Supporting Materials. Three founders for each mutation were backcrossed with C57BL/6J females. All mice were maintained on a 12/12-hour light/dark cycle and fed Teklad Mouse/Rat Diet 7001 (Harlan Teklad, Houston, TX) ad libitum. For the dietary challenge studies, mice were fed a high-sucrose diet (no. 901683; 74% kCal from sucrose, MP Biomedicals, Santa Ana, CA) or a high-fat diet (D12451; 45% kCal from fat, Research Diets, New Brunswick, NJ) for 4 weeks unless otherwise stated. Prior to each experiment the mice were metabolically synchronized for 3 days by withdrawing food from 8:00 am till 8:00 pm and providing food from 8:00 pm till 8:00 am and sacrificed at 8:00 am at the end of the third refeeding cycle. All animal experiments were performed with the approval of the Institutional Animal Care and Research Advisory Committee at University of Texas Southwestern Medical Center at Dallas.
Body Composition
Total body fat and lean body mass were determined by nuclear magnetic resonance using a Minispec analyzer (Bruker, Billerica, MA).
Liver and Plasma Chemistries
Lipids were extracted from livers (∼100 mg) using the method of Folch et al.20 The levels of TG, cholesterol, cholesteryl ester, free cholesterol, and phosphatidylcholine were measured using enzymatic assays (Infinity, Thermo Electron, Madison, WI, and Wako, Richmond, VA) and normalized to sample weight. Lipids were also separated into cholesterol esters, TGs, and phospholipids using thin-layer chromatography as previously described.21 Lipids in each fraction were hydrolyzed, derivatized with trimethylsilane, and separated by gas-liquid chromatography (GLC) using a Hewlett Packard 6890 Series gas chromatograph (Agilent, Santa Clara, CA). The fatty acids were identified by retention times established using purified fatty acid standards (GLC-744, NU-Chek Prep, Elysian, MN). The percentage of each fatty acid in each fraction was calculated using pentadecanoin (C15:0) as an internal standard.
Serum levels of liver enzymes, TG, cholesterol, and glucose were measured using the Vitros 250 system (GMI, Ramsey, MN). Enzymatic assay kits were used to determine serum levels of NEFA (Wako). Serum levels of insulin were measured using an enzyme-linked immunosorbent assay (ELISA) assay (Crystal Chem, Downers Grove, IL).
RNA Expression
Total RNA was isolated from tissues of four mice from each group using RNA STAT-60 (Tel-Test, Friendswood, TX), and real-time polymerase chain reaction (PCR) measurements were performed as previously described.22 Expression levels of mouse Pnpla3 hepatic mRNA were measured using oppositely oriented primers (5′-CGAGGCGAGCGGTACGT-3′ and 5′-TGACACCGTGATGGTGGTTT-3′). Mouse 36B4 mRNA was used as an internal control.
Lipid Droplet Isolation
Hepatic lipid droplets were isolated by centrifugation as described in the Supporting Methods.
Immunoblot Analysis of Lipid Droplet Proteins
Aliquots of resuspended lipid droplet protein (3 μg) were analyzed by quantitative immunoblot analysis as described in the Supporting Methods.
Tissue Morphology
Liver and white adipose tissue were examined by light microscopy as described in Supporting Methods.
Statistics
Data are presented as means ± SEM. Differences between group means were assessed by 2-tailed, unpaired Student t tests using GraphPad Prism statistical software (GraphPad Software, San Diego, CA). P-values below 0.05 were considered statistically significant.
Results
Development of Pnpla3148M and Pnpla347A Knockin (KI) Mice
To develop an animal model that more accurately recapitulates the human phenotype associated with PNPLA3-148M, we altered codon 148 of Pnpla3 in the mouse genome from ATT to ATG, substituting methionine for isoleucine (Pnpla3148M) (Fig. 1A). We also generated a second line of mice in which the catalytic serine at residue 47 was replaced with alanine (Pnpla347A mice). Sanger sequencing confirmed that the mutations were introduced without altering other sequences in the gene (Supporting Fig. 1) and RT-PCR indicated that expression of the mutant and wild-type alleles was similar in mice fed a chow diet and that transcript levels increased by similar amounts when the two lines were fed a high-sucrose diet (74% of kCal from sucrose) (Fig. 1B).
Fig. 1.
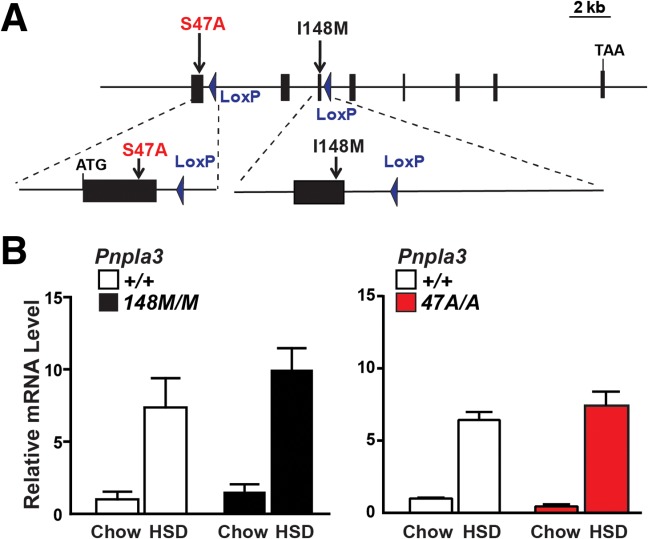
Generation of Pnpla3148M and Pnpla347A KI mice. (A) Diagram of Pnpla3 targeting strategy. Homologous recombination was used to replace either exon 1 (S47A) or exon 3 (I48M) with the corresponding mutant sequences. The selectable marker (Neo) located between two LoxP sites was then excised with Cre recombinase, leaving one LoxP site in the flanking intron. (B) Pnpla3 RNA levels in livers from female mice (n = 4/group) consuming a chow diet or a high-sucrose diet (HSD) for 4 weeks. Livers were collected at the end of the feeding cycle. Levels of Pnpla3 mRNA were determined using real-time PCR and normalized to values of chow-fed wild-type mice.
The KI mice were born in the expected Mendelian ratios and both litter sizes and gender distributions were unaltered by introduction of the mutations. No differences in body weight, liver weight, fat mass, or lean mass were detected between the two strains of KI mice and their wild-type littermates (Supporting Fig. 2). Morphological analysis of the adipose tissue revealed no differences in adipocyte size or conformation (Supporting Fig. 3)
Pnpla3148M and Pnpla347A KI Mice Develop Hepatic Steatosis on a High-Sucrose Diet
On a chow diet, the levels of TG, phosphatidylcholine (PC), cholesteryl esters, and free cholesterol in the livers of female mice that were heterozygous (Pnpla3148M/+) or homozygous (Pnpla3148M/M) for the Pnpla3148M allele did not differ significantly from those of wild-type littermates (Pnpla3+/+) (Fig. 2A, top). After 4 weeks on a high-sucrose diet, hepatic TG content was higher in female Pnpla3148M/+ and Pnpla3148M/M mice than in wild-type animals (Fig. 2A, bottom). Levels of hepatic TG in heterozygous mice were intermediate between those of wild-type and homozygous KI animals. Similar responses were observed in male mice (Supporting Fig. 4A). No differences in circulating lipid levels were seen between the two lines (Supporting Fig. 5A).
Fig. 2.
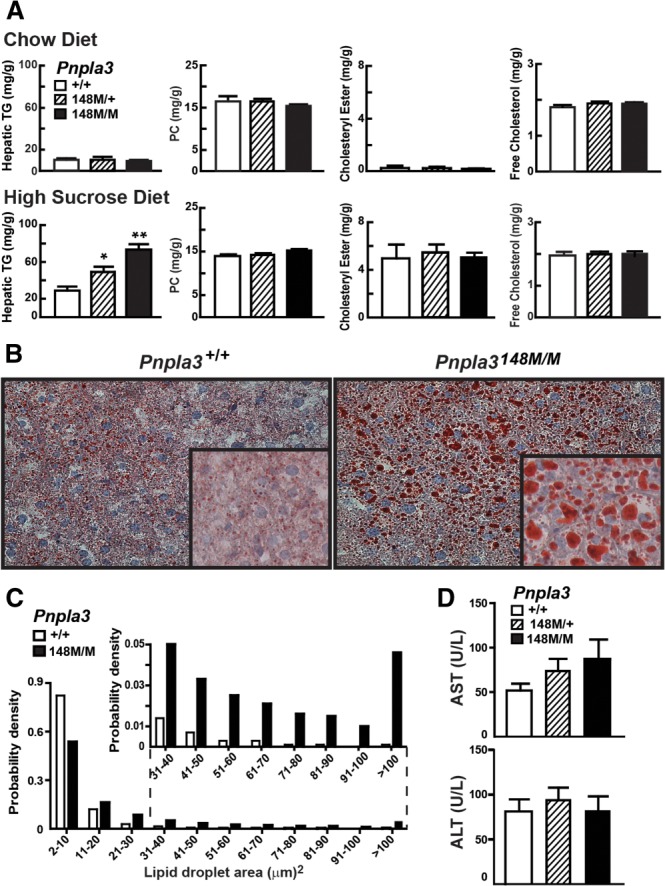
Hepatic TG content in Pnpla3I148M KI mice. Data in panels A-D are from the same experiment (n = 5 mice/group). Tissue and plasma were collected at the end of the feeding cycle. (A) Hepatic lipid levels were measured in 12-week-old female wild-type (+/+), heterozygous (148M/+), and homozygous (148M/M) KI mice fed a chow diet or high-sucrose diet (HSD) for 4 weeks. (B) Liver sections from wild-type (+/+), and 148M/M KI mice on high-sucrose diets were stained with Oil Red O and viewed using a Leica microscope (DM2000) (magnification: 20× and inset 64×). (C) Size distributions of hepatic lipid droplets in wild-type and Pnpla3148M/M mice. Oil Red O-stained slides were analyzed using ImageJ as described in Materials and Methods. (D) Plasma levels of aspartate aminotransferase (AST) and alanine aminotransaminase (ALT) in wild-type, 148M/+, and 148M/M KI mice. Values are means ± SEM. The experiment was performed twice with similar results. *P < 0.05, **P < 0.001.
Hematoxylin and eosin staining of liver sections from the Pnpla3148M/M mice revealed no differences in tissue architecture or cellular morphology (data not shown). Oil Red O staining of neutral lipids was markedly increased in Pnpla3148M/M mice (Fig. 2B) and the size distribution of their lipid droplets was shifted to the right (Fig. 2C), reflecting a striking increase in the number of large droplets (>20 μm2). In wild-type mice, the median area of the droplets was 4.3 μm2 and fewer than 5% were greater than 22 μm2. The median area of lipid droplets in the Pnpla3148M/M mice (8.7 μm2) was double that of their wild-type littermates, and 28% were greater than 22 μm2. Despite the increased lipid content of the livers in the Pnpla3148M/+ and Pnpla3148M/M mice, no changes in circulating liver enzymes were observed in these animals (Fig. 2D).
Similar results were obtained in Pnpla347A KI mice. On chow diets the lipid levels of Pnpla347A/+ and Pnpla347A/A KI mice were similar to those of their wild-type littermates in liver (Fig. 3A, top; Supporting Fig 4B, top) and in plasma (Supporting Fig. 5B). On the high-sucrose diet, hepatic TG content was significantly higher in both the female (Fig. 3A, bottom) and male (Supporting Fig. 4B, bottom) Pnpla347A/A mice than in their wild-type littermates. The hepatic TG content of mice heterozygous for the S47A substitution did not differ significantly from that of wild-type animals. Hepatic levels of PC, cholesteryl esters, and free cholesterol did not differ between the lines on either diet (Fig. 3A) and no differences in circulating liver enzymes (Fig. 3D) or plasma lipids (Supporting Figure 5B) were apparent on the high-sucrose diet. The increase in hepatic TG content was associated with an increase in the number of large, Oil Red O-positive lipid droplets in the livers of the Pnpla3S47A/A animals (Fig. 3B,C).
Fig. 3.
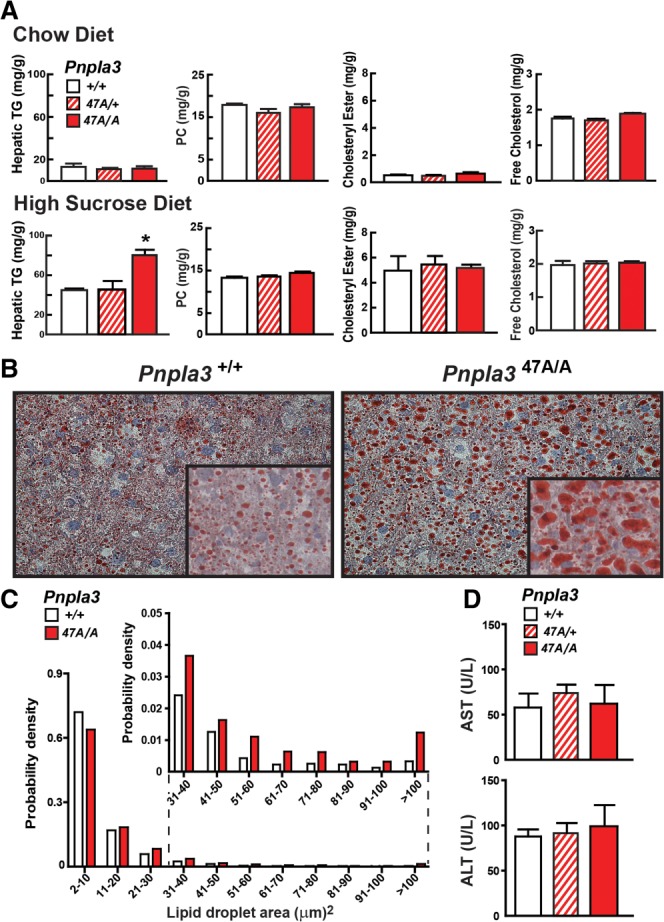
Hepatic TG content in Pnpla3S47A mice. Data in panels A-D are from the same experiment (n = 7 mice/group). Tissue and plasma samples were collected at the end of the feeding cycle. The experiment was repeated with similar results. (A) Hepatic lipid levels were measured in 13-week-old female wild-type (+/+), S47A heterozygous (S47A/+) and homozygous (S47A/A) KI mice fed a chow diet or high-sucrose diet (HSD) for 4 weeks. (B) Liver sections from sucrose-fed wild-type and S47A/A KI mice were stained with Oil Red O and viewed using a Leica microscope (DM2000) (magnification: 20×, inset 64×). (C) Size distributions of hepatic lipid droplets in wild-type and homozygous (S47A/A) KI mice. Oil Red O-stained slides were analyzed using ImageJ as described in the Materials and Methods. (D) Plasma levels of liver enzymes (AST and ALT). Values are means ± SEM. *P = 0.001.
No Change in Hepatic mRNA Profile in PNPLA3 KI Mice
High-level expression of human PNPLA3-148M in livers of transgenic mice was associated with increased mRNA levels of sterol regulatory element binding protein-1c (SREBP-1c), which orchestrates the up-regulation of the fatty acid synthesis pathway,23 and several of its target genes,17 suggesting that the elevated TG content associated with the PNPLA3-148M variant is due to accelerated TG synthesis. In mice consuming high-sucrose diets, hepatic mRNA levels of SREBP-1c and its target genes were similar in Pnpla3148M/M, Pnpla3 47A/A and wild-type animals (Fig. 4). We also found no strain-specific changes in genes involved in TG lipolysis (hormone sensitive lipase [HSL], adipocyte TG lipase [ATGL]), fatty acid oxidation, hepatic inflammation, or fibrosis.
Fig. 4.
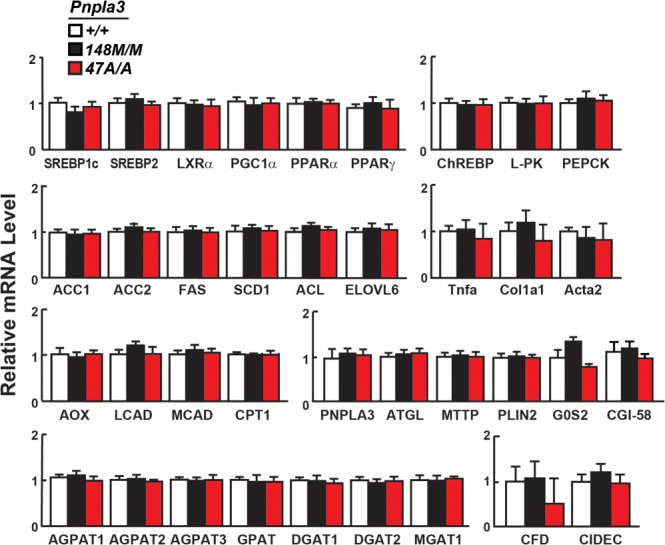
Relative mRNA levels of selected genes in livers of Pnpla3148M/M and Pnpla347A/A mice with wild-type littermates (n = 4/group). The mice used in this experiment are described in the legends to Figs. 2 and 3. mRNA levels were quantified by real-time PCR, normalized to levels of 36B4, and expressed relative to levels in wild-type mice. Values are means ± SEM. SREBP1c, sterol regulatory element binding protein 1 isoform C; SREBP2, sterol regulatory element binding protein 2; LXRα, liver X receptor alpha; PGC-1α, PPARγ coactivator 1α; PPARα, peroxisome proliferator-activated receptor alpha; PPARγ, peroxisome proliferator-activated receptor gamma; ChREBP, carbohydrate-responsive element-binding protein; L-PK, liver pyruvate kinase; PEPCK, phosphoenoylpyruvate carboxykinase; ACC1, acetyl-CoA carboxylase-1; ACC2, acetyl-CoA carboxylase 2; FAS, fatty acid synthase; SCD1, stearoyl-CoA desaturase-1; ACL, ATP citrate lyase; ELOVL6, ELOVL family member 6; TNFα, tumor necrosis factor alpha; COL1α1, collagen type-1α1; ACTA2, α-smooth muscle actin; AOX, acyl-CoA oxidase-1; LCAD, long-chain acyl-CoA dehydrogenase; MCAD, medium-chain acyl-CoA dehydrogenase; CPT1, carnitine palmitoyltransferase1; ATGL, adipose triglyceride lipase; MTTP, microsomal TG transfer protein; PLIN2, perilipin 2; G0S2, G0/G1switch 2; CGI-58, comparative gene identification 58; AGPAT1-3, 1-acylglycerol-3-phosphate O-acyltransferase 1-3; GPAT, glycerol-3-phosphate acyltransferase; DGAT1, diglyceride acyltransferase-1; DGAT2, diglyceride acyltransferase-2; MGAT1, monoacylglycerol O-acyltransferase 1; CFD, complement factor D (adipsin); CIDEC, cell death-inducing DFFA-like effector C. The experiment was repeated twice and the results were similar.
No Major Alterations in Fatty Acid Composition of Major Hepatic Lipids
Previously, we found a significant reduction in the proportion of long-chain polyunsaturated fatty acids in liver TG from mice expressing high levels of human PNPLA3-148M.17 The fatty acid profiles of hepatic TG, cholesteryl esters, and phospholipids were remarkably similar in the KI mice and their wild-type littermates (Fig. 5). Stearate (C18:0) was modestly decreased in the liver TG fraction of Pnpla3148M/M mice, while palmitoleate and linoleate were slightly higher in liver TGs of Pnpla3S47A/A animals, but no consistent differences were observed in the two KI strains. Thus, the increased hepatic TG content in the two KI strains was not associated with any obvious alteration in the spectrum of fatty acids of hepatic glycerolipids.
Fig. 5.
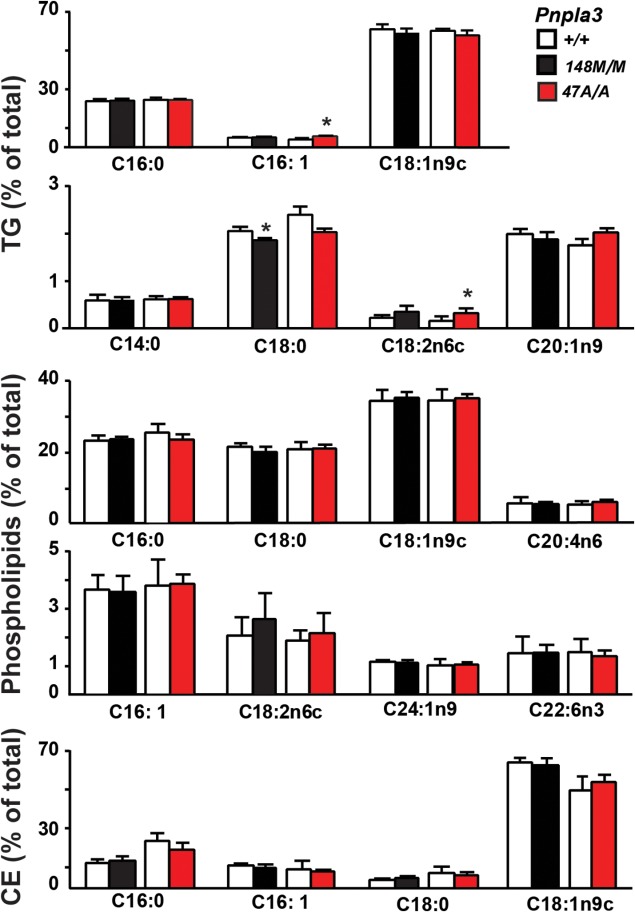
Fatty acid composition of hepatic lipids in wild-type, Pnpla3I148M/M, and Pnpla3S47A/A KI mice. Lipids were extracted from livers of 12-week-old female mice (n = 6/group) fed a high-sucrose diet for 4 weeks as described in the Materials and Methods. The TG, phospholipid, and cholesterol ester fractions were separated by TLC, hydrolyzed, and derivatized with trimethylsilane. The fatty acid methyl-esters were quantified by gas chromatography. Each value represents the mean ± SEM. *P < 0.05. The experiment was repeated and the results were similar.
Insulin Resistance Is Comparable in Wild-Type and PNPLA3148M Mice Fed a High-Fat Diet
To determine if the KI mice develop more severe insulin resistance than do wild-type animals, we challenged them with a high-fat diet (45% of kCal) for 12 weeks. High-fat feeding led to comparable increases in body weight and plasma levels of insulin in wild-type and KI mice (Supporting Fig. 6A). Fat feeding increased hepatic fat content 3 to 4-fold in the Pnpla3148M/M KI (41 ± 9 mg/g) and wild-type (51 ± 18 mg/g) mice compared to chow-fed animals (13 ± 5 mg/g). Fasting glucose (163 ± 25 mg/dL versus 154 ± 14 mg/dL) and insulin (1.6 ± 0.6 versus 2.1 ± 1.0) levels did not differ between the groups, and glucose tolerance testing (performed after 10 weeks of high-fat feeding) did not elicit any significant differences between the two groups of mice (Supporting Fig. 6B). A GTT was also performed in mice fed a chow or a high sucrose diet for 4 weeks and the results were similar (Supporting Fig. 6C). Thus, expression of PNPLA3-148M did not affect the development of insulin resistance in mice, which is congruent with the lack of association with either homeostasis model of assessment, insulin resistance (HOMA-IR) or diabetes in humans.24
Lipid Droplets From the Sucrose-Fed But Not Fat-Fed Pnpla3148M/M and Pnpla347A/A Mice Accumulate PNPLA3 and CGI-58 (ABHD5)
On a high-sucrose diet, levels of PNPLA3 mRNA were similar in the livers of wild-type and KI mice (Fig. 6A). Liver TG content was 2-fold higher in KI mice (Fig. 6B). Thus, the differences in hepatic TG associated with sucrose feeding were not due to differences arising from altered PNPLA3 mRNA levels. Quantitative immunoblotting revealed a dramatic (40-fold) increase in PNPLA3 levels in lipid droplets from Pnpla3148M/M and Pnpla347A/A mice compared to their wild-type littermates (Fig. 6C,D). No differences in levels of ATGL or HSL, the two major TG lipases in lipid droplets, were seen among the four groups. In contrast to these findings, the levels of CGI-58, a cofactor of ATGL that was barely detectable in the wild-type animals, were increased ∼5-fold in lipid droplets from both Pnpla3148M/M and Pnpla347A/A mice. The major differences in levels of both PNPLA3 and CGI-58 were not accompanied by changes in levels of the three perilipins that are most abundant in hepatic lipid droplets: PLIN2, PLIN3, and PLIN5. The selective accumulation of CGI-58 in the lipid droplets suggested that PNPLA3, which resembles ATGL, may sequester CGI-58 on the lipid droplet, resulting in a reduction in TG hydrolase activity by ATGL.
Fig. 6.
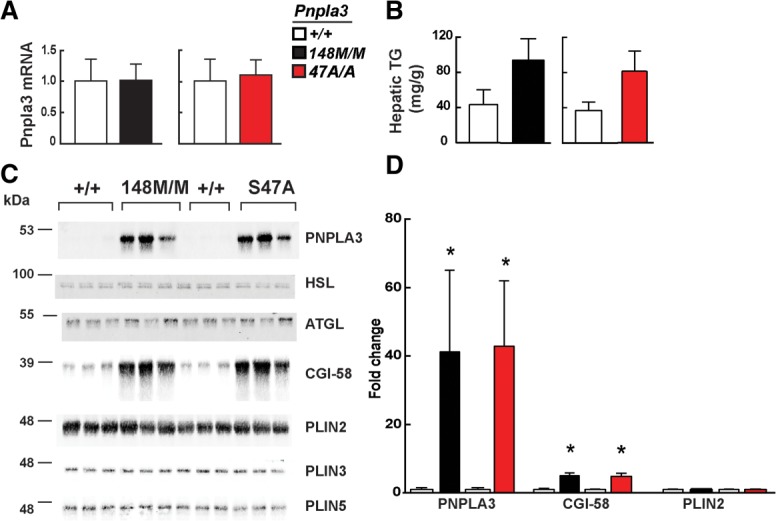
Accumulation of PNPLA3 and CGI-58 protein on hepatic lipid droplets of Pnpla3148M/M and Pnpla347A/A KI mice. Livers were harvested from 11-week-old female wild-type (+/+), Pnpla3148M/M, and Pnpla347A/A mice (n = 5/group) fed a high-sucrose diet for 4 weeks. (A) Hepatic PNPLA3 mRNA levels were determined using real-time PCR, as described in the Materials and Methods. (B) Hepatic TG levels were measured by enzymatic assay as described in the Materials and Methods. (C) Quantitative immunoblot analysis of lipid droplets (LD) isolated from 3 mice per group. A total of 3 μg of LD protein was size-fractionated on a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel and immunoblot analysis was performed as described in the Materials and Methods. (D) Immunoblots were quantitated using infrared fluorescent imaging as described in the Materials and Methods. Values are mean ± SEM. *P < 0.05. The experiment was repeated twice and the results were similar.
To determine if PNPLA3-148M accumulates secondary to the development of fatty liver, we measured PNPLA3 mRNA and protein in livers from mice fed a high-fat diet for 12 weeks (Supporting Fig. 6A). Hepatic levels of PNPLA3 mRNA were similar in wild-type and KI animals (Fig. 7A), but in both lines the levels were much lower on the high-fat diet (Ct = 24.5) than on the high-sucrose diet (Ct = 22). Despite the 3 to 4-fold increase in hepatic TG levels in fat-fed mice (Fig. 7B), PNPLA3 protein was barely detectable in the lipid droplets of wild-type or the KI animals (Fig. 7C). Quantitative immunoblotting of lipid droplets from the fat-fed mice indicated that PNPLA3 levels were 6-fold higher in the Pnpla3148M/M mice than in their wild-type littermates (Fig. 7C,D). Nonetheless, PNPLA3 levels in both strains were far lower in fat-fed than in sucrose-fed animals. Levels of CGI-58 were increased 2.5-fold in the KI mice.
Fig. 7.
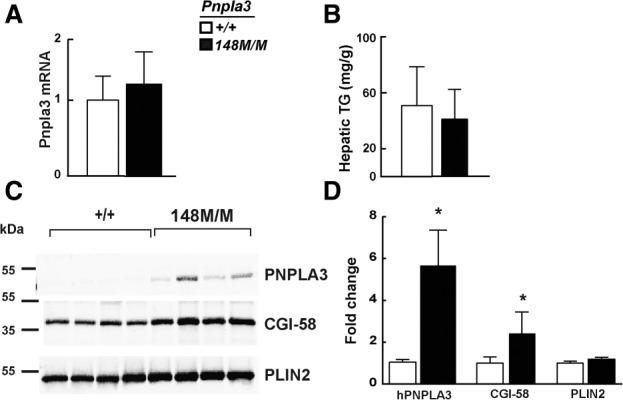
PNPLA3 on lipid droplets from fat-fed mice. Livers were harvested from 17-week-old female wild-type (+/+) and Pnpla3148M/M mice (n = 5/group) fed a high-fat diet for 12 weeks. (A) Hepatic PNPLA3 mRNA levels were determined using real-time PCR, as described in the Materials and Methods. (B) Hepatic TG levels were measured by enzymatic assay as described in the Materials and Methods. (C) Immunoblot analysis of lipid droplets isolated from 4 mice per group was performed as described in the legend to Fig. 6, and (D) quantified as described in the Materials and Methods. Values are means ± SEM. *P < 0.05.
PNPLA3, But Not CGI-58 Accumulates on Lipid Droplets of Liver-Specific PNPLA3148MTg Mice
To determine if the accumulation of CGI-58 on the lipid droplets of the KI was causally related to the accumulation of TG, we examined the levels of CGI-58 on the lipid droplets of mice expressing mutant human PNPLA3. If lipid accumulation in the mutant KI mice is due to sequestration of CGI-58 by PNPLA3, a similar increase in CGI-58 should be seen in the PNPLA3148MTg mice. In mice fed a high-sucrose diet for 4 weeks, levels of mouse PNPLA3 and CGI-58 mRNA were similar in livers from the transgenic and wild-type animals (Fig. 8A). The level of human PNPLA3 mRNA was ∼30% higher in the PNPLA3148MTg mice than in the mice expressing the wild-type human PNPLA3 transgene, although this difference was not statistically significant. The levels of human PNPLA3 were increased dramatically in liver lysates (Supporting Fig. 7) and in hepatic lipid droplets isolated from mice expressing the PNPLA3-148M transgene when compared to the mice expressing wild-type PNPLA3 (Fig. 8B). The levels of CGI-58 on hepatic lipid droplets did not differ significantly between the PNPLA3-148M and PNPLA3-WT transgenic mice. Thus, the accumulation of CGI-58 is not a prerequisite for the development of hepatic steatosis in association with expression of mutant PNPLA3.
Fig. 8.
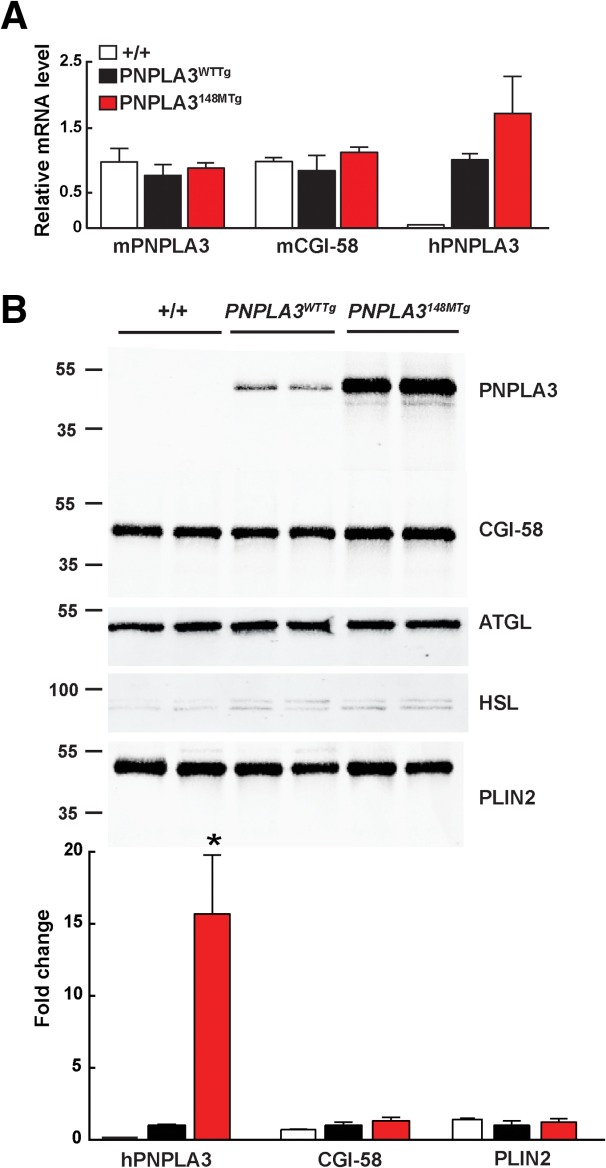
Accumulation of PNPLA3 and CGI-58 protein on hepatic lipid droplets of PNPLA3WTTg and PNPLA3148MTg mice. Lipid droplets and mRNA were prepared from livers of 11-week-old male mice after 4 weeks on a high-sucrose diet. (A) Relative levels of endogenous and human PNPLA3 and CGI-58 mRNA in wild-type (+/+), PNPLA3WTTg, and PNPLA3148MTg transgenic mice. (B) Immunoblot analysis of lipid droplet proteins from wild-type (+/+), PNPLA3WTTg, and PNPLA3148MTg transgenic mice. *P < 0.01. The experiment was repeated twice and the results were similar.
Discussion
The major finding of this study is that Pnpla3148M/M KI mice develop diet-dependent hepatic steatosis. The I148M substitution did not lead to increased hepatic TG levels in chow-fed mice, but elicited a 2 to 3-fold elevation in liver fat content in sucrose-fed animals (Fig. 2). This finding is congruent with the observation in humans that the PNPLA3-148M variant is associated with appreciably greater liver fat accumulation in obese than in lean individuals.24 Moreover, the increase in liver TG of KI mice was comparable in magnitude to that observed in humans homozygous for the PNPLA3-148M variant,10 and was not associated with any changes in circulating levels of lipids (Supporting Fig. 5) or in glucose homeostasis. Similarly, the variant has little effect on plasma lipid levels or glucose metabolism in humans,10 except in the extremely obese, where it is associated with lower plasma TG levels.25 Taken together, these data provide the first direct evidence that physiological expression of the PNPLA3-148M variant causes hepatic steatosis.
The phenotypic differences among PNPLA3 knockout, transgenic, and KI mice reiterate the limitations of genetically modified animals as models of human pathophysiology. Nonetheless, comparisons among these lines provide new insights into the biochemical sequelae of the I148M substitution and allow significant refinement of current models of how the 148M variant promotes steatosis. The accumulation of hepatic TG in Pnpla3148M mice indicates that the steatosis observed previously in PNPLA3148M transgenic mice17 is not an artifact of high-level expression of the human protein. Furthermore, the similar phenotypes of the Pnpla347A/A and PNPLA3148M KI indicate that the I148M substitution is equivalent to a loss of catalytic function, and that the association with steatosis is not a unique property of the 148M isoform. However, catalytic inactivation of PNPLA3 is not equivalent to loss of protein expression (as illustrated by the lack of steatosis in Pnpla3 knockout mice). Rather, PNPLA3-associated hepatic steatosis in mice requires the presence of the catalytically inactive protein, not simply the absence PNPLA3 activity.
Does the 148M variant also confer a gain-of-function? In the gain-of-function model proposed by Zechner and colleagues, the I148M substitution promotes TG synthesis by increasing the LPAAT activity of the enzyme.18 A limitation of this model is that it implies an enzymatic activity that is independent of the catalytic serine, since Pnpla3S47A mice also express the fatty liver phenotype. Moreover, transgenic mice overexpressing wild-type human PNPLA3 did not have increased hepatic fat content, as might have been expected if the protein has LPAAT activity. Furthermore, rates of phosphatidic acid turnover and TG synthesis in transgenic mice expressing the 148M isoform were similar to those of mice expressing wild-type PNPLA3.17
An alternative gain-of-function hypothesis is that PNPLA3-148M promotes formation of a signaling molecule that stimulates TG synthesis. In mice overexpressing human PNPLA3-148M protein in the liver, mRNA levels of several SREBP-1c target genes involved in fatty acid TG synthesis were significantly increased.17 However, no such changes were seen in mRNA expression profiles of the 148M KI mice. Thus, the steatosis in the KI mice appears not be due to the production of a signaling molecule that activates a transcriptional program promoting fatty acid and TG synthesis. We also found significant differences in the composition of hepatic TG-fatty acids in transgenic mice that could potentially alter SREBP-1c activity.17 These changes were not observed in KI mice and are therefore unlikely to be causally related to the steatosis associated with the 148M variant.
In both humans and mice, the PNPLA3-148M isoform promotes liver TG accumulation under dietary conditions that expose the liver to high levels of insulin: obesity and insulin resistance in humans, and sucrose-feeding in mice. Moreover, the effect of the 148M variant in humans is amplified by diets rich in carbohydrates.26 Sucrose feeding may promote hepatic steatosis by increasing fatty acid and TG synthesis in the liver and also by stimulating PNPLA3 expression.19 Increasing the availability of fatty acids to the liver is not sufficient to explain the effects of the 148M variant on liver TG content: Pnpla3148M KI mice fed a high-fat diet, which increases fatty acid flux to the liver but up-regulates PNPLA3 expression only modestly, did not develop hepatic steatosis. Conversely, high-level expression of PNPLA3-148M increased liver TG content even in chow-fed animals.12,17 Thus, the diet-dependence of PNPLA3-148M-associated steatosis may reflect the need for diet-induced stimulation of PNPLA3 expression. Additional studies will be required to fully define the relationship between PNPLA3 genotype, diet composition, and liver fat content in both mice and humans.
Kumashiro et al.27 recently reported that antisense oligonucleotide-mediated knockdown of PNPLA3 prevents hepatic steatosis, improves glucose tolerance, and increases hepatic insulin sensitivity in fat-fed rats. However, chronic inactivation (or overexpression) of PNPLA3 in mice was not associated with changes in glucose homeostasis.15–17 Nor did we find any association between the PNPLA3-148M isoform and insulin resistance in this study. Our results are similar to those of multiple studies in humans, including three population-based studies,10,29,28 which found no association between the I148M variant and either diabetes or HOMA-IR. Moreover, euglycemic clamp studies failed to identify defects in glucose homeostasis in individuals with the I148M variant.31,32,30
A striking and unanticipated consequence of sucrose feeding in the KI mice was a marked accumulation of mutant PNPLA3 on lipid droplets. Despite similar levels of hepatic PNPLA3 mRNA, lipid droplets from livers of both 148M and 47A KI animals had ∼40-fold more PNPLA3 protein than wild-type animals. The accumulation of inactive PNPLA3 on lipid droplets may cause hepatic TG accumulation associated with the I148M substitution. The inactive protein may limit TG hydrolysis, possibly by restricting droplet access to, or by sequestering, a factor required for lipolysis. Immunoblot analysis indicated that the accumulation of mutant PNPLA3 was not associated with a reduction in ATGL or hormone-sensitive lipase on lipid droplets, but CGI-58 (ABHD5), an essential cofactor of ATGL,33 was ∼5-fold more abundant on droplets from livers of 148M KI mice than on those of their wild-type littermates. Thus, we speculated that PNPLA3-148M promotes hepatic steatosis by sequestering CGI-58, thereby limiting hydrolysis of lipid droplet TG by ATGL. The marginal increase in CGI-58 accumulation on lipid droplets of PNPLA3148MTg mice despite a 2-fold increase in liver fat content and massive accumulation of the mutant PNPLA3 protein (Fig. 7) argues against this hypothesis.
The I148M substitution also resulted in a marked rightward shift in the size distribution of hepatic lipid droplets, such that very large droplets (>100 μm2) comprised less than 0.1% of the total in wild-type mice, but almost 5% in the Pnpla3148M KI animals. Since the volume of these very large droplets (∼750 μm3) is ∼40 times larger than that of the median-sized droplet (19 μm3), the major portion of hepatic TG in Pnpla3148M mice is contained in a small fraction of the droplets. This finding raises the possibility that the 148M isoform alters the partitioning of proteins among different-sized lipid droplets, rather than between the lipid droplets and cytosol. Partitioning of proteins among diverse lipid droplets would not be detected by the methods used in this study. Experiments are under way to further characterize the protein and lipid composition of the lipid droplets in which PNPLA3 resides.
We cannot exclude the possibility that the accumulation of mutant PNPLA3 on lipid droplets is a consequence, rather than a cause, of hepatic steatosis. Steatosis was not associated with high levels of the 148M isoform in fat-fed mice, but PNPLA3 mRNA was markedly reduced in livers of these animals. In cultured cells, addition of free fatty acids to the medium induces lipid droplet formation and stabilizes PNPLA3, leading to increased expression of the protein.19 In mice, expression of the 148M isoform is associated with an increase in large lipid droplets, which may stabilize the mutant allele. Further studies will be required to elucidate the relationships between PNPLA3 accumulation, lipid droplet size, and hepatic steatosis.
Acknowledgments
We thank Fang Xu for excellent technical assistance and John Shelton for helpful discussions. We thank Perrin Bickel for the polyclonal antibodies to PLIN2, PLIN3, and PLIN4.
Glossary
- ATGL
adipose triglyceride lipase
- CGI-58
comparative gene identification 58
- HSL
hormone sensitive lipase
- NAFLD
nonalcoholic fatty liver disease
- PC
phosphotidylcholine
- PLIN
perilipin
- PNPLA3
patatin phospholipase-domain containing protein
- TG
triglycerides
Supporting Information
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep.27242/suppinfo.
Supplementary Information
References
- 1.Singal AK, Guturu P, Hmoud B, Kuo YF, Salameh H, Wiesner RH. Evolving frequency and outcomes of liver transplantation based on etiology of liver disease. Transplantation. 2013;95:755–760. doi: 10.1097/TP.0b013e31827afb3a. [DOI] [PubMed] [Google Scholar]
- 2.Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol. 2013;10:686–690. doi: 10.1038/nrgastro.2013.171. [DOI] [PubMed] [Google Scholar]
- 3.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 4.Smits MM, Ioannou GN, Boyko EJ, Utzschneider KM. Non-alcoholic fatty liver disease as an independent manifestation of the metabolic syndrome: results of a US national survey in three ethnic groups. J Gastroenterol Hepatol. 2013;28:664–670. doi: 10.1111/jgh.12106. [DOI] [PubMed] [Google Scholar]
- 5.McCullough AJ. The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin Liver Dis. 2004;8:521–533, viii. doi: 10.1016/j.cld.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 6.Argo CK, Caldwell SH. Epidemiology and natural history of non-alcoholic steatohepatitis. Clin Liver Dis. 2009;13:511–531. doi: 10.1016/j.cld.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 7.Hashimoto E, Yatsuji S, Tobari M, Taniai M, Torii N, Tokushige K, et al. Hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. J Gastroenterol. 2009;44(Suppl 19):89–95. doi: 10.1007/s00535-008-2262-x. [DOI] [PubMed] [Google Scholar]
- 8.Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. 2010;51:1820–1832. doi: 10.1002/hep.23594. [DOI] [PubMed] [Google Scholar]
- 9.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dongiovanni P, Donati B, Fares R, Lombardi R, Mancina RM, Romeo S, et al. PNPLA3 I148M polymorphism and progressive liver disease. World J Gastroenterol. 2013;19:6969–6978. doi: 10.3748/wjg.v19.i41.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He S, McPhaul C, Li JZ, Garuti R, Kinch L, Grishin NV, et al. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem. 2010;285:6706–6715. doi: 10.1074/jbc.M109.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Y, Cohen JC, Hobbs HH. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J Biol Chem. 2011;286:37085–37093. doi: 10.1074/jbc.M111.290114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pingitore P, Pirazzi C, Mancina RM, Motta BM, Indiveri C, Pujia A, et al. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim Biophys Acta. 2014;184:574–580. doi: 10.1016/j.bbalip.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 15.Basantani MK, Sitnick MT, Cai L, Brenner DS, Gardner NP, Li JZ, et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J Lipid Res. 2011;52:318–329. doi: 10.1194/jlr.M011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen W, Chang B, Li L, Chan L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology. 2010;52:1134–1142. doi: 10.1002/hep.23812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, et al. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest. 2012;122:4130–4144. doi: 10.1172/JCI65179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumari M, Schoiswohl G, Chitraju C, Paar M, Cornaciu I, Rangrez AY, et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 2012;15:691–702. doi: 10.1016/j.cmet.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang Y, He S, Li JZ, Seo YK, Osborne TF, Cohen JC, et al. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc Natl Acad Sci U S A. 2010;107:7892–7897. doi: 10.1073/pnas.1003585107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 21.Moon YA, Hammer RE, Horton JD. Deletion of ELOVL5 leads to fatty liver through activation of SREBP-1c in mice. J Lipid Res. 2009;50:412–423. doi: 10.1194/jlr.M800383-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engelking LJ, Kuriyama H, Hammer RE, Horton JD, Brown MS, Goldstein JL, et al. Overexpression of Insig-1 in the livers of transgenic mice inhibits SREBP processing and reduces insulin-stimulated lipogenesis. J Clin Invest. 2004;113:1168–1175. doi: 10.1172/JCI20978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horton JD, Goldstein JL, Brown MS. SREBPs: transcriptional mediators of lipid homeostasis. Cold Spring Harb Symp Quant Biol. 2002;67:491–498. doi: 10.1101/sqb.2002.67.491. [DOI] [PubMed] [Google Scholar]
- 24.Graff M, North KE, Franceschini N, Reiner AP, Feitosa M, Carr JJ, et al. PNPLA3 gene-by-visceral adipose tissue volume interaction and the pathogenesis of fatty liver disease: the NHLBI family heart study. Int J Obes. 2013;37:432–438. doi: 10.1038/ijo.2012.65. (Lond) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palmer CN, Maglio C, Pirazzi C, Burza MA, Adiels M, Burch L, et al. Paradoxical lower serum triglyceride levels and higher type 2 diabetes mellitus susceptibility in obese individuals with the PNPLA3 148M variant. PLoS One. 2012;7:e39362. doi: 10.1371/journal.pone.0039362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davis JN, Le KA, Walker RW, Vikman S, Spruijt-Metz D, Weigensberg MJ, et al. Increased hepatic fat in overweight Hispanic youth influenced by interaction between genetic variation in PNPLA3 and high dietary carbohydrate and sugar consumption. Am J Clin Nutr. 92:1522–1527. doi: 10.3945/ajcn.2010.30185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumashiro N, Yoshimura T, Cantley JL, Majumdar SK, Guebre-Egziabher F, Kursawe R, et al. Role of patatin-like phospholipase domain-containing 3 on lipid-induced hepatic steatosis and insulin resistance in rats. Hepatology. 2013;57:1763–1772. doi: 10.1002/hep.26170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Speliotes EK, Butler JL, Palmer CD, Voight BF, Hirschhorn JN. PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology. 2010;52:904–912. doi: 10.1002/hep.23768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7:e1001324. doi: 10.1371/journal.pgen.1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kantartzis K, Peter A, Machicao F, Machann J, Wagner S, Konigsrainer I, et al. Dissociation between fatty liver and insulin resistance in humans carrying a variant of the patatin-like phospholipase 3 gene. Diabetes. 2009;58:2616–2623. doi: 10.2337/db09-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Santoro N, Kursawe R, D'Adamo E, Dykas DJ, Zhang CK, Bale AE, et al. A common variant in the patatin-like phospholipase 3 gene (PNPLA3) is associated with fatty liver disease in obese children and adolescents. Hepatology. 2010;52:1281–1290. doi: 10.1002/hep.23832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kotronen A, Johansson LE, Johansson LM, Roos C, Westerbacka J, Hamsten A, et al. A common variant in PNPLA3, which encodes adiponutrin, is associated with liver fat content in humans. Diabetologia. 2009;52:1056–1060. doi: 10.1007/s00125-009-1285-z. [DOI] [PubMed] [Google Scholar]
- 33.Lass A, Zimmermann R, Haemmerle G, Riederer M, Schoiswohl G, Schweiger M, et al. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab. 2006;3:309–319. doi: 10.1016/j.cmet.2006.03.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information