

Abstract
B-cell chronic lymphocytic leukemia (CLL) is the most common adult leukemia. The most common chromosomal abnormalities detectable by cytogenetics include deletion at 13q (55%), 11q (18%), trisomy 12 (12–16%) and 17p (8%). In 2002, we discovered that a microRNA cluster miR-15a/miR-16-1 (miR-15/16) is the target of 13q deletions in CLL. MicroRNAs encoded by the miR-15/16 locus (miR-15 and miR-16) function as tumor suppressors. Expression of these miRNAs downregulated in CLL, melanoma, colorectal cancer, bladder cancer and other solid tumors. miR-15/16 cluster targets multiple oncogenes, including BCL2, Cyclin D1, MCL1 and others. The most important target of miR-15/16 in CLL is arguably BCL2, as BCL2 is overexpressed in almost all CLLs. In this review, we discuss the discovery, functions, clinical relevance and treatment opportunities related to miR-15/16.
Facts
miR-15/16 is a first microRNA tumor suppressor located at 13q14 region deleted in CLL.
miR-15/16 exerts its tumor-suppressor function by targeting BCL2.
DLEU7 and miR-15/16 are two cooperating tumor suppressors at 13q14.
Open Questions
Is BCL2 the only important target of miR-15/16 in CLL?
Can miR-15/16 be used as a drug for CLL?
Will Bcl2 inhibitors cure CLL?
MiR-15/16 at 13q14, Gene Discovery
Chronic lymphocytic leukemia (CLL) is the most common human leukemia. It accounts for ∼12 000 newly cases diagnosed each year in the United States and represents one-third of all leukemia cases.1 Most CLL patients can survive for several of years and show relatively mild symptoms.1 Malignant CLL leukemic cells show morphologically mature appearance and typically do not proliferate in vitro.1, 2 CLL occurs in two forms, aggressive and indolent, and both forms show the clonal expansion of CD5-positive B cells.1, 2 In most cases, aggressive CLL is characterized by high zeta-chain-associated protein kinase 70 (ZAP-70) expression and unmutated IgH VH (immunoglobulin heavy chain variable region genes), while indolent CLLs express low ZAP-70 levels and show mutated IgH VH.1, 2
Genomic aberrations are detected in >80% of CLL cases. The most common chromosomal abnormalities detectable by cytogenetics include deletion at 13q (55%), 11q (18%), trisomy 12 (12–16%), and 17p (8%).3, 4
The most frequently deleted genomic region in CLL occurs at chromosome 13q14 and is associated with the indolent form of the disease.4 The same region is frequently found as the sole cytogenetic abnormality in other types of cancers: 50% of mantle cell lymphomas,5 ∼30% of multiple myeloma,6 and ∼60% of prostate cancers,7 indicating that 13q14 contains an important tumor suppressor involved in the pathogenesis of these malignancies. A number of years ago, several laboratories used positional cloning and sequencing of a region of >1 Mb at the deleted region to identify a tumor-suppressor gene at 13q14.8, 9 Detailed genetic analysis of protein-coding genes located in or close to the deleted region, including loss of heterozygosity (LOH) studies, mutation, and expression analysis, failed to show that any of these genes can function as tumor suppressors. And none of these known genes were found inactivated in CLL by mutations or deletions.8, 9, 10, 11 In 2001, we generated somatic cell hybrids between mouse and CLLs cells carrying 13q14 deletion and translocation. Analysis of these hybrids revealed that 13q14 tumor-suppressor gene lies within a 30-kb region between exons 2 and 5 of the DLEU2 gene (Figure 1). However, DLEU2 has been studied extensively and was excluded as a likely target of 13q14 deletions in CLL.2, 12, 13 Interestingly, the 13q14 translocation breakpoint in one of the CLL cases was mapped in the same region.12, 13 Thus we continued to investigate this 30-kb genomic region and finally found that a cluster of two microRNA genes miR-15a and miR-16-1 is located in the deleted region and very close to the translocation breakpoint.12 We then investigated the expression levels of miR-15/16 in CLL and found that miR-15/16 cluster was deleted or its expression of both miR-15 and miR-16 was downregulated in two-thirds of CLL cases.12 In contrast, expression of several protein-coding genes in the region in CLL was not affected by the 13q14 deletions.8, 9, 13 This was the first demonstration of the involvement of non-coding RNA in human disease.12
Figure 1.

13q14 genomic region deleted in CLL
The microRNAs are a large family of highly conserved non-coding genes thought to be involved in tissue-specific gene regulation.14 microRNAs represent an evolving class of gene products that are usually excised from 70- to 80-nt stem-loop RNA precursor structures.15 In mammals, single-stranded microRNA usually binds specific mRNAs (mainly in the 3′ untranslated (3′ UTR)) through sequences that are significantly, though not completely, complimentary to the target mRNA.15 By a mechanism that is not fully characterized, this binding causes a block of translation and/or degradation of target mRNAs, resulting in decreased levels of the corresponding protein.16 It is estimated that there could be up to 2000 microRNA genes in the human genome.17
Expression and Functions of miR-15/16 in CLL
Since our initial observation that miR-15/16 is deleted or downregulated in two-thirds of CLL samples, several other studies confirmed our results.18, 19 Tumor-suppressor genes in cancer are also frequent targets of mutations that can inactivate their function, and finding such mutations is a critical step in determining the contribution of microRNA or mRNA gene(s) in hematopoietic or solid malignancies. As miR-15/16 is a target of 13q14 deletions in CLL, it is possible that mutations could have a role in inactivation of its expression. Our subsequent study analyzed 75 CLL cases and 160 normal controls for mutations in microRNA genes.20 This study identified a germline mutation in the pri-miR-15a/miR-16-1 sequence in 2 of the 75 CLL patients (a C/T substitution, only 7-bp 3′ to miR-16-1 in the precursor), whereas no such mutation was found in 160 normal controls. Interestingly, this mutation significantly reduced the expression of both miR-16 and miR-15 in transfection experiments in vivo, indicating that this is the inactivating mutation.20 As CLL often shows a familial association (∼10% of patients have at least one first-degree relative with CLL), and CLL patients frequently have other cancers,21 mutations found in CLL may predispose patients to other associated cancers. Interestingly, one of the two CLL patients harboring the C/T miR-16-1 mutation was also diagnosed with breast cancer, her mother had CLL, and her sister had breast cancer.20 Thus miR-15/16 at 13q14 is deleted, mutated, and downregulated in a majority of CLL cases, indicating its contribution to the initiation and/or progression of CLL.
As we found that miR-15/16 is a target of 13q14 deletions in CLL, it is very important to understand its functions relevant to CLL pathogenesis. Using bioinformatic tools, we searched for targets of these microRNAs focusing on oncogenes implicated in CLL. We found that BCL2 oncogene was one of the top predicted targets of miR-15/16.22 By analyzing homology between miR-15/16 and the BCL2 mRNA, we found that the first 9 nucleotides from the 5′-ends of both miR-15 and miR-16 are complementary to bases 3287 to 3279 in the 3′-end of the BCL2 cDNA.22 BCL2 is a critical oncogene in a number of hematological malignancies as well as in solid tumors. It mostly functions by promoting survival and inhibiting cell death.23, 24 In B-cell malignancies, such as follicular lymphomas and diffuse B-cell lymphomas, BCL2 is activated by the translocation t(14;18)(q32;q21), which places the BCL2 oncogene in the proximity of immunoglobulin heavy-chain enhancers, resulting in overexpression of BCL2.25, 26 Interestingly, almost all CLLs overexpress Bcl-2,27 but t(14;18)(q32;q21) translocations are very rare in CLL,28 and no other mechanism for BCL2 overexpression in CLL has been reported. We thus hypothesized that miR-15/16 downregulation in CLL is responsible for the increased levels of BCL2. To demonstrate that overexpression regulation of BCL2 is a direct effect of loss of miR-15/16, a 536-bp fragment of human BCL2 interacting with the two microRNAs was fused to a luciferase reporter gene. Luciferase assay experiments showed that both miR-15 and miR-16 directly interact with 3′UTR of human BCL2 but not with the mutated sequence lacking the region of homology.22 This indicates that both microRNAs directly interact with and inhibit BCL2 expression. In addition, we found that expression of both miR-15 and miR-16 is inversely correlated with Bcl2 expression in CLL samples and that Bcl2 repression by miR-15/16 induced apoptosis in a leukemic cell line MEG-01 that carries alterations in both miR-15/16 alleles and does not express any miR-15/16. In these experiments, we found MEG-01 cells transfected with the wild-type miR-15/16 rapidly undergo apoptosis evidenced by cleavage of pro-caspase 9 and of poly (ADP-ribose) polymerase, in a sharp contrast with MEG-01 cells transfected with the miR-15/16 mutant found in CLL patients.22 We concluded that miR-15/16 targets BCL2 expression in CLL and that loss of miR-15/16 due to 13q14 deletions is the main reason of BCL2 overexpression in CLL.
To further investigate the tumor-suppressor action of miR-15/16 cluster, we tested its tumor-suppression function in vivo. MEG-01 cells, transfected miR-15/16 cluster were inoculated into ‘nude' mice. After 4 weeks days, tumor growth was completely suppressed in most of the mice inoculated with miR-15/16-transfected MEG-01, whereas we observed large tumors for the untreated and empty vector treated mice.29 Results of these experiments showed the tumor-suppressor function of miR-15/16 cluster in MEG-01 leukemia cells. Because most microRNAs inhibit translation of their target mRNAs, we also investigated the effects of miR-15/16 on MEG-01 proteome. Among differentially expressed proteins, BCL2 and WT1 were validated miR-15/16 targets. The targeted proteins have a variety of biological functions. Interestingly, 10 of the 27 proteins were involved in cell growth regulation or had anti-apoptotic function, and 8 of the 27 proteins were predicted targets of miR-15/16.29
We recently investigated transcriptional regulation of miR-15/16 in CLL. Several TP53-binding sites were found upstream of the miR-15/miR-16 chromosome 13q14 and on a homologous miR-15/16 cluster chromosome 3.30 Chromatin immunoprecipitation analysis revealed that TP53 directly binds to its predicted binding sites on both chromosome 13q14 and chromosome 3, both in cell lines and in primary CLLs with normal cytogenetic profiles. A luciferase reporter assay showed that TP53 significantly increased the luciferase reporter activity of all the binding site containing vectors.30 In MEG-01 cells, TP53 transactivation of the miR-15/16 cluster was also confirmed by real-time reverse transcription-PCR. Similarly, doxorubicin-mediated TP53 activation in MEG-01 cells increased the expression of miR-16, an effect that was abolished when TP53 was silenced by an anti-TP53 oligonucleotide.30 We also identified a binding site for both miR-15 and miR-16 inside the 3′UTR of TP53. A luciferase reporter assay showed that both miR-15 and miR-16 directly target the identified TP53-binding site and significantly reduced the luciferase reporter activity compared with a scrambled oligonucleotide-negative control. This effect was completely abolished when the binding site was either deleted or mutated. We concluded that miR-15/16 and TP53 are engaged in feedback circuitry loop in CLL: p53 transactivates miR-15/16, while increased levels of miR-15/16 target TP53 expression (Figure 2).30
Figure 2.
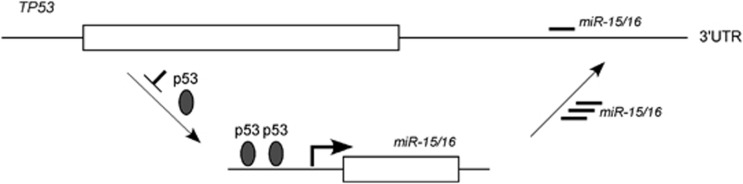
TP53–miR-15/16 circuitry in CLL
Although BCL2 is arguably the most important target of miR-15/16, several other oncogenes targeted by miR-15/16 were recently identified. For example, it has been shown that miR-15/16 inhibits MCL1 oncogene in CLL BMI1 oncogene in mantle call lymphoma.29, 31 Two other recent reports demonstrated that miR-15/16 directly target critical cell cycle regulator Cyclin D1 in bladder cancer and osteosarcoma.32, 33 Another study showed that miR-15/16 expression is downregulated in malignant pleural mesothelioma (MPM).34 Administration of synthetic mimics to restore miR-15/16 expression led to growth inhibition in MPM cell lines and in xenograft-bearing nude mice; administration of miR-15/16 mimics led to consistent inhibition of MPM tumor growth.34
miR-15/16 Cooperates with DLEU7 in CLL Pathogenesis
A recent study reported a high-resolution map of 13q14 deletions using 171 CLL samples.35 Interestingly, in addition to miR-15/16, this region also contained DLEU7 gene (that was previously identified as a candidate tumor-suppressor gene at 13q14) located telometic to miR-15/16.35, 36 We investigated whether DLEU7 can also function as a tumor suppressor and cooperate with miR-15/16,37 as DLEU7 is the only protein-coding gene located within reported deleted region. Among the 25 CLL samples examined for DLEU7 expression, 24 showed lower expression compared with normal CD19+ B cells, while 13 of the 25 samples showed decreases of ≥10-fold. Expression of miR-15/16 was also decreased in almost all CLL examined.37 Recent studies of CLL mouse demonstrated the importance of the NF-kB pathway. (reviewed in Pekarsky et al.38). For instance, transgenic mice expressing APRIL (a proliferation-inducing tumor necrosis factor (TNF) ligand), a member of the TNF superfamily involved in NF-kB activation showed significant expansions of CD5+ B cells. We thus investigated whether Dleu7 functions as an inhibitor of NF-kB, in particular if its function is related to APRIL. APRIL and BAFF are two members of the TNF superfamily and show abnormally high expression levels in a number of B-cell malignancies, including mantle cell lymphoma, diffuse large cell lymphoma, and CLL.39 APRIL binds to two of its receptors, TACI and BCMA (B-cell maturation antigen).39 TACI and BCMA interact with various TRAF family members and activate the NF-kB pathway.39 Thus, NF-kB activation through TACI and BCMA may be an important pathway and is important in CLL pathogenesis.37 We investigated whether DLEU7 expression has an effect on NF-kB activation by TACI and BCMA. Our results showed that indeed Dleu7 expression inhibited NF-kB activation by BCMA over fivefold and by TACI was inhibited over fourfold.37 We concluded that Dleu7 functions as an inhibitor of the NF-kB pathway, a critical pathway in CLL development (Figure 3). Nuclear factor of activated T-cells (NFAT) is a hallmark of unstimulated CLL cells and can also be activated by TACI and BCMA.40, 41, 42 NFAT is also involved in transactivation of CD5 promoter in B cells.41, 42 Since Dleu7 inhibits function of TACI and BCMA in NF-kB activation, we investigated whether Dleu7 can also inhibit NFAT activation by TACI and BCMA. Our results showed that Dleu7 expression can inhibit NFAT activation by TACI and BCMA approximately eightfold. To demonstrate tumor suppressor function of Dleu7 we used adenovirus expressing Dleu7 or empty adenovirus as a control. In these assays, Dleu7 expression resulted in over twofold decrease of cells in the S phase and in over threefold induction in apoptosis.37 Our current view is that 13q14 region deleted in CLL contains two tumor suppressors, miR-15/16 and DLEU7. DLEU7 inactivation results in NF-kB and NFAT activation, while miR-15/16 inactivation causes constitutive activation of Bcl2 and Mcl1.29, 37 Interestingly, transgenic mouse models confirmed that 13q14 region contains two tumor suppressors cooperating tumor suppressors. Transgenic mice expressing BCL2 or TRAF2DN (a molecule downstream of TACI and BCMA) do not develop CLL.43, 44 After the cross, TRAF2DN/BCL2 double transgenic mice developed CLL-like CD5-positive CLL phenotype.45 Thus, miR-15/16 and DLEU7 are inactivated by the 13q14 deletions. DLEU7 deletions cause the activtion of TNF signaling through TRAFs, while miR-15/16 deletions cause a constitutive increase of Bcl2 expression. These two events are very similar to the oncogenic mechanism causing CLL development in TRAF2DN/BCL2 transgenics (Figure 4).37
Figure 3.

DLEU7 inhibits NF-kB activation in CLL
Figure 4.

DLEU7 and miR-15/16 are two cooperating tumor suppressors at 13q14
miR-15/16 in CLL Mouse Models
There is only one natural mouse strain, as opposed to models of CLL that are genetically engineered, used as a model of disease presenting with a late-onset CLL due to slow progression of the leukemia induced by B-cell hyper-proliferation: the New Zealand Black (NZB) strain.46 Aging NZB mice are characterized by a sub-population of B-1 B cells that expands clonally and resembles that found in the human counterpart.46 An experiment of genome-wide linkage scan conducted in the laboratory of Dr. Raveche identified the loci associated with CLL development in this strain.47 Thirty-five percent of the animals generated from the backcross between NZB and the control strain DBA/2 presented with lymphoproliferative disease (LPD) of B cells. Three loci located on chromosomes 14, 18, and 19 were found to be associated with this disease. Half of all cases of human CLL show a deletion on chromosome 13q14.3 whose region is syntenic with the region on NZB chromosome 14 where the locus associated with LPD was found.47 The miR-15/16 cluster, contained in an intron of the gene DLEU2, is present in both human and mouse regions of synteny. Interestingly, by DNA sequencing of multiple NZB tissues, a point mutation in the 3′-flanking segment of the precursor miRNA, miR-16-1, was found to resemble the C→T point mutation previously identified in the miR-16-1 3′-flanking region of human patients.20, 47 No other strain of mice, including the NZW strain, the closest relative of NZB, was found to carry the same mutation. As expected, analysis of the levels of mature miRNA revealed a reduced expression of miR-16 in lymphoid tissues of NZB mice. In addition, cell cycle alterations such as G1 arrest and decrease in S phase cells were induced by exogenous miR-16 delivery into a NZB malignant cell line.47
To confirm the importance of miR-15/16 deletion in the pathogenesis of CLL, Dr. Dalla-Favera's laboratory generated the first genetically manipulated mice.48 This model was based on conditional alleles mimicking the loss of either the minimal deleted region (MDR) that spans the entire gene Dleu2, as was characterized in human CLL, or only the miR-15/16 cluster with no altered expression of Dleu2. At 1 year of age, while control animals displayed 15% of CD5+ B220+ B cells among mononuclear cells in the peritoneum, both Mdr and miR-15/16 knockout (KO) strains showed 50% of the same cell population.48 Overall, between 15 and 18 months, CLL affected 21% of miR-15a/16-1 KO and 27% of Mdr KO mice, but some type of clonal B-cell proliferation involved 26 and 42% of miR-15/16 KO and Mdr KO animals, respectively.48 To confirm that the Mdr KO phenotype was more severe than the miR-15/16 KO, the survival of the latter was not statistically different from that of wild-type siblings, while the former lived less than WT littermates, at the end succumbing to their leukemias.48 These findings likely point to other genetic elements with tumor-suppression function, including the gene DLEU2 itself, that are part of the MDR locus.48
Different approaches were also used to study B-cell proliferation mechanisms. For example, experiments of BrdU incorporation into DNA synthesis revealed that WT B cells started DNA synthesis later than miR-15/16 KO B cells.48 Expression levels of the phosphorylated retiniblastoma (p-Rb) protein indicate the entry into the cell cycle. In WT mitogen-stimulated B cells, p-Rb was produced at later time points than in B cells from miR-15/16 KO or Mdr KO animals. Investigators were also able to dissect the single contributions of miR-15/16 cluster and DLEU2 gene to the lympho-proliferation. In a human cell line derived from a 13q14 KO CLL, an inducible system was generated where the two genetic elements underwent separate in vitro re-expression. Results showed that, with higher fraction of cells in G0/G1 phase, in miR-15/16-expressing cells impaired proliferation occurred, whereas it did not in those expressing DLEU2.48 Thus a possible control of G0/G1 phase transition by miR-15/16 has been suggested.48
Treatment Implications
Due to high costs and difficulties in intracellular delivery, miR-15/16 itself cannot be easily used as a drug for CLL, at least currently. However, Bcl2, its arguably most important target in CLL, represents a very attractive molecular target. ABT-199 is a first-in-class orally bioavailable BCL2 selective inhibitor that was recently developed, and very recently, Souers et al.49 evaluated the ability of ABT-199 to suppress tumor growth in vivo in a broad spectrum of human hematological tumor xenografts established in immunocompromised mice. After a single oral dose of 12.5 mg/kg body weight in xenografts derived from RS(4;11) leukemia cells, ABT-199 caused a maximal tumor growth inhibition of 47% and tumor growth delay of 26%. The magnitude and durability of the response increased in a dose-dependent manner, with the highest dose of 100 mg/kg body weight tumor growth inhibition increased to 95% and a tumor growth delay to 152%.49 In addition to showing preclinical efficacy in BCL2-dependent cell lines and tumor xenograft models, ABT-199 demonstrated immediate anti-leukemic activity after a single dose in three patients with refractory CLL while causing only minor changes in platelet counts.49 Thus potent and selective BCL2 inhibitors such as ABT-199 could be efficiently used as CLL drugs.
Conclusions
Despite numerous studies of clinical features and genomic rearrangements in CLL, molecular mechanisms resulting in disease development are still not clearly understood. Our discovery that miR-15/16 is a main target of 13q14 deletions in CLL was the initial step in uncovering of one critical mechanism in CLL development as well as a first example of a major involvement of microRNAs in cancer pathogenesis. The fact that miR-15/16 targets BCL2 expression was a first link between major tumor-suppressor microRNA and major oncogene in CLL, and these findings increased interest in finding drugs inhibiting BCL2. These efforts recently resulted in the development of ABT-199, a very potent oral drug showing great promise in CLL treatment.
Acknowledgments
We thank Dr. Nicola Zanesi and Dr. Alexey Palamarchuk for helpful discussion.
Glossary
- TNF
tumor necrosis factor
- KO
knockout
- ZAP-70
zeta-chain-associated protein kinase 70
- IgH VH
immunoglobulin heavy chain variable region genes
- 3′UTR
3′ untranslated region
Footnotes
Edited by RA Knight
References
- Sgambati M, Linet M, Devesa S.Chronic Lymphocytic Leukemia, Epidemiological, Familial, and Genetic Aspects. Chronic Lymphocytic Leukemias, Revised and Expanded, 2nd ednIn: Cheson B, (ed).Marcel Dekker, Inc: New York, NY, USA; 200133–62. [Google Scholar]
- Bullrich F, Croce C.Molecular Biology of Chronic Lymphocytic Leukemia. Chronic Lymphocytic Leukemias, Revised and Expanded, 2nd ednIn: Cheson B, (ed). Marcel Dekker, Inc: New York, NY, USA; 20019–32. [Google Scholar]
- Edelmann J, Holzmann K, Miller F, Winkler D, Buhler A, Zenz T, et al. High-resolution genomic profiling of chronic lymphocytic leukemia reveals new recurrent genomic alterations. Blood. 2012;120:4783–4794. doi: 10.1182/blood-2012-04-423517. [DOI] [PubMed] [Google Scholar]
- Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343:1910–1916. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- Stilgenbauer S, Nickolenko J, Wilhelm J, Wolf S, Weitz S, Dohner K, et al. Expressed sequences as candidates for a novel tumor suppressor gene at band 13q14 in B-cell chronic lymphocytic leukemia and mantle cell lymphoma. Oncogene. 1998;16:1891–1897. doi: 10.1038/sj.onc.1201764. [DOI] [PubMed] [Google Scholar]
- Elnenaei MO, Hamoudi RA, Swansbury J, Gruszka-Westwood AM, Brito-Babapulle V, Matutes E, et al. Delineation of the minimal region of loss at 13q14 in multiple myeloma. Genes Chromosomes Cancer. 2003;36:99–106. doi: 10.1002/gcc.10140. [DOI] [PubMed] [Google Scholar]
- Dong JT, Boyd JC, Frierson HF., Jr Loss of heterozygosity at 13q14 and 13q21 in high grade, high stage prostate cancer. Prostate. 2001;49:166–171. doi: 10.1002/pros.1131. [DOI] [PubMed] [Google Scholar]
- Bullrich F, Fujii H, Calin G, Mabuchi H, Negrini M, Pekarsky Y, et al. Characterization of the 13q14 tumor suppressor locus in CLL: identification of ALT1, an alternative splice variant of the LEU2 gene. Cancer Res. 2001;61:6640–6648. [PubMed] [Google Scholar]
- Migliazza A, Bosch F, Komatsu H, Cayanis E, Martinotti S, Toniato E, et al. Nucleotide sequence, transcription map, and mutation analysis of the 13q14 chromosomal region deleted in B-cell chronic lymphocytic leukemia. Blood. 2001;97:2098–2104. doi: 10.1182/blood.v97.7.2098. [DOI] [PubMed] [Google Scholar]
- Rondeau G, Moreau I, Bezieau S, Petit JL, Heilig R, Fernandez S, et al. Comprehensive analysis of a large genomic sequence at the putative B-cell chronic lymphocytic leukaemia (B-CLL) tumour suppresser gene locus. Mutat Res. 2001;458:55–70. doi: 10.1016/s0027-5107(01)00219-6. [DOI] [PubMed] [Google Scholar]
- Mertens D, Wolf S, Schroeter P, Schaffner C, Dohner H, Stilgenbauer S, et al. Down-regulation of candidate tumor suppressor genes within chromosome band 13q14.3 is independent of the DNA methylation pattern in B-cell chronic lymphocytic leukemia. Blood. 2002;99:4116–4121. doi: 10.1182/blood.v99.11.4116. [DOI] [PubMed] [Google Scholar]
- Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99:15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekarsky Y, Calin GA, Aqeilan R. Chronic lymphocytic leukemia: molecular genetics and animal models. Curr Top Microbiol Immunol. 2005;294:51–70. doi: 10.1007/3-540-29933-5_4. [DOI] [PubMed] [Google Scholar]
- Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- Ambros V. MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell. 2003;113:673–676. doi: 10.1016/s0092-8674(03)00428-8. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42 (Database issue:D68–D73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allegra D, Bilan V, Garding A, Dohner H, Stilgenbauer S, Kuchenbauer F, et al. Defective DROSHA processing contributes to downregulation of MiR-15/-16 in chronic lymphocytic leukemia. Leukemia. 2014;28:98–107. doi: 10.1038/leu.2013.246. [DOI] [PubMed] [Google Scholar]
- Sampath D, Liu C, Vasan K, Sulda M, Puduvalli VK, Wierda WG, et al. Histone deacetylases mediate the silencing of miR-15a, miR-16, and miR-29b in chronic lymphocytic leukemia. Blood. 2012;119:1162–1172. doi: 10.1182/blood-2011-05-351510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin GA, Ferracin M, Cimmino A, Di Leva G, Shimizu M, Wojcik SE, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353:1793–1801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- Catovsky D.Definition and diagnosis of sporadic and familial chronic lymphocytic leukemia Hematol Oncol Clin North Am 200418783–794.vii. [DOI] [PubMed] [Google Scholar]
- Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Beato M, Sanchez-Aguilera A, Piris MA. Cell cycle deregulation in B-cell lymphomas. Blood. 2003;101:1220–1235. doi: 10.1182/blood-2002-07-2009. [DOI] [PubMed] [Google Scholar]
- Grabow S, Waring P, Happo L, Cook M, Mason KD, Kelly PN, et al. Pharmacological blockade of Bcl-2, Bcl-x(L) and Bcl-w by the BH3 mimetic ABT-737 has only minor impact on tumour development in p53-deficient mice. Cell Death Differ. 2012;19:623–632. doi: 10.1038/cdd.2011.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985;228:1440–1443. doi: 10.1126/science.3874430. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y, Yunis J, Onorato-Showe L, Erikson J, Nowell PC, Croce CM. Molecular cloning of the chromosomal breakpoint of B-cell lymphomas and leukemias with the t(11;14) chromosome translocation. Science. 1984;224:1403–1406. doi: 10.1126/science.6610211. [DOI] [PubMed] [Google Scholar]
- Kitada S, Andersen J, Akar S, Zapata JM, Takayama S, Krajewski S, et al. Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with In vitro and In vivo chemoresponses. Blood. 1998;91:3379–3389. [PubMed] [Google Scholar]
- Adachi M, Tefferi A, Greipp PR, Kipps TJ, Tsujimoto Y. Preferential linkage of bcl-2 to immunoglobulin light chain gene in chronic lymphocytic leukemia. J Exp Med. 1990;171:559–564. doi: 10.1084/jem.171.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin GA, Cimmino A, Fabbri M, Ferracin M, Wojcik SE, Shimizu M, et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Natl Acad Sci USA. 2008;105:5166–5171. doi: 10.1073/pnas.0800121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri M, Bottoni A, Shimizu M, Spizzo R, Nicoloso MS, Rossi S, et al. Association of a microRNA/TP53 feedback circuitry with pathogenesis and outcome of B-cell chronic lymphocytic leukemia. JAMA. 2011;305:59–67. doi: 10.1001/jama.2010.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teshima K, Nara M, Watanabe A, Ito M, Ikeda S, Hatano Y, et al. Dysregulation of BMI1 and microRNA-16 collaborate to enhance an anti-apoptotic potential in the side population of refractory mantle cell lymphoma. Oncogene. 2013;33:2191–2203. doi: 10.1038/onc.2013.177. [DOI] [PubMed] [Google Scholar]
- Cai CK, Zhao GY, Tian LY, Liu L, Yan K, Ma YL, et al. miR-15a and miR-16-1 downregulate CCND1 and induce apoptosis and cell cycle arrest in osteosarcoma. Oncol Rep. 2012;28:1764–1770. doi: 10.3892/or.2012.1995. [DOI] [PubMed] [Google Scholar]
- Jiang QQ, Liu B, Yuan T. MicroRNA-16 inhibits bladder cancer proliferation by targeting Cyclin D1. Asian Pac J Cancer Prev. 2013;14:4127–4130. doi: 10.7314/apjcp.2013.14.7.4127. [DOI] [PubMed] [Google Scholar]
- Reid G, Pel ME, Kirschner MB, Cheng YY, Mugridge N, Weiss J, et al. Restoring expression of miR-16: a novel approach to therapy for malignant pleural mesothelioma. Ann Oncol. 2013;24:3128–3135. doi: 10.1093/annonc/mdt412. [DOI] [PubMed] [Google Scholar]
- Ouillette P, Erba H, Kujawski L, Kaminski M, Shedden K, Malek SN. Integrated genomic profiling of chronic lymphocytic leukemia identifies subtypes of deletion 13q14. Cancer Res. 2008;68:1012–1021. doi: 10.1158/0008-5472.CAN-07-3105. [DOI] [PubMed] [Google Scholar]
- Hammarsund M, Corcoran MM, Wilson W, Zhu C, Einhorn S, Sangfelt O, et al. Characterization of a novel B-CLL candidate gene–DLEU7–located in the 13q14 tumor suppressor locus. FEBS Lett. 2004;556:75–80. doi: 10.1016/s0014-5793(03)01371-1. [DOI] [PubMed] [Google Scholar]
- Palamarchuk A, Efanov A, Nazaryan N, Santanam U, Alder H, Rassenti L, et al. 13q14 deletions in CLL involve cooperating tumor suppressors. Blood. 115:3916–3922. doi: 10.1182/blood-2009-10-249367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekarsky Y, Zanesi N, Aqeilan RI, Croce CM. Animal models for chronic lymphocytic leukemia. J Cell Biochem. 2007;100:1109–1118. doi: 10.1002/jcb.21147. [DOI] [PubMed] [Google Scholar]
- Haiat S, Billard C, Quiney C, Ajchenbaum-Cymbalista F, Kolb JP. Role of BAFF and APRIL in human B-cell chronic lymphocytic leukaemia. Immunology. 2006;118:281–292. doi: 10.1111/j.1365-2567.2006.02377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: a tutorial on B cell survival. Annu Rev Immunol. 2003;21:231–264. doi: 10.1146/annurev.immunol.21.120601.141152. [DOI] [PubMed] [Google Scholar]
- Schuh K, Avots A, Tony HP, Serfling E, Kneitz C. Nuclear NF-ATp is a hallmark of unstimulated B cells from B-CLL patients. Leuk Lymphoma. 1996;23:583–592. doi: 10.3109/10428199609054868. [DOI] [PubMed] [Google Scholar]
- Berland R, Wortis HH. An NFAT-dependent enhancer is necessary for anti-IgM-mediated induction of murine CD5 expression in primary splenic B cells. J Immunol. 1998;161:277–285. [PubMed] [Google Scholar]
- Katsumata M, Siegel RM, Louie DC, Miyashita T, Tsujimoto Y, Nowell PC, et al. Differential effects of Bcl-2 on T and B cells in transgenic mice. Proc Natl Acad Sci USA. 1992;89:11376–11380. doi: 10.1073/pnas.89.23.11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Reichlin A, Santana A, Sokol KA, Nussenzweig MC, Choi Y. TRAF2 is essential for JNK but not NF-kappaB activation and regulates lymphocyte proliferation and survival. Immunity. 1997;7:703–713. doi: 10.1016/s1074-7613(00)80390-8. [DOI] [PubMed] [Google Scholar]
- Zapata JM, Krajewska M, Morse HC, 3rd, Choi Y, Reed JC. TNF receptor-associated factor (TRAF) domain and Bcl-2 cooperate to induce small B cell lymphoma/chronic lymphocytic leukemia in transgenic mice. Proc Natl Acad Sci USA. 2004;101:16600–16605. doi: 10.1073/pnas.0407541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveche ES. Possible immunoregulatory role for CD5+B cells. Clin Immunol Immunopathol. 1990;56:135–150. doi: 10.1016/0090-1229(90)90136-e. [DOI] [PubMed] [Google Scholar]
- Raveche ES, Salerno E, Scaglione BJ, Manohar V, Abbasi F, Lin YC, et al. Abnormal microRNA-16 locus with synteny to human 13q14 linked to CLL in NZB mice. Blood. 2007;109:5079–5086. doi: 10.1182/blood-2007-02-071225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17:28–40. doi: 10.1016/j.ccr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]