

Abstract
Cells exposed to extreme physicochemical or mechanical stimuli die in an uncontrollable manner, as a result of their immediate structural breakdown. Such an unavoidable variant of cellular demise is generally referred to as ‘accidental cell death' (ACD). In most settings, however, cell death is initiated by a genetically encoded apparatus, correlating with the fact that its course can be altered by pharmacologic or genetic interventions. ‘Regulated cell death' (RCD) can occur as part of physiologic programs or can be activated once adaptive responses to perturbations of the extracellular or intracellular microenvironment fail. The biochemical phenomena that accompany RCD may be harnessed to classify it into a few subtypes, which often (but not always) exhibit stereotyped morphologic features. Nonetheless, efficiently inhibiting the processes that are commonly thought to cause RCD, such as the activation of executioner caspases in the course of apoptosis, does not exert true cytoprotective effects in the mammalian system, but simply alters the kinetics of cellular demise as it shifts its morphologic and biochemical correlates. Conversely, bona fide cytoprotection can be achieved by inhibiting the transduction of lethal signals in the early phases of the process, when adaptive responses are still operational. Thus, the mechanisms that truly execute RCD may be less understood, less inhibitable and perhaps more homogeneous than previously thought. Here, the Nomenclature Committee on Cell Death formulates a set of recommendations to help scientists and researchers to discriminate between essential and accessory aspects of cell death.
Defining life and death is more problematic than one would guess. In 1838, the work of several scientists including Matthias Jakob Schleiden, Theodor Schwann and Rudolf Carl Virchow culminated in the so-called ‘cell theory', postulating that: (1) all living organisms are composed of one or more cells; (2) the cell is the basic unit of life; and (3) all cells arise from pre-existing, living cells.1 Only a few decades later (in 1885), Walter Flemming described for the first time some of the morphologic features that have been largely (but often inappropriately) used to define apoptosis throughout the past four decades.2, 3, 4
A corollary of the cell theory is that viruses do not constitute bona fide living organisms.5 However, the discovery that the giant Acanthamoeba polyphaga mimivirus can itself be infected by other viral species has casted doubts on this point.6, 7, 8 Thus, the features that underlie the distinction between a living and an inert entity remain a matter of debate. Along similar lines, defining the transition between an organism's life and death is complex, even when the organism under consideration is the basic unit of life, a cell. From a conceptual standpoint, cell death can obviously be defined as the permanent degeneration of vital cellular functions. Pragmatically speaking, however, the precise boundary between a reversible alteration in homeostasis and an irreversible loss of cellular activities appears to be virtually impossible to identify. To circumvent this issue, the Nomenclature Committee on Cell Death (NCCD) previously proposed three criteria for the identification of dead cells: (1) the permanent loss of the barrier function of the plasma membrane; (2) the breakdown of cells into discrete fragments, which are commonly referred to as apoptotic bodies; or (3) the engulfment of cells by professional phagocytes or other cells endowed with phagocytic activity.9, 10, 11
However, the fact that a cell is engulfed by another via phagocytosis does not imply that the cell-containing phagosome fuses with a lysosome and that the phagosomal cargo is degraded by lysosomal hydrolases.12, 13, 14 Indeed, it has been reported that engulfed cells can be released from phagosomes as they preserve their viability, at least under some circumstances.15 Thus, the NCCD recommends here to consider as dead only cells that either exhibit irreversible plasma membrane permeabilization or have undergone complete fragmentation. A compendium of techniques that can be used to quantify these two markers of end-stage cell death in vitro and in vivo goes beyond the scope of this review and can be found in several recent articles.16, 17, 18, 19, 20, 21, 22, 23, 24, 25
Importantly, cell death instances can be operationally classified into two broad, mutually exclusive categories: ‘accidental' and ‘regulated'. Accidental cell death (ACD) is caused by severe insults, including physical (e.g., elevated temperatures or high pressures), chemical (e.g., potent detergents or extreme variations in pH) and mechanical (e.g., shearing) stimuli, is virtually immediate and is insensitive to pharmacologic or genetic interventions of any kind. The NCCD thinks that this reflects the structural disassembly of cells exposed to very harsh physicochemical conditions, which does not involve a specific molecular machinery. Although ACD can occur in vivo, for instance as a result of burns or traumatic injuries, it cannot be prevented or modulated and hence does not constitute a direct target for therapeutic interventions.23, 26, 27, 28 Nonetheless, cells exposed to extreme physicochemical or mechanical insults die while releasing elevated amounts of damage-associated molecular patterns (DAMPs), that is, endogenous molecules with immunomodulatory (and sometimes cytotoxic) activity. Some DAMPs can indeed propagate an unwarranted cytotoxic response (directly or upon the involvement of innate immune effectors) that promotes the demise of local cells surviving the primary insult.16, 19, 29, 30, 31 Intercepting DAMPs or blocking DAMP-ignited signaling pathways may mediate beneficial effects in a wide array of diseases involving accidental (as well as regulated) instances of cell death.19, 32
At odds with its accidental counterpart, regulated cell death (RCD) involves a genetically encoded molecular machinery.9, 33 Thus, the course of RCD can be altered by means of pharmacologic and/or genetic interventions targeting the key components of such a machinery. Moreover, RCD often occurs in a relatively delayed manner and is initiated in the context of adaptive responses that (unsuccessfully) attempt to restore cellular homeostasis.34, 35, 36, 37, 38 Depending on the initiating stimulus, such responses can preferentially involve an organelle, such as the reticular unfolded protein response, or operate at a cell-wide level, such as macroautophagy (hereafter referred to as autophagy).39, 40, 41, 42, 43, 44 Thus, while ACD is completely unpreventable, RCD can be modulated (at least to some extent, see below) not only by inhibiting the transduction of lethal signals but also by improving the capacity of cells to mount adaptive responses to stress.45, 46, 47, 48, 49, 50 Importantly, RCD occurs not only as a consequence of microenvironmental perturbations but also in the context of (post-)embryonic development, tissue homeostasis and immune responses.51, 52, 53, 54 Such completely physiologic instances of RCD are generally referred to as ‘programmed cell death' (PCD) (Figure 1).9, 33
Figure 1.

Types of cell death. Cells exposed to extreme physical, chemical or mechanical stimuli succumb in a completely uncontrollable manner, reflecting the immediate loss of structural integrity. We refer to such instances of cellular demise with the term ‘accidental cell death' (ACD). Alternatively, cell death can be initiated by a genetically encoded machinery. The course of such ‘regulated cell death' (RCD) variants can be influenced, at least to some extent, by specific pharmacologic or genetic interventions. The term ‘programmed cell death' (PCD) is used to indicate RCD instances that occur as part of a developmental program or to preserve physiologic adult tissue homeostasis
For the purpose of this discussion, it is useful to keep in mind the distinction that is currently made between the initiation of RCD and its execution. The term execution is generally used to indicate the ensemble of biochemical processes that truly cause the cellular demise. Conversely, initiation is commonly used to refer to the signal transduction events that activate executioner mechanisms. Thus, the activation of caspase-8 (CASP8) in the course of FAS ligand (FASL)-triggered apoptosis is widely considered as an initiator mechanism, whereas the consequent activation of caspase-3 (CASP3) is categorized as an executioner mechanism (see below).51, 55, 56, 57
Here, the NCCD formulates a set of recommendations to discriminate between essential and accessory aspects of RCD, that is, between those that etiologically mediate its occurrence and those that change its kinetics or morphologic and biochemical manifestations.
Morphologic Aspects of Cell Death
The early classifications of cell death were purely morphologic, owing to obvious technical limitations.18, 20 In 1964, the American biologist Richard A Lockshin was the first to thoroughly describe the demise of intersegmental muscles in developing silk moths, a seminal contribution to the modern understanding of PCD.58 A few years later, in 1972, the Australian pathologist John F Kerr together with his Scottish colleagues Andrew H Wyllie and Alastair R Currie coined the term ‘apoptosis' (from the ancient Greek 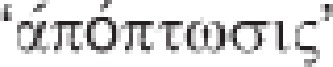 , meaning ‘falling off') to indicate a morphologically stereotyped form of cellular demise characterized by cytoplasmic shrinkage, chromatin condensation initiating at the nuclear membrane (marginalization) and then involving the whole nucleus (pyknosis), nuclear fragmentation (karyorrhexis), minimal alterations of other organelles and a peculiar ‘boiling-like' process (blebbing) culminating in the formation of a few discrete corpses that initially retain plasma membrane integrity (apoptotic bodies).10, 11, 59 Soon thereafter, the first ‘formal' classification of cell death differentiated between: (1) type I cell death (apoptosis), manifesting with the morphologic features described above; (2) type II cell death (autophagy), featuring an extensive vacuolization of the cytoplasm; and (3) type III cell death (necrosis), exhibiting neither apoptotic nor autophagic characteristics.3, 60 Such a ‘visual catalog' has dominated the field of cell death research for decades. Nonetheless, the NCCD views it as an oversimplification and considers it rather misleading, for several reasons.
, meaning ‘falling off') to indicate a morphologically stereotyped form of cellular demise characterized by cytoplasmic shrinkage, chromatin condensation initiating at the nuclear membrane (marginalization) and then involving the whole nucleus (pyknosis), nuclear fragmentation (karyorrhexis), minimal alterations of other organelles and a peculiar ‘boiling-like' process (blebbing) culminating in the formation of a few discrete corpses that initially retain plasma membrane integrity (apoptotic bodies).10, 11, 59 Soon thereafter, the first ‘formal' classification of cell death differentiated between: (1) type I cell death (apoptosis), manifesting with the morphologic features described above; (2) type II cell death (autophagy), featuring an extensive vacuolization of the cytoplasm; and (3) type III cell death (necrosis), exhibiting neither apoptotic nor autophagic characteristics.3, 60 Such a ‘visual catalog' has dominated the field of cell death research for decades. Nonetheless, the NCCD views it as an oversimplification and considers it rather misleading, for several reasons.
First, when this classification was formulated, necrosis (as defined by morphologic features) was considered as a strict equivalent of ACD, whereas apoptosis (as defined by morphologic features) was viewed as the sole programmed subroutine of cell death.11 Along similar lines, apoptosis was misconceived as an immunologically silent, if not tolerogenic, cell death modality.61, 62 It is now clear that PCD does not always manifest with an apoptotic morphotype and does not necessarily fail to induce inflammatory or immune responses. For instance, the degradation of Drosophila melanogaster salivary glands and larval midgut relies on PCD manifesting with a type II morphology,63, 64, 65, 66, 67, 68, 69, 70 whereas the remodeling of bones at the growth plates is associated with type III-like features.71 Moreover, specific inducers are capable of promoting a variant of RCD that displays an apoptotic morphotype, yet is capable of activating adaptive immune responses.72, 73 These observations indicate that morphologic and functional aspects of cell death are not necessarily linked to each other.
Second, the negative morphologic definition of necrosis as a cell death modality that fails to exhibit apoptotic or autophagic features has been reconsidered.22, 74 Indeed, necrosis can manifest with a stereotyped panel of features, including a generalized swelling of the cytoplasm, which acquires a translucent aspect, and organelles (oncosis), as well as a peculiar alteration of chromatin (condensation into small and irregular patches) and the nuclear membrane (dilatation).74 The evolution of the morphologic characterization of necrosis reflects the relatively recent discovery that RCD can also manifest with a necrotic aspect (see below).
Third, the use of the term ‘autophagic cell death' has been a matter of intense debate.75 Such an expression was coined based on morphologic considerations (i.e., the appearance of autophagic vacuoles in the course of type II cell death) only, but it soon became misused to imply that the molecular machinery of autophagy would actively contribute to the cellular demise.75 The NCCD strongly recommends the use of the expression ‘autophagic cell death' from a functional perspective only, that is, to indicate a cell death subroutine that is limited or delayed by the pharmacologic or genetic inhibition of the autophagic machinery (see below).9, 76
Fourth, many instances of RCD present both apoptotic and necrotic traits.10 Moreover, several pharmacologic agents and genetic interventions designed to inhibit the execution of cell death often fail to do so when administered in a therapeutic (as opposed to prophylactic)77 manner, at least in the mammalian system, yet efficiently change its morphology.78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91 This applies to N-benzyloxycarbonyl-Val-Ala-Asp(O-Me) fluoromethylketone (Z-VAD-fmk) and (3S)-5-(2,6-difluorophenoxy)-3-[[(2S)-3-methyl-1-oxo-2-[(2-quinolinylcarbonyl)amino]butyl]amino]-4-oxo-pentanoic acid hydrate (Q-VD-OPh), two broad-spectrum caspase inhibitors that have been widely investigated in the late 1990s as a means to mediate clinically relevant cytoprotection.92, 93, 94, 95 Z-VAD-fmk and Q-VD-OPh prevent the appearance of several morphologic markers of apoptosis, including nuclear pyknosis and blebbing (which rely on caspases),96, 97 yet fail to limit stimulus-dependent cell death if administered in therapeutic settings (i.e., after the cell death inducer).85, 98, 99, 100 Thus, caspase inhibition most often results in a shift from an overtly apoptotic to a mixed or necrotic cell death morphology.81, 91 Conversely, both necrostatin 1 (Nec-1), a highly specific small inhibitor of the enzymatic activity of receptor-interacting protein kinase 1 (RIPK1), and geldanamycin, which targets heat-shock protein 90 kDa alpha (cytosolic), class A member 1 (HSP90AA1), have been demonstrated to shift the necrotic morphotype of RCD induced by tumor necrosis factor (ligand) superfamily, member 10 (TNFSF10, best known as TNF-related apoptosis-inducing ligand, TRAIL) at slightly acidic pH to an apoptotic one.101 A similar morphologic shift has been observed in cells succumbing to tumor necrosis factor receptor superfamily, member 1A (TNFRSF1A, best known as tumor necrosis factor receptor 1, TNFR1) ligation in the presence of geldanamycin102 and in the absence of RIPK1 or one of its downstream targets, that is, receptor-interacting protein kinase 3 (RIPK3) and mixed lineage kinase domain-like (MLKL).103, 104 Also, cells exposed to DNA-alkylating agents in the presence of poly(ADP-ribose) polymerase 1 (PARP1) inhibitors die while manifesting an apoptotic, rather than a necrotic, morphology.105, 106 Conversely, the introduction of a non-cleavable PARP1 variant appears to convert the apoptotic phenotype of cells succumbing to FAS ligation into a necrotic one.107 Possibly, this reflects the ability of PARP1, an NAD+-dependent enzyme initially characterized for its role in DNA repair and the DNA damage response,108, 109 to provoke an abrupt decline in intracellular ATP levels (secondary to NAD+ depletion), hence blocking various morphologic manifestations of apoptosis.110, 111, 112, 113, 114, 115 Such a morphologic shift, however, does not appear to stem from the inhibition of caspases, because neither the catalytic functions nor the activation of these proteases require ATP (which should not be confounded with deoxy-ATP, see below).116, 117, 118, 119
In summary, the morphologic manifestations of cell death can easily be altered in the absence of bona fide cytoprotection, casting doubts on the actual value of morphology-based classifications of cell death.9
Biochemical Manifestations of Cell Death
In 2012, the NCCD proposed to abandon the morphologic catalog of cell death instances in favor of a new classification based on quantifiable biochemical parameters.9 In substitution, the NCCD identified the main molecular events associated with specific cell death subroutines as well as the pharmacologic and/or genetic interventions that may be used to discriminate between various instances of cell death in experimental settings, in vitro and in vivo.9
Since then, our comprehension of specific RCD modalities has progressed significantly. Thus, while no paradigm-breaking discoveries have been made on the regulation and execution of caspase-dependent RCD instances (which most often display an apoptotic morphology), profound insights have been obtained into the mechanisms underlying cases of RCD that do not depend on caspases and generally manifest with necrotic features.33, 74, 120, 121 This notion began to emerge in the late 1980s,122 but became widely accepted only two decades later, owing to the milestone discoveries of Peter Vandenabeele,123, 124, 125, 126, 127 Jurg Tschopp128 and Junying Yuan,129, 130, 131 and to the characterization of the key role played by peptidylprolyl isomerase F (PPIF, best known as cyclophilin D, CYPD) in necrotic instances of RCD.132, 133, 134, 135 The identification of a genetically encoded machinery that promotes RCD with necrotic features generated an intense wave of investigation that has not yet come to an end.33, 74, 120, 121
From a biochemical standpoint, apoptosis is defined as a caspase-dependent variant of RCD.9, 51 Other events commonly associated with apoptosis, such as the exposure of phosphatidylserine on the outer leaflet of the plasma membrane, are indeed less universal and more context-dependent136, 137, 138, 139 than previously thought.140 Apoptosis can be initiated by intracellular (intrinsic) or extracellular (extrinsic) stimuli. Intrinsic apoptosis critically relies on mitochondrial outer membrane permeabilization (MOMP), a process that results in the holocytochrome c (CYTC)-, deoxy-ATP- and apoptotic peptidase-activating factor 1 (APAF1)-dependent activation of caspase-9 (CASP9) and CASP3.117, 141, 142, 143, 144 MOMP obligatorily requires either of two Bcl-2 family members, namely, B-cell CLL/lymphoma 2 (BCL2)-associated X protein (BAX) and BCL2-antagonist/killer 1 (BAK1), whose pore-forming activity is inhibited (both directly and via indirect circuitries) by other components of the family, including BCL2 itself, BCL2-like 1 (BCL2L1, best known as BCL-XL) and myeloid cell leukemia 1 (MCL1).48, 145, 146, 147, 148 Importantly, the physical and functional interactions between pro- and antiapoptotic multidomain BCL2-like proteins are under the control of small components of the family known as BH3-only proteins, including (but not limited to) BCL2 binding component 3 (BBC3, best known as PUMA), BCL2-like 11 (BCL2L11, best known as BIM) and BH3-interacting domain death agonist (BID).149, 150, 151
Extrinsic apoptosis proceeds along with the activation of a CASP8/CASP3 signal transduction axis that in some cell types (including hepatocytes, pancreatic β cells and multiple neoplastic cells) also involves MOMP, owing to the CASP8-dependent activation of BID.152, 153, 154, 155, 156, 157 Whether the apoptotic response to extracellular cues requires MOMP or not reportedly depends on the expression levels of X-linked inhibitor of apoptosis (XIAP),158, 159 an ubiquitin ligase with multipronged cytoprotective functions.160, 161 High amounts of XIAP prevent indeed the direct activation of CASP3 by CASP8, a block that can be circumvented by the release of diablo, IAP-binding mitochondrial protein (DIABLO, best known as second mitochondria-derived activator of caspases, SMAC) and other XIAP inhibitors into the cytosol following MOMP.158, 159, 162, 163, 164
It recently became clear that the main players in the RCD subroutine commonly referred to as necroptosis, which we previously defined as a caspase-independent, RIPK1- and RIPK3-dependent lethal signaling pathway initiated by death receptors,9, 165 include not only RIPK1 and RIPK3, as initially thought,120, 121, 131, 166, 167, 168, 169 but also MLKL.104, 170, 171, 172, 173, 174, 175, 176, 177, 178, 179 The kinase activity of RIPK1 is required for necroptosis as induced by multiple stimuli, including death receptor ligation in the presence of caspase inhibitors.128, 129, 130, 131, 169, 180 Conversely, catalytically inactive and Nec-1-bound RIPK1 inhibits the necroptotic response of CASP8-incompetent cells to Toll-like receptor (TLR) agonists or type I interferons, which relies not only on RIPK3 but also on TLR adaptor molecule 1 (TICAM1, best known as TRIF).181, 182, 183, 184 In the absence of RIPK1, TLR agonists and type I interferons trigger necroptosis even in caspase-competent cells,182, 183 suggesting that RIPK1 can inhibit necroptotic RCD as induced by these stimuli at two distinct levels. Furthermore, RIPK1 tonically suppresses CASP8 and RIPK3 activation in developmental scenarios independent of its kinase activity.183, 185, 186 This explains why Ripk1−/− mice fail to survive to adulthood even in the absence of Fas (TNFRSF6)-associated via death domain (Fadd), which is required for CASP8 activation by extracellular cues, but mature normally in the absence of both Ripk3 and Fadd or Casp8.183 Other instances of necroptosis, such as those promoted by Z-DNA-binding protein 1 (ZBP1) in response to viral infection, appear to proceed independently of RIPK1.187 Upon phosphorylation by RIPK3, MLKL has a critical, non-redundant role in necroptosis.177, 179 Phosphorylated MLKL forms indeed oligomers that translocate to cellular membranes (including the plasma membrane) and bind specific phospholipids, resulting in the loss of barrier function.170, 171, 174, 175, 188
Recent data argue against an essential role for mitochondria in necroptosis. Indeed, parkin RBR E3 ubiquitin protein ligase (PARK2)-overexpressing cells depleted of the vast majority of mitochondria upon the induction of mitophagy (by means of a mitochondrial uncoupler) become resistant to inducers of MOMP-dependent RCD, but remain sensitive to TNFR1 ligation in the presence of Z-VAD-fmk (a conventional trigger of necroptosis).189 Moreover, contrary to initial beliefs,190 the lethal activity of RIPK3 is not influenced by the absence of phosphoglycerate mutase family member 5 (PGAM5) and dynamin 1 like (DNM1L, best known as dynamin-related protein 1, DRP1).104, 172, 176 Based on these results, the NCCD proposes here to redefine necroptosis as an RCD modality that critically depends on MLKL and on the kinase activity of RIPK1 (in some settings) and RIPK3. Of note, both RIPK1 and RIPK3 have been shown to regulate caspase activation, at least under some circumstances.186, 191, 192, 193 Taken together, these observations suggest that the signal transduction cascades responsible for the initiation of apoptosis and necroptosis are highly interconnected.
Necroptosis is actively inhibited by a supramolecular complex containing CASP8, FADD and the long isoform of CASP8 and FADD-like apoptosis regulator (CFLAR, best known as cellular FLICE inhibitor protein, c-FLIP),194, 195, 196, 197 three key components of caspase-dependent RCD initiated by death receptors.198, 199, 200, 201, 202, 203 Taken together with the notion that the absence of either Ripk3, Casp8 or Fadd fails to rescue Ripk1−/− mice from neonatal lethality,180, 185 these results pointed to the existence of a switch mechanism that regulates cell fate upon TNFR1 ligation.204, 205 Intriguingly, such switch may not operate in all cell types, as demonstrated by the fact that Ripk1−/− intestinal epithelial cells are fully rescued by the concomitant absence of Casp8 (Peter Vandenabeele, personal communication).
Recent data obtained with genetically engineered RIPK1 and RIPK3 variants indicate that the catalytic pathways activated in response to death receptor ligation depend on the availability of CASP8, FADD and MLKL.206 In comparatively more physiologic conditions, however, the fate of cells exposed to death receptor ligands may be determined by the activation kinetics of mitogen-activated protein kinase kinase kinase 7 (MAP3K7, best known as TGFβ-activated kinase 1, TAK1),192, 193, 207 which normally initiates a cytoprotective response centered around the transcription factor NF-κB and autophagy,208, 209, 210, 211, 212 or by the availability of baculoviral IAP repeat containing (BIRC) family members,192, 193 ubiquitin ligases with a central role in TNFR1 signaling.213 In line with this notion, cells treated with a SMAC mimetic (resulting in the depletion of BIRC2 and BIRC3) or a chemical TAK1 inhibitor (NP-009245) reportedly respond to TNFR1 ligation by activating caspases in a RIPK1-dependent manner.192 Taken together, these observations indicate that death receptors generate a lethal stimulus that can be propagated along several signal transduction cascades. Thus, caution should be used in evaluating necroptotic instances of cell death based on their sensitivity to Nec-1 only.
Another variant of RCD that often, although not always, manifests with a necrotic morphotype critically relies on CYPD.214 At present, CYPD is the sole genetically confirmed component of the permeability transition pore complex (PTPC) in the mammalian system.132, 133, 135, 215, 216, 217 The term PTPC generally refers to a supramolecular complex operating at the junctions between the inner and outer mitochondrial membrane to cause the so-called ‘mitochondrial permeability transition' (MPT), an abrupt increase in the permeability of the inner mitochondrial membrane to small solutes triggered by cytosolic Ca2+ overload or oxidative stress.214, 218, 219, 220, 221 Unlike MOMP,205, 222, 223, 224 MPT seals the cell fate independently of caspase activation.133, 225, 226 Nonetheless, MPT-driven RCD can manifest with (at least some) morphologic features associated with apoptosis,10, 227, 228 corroborating the limited informative value of cell death classifications solely based on morphology. The NCCD recommends the use of the term ‘MPT-driven RCD' for instances of cell death whose course can be influenced with the genetic or pharmacologic inhibition of CYPD or other components of the PTPC. Of note, CYPD surely does not constitute the long-sought pore-forming unit of the PTPC, which most likely involves subunits of the so-called ‘ATP synthasome', the supramolecular complex that imports ADP and inorganic phosphate into the mitochondrial matrix, catalyzes ATP synthesis and exports ATP back to the mitochondrial intermembrane space (from where it can easily reach the cytosol).229, 230, 231, 232, 233, 234 Perhaps, the central role of CYPD in MPT-driven RCD reflects its ability to control the Ca2+-buffering capacity of the mitochondrial network.235, 236 This hypothesis has not yet been formally addressed.
Two forms of RCD other than necroptosis and MPT-driven RCD have recently attracted attention as potential targets for the development of cytoprotective interventions, namely ‘parthanatos' and ‘ferroptosis'.33 The main molecular features of parthanatos are the hyperactivation of PARP1 and the release of apoptosis-inducing factor, mitochondrion-associated, 1 (AIFM1) from the mitochondria.237, 238, 239, 240 Interestingly, although TNFR1-driven necroptosis and parthanatos have been suggested to constitute completely independent RCD subroutines,241 this issue remains a matter of debate.101, 242 Possibly, such a controversy originates from the ability of some insults to simultaneously trigger necroptosis and parthanatos, at least in some model systems.243 Ferroptosis has been defined as an iron-dependent form of RCD under the control of glutathione peroxidase 4 (GPX4).244, 245, 246, 247 Both the pharmacologic and genetic inhibition of CYPD fail to prevent ferroptosis as triggered by erastin, a small molecule that is selectively lethal for cancer cells expressing oncogenic variants of Harvey rat sarcoma viral oncogene homolog (HRAS).246, 248 This suggests that ferroptosis and MPT-driven RCD constitute independent variants of RCD. Of note, erastin inhibits system xC−, an heterodimeric antiporter of the plasma membrane that normally exchanges intracellular glutamate for extracellular cysteine, resulting in glutathione depletion and iron-dependent accumulation of reactive oxygen species.247 A similar cascade of events contributes to (but is not the sole etiological determinant of) the death of neurons exposed to glutamate. This necrotic instance of RCD has previously been referred to as oxytosis.121, 249 Besides inhibiting system xC−, glutamate can trigger MPT-driven RCD upon the hyperactivation of ionotropic receptors, a neurotoxic process commonly known as excitotoxicity.250, 251
Caspase-unrelated variants of RCD include ‘autophagic cell death', which (among other processes) is biochemically associated with the lipidation of microtubule-associated protein 1 light chain 3 (MAP1LC3, best known as LC3) and the degradation of sequestosome 1 (SQSTM1, best known as p62).76 The NCCD recommends using this term only for RCD instances that can be influenced by the pharmacologic or genetic interventions targeting at least two distinct components of the molecular machinery for autophagy.9, 76 While autophagy accompanies RCD in a vast number of pathophysiologic settings,36, 50, 252 it truly contributes to the cellular demise only in a few of them.69, 70, 76, 253, 254, 255, 256, 257, 258, 259 Beth Levine's laboratory has recently discovered a bona fide instance of autophagic cell death that relies on the plasma membrane Na+/K+-ATPase, and dubbed it ‘autosis'.255 Of note, autosis occurs not only in vitro, in cells exposed to cell permeant autophagy-inducing peptides, but also in vivo, in the brain of rats subjected to an ischemic insult.255 It remains to be determined whether all cases of autophagic cell death require the Na+/K+-ATPase or not. If so, the terms ‘autosis' and ‘autophagic cell death' would be synonymous. If not, autosis would constitute a special instance of autophagic cell death.260
Importantly, a growing body of evidence indicates that the pharmacologic or genetic inhibition of the processes that are commonly considered as essential for cell death execution often does not avoid the demise of mammalian cells, but rather alters its kinetics and biochemical (and morphologic) manifestations. Thus, in many experimental paradigms (in vitro and in vivo), Z-VAD-fmk and more specific CASP3 inhibitors administered as therapeutic (as opposed to prophylactic)77 interventions fail to significantly limit primary RCD, in spite of the fact that they efficiently limit caspase activation.261, 262, 263, 264, 265 In some of these scenarios, RCD overtly manifests with alternative biochemical processes, including RIPK1, RIPK3 or PARP1 activation, and (at least in part) can be influenced by agents that interfere with these pathways, including Nec-1, 3-aminobenzamide (a PARP1-targeting agent) and necrosulfonamide (an inhibitor of human MLKL).261, 263, 264 However, the proportion of cells eventually succumbing to RCD does not change. The depletion of BIRC2/BIRC3, RIPK1, RIPK3 or MLKL, and the administration of NP-009245, Nec-1 or geldanamycin (which indirectly destabilizes RIPK1)266 reportedly changes the kinetics of necroptosis and its biochemical profile, that is, it allows for caspase activation, yet fails to block the cellular demise.101, 103, 104, 192, 267 Along similar lines, 3-aminobenzamide and 4-amino-1,8-naphthalimide (another PARP1 inhibitor) can convert the cytotoxic response to alkylating DNA damage or TNFR1 ligation from a caspase-independent one to apoptosis, manifesting with caspase activation.105, 106, 107
These observations indicate that, similar to their morphologic counterparts, the biochemical manifestations of cell death can be altered in the absence of efficient cytoprotection.
RCD and Stress Adaptation
Cells subjected to perturbations of intracellular or extracellular homeostasis almost invariably mount a tightly coordinated response aimed at (1) the removal of the initiating stimulus (when possible), (2) the repair of molecular and/or organelle damage, and (3) eventually, the re-establishment of physiologic conditions.34, 35, 36, 37, 38, 42, 57, 268 When these objectives cannot be attained, cells generally undergo RCD as a means to preserve the homeostasis of the whole organism (or colony, in the case of yeast cells). Two mutually exclusive models can be put forward to explain how adaptive stress responses promote RCD when unsuccessful. First, a ‘conversion model' postulates that RCD-inhibitory signals cease at some stage of the adaptive response and are replaced by RCD-promoting ones. Second, a ‘competition model' hypothesizes that RCD-inhibitory and -promoting signals coexist and counteract with each other starting from the detection of microenvironmental alterations, but at some stage the latter predominate over the former (Figure 2). Although data formally favoring one of these models over the other are lacking, circumstantial evidence suggests that RCD-promoting signals are activated when RCD-inhibitory mechanisms are still operational.36, 159, 252, 269, 270, 271, 272, 273
Figure 2.

Regulated cell death and adaptive stress responses. Regulated cell death (RCD) is often initiated in the context of unsuccessful responses to perturbations of intracellular or extracellular homeostasis. Two mutually exclusive models can be put forward to explain how failing responses to stress initiate RCD (which in many instances constitutes a means to preserve the homeostasis of the whole organism or colony). First, according to a ‘conversion model', RCD-inhibitory signals simply cease at some stage of the adaptive response and are substituted by RCD-promoting ones. Second, a ‘competition model' postulates that RCD-inhibitory and -promoting signals coexist and counteract each other starting from the very detection of microenvironmental alterations, and at some stage the latter predominate over the former. Circumstantial evidence favors the ‘competition model' in a majority of experimental scenarios
Based on this conceptual construction, the NCCD recommends to use the term ‘initiation' to indicate the RCD-causing events that are reversible, that is, that do not irrevocably commit cells to die as they occur when adaptive responses are still operational. In addition, we encourage the use of the expression ‘execution' for referring to the processes that irreversibly and causally seal the cell fate, and the term ‘propagation' to indicate the processes that link primary (stimulus-dependent) RCD to the stimulus-independent initiation of a secondary RCD wave, including the release of DAMPs and the consequent inflammatory response (Figure 3). The blockage of RCD-initiating mechanisms by either pharmacologic or genetic means has been associated with consistent degrees of cytoprotection in rodent models representing various human diseases linked to unwarranted cell death. For instance, this applies to the whole-body ablation of Ppif (the CYPD-coding gene),132, 133, 134, 274 Ripk3167, 176, 274, 275, 276, 277, 278 and Mlkl,176, 177 as well as to the administration of chemical CYPD inhibitors (i.e., cyclosporin A and sanglifehrin A)274 and Nec-1.274, 277 Conversely, pharmacologic and genetic interventions expected to interrupt RCD at late steps of the process (when cells are commonly considered as committed to die) generally fail to confer significant long-term cytoprotection in mammalian models, casting doubts on the actual etiological value of these steps for RCD. Thus, Casp3−/−, Casp9−/− and Apaf1−/− mice display a consistent hyperplasia of the central nervous system associated with reduced amounts of PCD in specific cerebral areas, resulting in embryonic or perinatal lethality.279, 280, 281, 282, 283, 284 However, the neuronal phenotype of Casp3−/− mice does not develop in all genetic backgrounds,89, 285, 286 and the penetrance of the perinatal lethality associated with the Apaf1−/− genotype is incomplete (as some animals survive through 10 months of age).88, 287 Moreover, the developmental death of interdigital cells (which generally manifests with biochemical correlates of apoptosis) occurs close to normally (allowing for normal morphogenesis) in mice bearing a homozygous loss-of-function mutation in Apaf1, and in mice exposed to broad-spectrum caspase inhibitors.80 In these settings, however, the demise of interdigital cells cannot be detected by the terminal-deoxynucleotidyl-mediated dUTP nick end-labeling (TUNEL) assay, measuring caspase-dependent DNA fragmentation.80 Conversely, the simultaneous ablation of Bax and Bak1288 or Bcl2l11 and Bmf (encoding two BH3-only proteins involved in MOMP initiation)289, 290, 291 truly prevents the programmed demise of several cell types, causing their persistence throughout adult life.292, 293 These observations are compatible with the hypothesis that the phenotype associated with some defects in the molecular cascades linking MOMP to caspase activation originates from a delay, rather than from bona fide inhibition, of PCD. Moreover they suggest that RCD, be it programmed or caused by microenvironmental perturbations, can only be avoided by interventions that target upstream steps of the process.
Figure 3.

Initiation, execution and propagation of regulated cell death. The term ‘execution' has largely been used to indicate the processes that (were thought to) mediate regulated cell death (RCD), such as the massive activation of CASP3 in the course of apoptosis. Conversely, the word ‘initiation' has generally been used to refer to the signal transduction events that trigger executioner mechanisms, such as the activation of CASP8 or CASP9, both of which normally impinge on CASP3. Upon an attentive re-evaluation of the available literature, the NCCD recommends caution in attributing a specific process a bona fide causative value in the execution of cell death. In addition, the NCCD proposes to use the term ‘initiation' with a pragmatic connotation, that is, to indicate the steps in the cascades of events leading to RCD that are truly reversible, and the term ‘propagation' to indicate the processes that link primary RCD to the insult-independent initiation of a secondary wave of RCD, that is, the release of cytotoxic and proinflammatory factors, including damage-associated molecular patterns (DAMPs), by dying cells and their consequences. Based on this conceptual construction, only pharmacologic and genetic interventions that target the initiation phase exert bona fide cytoprotective effects, that is, truly inhibit primary RCD rather than just delaying its course or changing its morphologic or biochemical correlates. Robust cytoprotection can also be achieved in vivo by the administration of anti-inflammatory agents and by measures that block DAMPs or their receptors. These maneuvers, however, appear to be efficient as they prevent the propagation of primary RCD or the initiation of secondary RCD
Caution should also be taken in inferring the actual etiological value of caspases in RCD based on the therapeutic administration of Z-VAD-fmk or other broad-spectrum caspase inhibitors. Caspase blockers have indeed been associated with (at least some degree of) cytoprotection in rodent models of various human diseases linked to the excessive loss of parenchymal cells. These pathologies include, but are not limited to, neurodegenerative disorders,294, 295, 296, 297, 298 traumatic events299, 300 and ischemia/reperfusion injuries of the central nervous system, heart and kidney.301, 302, 303, 304 Nonetheless, Z-VAD-fmk and similar compounds inhibit not only several caspases but also a wide panel of non-caspase proteases that participate in the initiation of RCD, such as calpains.305, 306 Moreover, CASP3, caspase-6 (CASP6) and caspase-7 (CASP7) (i.e., the putative executioners of apoptosis) have been involved in feedforward circuitries that amplify lethal cues leading to MOMP,307, 308, 309, 310 implying that their inhibition may also counteract the initiation of RCD. Finally, in models of this type it is difficult to discriminate between the primary wave of RCD (promoted by experimental interventions) and the delayed, secondary demise of parenchymal cells caused by DAMPs (directly or upon the establishment of inflammation).73, 311, 312, 313 The cytoprotective effects that Z-VAD-fmk-like chemicals exert in similar scenarios, which are most reliably evaluated by histological determinations or functional tests, might therefore reflect their ability to block the initiation of DAMP- and inflammation-driven secondary RCD rather than the execution of stimulus-induced, primary RCD. In line with this notion, consistent cytoprotection has also been achieved in vivo by means of anti-inflammatory agents, even when these compounds do not directly influence RCD.314, 315, 316, 317 Taken together, these observations reinforce the notion that caspases may not mediate RCD but simply accelerate its course, at least in the mammalian system.
Apparently at odds with the role of PARP1 in the execution of parthanatos, both the Parp1−/− genotype and the administration of (relatively unselective) PARP inhibitors have been associated with bona fide cytoprotection in rodent models of ischemia/reperfusion injury and retinal degeneration.318, 319, 320, 321 These observations suggest that PARP1 and/or other members of the PARP family also participate in the initiation of RCD. Alternatively, the inhibition of PARP1 may limit the release of DAMPs or the consequent inflammatory response, at least in some pathophysiologic settings. Further experiments are required to clarify these possibilities.
Concluding Remarks
As discussed above, the processes that until now were thought to mediate RCD most often do not causally underpin the cellular demise but represent biochemical manifestations of it. A growing body of data indicates indeed that the bona fide executioners of RCD, that is, the processes that directly drive cells across the boundary between life and death are less characterized, less inhibitable and perhaps more homogeneous than previously thought.322 In line with this theoretical construction, here the NCDD proposes to use the term ‘initiation' to refer to all the steps in the RCD cascade that are reversible, that is, which occur before cells make an irrevocable commitment to die. An attentive reinterpretation of the literature suggests that actual cytoprotection can only be achieved with pharmacologic or genetic interventions that inhibit or outcompete lethal signals at this stage, when adaptive responses to stress are still operational. Interestingly, some cells manifesting biochemical and morphologic features associated with late-stage RCD (including partial MOMP, caspase activation and blebbing) appear to recover (upon removal of the RCD-initiating insult) and replicate, a process that has been dubbed ‘anastasis' (from the ancient Greek ‘ανστασις', meaning ‘raising to life').323, 324 This suggests that the actual point of no return in the signal-transduction cascades leading to RCD may exhibit at least some degree of context dependency.
In the vast majority of scientific reports, RCD is measured in vitro 24–96 h after stimulation, whereas the most reliable assessment of RCD in vivo is based upon histologic determinations or functional tests performed days, if not weeks or months, after such experimental interventions. In the former scenario, investigators can collect valuable kinetic data but are unable to estimate the true long-term survival of cells exposed to perturbations of homeostasis. In the second scenario, it is difficult to discriminate between the interruption of primary RCD and the inhibition of DAMP-driven inflammatory reactions and secondary RCD. This may have profound mechanistic and therapeutic implications. Indeed, retarding the demise of a cell that has already committed to die in an irreversible manner, and the biochemical manifestations of such death, may have limited cytoprotective effects for the cell in question, but may impact on the emission of DAMPs and hence significantly influence RCD propagation. Thus, considerable degrees of cytoprotection might be attained by means of agents that interrupt lethal cues at (or before) RCD initiation (when adaptive stress responses are still functional) combined with strategies that inhibit propagation (e.g., chemicals that favor RCD instances associated with a limited release of DAMPs, DAMP-neutralizing measures, anti-inflammatory agents). The superior beneficial effects of cyclosporin A may indeed stem from its ability to inhibit MPT-driven RCD and simultaneously exert a robust anti-inflammatory activity.325, 326
If caspases and other enzymes commonly thought to mediate RCD in mammalian cells only underpin its manifestations, what are the true causes of cell death? Although the concentration of ATP is preserved (or even increases to some extent) in the course of adaptive stress responses,247, 327 circumstantial evidence points to dropping ATP levels, which at some point abolish the activity of all ATP-dependent enzymes (including various transporters that maintain ionic balance at the plasma membrane) and a compromised redox balance (which inactivates various enzymes and causes oxidative molecular damage to organelles and membranes) as central players in the execution of RCD (Figure 4).328 Alternatively, one or more hitherto uncharacterized mechanism(s) may causally underpin RCD in all its manifestations. Further experiments are required to explore these possibilities.
Figure 4.

Declining ATP levels and redox alterations as a potential cause of regulated cell death. A growing amount of evidence indicates that the pharmacologic or genetic inhibition of the mechanisms that are commonly regarded as the executioners of regulated cell death (RCD) changes the kinetics of the process while altering its morphologic and biochemical manifestations, but fails to mediate bona fide long-term cytoprotection. It is therefore difficult to evaluate the actual causes that push cells beyond the point-of-no-return between life and death, especially as it remains to be formally demonstrated where the frontier between reversible alterations of homeostasis and the irreversible degeneration of cellular functions stands. ATP is required for a wide panel of vital activities, including the maintenance of the ionic equilibrium across the plasma membrane, implying that the drop of ATP concentrations below a specific threshold level may irremediably compromise the ability of cells to maintain structural integrity (which is the most reliable marker of cell death currently available). Along similar lines, variations in the oxidative potential of the intracellular milieu not only inhibit several enzymatic activities, including mitochondrial ATP synthesis, but also cause direct structural damage to organelles and membranes. We therefore hypothesize that declining ATP levels and a compromised redox homeostasis may constitute common causes of cell death in many RCD models. ROS, reactive oxygen species
At odds with mammalian models, Caenorhabditis elegans and D. melanogaster are truly protected by Z-VAD-fmk and by the genetic inhibition of caspase orthologs and other proteins involved in the postmitochondrial phase of apoptosis.329, 330, 331, 332, 333, 334, 335 This may indicate that the signal transduction cascades underlying RCD are interconnected in a different manner in mammals and non-mammalian organisms. Alternatively, the actual requirement of caspases for (at least some instances of) RCD might have been concealed by the evolutionary expansion of the caspase family. Both the human and murine genome encode indeed 14 distinct caspases,336, 337 and it seems unlikely that Z-VAD-fmk and other pharmacologic or genetic interventions may simultaneously inhibit all of them in an efficient manner.
Until these uncertainties have been resolved, the NCCD recommends that investigators focus on essential aspects of cell death; first of all its actual occurrence. It appears indeed that measuring the functional status or subcellular localization of RCD-relevant proteins including (but not limited to) caspases, RIPK1, RIPK3, MLKL, CYPD, PARP1 and GPX4, can provide insights into the mechanisms that accompany (and regulate the kinetics of) cellular demise, but not into those that truly push cells beyond the point-of-no-return separating life and death. Precisely defining where this border stands from a bioenergetic and metabolic perspective may facilitate the development of novel and efficient cytoprotective agents for clinical use.
Glossary
- ACD
accidental cell death
- APAF1
apoptotic peptidase-activating factor 1
- BAX
BCL2-associated X protein
- BAK1
BCL2-antagonist/killer 1
- BCL2
B-cell CLL/lymphoma 2
- BID
BH3-interacting domain death agonist
- BIRC
baculoviral IAP repeat containing
- CASP3
caspase-3
- CASP8
caspase-8
- CASP9
caspase-9
- CYPD
cyclophilin D
- DAMP
damage-associated molecular pattern
- FADD
Fas (TNFRSF6)-associated via death domain
- GPX4
glutathione peroxidase 4
- MLKL
mixed lineage kinase domain-like
- MOMP
mitochondrial outer membrane permeabilization
- MPT
mitochondrial permeability transition
- NCCD
Nomenclature Committee on Cell Death
- Nec-1
necrostatin 1
- PARP1
poly(ADP-ribose) polymerase 1
- PCD
programmed cell death
- PPIF
peptidylprolyl isomerase F
- PTPC
permeability transition pore complex
- Q-VD-OPh
(3S)-5-(2,6-difluorophenoxy)-3-[[(2S)-3-methyl-1-oxo-2-[(2-quinolinylcarbonyl)amino]butyl]amino]-4-oxo-pentanoic acid hydrate
- RCD
regulated cell death
- RIPK1
receptor-interacting protein kinase 1
- RIPK3
receptor-interacting protein kinase 3
- SMAC
second mitochondria-derived activator of caspases
- TAK1
TGFβ-activated kinase 1
- TLR
Toll-like receptor
- TNFR1
tumor necrosis factor receptor 1
- XIAP
X-linked inhibitor of apoptosis
- Z-VAD-fmk
N-benzyloxycarbonyl-Val-Ala-Asp(O-Me) fluoromethylketone
The authors declare no conflict of interest.
Footnotes
Edited by A Stephanou
References
- Mazzarello P. A unifying concept: the history of cell theory. Nat Cell Biol. 1999;1:E13–E15. doi: 10.1038/8964. [DOI] [PubMed] [Google Scholar]
- Cotter TG. Apoptosis and cancer: the genesis of a research field. Nat Rev Cancer. 2009;9:501–507. doi: 10.1038/nrc2663. [DOI] [PubMed] [Google Scholar]
- Schweichel JU, Merker HJ. The morphology of various types of cell death in prenatal tissues. Teratology. 1973;7:253–266. doi: 10.1002/tera.1420070306. [DOI] [PubMed] [Google Scholar]
- Vaux DL. Apoptosis timeline. Cell Death Differ. 2002;9:349–354. doi: 10.1038/sj.cdd.4400990. [DOI] [PubMed] [Google Scholar]
- Pearson H. ‘Virophage' suggests viruses are alive. Nature. 2008;454:677. doi: 10.1038/454677a. [DOI] [PubMed] [Google Scholar]
- La Scola B, Audic S, Robert C, Jungang L, de Lamballerie X, Drancourt M, et al. A giant virus in amoebae. Science. 2003;299:2033. doi: 10.1126/science.1081867. [DOI] [PubMed] [Google Scholar]
- La Scola B, Desnues C, Pagnier I, Robert C, Barrassi L, Fournous G, et al. The virophage as a unique parasite of the giant mimivirus. Nature. 2008;455:100–104. doi: 10.1038/nature07218. [DOI] [PubMed] [Google Scholar]
- Raoult D, Forterre P. Redefining viruses: lessons from Mimivirus. Nat Rev Microbiol. 2008;6:315–319. doi: 10.1038/nrmicro1858. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, et al. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007;14:1237–1243. doi: 10.1038/sj.cdd.4402148. [DOI] [PubMed] [Google Scholar]
- Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. 2014;14:166–180. doi: 10.1038/nri3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochreiter-Hufford A, Ravichandran KS. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol. 2013;5:a008748. doi: 10.1101/cshperspect.a008748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill DM, Goodridge HS. Information processing during phagocytosis. Nat Rev Immunol. 2012;12:492–502. doi: 10.1038/nri3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overholtzer M, Mailleux AA, Mouneimne G, Normand G, Schnitt SJ, King RW, et al. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell. 2007;131:966–979. doi: 10.1016/j.cell.2007.10.040. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Kroemer G. Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol. 2012;13:780–788. doi: 10.1038/nrm3479. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepp O, Galluzzi L, Lipinski M, Yuan J, Kroemer G. Cell death assays for drug discovery. Nat Rev Drug Discov. 2011;10:221–237. doi: 10.1038/nrd3373. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009;16:1093–1107. doi: 10.1038/cdd.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdemir E, Galluzzi L, Maiuri MC, Criollo A, Vitale I, Hangen E, et al. Methods for assessing autophagy and autophagic cell death. Methods Mol Biol. 2008;445:29–76. doi: 10.1007/978-1-59745-157-4_3. [DOI] [PubMed] [Google Scholar]
- Krysko DV, Vanden Berghe T, D'Herde K, Vandenabeele P. Apoptosis and necrosis: detection, discrimination and phagocytosis. Methods. 2008;44:205–221. doi: 10.1016/j.ymeth.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G. Pharmacological manipulation of cell death: clinical applications in sight. J Clin Invest. 2005;115:2610–2617. doi: 10.1172/JCI26321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe T, Grootjans S, Goossens V, Dondelinger Y, Krysko DV, Takahashi N, et al. Determination of apoptotic and necrotic cell death in vitro and in vivo. Methods. 2013;61:117–129. doi: 10.1016/j.ymeth.2013.02.011. [DOI] [PubMed] [Google Scholar]
- Grootjans S, Goossens V, Vandenabeele P, Vanden Berghe T. Cell Death in Biology and Diseases: Necrotic Cell Death. Humana Press, Springer; New York, NY, USA; 2014. Methods to study and distinguish necroptosis; pp. 335–361. [Google Scholar]
- Long JS, Ryan KM. New frontiers in promoting tumour cell death: targeting apoptosis, necroptosis and autophagy. Oncogene. 2012;31:5045–5060. doi: 10.1038/onc.2012.7. [DOI] [PubMed] [Google Scholar]
- MacFarlane M. Cell death pathways – potential therapeutic targets. Xenobiotica. 2009;39:616–624. doi: 10.1080/00498250903137990. [DOI] [PubMed] [Google Scholar]
- Reed JC. Drug insight: cancer therapy strategies based on restoration of endogenous cell death mechanisms. Nat Clin Pract Oncol. 2006;3:388–398. doi: 10.1038/ncponc0538. [DOI] [PubMed] [Google Scholar]
- Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. 2013;59:583–594. doi: 10.1016/j.jhep.2013.03.033. [DOI] [PubMed] [Google Scholar]
- Zelenay S, Reis e Sousa C. Adaptive immunity after cell death. Trends Immunol. 2013;34:329–335. doi: 10.1016/j.it.2013.03.005. [DOI] [PubMed] [Google Scholar]
- Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–388. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell. 2010;140:798–804. doi: 10.1016/j.cell.2010.02.015. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Krautwald S, Kroemer G, Linkermann A.Molecular mechanisms of regulated necrosis Semin Cell Dev Biol 2014. doi: 10.1016/j.semcdb.2014.02.006 [DOI] [PubMed]
- Bhattacharyya S, Yu H, Mim C, Matouschek A. Regulated protein turnover: snapshots of the proteasome in action. Nat Rev Mol Cell Biol. 2014;15:122–133. doi: 10.1038/nrm3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda S, Gorman AM, Hori O, Samali A. Cellular stress responses: cell survival and cell death. Int J Cell Biol. 2010;2010:214074. doi: 10.1155/2010/214074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter K, Haslbeck M, Buchner J. The heat shock response: life on the verge of death. Mol Cell. 2010;40:253–266. doi: 10.1016/j.molcel.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Spriggs KA, Bushell M, Willis AE. Translational regulation of gene expression during conditions of cell stress. Mol Cell. 2010;40:228–237. doi: 10.1016/j.molcel.2010.09.028. [DOI] [PubMed] [Google Scholar]
- Haynes CM, Fiorese CJ, Lin YF. Evaluating and responding to mitochondrial dysfunction: the mitochondrial unfolded-protein response and beyond. Trends Cell Biol. 2013;23:311–318. doi: 10.1016/j.tcb.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- Jensen MB, Jasper H. Mitochondrial proteostasis in the control of aging and longevity. Cell Metab. 2014;20:214–225. doi: 10.1016/j.cmet.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Bravo-San Pedro JM, Kroemer G. Organelle-specific initiation of cell death. Nat Cell Biol. 2014;16:728–736. doi: 10.1038/ncb3005. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amelio I, Melino G, Knight RA. Cell death pathology: cross-talk with autophagy and its clinical implications. Biochem Biophys Res Commun. 2011;414:277–281. doi: 10.1016/j.bbrc.2011.09.080. [DOI] [PubMed] [Google Scholar]
- Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria as targets for cancer chemotherapy. Semin Cancer Biol. 2009;19:57–66. doi: 10.1016/j.semcancer.2008.11.007. [DOI] [PubMed] [Google Scholar]
- Lindner AU, Concannon CG, Boukes GJ, Cannon MD, Llambi F, Ryan D, et al. Systems analysis of BCL2 protein family interactions establishes a model to predict responses to chemotherapy. Cancer Res. 2013;73:519–528. doi: 10.1158/0008-5472.CAN-12-2269. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- Lettre G, Hengartner MO. Developmental apoptosis in C. elegans: a complex CEDnario. Nat Rev Mol Cell Biol. 2006;7:97–108. doi: 10.1038/nrm1836. [DOI] [PubMed] [Google Scholar]
- Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–758. doi: 10.1016/j.cell.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbridge AR, Valente LJ, Strasser A.The role of the apoptotic machinery in tumor suppression Cold Spring Harb Perspect Biol 20124doi: 10.1101/cshperspect.a008789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser A, Jost PJ, Nagata S. The many roles of FAS receptor signaling in the immune system. Immunity. 2009;30:180–192. doi: 10.1016/j.immuni.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter ME, Budd RC, Desbarats J, Hedrick SM, Hueber AO, Newell MK, et al. The CD95 receptor: apoptosis revisited. Cell. 2007;129:447–450. doi: 10.1016/j.cell.2007.04.031. [DOI] [PubMed] [Google Scholar]
- Walczak H. Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harb Perspect Biol. 2013;5:a008698. doi: 10.1101/cshperspect.a008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockshin RA, Williams CM. Programmed cell death II. Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths. J Insect Phys. 1964;10:643–649. [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- Igney FH, Krammer PH. Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer. 2002;2:277–288. doi: 10.1038/nrc776. [DOI] [PubMed] [Google Scholar]
- Bellamy CO, Malcomson RD, Harrison DJ, Wyllie AH. Cell death in health and disease: the biology and regulation of apoptosis. Semin Cancer Biol. 1995;6:3–16. doi: 10.1006/scbi.1995.0002. [DOI] [PubMed] [Google Scholar]
- Tracy K, Baehrecke EH. The role of autophagy in Drosophila metamorphosis. Curr Top Dev Biol. 2013;103:101–125. doi: 10.1016/B978-0-12-385979-2.00004-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nezis IP, Shravage BV, Sagona AP, Lamark T, Bjorkoy G, Johansen T, et al. Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis. J Cell Biol. 2010;190:523–531. doi: 10.1083/jcb.201002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton D, Shravage B, Simin R, Baehrecke EH, Kumar S. Larval midgut destruction in Drosophila: not dependent on caspases but suppressed by the loss of autophagy. Autophagy. 2010;6:163–165. doi: 10.4161/auto.6.1.10601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, et al. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol. 2009;19:1741–1746. doi: 10.1016/j.cub.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131:1137–1148. doi: 10.1016/j.cell.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baehrecke EH. Autophagy: dual roles in life and death. Nat Rev Mol Cell Biol. 2005;6:505–510. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- Denton D, Aung-Htut MT, Lorensuhewa N, Nicolson S, Zhu W, Mills K, et al. UTX coordinates steroid hormone-mediated autophagy and cell death. Nat Commun. 2013;4:2916. doi: 10.1038/ncomms3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton D, Chang TK, Nicolson S, Shravage B, Simin R, Baehrecke EH, et al. Relationship between growth arrest and autophagy in midgut programmed cell death in Drosophila. Cell Death Differ. 2012;19:1299–1307. doi: 10.1038/cdd.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach HI, Clarke NM. Physiological cell death of chondrocytes in vivo is not confined to apoptosis. New observations on the mammalian growth plate. J Bone Joint Surg Br. 2000;82:601–613. doi: 10.1302/0301-620x.82b4.9846. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–875. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–1010. doi: 10.1038/nrm2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 2012;19:87–95. doi: 10.1038/cdd.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinou I, Desagher S, Eskes R, Antonsson B, Andre E, Fakan S, et al. The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J Cell Biol. 1999;144:883–889. doi: 10.1083/jcb.144.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenabeele P, Vanden Berghe T, Festjens N. Caspase inhibitors promote alternative cell death pathways. Sci STKE. 2006;2006:pe44. doi: 10.1126/stke.3582006pe44. [DOI] [PubMed] [Google Scholar]
- Scheller C, Knoferle J, Ullrich A, Prottengeier J, Racek T, Sopper S, et al. Caspase inhibition in apoptotic T cells triggers necrotic cell death depending on the cell type and the proapoptotic stimulus. J Cell Biochem. 2006;97:1350–1361. doi: 10.1002/jcb.20670. [DOI] [PubMed] [Google Scholar]
- Chautan M, Chazal G, Cecconi F, Gruss P, Golstein P. Interdigital cell death can occur through a necrotic and caspase-independent pathway. Curr Biol. 1999;9:967–970. doi: 10.1016/s0960-9822(99)80425-4. [DOI] [PubMed] [Google Scholar]
- Hirsch T, Marchetti P, Susin SA, Dallaporta B, Zamzami N, Marzo I, et al. The apoptosis-necrosis paradox. Apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene. 1997;15:1573–1581. doi: 10.1038/sj.onc.1201324. [DOI] [PubMed] [Google Scholar]
- Lockshin RA, Zakeri Z. Caspase-independent cell deaths. Curr Opin Cell Biol. 2002;14:727–733. doi: 10.1016/s0955-0674(02)00383-6. [DOI] [PubMed] [Google Scholar]
- Mailleux AA, Overholtzer M, Schmelzle T, Bouillet P, Strasser A, Brugge JS. BIM regulates apoptosis during mammary ductal morphogenesis, and its absence reveals alternative cell death mechanisms. Dev Cell. 2007;12:221–234. doi: 10.1016/j.devcel.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashmi R, Pillai SG, Vijayalingam S, Ryerse J, Chinnadurai G. BH3-only protein BIK induces caspase-independent cell death with autophagic features in Bcl-2 null cells. Oncogene. 2008;27:1366–1375. doi: 10.1038/sj.onc.1210783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M, Adachi S, Imai T, Watanabe K, Toyokuni SY, Ueno M, et al. A novel mechanism for imatinib mesylate-induced cell death of BCR-ABL-positive human leukemic cells: caspase-independent, necrosis-like programmed cell death mediated by serine protease activity. Blood. 2004;103:2299–2307. doi: 10.1182/blood-2003-05-1605. [DOI] [PubMed] [Google Scholar]
- Volbracht C, Leist M, Kolb SA, Nicotera P. Apoptosis in caspase-inhibited neurons. Mol Med. 2001;7:36–48. [PMC free article] [PubMed] [Google Scholar]
- Miyazaki K, Yoshida H, Sasaki M, Hara H, Kimura G, Mak TW, et al. Caspase-independent cell death and mitochondrial disruptions observed in the Apaf1-deficient cells. J Biochem. 2001;129:963–969. doi: 10.1093/oxfordjournals.jbchem.a002944. [DOI] [PubMed] [Google Scholar]
- Nagasaka A, Kawane K, Yoshida H, Nagata S. Apaf-1-independent programmed cell death in mouse development. Cell Death Differ. 2010;17:931–941. doi: 10.1038/cdd.2009.186. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Flavell RA, Vinsant S, Prevette D, Kuan CY, Rakic P. Programmed cell death of developing mammalian neurons after genetic deletion of caspases. J Neurosci. 2001;21:4752–4760. doi: 10.1523/JNEUROSCI.21-13-04752.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin SA, Daugas E, Ravagnan L, Samejima K, Zamzami N, Loeffler M, et al. Two distinct pathways leading to nuclear apoptosis. J Exp Med. 2000;192:571–580. doi: 10.1084/jem.192.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golstein P, Kroemer G. Redundant cell death mechanisms as relics and backups. Cell Death Differ. 2005;12 (Suppl 2:1490–1496. doi: 10.1038/sj.cdd.4401607. [DOI] [PubMed] [Google Scholar]
- Caserta TM, Smith AN, Gultice AD, Reedy MA, Brown TL. Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis. 2003;8:345–352. doi: 10.1023/a:1024116916932. [DOI] [PubMed] [Google Scholar]
- Zhu H, Fearnhead HO, Cohen GM. An ICE-like protease is a common mediator of apoptosis induced by diverse stimuli in human monocytic THP.1 cells. FEBS Lett. 1995;374:303–308. doi: 10.1016/0014-5793(95)01116-v. [DOI] [PubMed] [Google Scholar]
- Chow SC, Weis M, Kass GE, Holmstrom TH, Eriksson JE, Orrenius S. Involvement of multiple proteases during Fas-mediated apoptosis in T lymphocytes. FEBS Lett. 1995;364:134–138. doi: 10.1016/0014-5793(95)00370-o. [DOI] [PubMed] [Google Scholar]
- Slee EA, Zhu H, Chow SC, MacFarlane M, Nicholson DW, Cohen GM. Benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (Z-VAD.FMK) inhibits apoptosis by blocking the processing of CPP32. Biochem J. 1996;315 (Part 1:21–24. doi: 10.1042/bj3150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicke RU, Sprengart ML, Wati MR, Porter AG. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem. 1998;273:9357–9360. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol. 2001;3:339–345. doi: 10.1038/35070009. [DOI] [PubMed] [Google Scholar]
- Enzenmuller S, Gonzalez P, Karpel-Massler G, Debatin KM, Fulda S. GDC-0941 enhances the lysosomal compartment via TFEB and primes glioblastoma cells to lysosomal membrane permeabilization and cell death. Cancer Lett. 2013;329:27–36. doi: 10.1016/j.canlet.2012.09.007. [DOI] [PubMed] [Google Scholar]
- Mediavilla-Varela M, Pacheco FJ, Almaguel F, Perez J, Sahakian E, Daniels TR, et al. Docetaxel-induced prostate cancer cell death involves concomitant activation of caspase and lysosomal pathways and is attenuated by LEDGF/p75. Mol Cancer. 2009;8:68. doi: 10.1186/1476-4598-8-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbaa A, Zheng X, Chou PM, Mirkin BL. Caspase inhibition switches doxorubicin-induced apoptosis to senescence. Oncogene. 2003;22:2805–2811. doi: 10.1038/sj.onc.1206366. [DOI] [PubMed] [Google Scholar]
- Jouan-Lanhouet S, Arshad MI, Piquet-Pellorce C, Martin-Chouly C, Le Moigne-Muller G, Van Herreweghe F, et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ. 2012;19:2003–2014. doi: 10.1038/cdd.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe T, Kalai M, van Loo G, Declercq W, Vandenabeele P. Disruption of HSP90 function reverts tumor necrosis factor-induced necrosis to apoptosis. J Biol Chem. 2003;278:5622–5629. doi: 10.1074/jbc.M208925200. [DOI] [PubMed] [Google Scholar]
- Vanlangenakker N, Bertrand MJ, Bogaert P, Vandenabeele P, Vanden Berghe T. TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis. 2011;2:e230. doi: 10.1038/cddis.2011.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remijsen Q, Goossens V, Grootjans S, Van den Haute C, Vanlangenakker N, Dondelinger Y, et al. Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis. 2014;5:e1004. doi: 10.1038/cddis.2013.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehe K, Raithel K, Kreppel H, Jochum M, Worek F, Thiermann H. Inhibition of poly(ADP-ribose) polymerase (PARP) influences the mode of sulfur mustard (SM)-induced cell death in HaCaT cells. Arch Toxicol. 2008;82:461–470. doi: 10.1007/s00204-007-0265-7. [DOI] [PubMed] [Google Scholar]
- Pogrebniak A, Schemainda I, Pelka-Fleischer R, Nussler V, Hasmann M. Poly ADP-ribose polymerase (PARP) inhibitors transiently protect leukemia cells from alkylating agent induced cell death by three different effects. Eur J Med Res. 2003;8:438–450. [PubMed] [Google Scholar]
- Los M, Mozoluk M, Ferrari D, Stepczynska A, Stroh C, Renz A, et al. Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol Biol Cell. 2002;13:978–988. doi: 10.1091/mbc.01-05-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol. 2012;13:411–424. doi: 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- Michels J, Vitale I, Saparbaev M, Castedo M, Kroemer G. Predictive biomarkers for cancer therapy with PARP inhibitors. Oncogene. 2013;33:3894–3907. doi: 10.1038/onc.2013.352. [DOI] [PubMed] [Google Scholar]
- Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, et al. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med. 1997;185:1481–1486. doi: 10.1084/jem.185.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicotera P, Leist M, Ferrando-May E. Intracellular ATP, a switch in the decision between apoptosis and necrosis. Toxicol Lett. 1998;102-103:139–142. doi: 10.1016/s0378-4274(98)00298-7. [DOI] [PubMed] [Google Scholar]
- Volbracht C, Leist M, Nicotera P. ATP controls neuronal apoptosis triggered by microtubule breakdown or potassium deprivation. Mol Med. 1999;5:477–489. [PMC free article] [PubMed] [Google Scholar]
- Eguchi Y, Shimizu S, Tsujimoto Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997;57:1835–1840. [PubMed] [Google Scholar]
- Nicotera P, Melino G. Regulation of the apoptosis-necrosis switch. Oncogene. 2004;23:2757–2765. doi: 10.1038/sj.onc.1207559. [DOI] [PubMed] [Google Scholar]
- Saikumar P. Differential energy requirements for caspase activation and apoptosis. FASEB J. 2007;21:A258. [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Chandra D, Bratton SB, Person MD, Tian Y, Martin AG, Ayres M, et al. Intracellular nucleotides act as critical prosurvival factors by binding to cytochrome C and inhibiting apoptosome. Cell. 2006;125:1333–1346. doi: 10.1016/j.cell.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Bratton SB, Salvesen GS. Regulation of the Apaf-1-caspase-9 apoptosome. J Cell Sci. 2010;123:3209–3214. doi: 10.1242/jcs.073643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370:455–465. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- Laster SM, Wood JG, Gooding LR. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol. 1988;141:2629–2634. [PubMed] [Google Scholar]
- Denecker G, Vercammen D, Steemans M, Vanden Berghe T, Brouckaert G, Van Loo G, et al. Death receptor-induced apoptotic and necrotic cell death: differential role of caspases and mitochondria. Cell Death Differ. 2001;8:829–840. doi: 10.1038/sj.cdd.4400883. [DOI] [PubMed] [Google Scholar]
- Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W, et al. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med. 1998;188:919–930. doi: 10.1084/jem.188.5.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med. 1998;187:1477–1485. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercammen D, Vandenabeele P, Beyaert R, Declercq W, Fiers W. Tumour necrosis factor-induced necrosis versus anti-Fas-induced apoptosis in L929 cells. Cytokine. 1997;9:801–808. doi: 10.1006/cyto.1997.0252. [DOI] [PubMed] [Google Scholar]
- Fiers W, Beyaert R, Boone E, Cornelis S, Declercq W, Decoster E, et al. TNF-induced intracellular signaling leading to gene induction or to cytotoxicity by necrosis or by apoptosis. J Inflamm. 1995;47:67–75. [PubMed] [Google Scholar]
- Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–1323. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, et al. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci USA. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of cyclophilin D. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- Mellen MA, de la Rosa EJ, Boya P. Autophagy is not universally required for phosphatidyl-serine exposure and apoptotic cell engulfment during neural development. Autophagy. 2009;5:964–972. doi: 10.4161/auto.5.7.9292. [DOI] [PubMed] [Google Scholar]
- Mellen MA, de la Rosa EJ, Boya P. The autophagic machinery is necessary for removal of cell corpses from the developing retinal neuroepithelium. Cell Death Differ. 2008;15:1279–1290. doi: 10.1038/cdd.2008.40. [DOI] [PubMed] [Google Scholar]
- Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science. 2014;344:1164–1168. doi: 10.1126/science.1252809. [DOI] [PubMed] [Google Scholar]
- Kenis H, Zandbergen HR, Hofstra L, Petrov AD, Dumont EA, Blankenberg FD, et al. Annexin A5 uptake in ischemic myocardium: demonstration of reversible phosphatidylserine externalization and feasibility of radionuclide imaging. J Nucl Med. 2010;51:259–267. doi: 10.2967/jnumed.109.068429. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, et al. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- Ow YP, Green DR, Hao Z, Mak TW. Cytochrome c: functions beyond respiration. Nat Rev Mol Cell Biol. 2008;9:532–542. doi: 10.1038/nrm2434. [DOI] [PubMed] [Google Scholar]
- Hardwick JM, Chen YB, Jonas EA. Multipolar functions of BCL-2 proteins link energetics to apoptosis. Trends Cell Biol. 2012;22:318–328. doi: 10.1016/j.tcb.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN. BCL-2 family proteins: critical checkpoints of apoptotic cell death. Clin Cancer Res. 2007;13:7254–7263. doi: 10.1158/1078-0432.CCR-07-1598. [DOI] [PubMed] [Google Scholar]
- Rong Y, Distelhorst CW. Bcl-2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu Rev Physiol. 2008;70:73–91. doi: 10.1146/annurev.physiol.70.021507.105852. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization. Trends Cell Biol. 2008;18:157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, et al. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell. 2009;36:487–499. doi: 10.1016/j.molcel.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, et al. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science. 2010;330:1390–1393. doi: 10.1126/science.1190217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C, et al. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem. 1999;274:1156–1163. doi: 10.1074/jbc.274.2.1156. [DOI] [PubMed] [Google Scholar]
- Yin XM, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, et al. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 1999;400:886–891. doi: 10.1038/23730. [DOI] [PubMed] [Google Scholar]
- Scaffidi C, Schmitz I, Zha J, Korsmeyer SJ, Krammer PH, Peter ME. Differential modulation of apoptosis sensitivity in CD95 type I and type II cells. J Biol Chem. 1999;274:22532–22538. doi: 10.1074/jbc.274.32.22532. [DOI] [PubMed] [Google Scholar]
- Gonzalvez F, Schug ZT, Houtkooper RH, MacKenzie ED, Brooks DG, Wanders RJ, et al. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J Cell Biol. 2008;183:681–696. doi: 10.1083/jcb.200803129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaltsman Y, Shachnai L, Yivgi-Ohana N, Schwarz M, Maryanovich M, Houtkooper RH, et al. MTCH2/MIMP is a major facilitator of tBID recruitment to mitochondria. Nat Cell Biol. 2010;12:553–562. doi: 10.1038/ncb2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, Huang DC, et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature. 2009;460:1035–1039. doi: 10.1038/nature08229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albeck JG, Burke JM, Aldridge BB, Zhang M, Lauffenburger DA, Sorger PK. Quantitative analysis of pathways controlling extrinsic apoptosis in single cells. Mol Cell. 2008;30:11–25. doi: 10.1016/j.molcel.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schile AJ, Garcia-Fernandez M, Steller H. Regulation of apoptosis by XIAP ubiquitin-ligase activity. Genes Dev. 2008;22:2256–2266. doi: 10.1101/gad.1663108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, et al. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature. 2001;410:112–116. doi: 10.1038/35065125. [DOI] [PubMed] [Google Scholar]
- Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Van Loo G, Demol H, van Gurp M, Hoorelbeke B, Schotte P, Beyaert R, et al. A matrix-assisted laser desorption ionization post-source decay (MALDI-PSD) analysis of proteins released from isolated liver mitochondria treated with recombinant truncated Bid. Cell Death Differ. 2002;9:301–308. doi: 10.1038/sj.cdd.4400966. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Kroemer G. Necroptosis: a specialized pathway of programmed necrosis. Cell. 2008;135:1161–1163. doi: 10.1016/j.cell.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1–RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–1360. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7:971–981. doi: 10.1016/j.celrep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133–146. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- Moujalled DM, Cook WD, Murphy JM, Vaux DL. Necroptosis induced by RIPK3 requires MLKL but not Drp1. Cell Death Dis. 2014;5:e1086. doi: 10.1038/cddis.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Kroemer G. MLKL regulates necrotic plasma membrane permeabilization. Cell Res. 2014;24:139–140. doi: 10.1038/cr.2014.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Li W, Ren J, Huang D, He WT, Song Y, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24:105–121. doi: 10.1038/cr.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443–453. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013;23:994–1006. doi: 10.1038/cr.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci USA. 2012;109:5322–5327. doi: 10.1073/pnas.1200012109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- Orozco S, Yatim N, Werner MR, Tran H, Gunja SY, Tait SW, et al. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis Cell Death Differ 2014. e-pub ahead of print 6 June 2014doi: 10.1038/cdd.2014.76 [DOI] [PMC free article] [PubMed]
- Moujalled DM, Cook WD, Okamoto T, Murphy J, Lawlor KE, Vince JE, et al. TNF can activate RIPK3 and cause programmed necrosis in the absence of RIPK1. Cell Death Dis. 2013;4:e465. doi: 10.1038/cddis.2012.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288:31268–31279. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. 2014;157:1189–1202. doi: 10.1016/j.cell.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011;43:432–448. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Rickard JA, O'Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell. 2014;157:1175–1188. doi: 10.1016/j.cell.2014.04.019. [DOI] [PubMed] [Google Scholar]
- Kaiser WJ, Daley-Bauer LP, Thapa RJ, Mandal P, Berger SB, Huang C, et al. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc Natl Acad Sci USA. 2014;111:7753–7758. doi: 10.1073/pnas.1401857111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11:290–297. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basit F, Cristofanon S, Fulda S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ. 2013;20:1161–1173. doi: 10.1038/cdd.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SW, Oberst A, Quarato G, Milasta S, Haller M, Wang R, et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 2013;5:878–885. doi: 10.1016/j.celrep.2013.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228–243. doi: 10.1016/j.cell.2011.11.030. [DOI] [PubMed] [Google Scholar]
- Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O'Reilly L, et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity. 2012;36:215–227. doi: 10.1016/j.immuni.2012.01.012. [DOI] [PubMed] [Google Scholar]
- Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E, et al. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 2013;20:1381–1392. doi: 10.1038/cdd.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlangenakker N, Vanden Berghe T, Bogaert P, Laukens B, Zobel K, Deshayes K, et al. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ. 2011;18:656–665. doi: 10.1038/cdd.2010.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, et al. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol. 2011;13:1437–1442. doi: 10.1038/ncb2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Oberst A, Dillon CP, Weinlich R, Salvesen GS. RIPK-dependent necrosis and its regulation by caspases: a mystery in five acts. Mol Cell. 2011;44:9–16. doi: 10.1016/j.molcel.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinlich R, Dillon CP, Green DR. Ripped to death. Trends Cell Biol. 2011;21:630–637. doi: 10.1016/j.tcb.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase-8–FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajant H. The Fas signaling pathway: more than a paradigm. Science. 2002;296:1635–1636. doi: 10.1126/science.1071553. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, Tepper CG, Seldin MF, O'Rourke K, Kischkel FC, Hellbardt S, et al. FADD/MORT1 is a common mediator of CD95 (Fas/APO-1) and tumor necrosis factor receptor-induced apoptosis. J Biol Chem. 1996;271:4961–4965. doi: 10.1074/jbc.271.9.4961. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, et al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, et al. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenabeele P, Melino G. The flick of a switch: which death program to choose. Cell Death Differ. 2012;19:1093–1095. doi: 10.1038/cdd.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SW, Ichim G, Green DR. Die another way – non-apoptotic mechanisms of cell death. J Cell Sci. 2014;127:2135–2144. doi: 10.1242/jcs.093575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook WD, Moujalled DM, Ralph TJ, Lock P, Young SN, Murphy JM, et al. RIPK1- and RIPK3-induced cell death mode is determined by target availability Cell Death Differ 2014. e-pub ahead of print 6 June 2014doi: 10.1038/cdd.2014.70 [DOI] [PMC free article] [PubMed]
- Morioka S, Broglie P, Omori E, Ikeda Y, Takaesu G, Matsumoto K, et al. TAK1 kinase switches cell fate from apoptosis to necrosis following TNF stimulation. J Cell Biol. 2014;204:607–623. doi: 10.1083/jcb.201305070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrocola F, Izzo V, Niso-Santano M, Vacchelli E, Galluzzi L, Maiuri MC, et al. Regulation of autophagy by stress-responsive transcription factors. Semin Cancer Biol. 2013;23:310–322. doi: 10.1016/j.semcancer.2013.05.008. [DOI] [PubMed] [Google Scholar]
- Shin JH, Min SH, Kim SJ, Kim YI, Park J, Lee HK, et al. TAK1 regulates autophagic cell death by suppressing the phosphorylation of p70 S6 kinase 1. Sci Rep. 2013;3:1561. doi: 10.1038/srep01561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criollo A, Niso-Santano M, Malik SA, Michaud M, Morselli E, Marino G, et al. Inhibition of autophagy by TAB2 and TAB3. EMBO J. 2011;30:4908–4920. doi: 10.1038/emboj.2011.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol. 2011;12:715–723. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Kroemer G. Mitochondrial apoptosis without VDAC. Nat Cell Biol. 2007;9:487–489. doi: 10.1038/ncb0507-487. [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. P53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012;149:1536–1548. doi: 10.1016/j.cell.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrier F, Mignotte B, Jan G, Brenner C. Study of PTPC composition during apoptosis for identification of viral protein target. Ann NY Acad Sci. 2003;1010:126–142. doi: 10.1196/annals.1299.022. [DOI] [PubMed] [Google Scholar]
- Brenner C, Moulin M. Physiological roles of the permeability transition pore. Circ Res. 2012;111:1237–1247. doi: 10.1161/CIRCRESAHA.112.265942. [DOI] [PubMed] [Google Scholar]
- Brenner C, Grimm S. The permeability transition pore complex in cancer cell death. Oncogene. 2006;25:4744–4756. doi: 10.1038/sj.onc.1209609. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta. 2009;1787:1395–1401. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SW, Green DR.Mitochondrial regulation of cell death Cold Spring Harb Perspect Biol 20135doi: 10.1101/cshperspect.a008706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SW, Green DR. Mitochondria and cell signalling. J Cell Sci. 2012;125:807–815. doi: 10.1242/jcs.099234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- Tait SW, Parsons MJ, Llambi F, Bouchier-Hayes L, Connell S, Munoz-Pinedo C, et al. Resistance to caspase-independent cell death requires persistence of intact mitochondria. Dev Cell. 2010;18:802–813. doi: 10.1016/j.devcel.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Johnson N, Capano M, Edwards M, Crompton M. Cyclophilin-D promotes the mitochondrial permeability transition but has opposite effects on apoptosis and necrosis. Biochem J. 2004;383:101–109. doi: 10.1042/BJ20040669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Martin SJ. Caspase-independent cell death. Nat Med. 2005;11:725–730. doi: 10.1038/nm1263. [DOI] [PubMed] [Google Scholar]
- Dam AD, Mitchell AS, Quadrilatero J. Induction of mitochondrial biogenesis protects against caspase-dependent and caspase-independent apoptosis in L6 myoblasts. Biochim Biophys Acta. 2013;1833:3426–3435. doi: 10.1016/j.bbamcr.2013.04.014. [DOI] [PubMed] [Google Scholar]
- Bonora M, Wieckowski MR, Chinopoulos C, Kepp O, Kroemer G, Galluzzi L, et al. Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition Oncogene 2014. e-pub ahead of print 14 April 2014doi: 10.1038/onc.2014.96 [DOI] [PubMed]
- Zhivotovsky B, Galluzzi L, Kepp O, Kroemer G. Adenine nucleotide translocase: a component of the phylogenetically conserved cell death machinery. Cell Death Differ. 2009;16:1419–1425. doi: 10.1038/cdd.2009.118. [DOI] [PubMed] [Google Scholar]
- Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci USA. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle. 2013;12:674–683. doi: 10.4161/cc.23599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanella M, Parker N, Tan CH, Hall AM, Duchen MR. IF(1): setting the pace of the F(1)F(o)-ATP synthase. Trends Biochem Sci. 2009;34:343–350. doi: 10.1016/j.tibs.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci USA. 2014;111:10580–10585. doi: 10.1073/pnas.1401591111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–1472. doi: 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Pan X, Nguyen T, Liu J, Holmstrom KM, Finkel T. Unresolved questions from the analysis of mice lacking MCU expression. Biochem Biophys Res Commun. 2014;449:384–385. doi: 10.1016/j.bbrc.2014.04.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatokun AA, Dawson VL, Dawson TM. Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br J Pharmacol. 2014;171:2000–2016. doi: 10.1111/bph.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kim NS, Haince JF, Kang HC, David KK, Andrabi SA, et al. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos) Sci Signal. 2011;4:ra20. doi: 10.1126/scisignal.2000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, et al. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci USA. 2006;103:18314–18319. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- Sosna J, Voigt S, Mathieu S, Lange A, Thon L, Davarnia P, et al. TNF-induced necroptosis and PARP-1-mediated necrosis represent distinct routes to programmed necrotic cell death. Cell Mol Life Sci. 2014;71:331–348. doi: 10.1007/s00018-013-1381-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EJ, Min KJ, Lee TJ, Yoo YH, Kim YS, Kwon TK. beta-Lapachone induces programmed necrosis through the RIP1-PARP-AIF-dependent pathway in human hepatocellular carcinoma SK-Hep1 cells. Cell Death Dis. 2014;5:e1230. doi: 10.1038/cddis.2014.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlangenakker N, Vanden Berghe T, Vandenabeele P. Many stimuli pull the necrotic trigger, an overview. Cell Death Differ. 2012;19:75–86. doi: 10.1038/cdd.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136:4551–4556. doi: 10.1021/ja411006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9–17. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3:285–296. doi: 10.1016/s1535-6108(03)00050-3. [DOI] [PubMed] [Google Scholar]
- Tan S, Schubert D, Maher P. Oxytosis: a novel form of programmed cell death. Curr Top Med Chem. 2001;1:497–506. doi: 10.2174/1568026013394741. [DOI] [PubMed] [Google Scholar]
- Noch E, Khalili K. Molecular mechanisms of necrosis in glioblastoma: the role of glutamate excitotoxicity. Cancer Biol Ther. 2009;8:1791–1797. doi: 10.4161/cbt.8.19.9762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arundine M, Tymianski M. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium. 2003;34:325–337. doi: 10.1016/s0143-4160(03)00141-6. [DOI] [PubMed] [Google Scholar]
- Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grander D, Kharaziha P, Laane E, Pokrovskaja K, Panaretakis T. Autophagy as the main means of cytotoxicity by glucocorticoids in hematological malignancies. Autophagy. 2009;5:1198–1200. doi: 10.4161/auto.5.8.10122. [DOI] [PubMed] [Google Scholar]
- Laane E, Tamm KP, Buentke E, Ito K, Kharaziha P, Oscarsson J, et al. Cell death induced by dexamethasone in lymphoid leukemia is mediated through initiation of autophagy. Cell Death Differ. 2009;16:1018–1029. doi: 10.1038/cdd.2009.46. [DOI] [PubMed] [Google Scholar]
- Liu Y, Shoji-Kawata S, Sumpter RM, Jr, Wei Y, Ginet V, Zhang L, et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia–ischemia. Proc Natl Acad Sci USA. 2013;110:20364–20371. doi: 10.1073/pnas.1319661110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma H, Gangadhar NM, Letso RR, Wolpaw AJ, Sriramaratnam R, Stockwell BR. Identification of a small molecule that induces ATG5-and-cathepsin-l-dependent cell death and modulates polyglutamine toxicity. Exp Cell Res. 2013;319:1759–1773. doi: 10.1016/j.yexcr.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell. 2011;42:23–35. doi: 10.1016/j.molcel.2011.02.009. [DOI] [PubMed] [Google Scholar]
- Nihira K, Miki Y, Ono K, Suzuki T, Sasano H. An inhibition of p62/SQSTM1 caused autophagic cell death of several human carcinoma cells. Cancer Sci. 2014;105:568–575. doi: 10.1111/cas.12396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SY, Chiu LY, Maa MC, Wang JS, Chien CL, Lin WW. zVAD-induced autophagic cell death requires c-Src-dependent ERK and JNK activation and reactive oxygen species generation. Autophagy. 2011;7:217–228. doi: 10.4161/auto.7.2.14212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Pinedo C, Martin SJ. Autosis: a new addition to the cell death tower of babel. Cell Death Dis. 2014;5:e1319. doi: 10.1038/cddis.2014.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakaran K, Li L, Borowitz JL, Isom GE. Caspase inhibition switches the mode of cell death induced by cyanide by enhancing reactive oxygen species generation and PARP-1 activation. Toxicol Appl Pharmacol. 2004;195:194–202. doi: 10.1016/j.taap.2003.11.012. [DOI] [PubMed] [Google Scholar]
- McCarthy NJ, Whyte MK, Gilbert CS, Evan GI. Inhibition of Ced-3/ICE-related proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J Cell Biol. 1997;136:215–227. doi: 10.1083/jcb.136.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhart L, Belz K, Fulda S. Smac mimetic and demethylating agents synergistically trigger cell death in acute myeloid leukemia cells and overcome apoptosis resistance by inducing necroptosis. Cell Death Dis. 2013;4:e802. doi: 10.1038/cddis.2013.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunai ZA, Imre G, Barna G, Korcsmaros T, Petak I, Bauer PI, et al. Staurosporine induces necroptotic cell death under caspase-compromised conditions in U937 cells. PLoS One. 2012;7:e41945. doi: 10.1371/journal.pone.0041945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minina EA, Filonova LH, Fukada K, Savenkov EI, Gogvadze V, Clapham D, et al. Autophagy and metacaspase determine the mode of cell death in plants. J Cell Biol. 2013;203:917–927. doi: 10.1083/jcb.201307082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WW, Yu H, Fan HB, Zhang CC, Zhang M, Zhang C, et al. RIP1 mediates the protection of geldanamycin on neuronal injury induced by oxygen-glucose deprivation combined with zVAD in primary cortical neurons. J Neurochem. 2012;120:70–77. doi: 10.1111/j.1471-4159.2011.07526.x. [DOI] [PubMed] [Google Scholar]
- Han W, Xie J, Fang Y, Wang Z, Pan H. Nec-1 enhances shikonin-induced apoptosis in leukemia cells by inhibition of RIP-1 and ERK1/2. Int J Mol Sci. 2012;13:7212–7225. doi: 10.3390/ijms13067212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death – a new approach to cancer therapy. J Clin Invest. 2005;115:2625–2632. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes MA, Harper N, Butterworth M, Cain K, Cohen GM, MacFarlane M. Reconstitution of the death-inducing signaling complex reveals a substrate switch that determines CD95-mediated death or survival. Mol Cell. 2009;35:265–279. doi: 10.1016/j.molcel.2009.06.012. [DOI] [PubMed] [Google Scholar]
- Ide T, Brown-Endres L, Chu K, Ongusaha PP, Ohtsuka T, El-Deiry WS, et al. GAMT, a p53-inducible modulator of apoptosis, is critical for the adaptive response to nutrient stress. Mol Cell. 2009;36:379–392. doi: 10.1016/j.molcel.2009.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Khalil H, Bertrand MJ, Vandenabeele P, Widmann C. Caspase-3 and RasGAP: a stress-sensing survival/demise switch. Trends Cell Biol. 2014;24:83–89. doi: 10.1016/j.tcb.2013.08.002. [DOI] [PubMed] [Google Scholar]
- Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta. 2013;1833:3448–3459. doi: 10.1016/j.bbamcr.2013.06.001. [DOI] [PubMed] [Google Scholar]
- Albeck JG, Burke JM, Spencer SL, Lauffenburger DA, Sorger PK. Modeling a snap-action, variable-delay switch controlling extrinsic cell death. PLoS Biol. 2008;6:2831–2852. doi: 10.1371/journal.pbio.0060299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A, Brasen JH, Darding M, Jin MK, Sanz AB, Heller JO, et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2013;110:12024–12029. doi: 10.1073/pnas.1305538110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A, Brasen JH, De Zen F, Weinlich R, Schwendener RA, Green DR, et al. Dichotomy between RIP1- and RIP3-mediated necroptosis in tumor necrosis factor-alpha-induced shock. Mol Med. 2012;18:577–586. doi: 10.2119/molmed.2011.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35:908–918. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- Trichonas G, Murakami Y, Thanos A, Morizane Y, Kayama M, Debouck CM, et al. Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc Natl Acad Sci USA. 2010;107:21695–21700. doi: 10.1073/pnas.1009179107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Li J. Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis. 2013;4:e716. doi: 10.1038/cddis.2013.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, et al. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–737. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, et al. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–352. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, et al. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- Woo M, Hakem R, Soengas MS, Duncan GS, Shahinian A, Kagi D, et al. Essential contribution of caspase 3/CPP32 to apoptosis and its associated nuclear changes. Genes Dev. 1998;12:806–819. doi: 10.1101/gad.12.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, et al. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- Li J, Yuan J. Caspases in apoptosis and beyond. Oncogene. 2008;27:6194–6206. doi: 10.1038/onc.2008.297. [DOI] [PubMed] [Google Scholar]
- Degterev A, Boyce M, Yuan J. A decade of caspases. Oncogene. 2003;22:8543–8567. doi: 10.1038/sj.onc.1207107. [DOI] [PubMed] [Google Scholar]
- Honarpour N, Du C, Richardson JA, Hammer RE, Wang X, Herz J. Adult Apaf-1-deficient mice exhibit male infertility. Dev Biol. 2000;218:248–258. doi: 10.1006/dbio.1999.9585. [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthalakath H, Strasser A. Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ. 2002;9:505–512. doi: 10.1038/sj.cdd.4400998. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, Villunger A, O'Reilly LA, Beaumont JG, Coultas L, Cheney RE, et al. Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science. 2001;293:1829–1832. doi: 10.1126/science.1062257. [DOI] [PubMed] [Google Scholar]
- Bouillet P, Purton JF, Godfrey DI, Zhang LC, Coultas L, Puthalakath H, et al. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature. 2002;415:922–926. doi: 10.1038/415922a. [DOI] [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labi V, Woess C, Tuzlak S, Erlacher M, Bouillet P, Strasser A, et al. Deregulated cell death and lymphocyte homeostasis cause premature lethality in mice lacking the BH3-only proteins Bim and Bmf. Blood. 2014;123:2652–2662. doi: 10.1182/blood-2013-11-537217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. Annu Rev Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amelio M, Sheng M, Cecconi F. Caspase-3 in the central nervous system: beyond apoptosis. Trends Neurosci. 2012;35:700–709. doi: 10.1016/j.tins.2012.06.004. [DOI] [PubMed] [Google Scholar]
- Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature. 2006;443:796–802. doi: 10.1038/nature05293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Wang L, Wong CC, Scott FL, Eckelman BP, Han X, et al. Transnitrosylation of XIAP regulates caspase-dependent neuronal cell death. Mol Cell. 2010;39:184–195. doi: 10.1016/j.molcel.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burguillos MA, Deierborg T, Kavanagh E, Persson A, Hajji N, Garcia-Quintanilla A, et al. Caspase signalling controls microglia activation and neurotoxicity. Nature. 2011;472:319–324. doi: 10.1038/nature09788. [DOI] [PubMed] [Google Scholar]
- Barut S, Unlu YA, Karaoglan A, Tuncdemir M, Dagistanli FK, Ozturk M, et al. The neuroprotective effects of z-DEVD.fmk, a caspase-3 inhibitor, on traumatic spinal cord injury in rats Surg Neurol 200564213–220.discussion 220. [DOI] [PubMed] [Google Scholar]
- Knoblach SM, Alroy DA, Nikolaeva M, Cernak I, Stoica BA, Faden AI. Caspase inhibitor z-DEVD-fmk attenuates calpain and necrotic cell death in vitro and after traumatic brain injury. J Cereb Blood Flow Metab. 2004;24:1119–1132. doi: 10.1097/01.WCB.0000138664.17682.32. [DOI] [PubMed] [Google Scholar]
- Renolleau S, Fau S, Goyenvalle C, Joly LM, Chauvier D, Jacotot E, et al. Specific caspase inhibitor Q-VD-OPh prevents neonatal stroke in P7 rat: a role for gender. J Neurochem. 2007;100:1062–1071. doi: 10.1111/j.1471-4159.2006.04269.x. [DOI] [PubMed] [Google Scholar]
- Endres M, Namura S, Shimizu-Sasamata M, Waeber C, Zhang L, Gomez-Isla T, et al. Attenuation of delayed neuronal death after mild focal ischemia in mice by inhibition of the caspase family. J Cereb Blood Flow Metab. 1998;18:238–247. doi: 10.1097/00004647-199803000-00002. [DOI] [PubMed] [Google Scholar]
- Han BH, Xu D, Choi J, Han Y, Xanthoudakis S, Roy S, et al. Selective, reversible caspase-3 inhibitor is neuroprotective and reveals distinct pathways of cell death after neonatal hypoxic–ischemic brain injury. J Biol Chem. 2002;277:30128–30136. doi: 10.1074/jbc.M202931200. [DOI] [PubMed] [Google Scholar]
- Han W, Sun Y, Wang X, Zhu C, Blomgren K. Delayed, long-term administration of the caspase inhibitor Q-VD-OPh reduced brain injury induced by neonatal hypoxia–ischemia. Dev Neurosci. 2014;36:64–72. doi: 10.1159/000357939. [DOI] [PubMed] [Google Scholar]
- Wolf BB, Goldstein JC, Stennicke HR, Beere H, Amarante-Mendes GP, Salvesen GS, et al. Calpain functions in a caspase-independent manner to promote apoptosis-like events during platelet activation. Blood. 1999;94:1683–1692. [PubMed] [Google Scholar]
- Schotte P, Declercq W, Van Huffel S, Vandenabeele P, Beyaert R. Non-specific effects of methyl ketone peptide inhibitors of caspases. FEBS Lett. 1999;442:117–121. doi: 10.1016/s0014-5793(98)01640-8. [DOI] [PubMed] [Google Scholar]
- Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, Kroemer G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006;13:1423–1433. doi: 10.1038/sj.cdd.4401950. [DOI] [PubMed] [Google Scholar]
- Mondragon L, Galluzzi L, Mouhamad S, Orzaez M, Vicencio JM, Vitale I, et al. A chemical inhibitor of Apaf-1 exerts mitochondrioprotective functions and interferes with the intra-S-phase DNA damage checkpoint. Apoptosis. 2009;14:182–190. doi: 10.1007/s10495-008-0310-x. [DOI] [PubMed] [Google Scholar]
- Slee EA, Keogh SA, Martin SJ. Cleavage of BID during cytotoxic drug and UV radiation-induced apoptosis occurs downstream of the point of Bcl-2 action and is catalysed by caspase-3: a potential feedback loop for amplification of apoptosis-associated mitochondrial cytochrome c release. Cell Death Differ. 2000;7:556–565. doi: 10.1038/sj.cdd.4400689. [DOI] [PubMed] [Google Scholar]
- Lakhani SA, Masud A, Kuida K, Porter GA, Jr, Booth CJ, Mehal WZ, et al. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–851. doi: 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysko O, Love Aaes T, Bachert C, Vandenabeele P, Krysko DV. Many faces of DAMPs in cancer therapy. Cell Death Dis. 2013;4:e631. doi: 10.1038/cddis.2013.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Hou W, Zhang Q, Yan Z, Chen R, Zeh Iii HJ, Kang R, et al. Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell Death Dis. 2013;4:e966. doi: 10.1038/cddis.2013.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight KR, Zhang B, Morrison WA, Stewart AG. Ischaemia-reperfusion injury in mouse skeletal muscle is reduced by N omega-nitro-L-arginine methyl ester and dexamethasone. Eur J Pharmacol. 1997;332:273–278. doi: 10.1016/s0014-2999(97)01101-1. [DOI] [PubMed] [Google Scholar]
- Kumar S, Allen DA, Kieswich JE, Patel NS, Harwood S, Mazzon E, et al. Dexamethasone ameliorates renal ischemia–reperfusion injury. J Am Soc Nephrol. 2009;20:2412–2425. doi: 10.1681/ASN.2008080868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Xing J, Liu D, Gan X, Gao W, Hei Z. Dexamethasone pretreatment alleviates intestinal ischemia–reperfusion injury. J Surg Res. 2013;185:851–860. doi: 10.1016/j.jss.2013.07.049. [DOI] [PubMed] [Google Scholar]
- Kraft P, Gob E, Schuhmann MK, Gobel K, Deppermann C, Thielmann I, et al. FTY720 ameliorates acute ischemic stroke in mice by reducing thrombo-inflammation but not by direct neuroprotection. Stroke. 2013;44:3202–3210. doi: 10.1161/STROKEAHA.113.002880. [DOI] [PubMed] [Google Scholar]
- Pacher P, Szabo C. Role of poly(ADP-ribose) polymerase 1 (PARP-1) in cardiovascular diseases: the therapeutic potential of PARP inhibitors. Cardiovasc Drug Rev. 2007;25:235–260. doi: 10.1111/j.1527-3466.2007.00018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Moral RM, Gomez-Morales M, Hernandez-Cortes P, Aguilar D, Caballero T, Aneiros-Fernandez J, et al. PARP inhibition attenuates histopathological lesion in ischemia/reperfusion renal mouse model after cold prolonged ischemia. Scientific World J. 2013;2013:486574. doi: 10.1155/2013/486574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Klaus JA, Zhang J, Xu Z, Kibler KK, Andrabi SA, et al. Contributions of poly(ADP-ribose) polymerase-1 and -2 to nuclear translocation of apoptosis-inducing factor and injury from focal cerebral ischemia. J Neurochem. 2010;113:1012–1022. doi: 10.1111/j.1471-4159.2010.06667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin NJ, Szabo C. Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Mol Aspects Med. 2013;34:1217–1256. doi: 10.1016/j.mam.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe T, Vanlangenakker N, Parthoens E, Deckers W, Devos M, Festjens N, et al. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010;17:922–930. doi: 10.1038/cdd.2009.184. [DOI] [PubMed] [Google Scholar]
- Tang HL, Tang HM, Mak KH, Hu S, Wang SS, Wong KM, et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol Biol Cell. 2012;23:2240–2252. doi: 10.1091/mbc.E11-11-0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang HL, Tang HM, Hardwick JM, Fung MC. Strategies for tracking anastasis, a cell survival phenomenon that reverses apoptosis. J Vis Exp. 2014;2014:e51964. doi: 10.3791/51964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Cardenas ME, Heitman J. Calcineurin mutants render T lymphocytes resistant to cyclosporin A. Mol Pharmacol. 1996;50:506–511. [PubMed] [Google Scholar]
- Fruman DA, Klee CB, Bierer BE, Burakoff SJ. Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc Natl Acad Sci USA. 1992;89:3686–3690. doi: 10.1073/pnas.89.9.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love–hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–C833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- Zimmermann KC, Ricci JE, Droin NM, Green DR. The role of ARK in stress-induced apoptosis in Drosophila cells. J Cell Biol. 2002;156:1077–1087. doi: 10.1083/jcb.20112068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanuka H, Sawamoto K, Inohara N, Matsuno K, Okano H, Miura M. Control of the cell death pathway by Dapaf-1, a Drosophila Apaf-1/CED-4-related caspase activator. Mol Cell. 1999;4:757–769. doi: 10.1016/s1097-2765(00)80386-x. [DOI] [PubMed] [Google Scholar]
- Xue D, Horvitz HR. Inhibition of the Caenorhabditis elegans cell-death protease CED-3 by a CED-3 cleavage site in baculovirus p35 protein. Nature. 1995;377:248–251. doi: 10.1038/377248a0. [DOI] [PubMed] [Google Scholar]
- Daish TJ, Mills K, Kumar S. Drosophila caspase DRONC is required for specific developmental cell death pathways and stress-induced apoptosis. Dev Cell. 2004;7:909–915. doi: 10.1016/j.devcel.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Fraser AG, McCarthy NJ, Evan GI. drICE is an essential caspase required for apoptotic activity in Drosophila cells. EMBO J. 1997;16:6192–6199. doi: 10.1093/emboj/16.20.6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, McCall K, Steller H. DCP-1, a Drosophila cell death protease essential for development. Science. 1997;275:536–540. doi: 10.1126/science.275.5299.536. [DOI] [PubMed] [Google Scholar]
- Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- Fan TJ, Han LH, Cong RS, Liang J. Caspase family proteases and apoptosis. Acta Biochim Biophys Sin (Shanghai) 2005;37:719–727. doi: 10.1111/j.1745-7270.2005.00108.x. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Henry CM, Cullen SP. A perspective on mammalian caspases as positive and negative regulators of inflammation. Mol Cell. 2012;46:387–397. doi: 10.1016/j.molcel.2012.04.026. [DOI] [PubMed] [Google Scholar]