

Abstract
Purpose:
Expression of imprinted genes is regulated by DNA methylation of differentially methylated regions (DMRs). Beckwith–Wiedemann syndrome is an imprinting disorder caused by epimutations of DMRs at 11p15.5. To date, multiple methylation defects have been reported in Beckwith–Wiedemann syndrome patients with epimutations; however, limited numbers of DMRs have been analyzed. The susceptibility of DMRs to aberrant methylation, alteration of gene expression due to aberrant methylation, and causative factors for multiple methylation defects remain undetermined.
Methods:
Comprehensive methylation analysis with two quantitative methods, matrix-assisted laser desorption/ionization mass spectrometry and bisulfite pyrosequencing, was conducted across 29 DMRs in 54 Beckwith–Wiedemann syndrome patients with epimutations. Allelic expressions of three genes with aberrant methylation were analyzed. All DMRs with aberrant methylation were sequenced.
Results:
Thirty-four percent of KvDMR1–loss of methylation patients and 30% of H19DMR–gain of methylation patients showed multiple methylation defects. Maternally methylated DMRs were susceptible to aberrant hypomethylation in KvDMR1–loss of methylation patients. Biallelic expression of the genes was associated with aberrant methylation. Cis-acting pathological variations were not found in any aberrantly methylated DMR.
Conclusion:
Maternally methylated DMRs may be vulnerable to DNA demethylation during the preimplantation stage, when hypomethylation of KvDMR1 occurs, and aberrant methylation of DMRs affects imprinted gene expression. Cis-acting variations of the DMRs are not involved in the multiple methylation defects.
Keywords: Beckwith–Wiedemann syndrome, DNA methylation, differentially methylated region, genomic imprinting, multiple methylation defects
Introduction
Genomic imprinting is an epigenetic phenomenon that leads to parent-specific differential expression of a subset of mammalian genes. Most imprinted genes are clustered in regions called imprinting domains, and the expression of imprinted genes within these domains is regulated by imprinting control regions.1,2 Differentially methylated regions (DMRs), which are defined as having DNA methylation on only one of the two parental alleles, play critical roles in the regulation of imprinting. There are two kinds of DMRs: maternally methylated DMRs (matDMRs) and paternally methylated DMRs (patDMRs). In addition, there is another classification, gametic DMRs and somatic DMRs, based on the timing of the establishment of differential methylation. Gametic DMRs acquire DNA methylation during gametogenesis, and the methylation is maintained from zygote to somatic cells during all developmental stages. Most gametic DMRs are identical to imprinting control regions. On the other hand, somatic DMRs are established during early embryogenesis after fertilization under the control of nearby imprinting control regions.1,2 Because imprinted genes play an important role in the growth and development of embryos, placental formation, and metabolism, aberrant expression of imprinted genes due to epigenetic or genetic abnormalities is implicated in the pathogenesis of some human disorders, such as congenital anomalies and tumors.1,2
Beckwith–Wiedemann syndrome (BWS; Online Mendelian Inheritance in Man (OMIM) #130650) is an imprinting disease that is characterized by prenatal and postnatal macrosomia, macroglossia, abdominal wall defects, and variable minor features. The relevant imprinted chromosomal region in BWS is 11p15.5, which consists of two imprinted domains, IGF2/H19 and CDKN1C/KCNQ1OT1, H19DMR and KvDMR1 being the respective imprinting control regions.3,4,5 Among several causative alterations identified so far, loss of methylation (LOM) at KvDMR1 and gain of methylation (GOM) at H19DMR are isolated epimutations. Hypomethylation at multiple imprinted DMRs has been reported in patients with transient neonatal diabetes mellitus type 1,6 and the same phenomenon, referred to as multiple methylation defects (MMDs), has been reported in BWS patients with KvDMR1-LOM.7,8,9,10,11,12,13 However, although the human genome contains more than 30 imprinting domains (http://www.geneimprint.com), a limited number of imprinted DMRs have been analyzed so far, with the exception of a report by Court et al.12 In addition, methods used for methylation analysis have ranged from nonquantitative to quantitative approaches, and although some studies have used only one method for methylation analysis,8,9,11 others have used two or more in conjuction.7,10,11,12,13 Furthermore, the questions of whether susceptibility to aberrant methylation is different in each type of DMR, whether aberrant methylation indeed affects imprinted gene expression, and what causative factors are responsible for MMDs still remain unanswered. To clarify these issues, we have conducted a comprehensive methylation screening in BWS patients with KvDMR1-LOM or H19DMR-GOM with a quantitative method, matrix-assisted laser desorption/ionization mass spectrometry (MALDI-TOF MS), on 29 imprinted DMRs, which represents the largest number of DMRs analyzed to date, followed by confirmation with another quantitative method, bisulfite pyrosequencing. We also performed gene expression analysis and sequencing of aberrantly methylated DMRs. We found that matDMRs are susceptible to aberrant methylation. We also found alterations in imprinted gene expression due to the aberrant methylation and no cis-acting pathological variations in DMRs with MMDs.
Materials and Methods
Patients
Fifty-four BWS patients (25 boys, 26 girls, 3 gender-unspecified patients; average age: 3.0 years (0–13.9 years)) and their parents were enrolled in this study. Among them, 46 patients met clinical criteria for BWS as described by Weksberg et al.3 and 6 patients met clinical criteria as described by DeBaun et al.14 (Supplementary Table S1 online). Because two patients were clinically diagnosed more than 20 years ago, their specific diagnostic criteria were unknown. The methylation statuses of H19DMR and KvDMR1, paternal uniparental disomy of chromosome 11 (upd(11)pat), and CDKN1C mutations were screened as described previously.15,16,17 Peripheral blood samples of most patients were subjected to standard G-banding chromosome analysis and/or high-resolution G-band patterning of human chromosome 11, but neither assay showed any abnormalities in any patient (data not shown). Among the 54 patients, 44 displayed KvDMR1-LOM but did not show other causative alterations, including H19DMR-GOM, upd(11)pat, and CDKN1C mutations (data not shown). The remaining 10 patients displayed H19DMR-GOM but did not show other causative alterations (data not shown). We sequenced the entire H19DMR in H19DMR-GOM patients and found no mutations.18 We used the peripheral blood samples of 24 children (11 boys, 13 girls; average age: 3.8 years (range of 0–8 years)) who visited the Department of Pediatrics, Saga University Hospital, as normal controls having only mild illness such as common cold. This study was approved by the Ethics Committee for Human Genome and Gene Analyses of the Faculty of Medicine, Saga University. Written informed consent was obtained from the parents or the guardians of the patients and participants.
DNA isolation and bisulfite conversion
Genomic DNA was extracted from the peripheral blood of patients using the FlexiGene DNA Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. A total of 1 µg of genomic DNA was subjected to bisulfite conversion using the EZ DNA Methylation Kit (Zymo Research, Irvine, CA), and then the converted DNA was eluted in 100 µl of water. Unmethylated control DNA was created by whole-genome amplification using the REPLI-g Mini Kit (Qiagen). To prepare fully methylated control DNA, the unmethylated DNA created by whole-genome amplification was treated twice with SssI methylase.
Methylation analysis by MALDI-TOF MS
The DNA methylation status of imprinted DMRs was analyzed by MALDI-TOF MS analysis with a MassARRAY system (Sequenom, San Diego, CA) as previously described.19,20 Briefly, each DMR was amplified by bisulfite-mediated polymerase chain reaction (PCR) using a primer set containing a primer carrying the T7 promoter sequence at the 5′ end. In vitro transcription of the PCR product was performed with T7 RNA polymerase, and the transcript was subjected to uracil-specific cleavage with RNase A. MALDI-TOF MS analysis of the cleaved fragments produced signal pattern pairs indicative of nonmethylated and methylated DNA. Epityper software (Sequenom) analysis of the signals yielded a methylation index (MI) ranging from 0 (no methylation) to 1 (full methylation) for each CpG unit, which contained one or more CpG sites. Aberrant methylation of a CpG unit was defined as the condition in which the difference of MIs between each patient and the average of normal controls exceeded 0.15. This definition was based on our finding in methylation-sensitive Southern blots, which revealed that the differences in MI for KvDMR1-LOM or H19DMR-GOM in BWS patients were ≥0.15 (data not shown). Because the analyzed DMRs included several CpG units, aberrant methylation of each DMR was defined as the situation in which more than 60% of the total number of analyzed CpG units showed aberrant methylation (with the MI difference exceeding 0.15). In the case of IGF2-DMR0, the three CpG sites were analyzed based on previous reports.21,22 All primers used in this study are shown in Supplementary Table S2 online.
Methylation analysis by bisulfite pyrosequencing
The aberrant methylation status of DMRs identified by MALDI-TOF MS was confirmed by bisulfite pyrosequencing using QIAGEN PyroMark Q24 according to the manufacturer's instructions (Qiagen). Primers for bisulfite-mediated PCR and pyrosequencing were designed using PyroMark Assay Design 2.0 (Qiagen). In analogy with MALDI-TOF MS analysis, aberrant methylation of a CpG site was defined as the situation in which the difference of MIs between each patient and the average of normal controls exceeded 0.15. Aberrant methylation of each DMR was defined as the condition in which more than 60% of the total number of analyzed CpG sites showed aberrant methylation (with the MI difference exceeding 0.15).
Bisulfite sequencing
Bisulfite sequencing was performed to analyze allelic methylation of ZDBF2-DMR. After PCR amplification, the PCR products were cloned into a pT7Blue T-Vector (Novagen, Darmstadt, Germany), and individual clones were sequenced. Parental alleles were distinguished by a single-nucleotide polymorphism (SNP, rs1861437) within the DMR.
Expression analysis of ZDBF2, FAM50B, and GNAS1A
Total RNA was extracted from the peripheral blood of patients using the QIAamp RNA Blood Mini Kit (Qiagen). The RNA was treated with RNase-free DNase I, and reverse transcription was performed with random primers. We used SNPs for allelic expression to distinguish between the two parental alleles: rs10932150 in exon 5 of ZDBF2; rs6597007 in exon 2 of FAM50B; and rs143800311, which is a 5-bp deletion/insertion variation in exon 1A of GNAS1A. Reverse transcription–PCR (RT-PCR) products encompassing the SNPs of ZDBF2 and FAM50B were directly sequenced. The products encompassing the deletion/insertion variation of GNAS1A were separated by electrophoresis on an Applied Biosystems 3130 genetic analyzer (Applied Biosystems, Foster City, CA) and then analyzed with GeneMapper software (Applied Biosystems). Total expression levels of ZDBF2 and FAM50B were quantitated by real-time PCR with TaqMan probes (Applied Biosystems). The expression level of each gene was normalized against that of the housekeeping genes encoding hydroxymethylbilane synthase (HMBS) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). All quantitative RT-PCRs were performed in triplicate.
Sequencing of aberrantly methylated DMRs
Direct sequencing of all DMRs showing aberrant methylation in KvDMR1-LOM patients was performed to determine whether there was any pathological variation.
Statistical analyses
Fisher's exact test was used for the comparison of aberrant methylated DMRs. Fisher's exact test or Mann–Whitney U-test was used for statistical analyses of clinical features between MMDs and monolocus methylation defects in KvDMR1-LOM patients. A P value < 0.05 was considered statistically significant.
Results
Validation of methylation analyses, MALDI-TOF MS, and bisulfite pyrosequencing
First, we selected 37 regions reported previously as imprinted DMRs in the human genome16,20,23 (refer to http://www.geneimprint.com/). To validate the quantitative capability of MALDI-TOF MS methylation analysis, mixtures of the unmethylated control DNA and the fully methylated control DNA (0, 25, 50, 75, and 100% methylated DNA) were subjected to bisulfite conversion and analyzed. We found a significant correlation between the measured MIs and predicted MIs in all DMRs, except for GRB10, PEG13, and IG-DMR-CG4 (Supplementary Figure S1 online). Furthermore, in normal leukocytes, two regions (TCEB3C, USP29) showed mostly full methylation and three regions (TP73, SPTBN1, WT1-AS) showed mostly no methylation, suggesting that these regions were not differentially methylated in leukocytes (data not shown). Therefore, we excluded these eight regions and decided to analyze the remaining 29 DMRs by MALDI-TOF MS. Second, we obtained MIs from 24 normal controls using MALDI-TOF MS and calculated the average and SD of each CpG unit. We excluded CpG units in which SDs were >0.1 from further analysis. Averages and SDs of all CpG units analyzed in this study are shown in Supplementary Table S3 online. After the MALDI-TOF MS analysis, we used bisulfite pyrosequencing to confirm the aberrant methylation uncovered. We also obtained MIs from the 24 controls using bisulfite pyrosequencing and calculated the average and SD of each CpG site. We excluded one CpG site in H19DMR because its SD was >0.1 due to a known SNP (rs10732516). Averages and SDs of control CpG sites are shown in Supplementary Table S3 online. Finally, we compared the MIs of MALDI-TOF MS and bisulfite pyrosequencing of each DMR and found a significant correlation (Supplementary Figure S2 online).
Multilocus methylation defects in BWS patients with epimutations
Among the 44 KvDMR1-LOM patients, 15 (34.1%) showed aberrantly methylated DMRs outside of KvDMR1: six showed aberrant methylation at only one DMR, and the other nine showed two or more methylated DMRs (Figure 1a and Supplementary Figure S3 online). The greatest number of aberrantly methylated DMRs was found in patient BWS-s113, who exhibited 12 DMRs. Most of the aberrantly methylated DMRs demonstrated LOM, which was seen at ARHI-CG1, ARHI-CG2, ARHI-CG3, FAM50B, ZAC, IGF2R-DMR2, MEST, NNAT, L3MBTL1, NESPAS, GNASXL, and GNAS1A. Among them, the most frequently hypomethylated DMRs were ARHI-CG1 and ARHI-CG3, found in nine (20.5%) and eight (18.2%) patients, respectively. By contrast, three DMRs, located at ZDBF2, NESP, and MCTS2, showed GOM, which was found in six (13.6%), two (4.5%), and one (2.3%) patients, respectively. GNASXL-DMR showed GOM in one patient (2.3%), whereas four patients (9.1%) showed LOM. The other 13 DMRs were not aberrantly methylated in any KvDMR1-LOM patient.
Figure 1.

Results of methylation analyses of 29 imprinted differentially methylated regions (DMRs) in Beckwith–Wiedemann syndrome patients with epimutations. (a) Results of patients with KvDMR1-LOM. Only the results of multiple methylation defects are shown. Aberrant methylation was confirmed by two quantitative methods: matrix-assisted laser desorption/ionization mass spectrometry and bisulfite pyrosequencing. The definition of aberrant methylation used here is described in the Materials and Methods section. Shaded rectangle: aberrant hypomethylation; dark gray rectangle: aberrant hypermethylation. (b) Results of all patients with H19DMR-GOM. GOM, gain of methylation; LOM, loss of methylation.
Among the 10 H19DMR-GOM patients, all patients showed GOM at the H19 promoter DMR, which was usually observed with loss of imprinting of IGF2 (Figure 1b).24 Four patients showed GOM at either IGF2-DMR0 or IGF2-DMR2; two patients showed GOM at both. Moreover, both LOM and GOM at other DMRs were found: LOM was found at INPP5Fv2-DMR in patients BWS-s015 and BWS-s064, and GOM was found at NESP-DMR in patient BWS-s012.
In addition, to exclude aberrantly methylated DMRs resulting from chromosome abnormalities such as uniparental disomy and copy number abnormality, microsatellite analyses using patients' and their parents' DNA were performed on all DMRs showing aberrant methylation. For quantitative analyses, tetranucleotide repeat markers near the imprinted DMRs were used (Supplementary Materials and Methods online). We found that no DMRs, except for six DMRs in three patients, exhibited any chromosome abnormalities (summarized in Supplementary Figure S4 online). These results strongly suggest that the aberrant methylation of DMRs observed was an isolated epimutation and was not due to chromosome abnormalities.
Comparison of aberrantly methylated DMRs
We found that 34.1% (15 of 44) of KvDMR1-LOM patients and 30.0% (3 of 10) of H19DMR-GOM patients showed MMDs (Figure 1a). There was no statistical difference between them (P > 0.99, Fisher's exact test).
Among the 29 DMRs analyzed, there were 20 gametic DMRs and 8 somatic DMRs (Figure 1a). The timing of methylation establishment of one DMR (FAM50B-DMR) has not yet been determined. On the other hand, there were 20 matDMRs and 9 patDMRs. We investigated whether susceptibility to aberrant methylation differed for each type of DMR in KvDMR1-LOM patients. KvDMR1 itself, a gametic and matDMR, was excluded from this analysis. Several DMRs were mapped to certain imprinted domains, e.g., three DMRs in the ARHI domain and four in the GNAS domain. However, these DMRs differed by type, and aberrant methylations of these DMRs were not always linked. We also had previously found that DMRs in the GNAS domain were independently aberrantly methylated in hepatoblastoma.20 Therefore, we decided to perform statistical analyses assuming the independence of each DMR.
We first compared gametic DMRs with somatic DMRs and found no significant difference in susceptibility (P = 0.42, Fisher's exact test; Figure 2a). FAM50B-DMR was excluded from this comparison. By contrast, matDMRs were aberrantly methylated more frequently than patDMRs (P = 0.042, Fisher's exact test; Figure 2b). In addition, among the aberrantly methylated DMRs, 12 showed LOM and 4 showed GOM. When we compared LOM with GOM, LOM preferentially occurred on matDMRs (P = 0.050, Fisher's exact test; Figure 2c). In this subanalysis, GNASXL-DMR was counted as having both GOM and LOM (Figure 1a). Furthermore, among the 12 DMRs with LOM, most of them (10) were gametic DMRs. These results suggest that matDMRs are susceptible to aberrant methylation and that gametic maternally methylated DMRs tend to be susceptible to LOM in KvDMR1-LOM patients.
Figure 2.
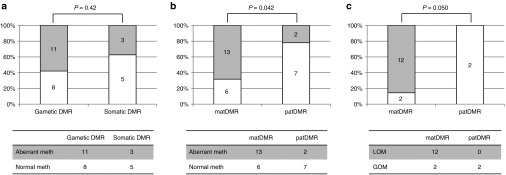
Statistical analyses of aberrantly methylated differentially methylated region (DMRs). (a) Comparison of the number of aberrantly methylated DMRs between gametic DMRs and somatic DMRs in KvDMR1-LOM patients. There was no statistical difference between the two DMRs (P = 0.42, Fisher's exact test). (b) Comparison of the number of aberrantly methylated DMRs between matDMRs and patDMRs in KvDMR1-LOM patients. matDMRs were aberrantly methylated more frequently than patDMRs (P = 0.042, Fisher's exact test). (c) Comparison of the number of LOMs and GOMs between matDMRs and patDMRs among the aberrantly methylated DMRs in KvDMR1-LOM patients. LOM preferentially occurred on matDMRs (P = 0.050, Fisher's exact test). GOM, gain of methylation; LOM, loss of methylation; matDMR, maternally methylated DMR; patDMR, paternally methylated DMR.
Biallelic expression of imprinted genes induced by aberrant methylation at their corresponding DMRs
We continued our investigation by determining whether allelic expression was associated with the methylation status of the corresponding DMR. We selected three genes (ZDBF2, FAM50B, and GNAS1A) expressed in lymphocytes.25,26,27 In the case of ZDBF2, bisulfite sequencing of ZDBF2-DMR showed paternal monoallelic methylation in normal controls heterozygous for a specific SNP (rs1861437), whereas four BWS patients with GOM showed biallelic methylation: these findings were consistent with the results of MALDI-TOF MS and bisulfite pyrosequencing (Figure 3a,b and Supplementary Figure S5 online). Because paternal expression of the ZDBF2 gene is coupled with methylation of ZDBF2-DMR on the paternal allele,25 biallelic expression due to biallelic methylation was expected. Indeed, three BWS patients heterozygous for a coding SNP (rs10932150) with hypermethylated DMRs clearly showed biallelic expression, in contrast with the paternal monoallelic expression in patients with normally methylated DMRs (Figure 3c). FAM50B and GNAS1A were paternally expressed and were coupled with maternal methylation of corresponding DMRs. RT-PCR using coding SNPs (rs6597007 for FAM50B and rs143800311 for GNAS1A) revealed that both genes were expressed biallelically with LOM of each corresponding DMR, which was in contrast with monoallelic expression in the patients with normally methylated DMRs (Figure 4 and Supplementary Figure S5 online). It is intriguing that FAM50B in patient BWS-s096 and GNAS1A in patient BWS-s062 were expressed from the maternal allele despite low-grade LOM, which suggests that our definition of aberrant methylation is appropriate. In addition, we investigated the expression levels of ZDBF2 and FAM50B by quantitative RT-PCR. The expression levels in patients with aberrantly methylated DMRs were higher than those in patients with normally methylated DMRs (Supplementary Figure S6 online). These results indicate that allelic expression and expression levels were indeed associated with the methylation status of the corresponding DMR in patients with MMDs.
Figure 3.

Methylation analysis of ZDBF2-DMR and expression analysis of the ZDBF2 gene. (a) Results of matrix-assisted laser desorption/ionization mass spectrometry analysis. Averages with SD of 24 normal controls are shown in blue. Methylation indexes of the patients showing GOM are indicated in different colors. Units 1 and 2 included two and one CpG sites, respectively. (b) Results of bisulfite sequencing. Normal controls show monoallelic differential methylation, whereas four Beckwith–Wiedemann syndrome (BWS) patients (BWS-s001, BWS-s011, BWS-s023, and BWS-s060) show biallelic methylation. Two parental alleles were distinguished by a SNP (rs1861437). Mat, maternal allele; Pat, paternal allele. (c) Results of expression analysis of the ZDBF2 gene. Three BWS patients (BWS-s001, BWS-s011, and BWS-s113) heterozygous for a coding SNP (rs10932150) with GOM clearly showed biallelic expression; by contrast, two patients with normally methylated differentially methylated region (DMRs) exhibited paternal monoallelic expression (patients BWS-s004 and BWS-s060). gDNA, genomic DNA; GOM, gain of methylation; SNP, single-nucleotide polymorphism.
Figure 4.
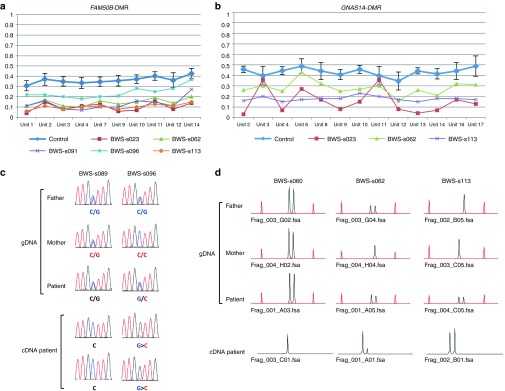
Methylation analysis of FAM50B- and GNAS1A-DMRs and expression analysis of the FAM50B and GNAS1A genes. (a,b) Results of matrix-assisted laser desorption/ionization mass spectrometry analysis. Averages with SD of 24 normal controls are shown in blue. Methylation indexes of patients showing LOM are indicated in different colors. Ten CpG units analyzed for FAM50B-DMR covered 13 CpG sites, and 13 CpG units analyzed for GNAS1A-DMR covered 18 CpG sites. (c) Results of expression analysis of the FAM50B gene. Beckwith–Wiedemann syndrome (BWS) patient BWS-s096 was heterozygous for a coding SNP (rs6597007) with LOM and showed biallelic expression with a low peak of maternal expression, whereas monoallelic expression was seen in a patient with normally methylated differentially methylated regions (DMRs) (patient BWS-s089). In patient BWS-s096, maternal expression was noted in two independent analyses despite low-grade LOM. gDNA, genomic DNA. (d) Results of expression analysis of the GNAS1A gene. Patients BWS-s062 and BWS-s113, heterozygous for a deletion/insertion variation (rs143800311) with LOM, showed biallelic expression, whereas patient BWS-s060 possessed normally methylated DMRs and exhibited monoallelic expression. Maternal expression was noted despite low-grade LOM in patient BWS-s062. Red peaks are molecular markers. GOM, gain of methylation; LOM, loss of methylation.
Lack of pathological variation in all aberrantly methylated DMRs in KvDMR1-LOM patients
Because the genetic aberrations of H19DMR explained only ~20% of BWS patients with H19DMR-GOM,28 we hypothesized the existence of cis-acting variations within aberrantly methylated DMRs. Therefore, we sequenced all aberrantly methylated DMRs, including KvDMR1, in KvDMR1-LOM patients. However, no variations were found in any aberrantly hypomethylated DMRs, except for four known SNPs (summarized in Supplementary Figure S7 online), suggesting that cis-acting pathological variations are not involved in aberrant methylation of these DMRs.
No difference in clinical features between MMDs and monolocus methylation defects
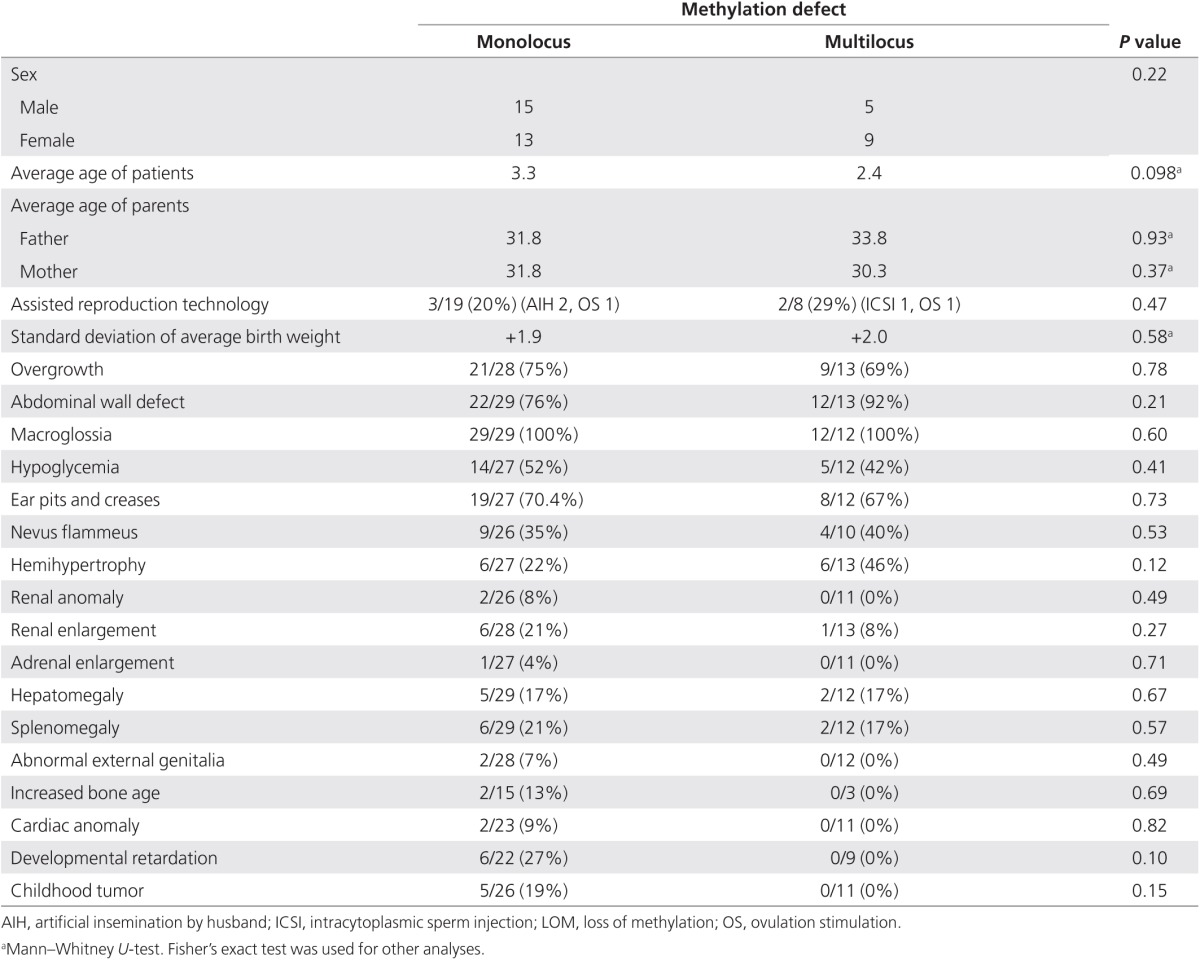
In KvDMR1-LOM patients, there was no significant difference in clinical features between MMDs and monolocus methylation defects, which demonstrated LOM only at KvDMR1 (Table 1). Among 27 patients with KvDMR1-LOM for whom information on conception was available, one patient was conceived using intracytoplasmic sperm injection, two were from artificial insemination by the husband, and two were from ovulation stimulation. We searched for a link between assisted reproductive technology and MMD but could find no relationship (Table 1). The average age of neither the mother nor the father differed between patients with MMDs versus those with monolocus methylation defects (Table 1). The fact that monozygotic twins discordant for BWS were found predominantly for females suggests an insufficient amount of DNA methyltransferase 1 (DNMT1) to maintain KvDMR1 methylation during the overlap in timing with X-chromosome inactivation and twinning.29 This hypothesis suggests that females might tend to suffer from MMDs. We compared the frequency of female patients with MMDs with the frequency of those with monolocus methylation defects, but no significant difference could be found (Table 1).
Table 1. Clinical features of KvDMR-LOM patients with monolocus methylation defect and those with multilocus methylation defects.
Discussion
Currently, most reports have studied 3–10 imprinted DMRs in BWS patients,7,8,9,10,13 with the exception of two reports in which 16 and 27 DMRs were analyzed.11,12 In addition, the quantitative capability of methods used for multiple methylation analyses has been variable, and few studies have conducted multiple checks to confirm the methylation statuses of all DMRs showing aberrant methylation.7,8,9,10,11,12,13 To resolve these matters, we analyzed 29 DMRs and confirmed all aberrantly methylated DMRs using MALDI-TOF MS and bisulfite pyrosequencing, which are the most reliable quantitative methods of methylation analysis available at present.19,30,31 We found that 34.1% of KvDMR1-LOM patients exhibited MMDs. The frequency was higher than that in previous reports, which can be summarized as reporting an overall frequency of 20.6% (102 of 495 patients).7,8,9,10,11,12,13 However, within these reports, the frequency in studies that analyzed 10 or fewer DMRs is 19.0% (82 of 431),7,8,9,10,13 and the frequency in studies that analyzed more than 10 DMRs is 31.3% (20 of 64).11,12 In addition, we found that 30.0% of H19DMR-GOM patients showed MMDs, which is surprising considering that no MMDs were found in two previous reports in which 10 and 16 DMRs were analyzed.8,11 These data suggest that the greater the number of DMRs analyzed, the higher the frequency of MMDs observed. In future, all DMRs in the genome should be analyzed to understand the precise frequency of MMDs, which DMRs become preferentially aberrantly methylated, and the mechanism by which MMDs occur.
In both KvDMR1-LOM patients and H19DMR-GOM patients, we found MMDs in which not only LOM but also GOM were seen. We also found that both matDMRs and patDMRs were aberrantly methylated in both patient groups. It is noteworthy that matDMRs, probably gametic maternally methylated DMRs, were more susceptible to aberrant methylation than patDMRs in KvDMR1-LOM patients, although no particular parent-based pattern of aberrant methylation has been reported previously.12 This suggests that gametic maternally methylated DMRs are vulnerable to DNA demethylation during the preimplantation stage of early embryogenesis when KvDMR1-LOM occurs.
Although it has not been reported that aberrant methylation of the corresponding DMR affects imprinted gene expression in MMD patients, we found biallelic expression of three imprinted genes (ZDBF2, FAM50B, and GNAS1A) to be associated with the aberrant methylation of their respective DMRs. Because biallelic expression increased the total expression levels of ZDBF2 and FAM50B, we expect that had we measured the expression levels of GNAS1A, we would have observed an increase. Therefore, alteration of gene expression levels due to MMDs might affect the phenotype; however, clinical features between MMDs and monolocus methylation defects were not different in our study. This lack of difference has been previously reported,7,9,10,13 although a few groups have reported a difference in clinical features.8,11,12 Two reasons for this similarity in terms of clinical features could be suggested. First, the mosaic ratio might be different in each organ. Because aberrant methylation was generally partial, it would occur after fertilization, and the patients would be mosaic. A high mosaic ratio would be a critical factor in the emergence of a distinct phenotype in BWS patients with monolocus methylation defects. Second, the imprinted locus at 11p15 might be epidominant over other imprinted loci because all MMD patients were clinically diagnosed as BWS.
Regarding the causative factor(s) for MMD, we could not find any pathological variation in any aberrantly methylated DMR, including KvDMR1, suggesting that cis-acting variations of each specific DMR itself were not involved in the genesis of MMDs. On the other hand, the involvement of trans-acting factors has been advocated in other reports because mutations of ZFP57 (which are required for the postfertilization maintenance of maternal and paternal methylation imprinting at multiple loci) have been found in transient neonatal diabetes mellitus type 1 patients with multilocus hypomethylation.32 Mutations of NLRP2 were also identified in a BWS patient with KvDMR1-LOM and MEST-LOM in a family with complex consanguinity and in a Silver–Russell syndrome patient with multilocus hypomethylation.12,33 In addition, TRIM28, NLRP7, KHDC3L, and DNMT3L have been considered to be candidate trans-acting factors. However, no mutations in any of these candidates or other genes, such as DNMT1, DNMT3A, and DNMT3B, were found in our BWS patients with MMDs, as determined by exome sequencing (K. Sasaki and K. Hata, personal communication). Recently, Lorthongpanich et al.34 reported that the absence of maternal Trim28 until zygotic gene activation at the two-cell late stage caused mosaicism of MMDs randomly, suggesting that insufficient expression of the candidate gene(s) at very early embryogenesis is an important event in the generation of MMDs in human imprinted diseases. Whole-genome sequencing and whole-genome bisulfite sequencing, including the regulatory regions of the candidate genes, and transcriptome analysis in early embryogenesis would be useful to identify the cause(s) of MMDs.
In our H19DMR-GOM patients, we also found GOM of IGF2-DMR0 and IGF2-DMR2 to be associated with GOM of H19DMR and H19promoter DMR, in agreement with previous reports.22,35,36 Two patients showed simultaneous GOM at both IGF2-DMRs. Because Igf2-DMRs were established at the postimplantation stage under the control of H19DMR in mice,37 GOM of IGF2-DMRs in BWS is likely to occur at the same stage. Although the function of IGF2-DMR0 is still unknown, methylated Igf2-DMR2 plays a role in transcription initiation of Igf2 in mice.38 GOM of the DMRs might change the high-order chromatin structure of the maternal allele and increase the expression of IGF2 in cooperation with H19DMR-GOM in BWS patients.
In conclusion, our comprehensive and quantitative methylation analysis of multiple imprinted DMRs revealed several new findings: (i) matDMRs, probably gametic maternally methylated DMRs, are more susceptible to aberrant methylation during the preimplantation stage, when KvDMR1-LOM occurs; (ii) aberrant methylation indeed alters imprinted gene expression; and (iii) cis-acting pathological variations of each DMR are not involved in the MMDs analyzed. Moreover, our study confirmed the simultaneous aberrant hypermethylation of IGF2-DMR0 and/or -DMR2 with isolated H19DMR-GOM. These findings may help us to understand the molecular mechanisms and pathophysiological features of MMDs.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
We thank all the participants and their families who provided samples and all the doctors who referred patients to us. This study was supported, in part, by a Grant for Research on Intractable Diseases from the Ministry of Health, Labor, and Welfare; a Grant for Child Health and Development from the National Center for Child Health and Development; a Grant-in-Aid for Challenging Exploratory Research; and a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science.
Supplementary Material
References
- Abramowitz LK, Bartolomei MS. Genomic imprinting: recognition and marking of imprinted loci. Curr Opin Genet Dev. 2012;22:72–78. doi: 10.1016/j.gde.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomizawa S, Sasaki H. Genomic imprinting and its relevance to congenital disease, infertility, molar pregnancy and induced pluripotent stem cell. J Hum Genet. 2012;57:84–91. doi: 10.1038/jhg.2011.151. [DOI] [PubMed] [Google Scholar]
- Weksberg R, Shuman C, Beckwith JB. Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2010;18:8–14. doi: 10.1038/ejhg.2009.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choufani S, Shuman C, Weksberg R. Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:343–354. doi: 10.1002/ajmg.c.30267. [DOI] [PubMed] [Google Scholar]
- Soejima H, Higashimoto K. Epigenetic and genetic alterations of the imprinting disorder Beckwith-Wiedemann syndrome and related disorders. J Hum Genet. 2013;58:402–409. doi: 10.1038/jhg.2013.51. [DOI] [PubMed] [Google Scholar]
- Mackay DJ, Boonen SE, Clayton-Smith J, et al. A maternal hypomethylation syndrome presenting as transient neonatal diabetes mellitus. Hum Genet. 2006;120:262–269. doi: 10.1007/s00439-006-0205-2. [DOI] [PubMed] [Google Scholar]
- Rossignol S, Steunou V, Chalas C, et al. The epigenetic imprinting defect of patients with Beckwith-Wiedemann syndrome born after assisted reproductive technology is not restricted to the 11p15 region. J Med Genet. 2006;43:902–907. doi: 10.1136/jmg.2006.042135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliek J, Verde G, Callaway J, et al. Hypomethylation at multiple maternally methylated imprinted regions including PLAGL1 and GNAS loci in Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2009;17:611–619. doi: 10.1038/ejhg.2008.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzi S, Rossignol S, Steunou V, et al. Multilocus methylation analysis in a large cohort of 11p15-related foetal growth disorders (Russell Silver and Beckwith Wiedemann syndromes) reveals simultaneous loss of methylation at paternal and maternal imprinted loci. Hum Mol Genet. 2009;18:4724–4733. doi: 10.1093/hmg/ddp435. [DOI] [PubMed] [Google Scholar]
- Lim D, Bowdin SC, Tee L, et al. Clinical and molecular genetic features of Beckwith-Wiedemann syndrome associated with assisted reproductive technologies. Hum Reprod. 2009;24:741–747. doi: 10.1093/humrep/den406. [DOI] [PubMed] [Google Scholar]
- Poole RL, Docherty LE, Al Sayegh A, et al. International Clinical Imprinting Consortium Targeted methylation testing of a patient cohort broadens the epigenetic and clinical description of imprinting disorders. Am J Med Genet A. 2013;161:2174–2182. doi: 10.1002/ajmg.a.36049. [DOI] [PubMed] [Google Scholar]
- Court F, Martin-Trujillo A, Romanelli V, et al. Genome-wide allelic methylation analysis reveals disease-specific susceptibility to multiple methylation defects in imprinting syndromes. Hum Mutat. 2013;34:595–602. doi: 10.1002/humu.22276. [DOI] [PubMed] [Google Scholar]
- Tee L, Lim DH, Dias RP, et al. Epimutation profiling in Beckwith-Wiedemann syndrome: relationship with assisted reproductive technology. Clin Epigenetics. 2013;5:23. doi: 10.1186/1868-7083-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBaun MR, Tucker MA. Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. J Pediatr. 1998;132 3 Pt 1:398–400. doi: 10.1016/s0022-3476(98)70008-3. [DOI] [PubMed] [Google Scholar]
- Soejima H, Nakagawachi T, Zhao W, et al. Silencing of imprinted CDKN1C gene expression is associated with loss of CpG and histone H3 lysine 9 methylation at DMR-LIT1 in esophageal cancer. Oncogene. 2004;23:4380–4388. doi: 10.1038/sj.onc.1207576. [DOI] [PubMed] [Google Scholar]
- Higashimoto K, Nakabayashi K, Yatsuki H, et al. Aberrant methylation of H19-DMR acquired after implantation was dissimilar in soma versus placenta of patients with Beckwith-Wiedemann syndrome. Am J Med Genet A. 2012;158A:1670–1675. doi: 10.1002/ajmg.a.35335. [DOI] [PubMed] [Google Scholar]
- Yatsuki H, Higashimoto K, Jozaki K, et al. Novel mutations of CDKN1C in Japanese patients with Beckwith-Wiedemann syndrome. Genes & Genomics. 2013;35:141–147. [Google Scholar]
- Higashimoto K, Jozaki K, Kosho T, et al. Clin Genet. e-pub ahead of print 8 November 2013; 2013. A novel de novo point mutation of the OCT-binding site in the IGF2/H19-imprinting control region in a Beckwith-Wiedemann syndrome patient. [DOI] [PubMed] [Google Scholar]
- Ehrich M, Nelson MR, Stanssens P, et al. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci USA. 2005;102:15785–15790. doi: 10.1073/pnas.0507816102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbajan JM, Maeda T, Souzaki R, et al. Comprehensive analyses of imprinted differentially methylated regions reveal epigenetic and genetic characteristics in hepatoblastoma. BMC Cancer. 2013;13:608. doi: 10.1186/1471-2407-13-608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H, Onyango P, Brandenburg S, Wu Y, Hsieh CL, Feinberg AP. Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res. 2002;62:6442–6446. [PubMed] [Google Scholar]
- Murrell A, Ito Y, Verde G, et al. Distinct methylation changes at the IGF2-H19 locus in congenital growth disorders and cancer. PLoS One. 2008;3:e1849. doi: 10.1371/journal.pone.0001849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodfine K, Huddleston JE, Murrell A. Quantitative analysis of DNA methylation at all human imprinted regions reveals preservation of epigenetic stability in adult somatic tissue. Epigenetics Chromatin. 2011;4:1. doi: 10.1186/1756-8935-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reik W, Maher ER. Imprinting in clusters: lessons from Beckwith-Wiedemann syndrome. Trends Genet. 1997;13:330–334. doi: 10.1016/s0168-9525(97)01200-6. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Yamada K, Morita S, et al. Identification of the mouse paternally expressed imprinted gene Zdbf2 on chromosome 1 and its imprinted human homolog ZDBF2 on chromosome 2. Genomics. 2009;93:461–472. doi: 10.1016/j.ygeno.2008.12.012. [DOI] [PubMed] [Google Scholar]
- Nakabayashi K, Trujillo AM, Tayama C, et al. Methylation screening of reciprocal genome-wide UPDs identifies novel human-specific imprinted genes. Hum Mol Genet. 2011;20:3188–3197. doi: 10.1093/hmg/ddr224. [DOI] [PubMed] [Google Scholar]
- Liu J, Litman D, Rosenberg MJ, Yu S, Biesecker LG, Weinstein LS. A GNAS1 imprinting defect in pseudohypoparathyroidism type IB. J Clin Invest. 2000;106:1167–1174. doi: 10.1172/JCI10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demars J, Shmela ME, Rossignol S, et al. Analysis of the IGF2/H19 imprinting control region uncovers new genetic defects, including mutations of OCT-binding sequences, in patients with 11p15 fetal growth disorders. Hum Mol Genet. 2010;19:803–814. doi: 10.1093/hmg/ddp549. [DOI] [PubMed] [Google Scholar]
- Weksberg R, Shuman C, Caluseriu O, et al. Discordant KCNQ1OT1 imprinting in sets of monozygotic twins discordant for Beckwith-Wiedemann syndrome. Hum Mol Genet. 2002;11:1317–1325. doi: 10.1093/hmg/11.11.1317. [DOI] [PubMed] [Google Scholar]
- Tost J, Dunker J, Gut IG. Analysis and quantification of multiple methylation variable positions in CpG islands by Pyrosequencing. Biotechniques. 2003;35:152–156. doi: 10.2144/03351md02. [DOI] [PubMed] [Google Scholar]
- Claus R, Wilop S, Hielscher T, et al. A systematic comparison of quantitative high-resolution DNA methylation analysis and methylation-specific PCR. Epigenetics. 2012;7:772–780. doi: 10.4161/epi.20299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay DJ, Callaway JL, Marks SM, et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet. 2008;40:949–951. doi: 10.1038/ng.187. [DOI] [PubMed] [Google Scholar]
- Meyer E, Lim D, Pasha S, et al. Germline mutation in NLRP2 (NALP2) in a familial imprinting disorder (Beckwith-Wiedemann Syndrome) PLoS Genet. 2009;5:e1000423. doi: 10.1371/journal.pgen.1000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorthongpanich C, Cheow LF, Balu S, et al. Single-cell DNA-methylation analysis reveals epigenetic chimerism in preimplantation embryos. Science. 2013;341:1110–1112. doi: 10.1126/science.1240617. [DOI] [PubMed] [Google Scholar]
- Reik W, Brown KW, Schneid H, Le Bouc Y, Bickmore W, Maher ER. Imprinting mutations in the Beckwith-Wiedemann syndrome suggested by altered imprinting pattern in the IGF2-H19 domain. Hum Mol Genet. 1995;4:2379–2385. doi: 10.1093/hmg/4.12.2379. [DOI] [PubMed] [Google Scholar]
- Sparago A, Russo S, Cerrato F, et al. Mechanisms causing imprinting defects in familial Beckwith-Wiedemann syndrome with Wilms' tumour. Hum Mol Genet. 2007;16:254–264. doi: 10.1093/hmg/ddl448. [DOI] [PubMed] [Google Scholar]
- Lopes S, Lewis A, Hajkova P, et al. Epigenetic modifications in an imprinting cluster are controlled by a hierarchy of DMRs suggesting long-range chromatin interactions. Hum Mol Genet. 2003;12:295–305. doi: 10.1093/hmg/ddg022. [DOI] [PubMed] [Google Scholar]
- Murrell A, Heeson S, Bowden L, et al. An intragenic methylated region in the imprinted Igf2 gene augments transcription. EMBO Rep. 2001;2:1101–1106. doi: 10.1093/embo-reports/kve248. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.