

Abstract
Intrinsic or acquired resistance to the HER2-targeted therapy trastuzumab is a clinical concern in the treatment of patients with HER2-over-expressing metastatic breast cancers. We demonstrate here that multiple models of intrinsic and acquired resistance exhibit increased phosphorylation of p38 MAPK. Kinase inhibition of p38 rescued trastuzumab sensitivity in cells with acquired resistance. In addition, knockdown of p38 increased sensitivity to trastuzumab in an intrinsically resistant cell line. We previously reported that expression of growth differentiation factor 15 (GDF15) is increased in trastuzumab-resistant HER2-overexpressing breast cancer cells. In this study, we found that exogenous GDF15 or stable overexpression of GDF15 stimulated p38 phosphorylation in HER2-positive cells, suggesting a possible mechanism by which p38 is activated in resistant cells. GDF15 stable clones showed significantly increased invasiveness, which was rescued by p38 kinase inhibition, suggesting that p38 plays a role in the pro-invasive phenotype conferred by GDF15. Importantly, immunohistochemical analysis of a breast tumor tissue array indicated a significant (p=0.0053) correlation between HER2 and phosphorylated p38 specifically in GDF15-positive tissues. Our results suggest that p38 signaling drives trastuzumab resistance and invasiveness in HER2-overexpressing breast cancer. Upstream growth factor signals that have previously been implicated in trastuzumab resistance, such as GDF15, may contribute to the increased phosphorylation of p38 found in resistant cells.
Keywords: breast cancer, HER2, invasion, p38, trastuzumab
INTRODUCTION
The HER2 (erbB2/neu) receptor kinase is overexpressed in approximately 15% to 20% of primary breast cancers due to gene amplification [1]. HER2-overexpressing breast cancers tend to be more aggressive and likely to metastasize. However, the introduction of specific HER2-targeted therapies has dramatically improved the course of this disease subtype. The first-line treatment for HER2-overexpressing breast cancers is the HER2 monoclonal antibody trastuzumab. Response rates, duration of response, and time to progression are all improved in patients treated with combined trastuzumab plus chemotherapy. However, response to single-agent trastuzumab ranges from 18% to 35% depending on whether trastuzumab is administered in a heavily pre-treated versus front-line setting [2]. Furthermore, despite an initial response to trastuzumab-based therapy, many patients eventually develop resistance.
Many molecular mechanisms have been proposed to cause resistance to trastuzumab. These mechanisms include changes at the level of the receptor. For example, drug-target interactions may be affected by increased expression of cell surface proteins, such as MUC4, which blocks the ability of trastuzumab to recognize and bind to its specific epitope on HER2. Additional mechanisms of resistance exist in the form of compensatory lateral signaling by other receptor kinases, such as MET or insulin-like growth factor-I receptor (IGF-IR). Mutations or hyper-activation of downstream signaling molecules also reduces sensitivity to trastuzumab. For example, PIK3CA mutations or PTEN deletion can result in hyper-activation of PI3K signaling and have been strongly correlated with resistance to trastuzumab by multiple investigators.
We previously showed that the cytokine growth differentiation factor 15 (GDF15) was over-expressed in breast cancer cell lines that had acquired or intrinsic resistance to trastuzumab [3]. GDF15 shares sequence similarity with TGF-beta [4] and has been shown to activate phosphorylation of the TGF-beta receptor substrates, Smad2/3, in some cell lines [3, 5]. Thus, GDF15 is generally considered to be a member of the TGF-beta superfamily. However, a specific receptor has not yet been identified for circulating GDF15. We [3, 6] and others [7, 8] have shown that GDF15 induces phosphorylation of multiple signaling molecules, including human epidermal growth factor receptor 2 (HER2/erbB2), epidermal growth factor receptor (EGFR), Src, mitogen-activated kinases (MAPKs), and Akt. Pleiotropic biological activities have been shown to be activated by GDF15. For example, GDF15 mediates apoptosis in response to multiple anti-inflammatory and anti-cancer agents [9, 10], such that loss of GDF15 may reduce the efficacy of these agents. However, increased GDF15 levels have been detected in the sera or tumor tissues of patients with breast, prostate, ovarian, and colorectal cancers [11–15]. The current thought is that GDF15 may promote apoptosis in pre-malignant stages of disease, but may activate cell survival and anti-apoptotic pathways in advanced stages of disease, similar to what has been observed for TGF-beta.
In addition to observing an association between GDF15 and trastuzumab resistance in breast cancer, we found that a majority of ovarian tumor tissues expressed high levels of GDF15 based on immunohistochemical staining results [6]. Furthermore, stimulation of breast or ovarian cancer cells with exogenous recombinant human GDF15 increased phosphorylation of p38 MAPK, p42/p44 MAPKs, and Akt [3, 6]. Interestingly, stable over-expression of GDF15 or exogenous stimulation with GDF15 significantly increased the invasiveness of ovarian cancer cells through matrigel-coated Boyden chambers in an mTOR-dependent manner [6]. However, the pro-invasive potential of GDF15 in HER2-positive breast cancer has not been examined. Furthermore, the biological consequences of p38 induction by GDF15 have not been fully examined, particularly in the context of trastuzumab resistance. In the current study, we provide evidence that p38 phosphorylation is increased in trastuzumab-resistant lines, and that p38 inhibition restores sensitivity. We also demonstrate that GDF15 induces p38 phosphorylation, resulting in increased invasiveness of HER2-positive breast cancers. Finally, we show that phosphorylation of p38 and HER2 overexpression significantly correlate in GDF15-positive breast cancers.
2 MATERIALS AND METHODS
Materials
Trastuzumab (Herceptin™, Genentech, South San Francisco, CA) was dissolved in sterile water, which was included in the box from the manufacturer, at a stock concentration of 20 mg/mL, and was purchased from the Winship Cancer Institute pharmacy. Recombinant human GDF15 (rhGDF15; R&D Systems, Minneapolis, MN) was dissolved at a final stock concentration of 200 μg/mL in 4 mM HCl. SB203580 (Sigma-Aldrich; St. Louis, MO), a small-molecule inhibitor of the p38 MAPK isoforms alpha and beta [16], was dissolved in DMSO at a final concentration of 10 mM.
Cell culture
SKBR3, BT474, and MDA-MB-453 breast cancer cells over-express HER2/erbB2 and were maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). The HCC1419 and HCC1954 breast cancer cells also over-express HER2 and were maintained in RPMI with 10% FBS and 1% P/S. MDA-MB-361 HER2-over-expressing cells were maintained in RPMI with 20% FBS and 1% P/S. All cell lines were purchased from American Type Culture Collection, Manassas, VA. We created trastuzumab-resistant cells by maintaining SKBR3 and BT474 cells in 4 μg/ml trastuzumab for three months [17, 18], at which point surviving pools or clones were selected. All of the acquired resistant cells were maintained on 4 μg/ml trastuzumab, but the drug was removed from culture for 24 hours prior to performing experiments. GDF15 stable clones were created by transfecting BT474 cells with 3 μg of pCMV empty vector or pCMV myc-GDF15, both from Origene (Rockville, MD), using Lipofectamine (Invitrogen, Carlsbad, CA) combined with DMSO shock and selecting cells in 200 μg/mL G418.
Western blotting
Cells were lysed in RIPA buffer (Cell Signaling, Danvers, MA) supplemented with protease and phosphatase inhibitors. Total protein extracts were separated on SDS-PAGE and transferred onto nitrocellulose membrane. Blots were probed overnight with the desired antibodies at indicated dilutions as follows: p-p38 (Thr180/Tyr182), Cell Signaling, 4511s, 1:1000; total p38, EMD Chemicals, 506123, 1:1000; actin mAb AC-15, Sigma, A5441, 1:10,000. Appropriate secondary antibodies were used. Protein bands were detected with the Odyssey Imaging System (Li-Cor Biosciences, Lincoln, NE) and quantified with Image J. Blots were repeated at least three times.
Cell proliferation assays
Cells were plated at 3 × 103 per well in 96-well format and treated with vehicle control (DMSO), SB203580 (at indicated dose), trastuzumab (20 μg/mL), or a combination of trastuzumab and SB203580. DMSO concentration was maintained at the same level in all treatment groups. Each group was performed with six replicates, and experiments were repeated three times. MTS colorimetric assays (Promega, Madison, WI) were performed after 72 hours as directed by the manufacturer. Combination index (C.I.) values were determined with the commercial software package Calcusyn (Biosoft, Cambridge, United Kingdom) by the method of Chou and Talalay [19].
Transfection
Cells were transfected with 100 nM p38 siRNA or 100 nM control siRNA (Santa Cruz Biotechnology; Santa Cruz, CA) in antibiotic-free media using Lipofectamine 2000 (Invitrogen). After 24 hours, cells were treated with 20 μg/mL trastuzumab for an additional 72 hours; viable cells were then counted by trypan blue exclusion. Alternatively, after 48 hours of transfection, protein lysates were analyzed by Western blotting for total p38.
Invasion chamber assays
BT474 control clone or stable GDF15-expressing cells (5×104 cells/chamber), were plated in serum-free media in Trevigen Culture Coat R 24-well Low-BME Cell Invasion Optimization assay chambers (Trevigen Inc., Gaithersburg, MD) with 0.75 mL of culture media containing 10% FBS as the chemoattractant. Depending on the experiment, p38 MAPK inhibitor, SB203580 was added directly to upper chambers. After 48 hours, non-invading cells were removed from the interior surface of the membrane by scrubbing gently with a dry cotton-tipped swab. Each insert was then transferred into 100% methanol for two minutes followed by staining. The chambers were incubated with hematoxylin for six minutes followed by acid alcohol and nuclear stain. Chambers were then incubated in eosin for two minutes to stain the cytoplasm. The chambers were washed in water and dehydrated by passing through alcohol grades. The membranes were dried completely, separated from the chambers, and mounted with Permount. Photographs were taken of 10 fields at 10× magnification per sample with triplicates per group. The total number of cells in 10 fields was calculated for each sample. Experiments were performed twice.
Immunohistochemistry (IHC)
IHC was performed on a human breast tumor tissue microarray (TMA) (catalog # Z7020004, Biochain Institute; Hayward, CA) with a standard immunoperoxidase procedure as previously described [6]. The breast TMA consisted of 140 tumor tissues, including 83 HER2-overexpressing samples. The tissue sections on glass slides were deparaffinized by heating at 60°C followed by passages through xylene and alcohol grades and ultimately to water. Antigen retrieval was performed by boiling the slides in 10mM citrate buffer, pH 6.0 for 10 minutes followed by cooling in same buffer for 30 minutes. The endogenous peroxidase was quenched by incubating slides with 0.3% H2O2 in methanol for 15 minutes. Following washes with water and PBS/TBS, slides were incubated in 10% swine serum (Dako) for one hour to eliminate any non-specific background staining. Tissue sections were stained for human GDF15 (Cell Signaling #3249; dilution 1:100) or phosphorylated p38 (Cell Signaling #4511; dilution 1:800) overnight at 4°C. Biotinylated anti-rabbit antibody (Dako) was used as secondary antibody, and positive staining was detected by incubation with 3, 3-diaminobenzidine solution (DAB+ chromogen, Dako) with hematoxylin as a counterstain. The slides were mounted with the permanent mounting medium, Permount (Fisher Scientific). The slides were completely dried and viewed under a light microscope; pictures were taken at 10×/20×.
Statistics
P-values were determined for experimental versus control treatments by two-tailed student’s t-test, *p<0.05, **p<0.005. For IHC experiments on the tumor array, the significance of each correlation was determined by the Chi-square (p-p38 and GDF15+/HER2+) or two-tailed Fisher’s exact test (HER2 and GDF15, HER2 and p-p38); significance was viewed as p<0.05.
3. RESULTS
3.1 Increased p38 phosphorylation contributes to trastuzumab resistance
Total and phosphorylated p38 were examined in BT474 and SKBR3-derived clones with acquired trastuzumab resistance and intrinsically trastuzumab-resistant HCC1419, HCC1954, MDA-MB361, and MDA-MB 453 cells. Intrinsically resistant cells showed 1.5- to 2-fold increased p38 phosphorylation relative to parental BT474 and SKBR3 cell lines (Figure 1A). Similarly, acquired resistant cells exhibited 1.5- to 3-fold increased p38 phosphorylation relative to the corresponding parental line (Figure 1B).
Figure 1. Trastuzumab-resistant cells exhibit increased phosphorylation of p38 MAPK.
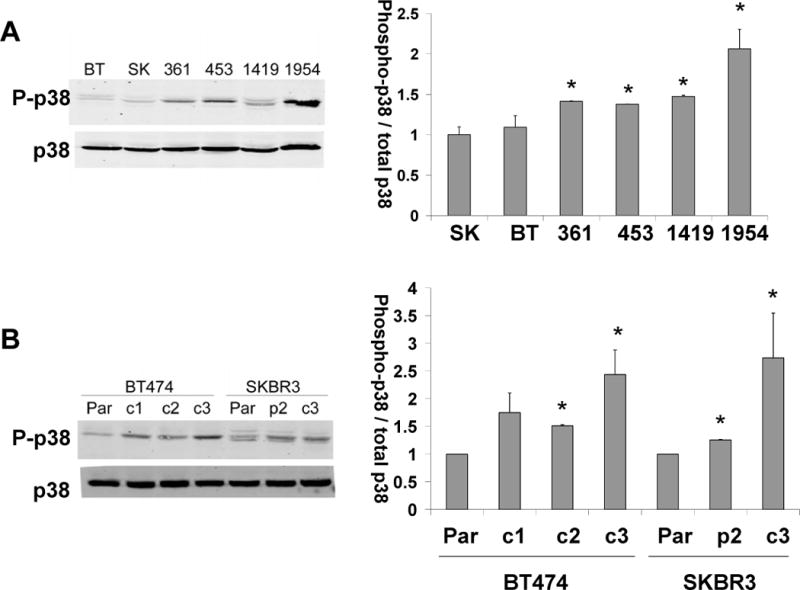
(A) Total protein lysates from BT474, SKBR3, MDA-MB-361, MDA-MB-453, HCC1419, and HCC1954 cells; (B) Total protein lysates from BT474 and SKBR3 parental and acquired trastuzumab-resistant cells derived from BT474 (c1, c2, c3) and SKBR3 (p2, c3). Lysates were Western blotted for phosphorylated and total p38. Representative blots are shown. Bands were quantified, and the average intensity of phosphorylated p38 normalized to total p38 is shown for (A) triplicate runs for each intrinsically resistant cell line relative to SKBR3 cells or (B) duplicate runs for acquired resistant lines relative to the corresponding parental line. Error bars represent standard deviations between duplicate experiments; *p<0.05, student’s t-test.
To determine if p38 phosphorylation directly contributes to resistance, we treated BT474 parental (BT-par) and BT474-derived acquired resistant clone 3 (BT-c3) with increasing concentrations of p38 kinase inhibitor SB203580 in combination with a steady, clinically relevant dose of trastuzumab (20 μg/mL) (Figure 2A). Addition of SB203580 showed a slight benefit in combination with trastuzumab in parental cells, as there were statistically significant reductions in cell proliferation. As expected, trastuzumab alone did not affect the proliferation of trastuzumab-resistant BT-c3 cells. However, addition of SB203580 to trastuzumab inhibited proliferation of resistant cells by 50%.
Figure 2. p38 inhibition improves trastuzumab sensitivity.
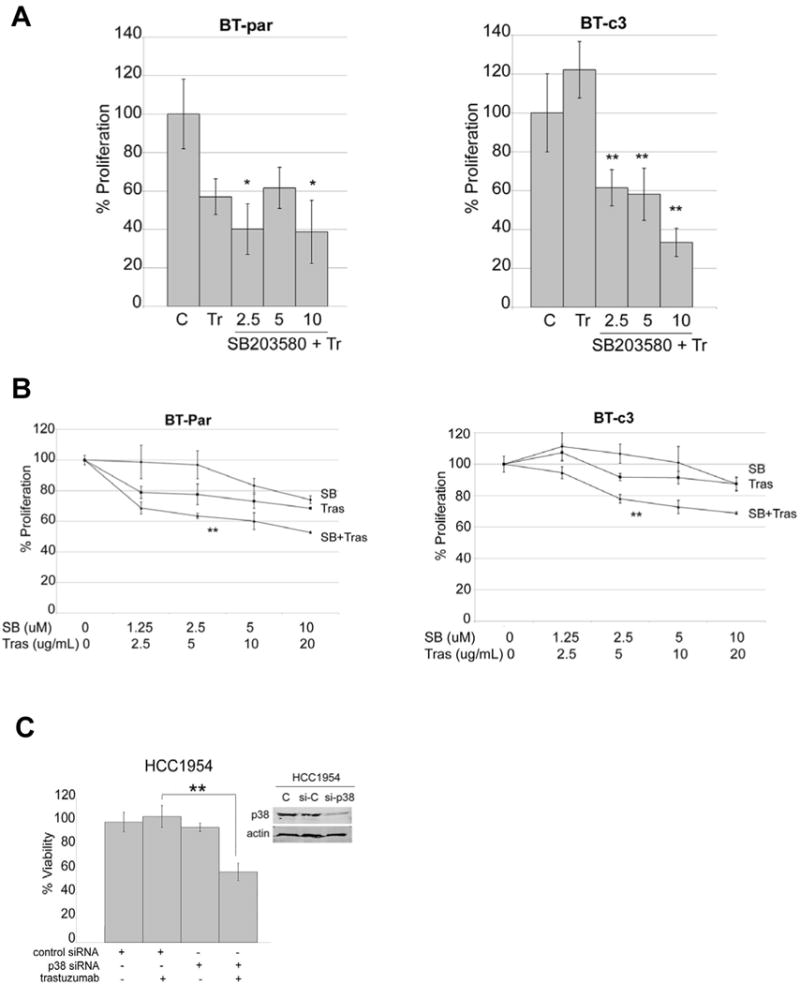
(A) BT474 parental and acquired trastuzumab-resistant clone 3 (BT-c3) cells were treated with vehicle control, 20 μg/mL trastuzumab, or a combination of 20 μg/mL trastuzumab plus p38 inhibitor SB203580 at either 2.5, 5, or 10 μM. After 72 hours, proliferation was measured by MTS assay. Values represent the average proliferation calculated as a percentage of vehicle control. Error bars represent the standard deviation between 6 replicates per group; compared to trastuzumab alone, *p<0.05, **p<0.005, student’s t-test. (B) BT474 parental and acquired trastuzumab-resistant clone 3 (BT-c3) cells were treated with vehicle control or two-fold serial dilutions of trastuzumab (20, 10, 5, 2.5 μg/mL tras), SB203580 (10, 5, 2.5, 1.25 μM SB), or a combination of SB + tras. After 72 hours, proliferation was measured by MTS assay. Values represent the average proliferation calculated as a percentage of vehicle control. Error bars represent the standard deviation between 6 replicates per group; compared to trastuzumab alone, **p<0.005, student’s t-test. (C) HCC1954 cells were transfected with 100 nM control siRNA (si-C) or p38 siRNA (si-p38) for 48 hours and then untreated or treated with 20 μg/mL trastuzumab for an additional 72 hours. Viable cells were counted by trypan blue exclusion and are shown relative to untreated cells transfected with control siRNA. Error bars represent standard deviations between triplicates; **p<0.005, student’s t-test. Inset, Western blot confirms knockdown of p38.
Next, we treated cells with two-fold serial dilutions of SB203580 and trastuzumab (Figure 2B). Statistical analysis showed that the combination index values were less than 1.0 at specific dose-combinations in both parental and resistant cells (Table 1), indicating potential synergy between trastuzumab and SB203580. Finally, knockdown of total p38 with a small oligonucleotide resulted in a significantly increased sensitivity to trastuzumab in the intrinsically-resistant cell line HCC1954 (Figure 2C). These results suggest that p38 kinase inhibition may improve response to trastuzumab in HER2-positive cells, including those that have progressed on prior trastuzumab therapy.
Table 1.
Drug combination analysis of SB203580 plus trastuzumab
| Cell line
|
ED50
|
ED75
|
ED90
|
Dm
|
m
|
r
|
|---|---|---|---|---|---|---|
| BT-parental | 1.10078 | 0.20055 | 0.22829 | 15.62410 | −0.31259 | 0.99041 |
| BT-HRc3 | 1.47908 | 0.70843 | 0.33931 | 18.30336 | −0.88369 | 0.89201 |
Cells were treated with SB203580 (1.25, 2.5, 5, or 10 μM), trastuzumab (2.5, 5, 10, or 20 μg/mL), or combination SB203580 plus trastuzumab. Proliferation was measured after six days with an MTS assay. The fraction of cells proliferating relative to DMSO control-treated cells was determined, and C.I. values were determined with CalcuSyn software. C.I. values are listed for effective doses at which 50%, 75%, or 90% (ED50, ED75, and ED90, respectively) of cells were killed. Statistically, drug synergy is defined by C.I. values less than 1.0, while drug additivity is defined by C.I. values that are approximately 1.0. (Dm, the median-effect (ED50) drug concentration; m < 1 indicates a negative sigmoidal shape to the dose-effect curve; r states the linear correlation coefficient.)
3.2 GDF15 promotes trastuzumab resistance and invasiveness via p38 signaling
We [3, 6] and others [7, 8] have previously shown that GDF15 induces phosphorylation of multiple signaling molecules, including p38. Stimulation of parental BT474 and SKBR3 cells with a physiologically relevant concentration (2 ng/mL) of recombinant human GDF15 (rhGDF15) rapidly and transiently induced phosphorylation of p38 (Figure 3A). Stable GDF15 transfectants derived from BT474 also showed increased p38 phosphorylation (Figure 3B). To determine if p38 signaling drives trastuzumab resistance or invasiveness of GDF15-overexpressing cells, we treated GDF15 stable clones with the p38 kinase inhibitor SB203580. BT474 GDF15 stable transfectants were significantly more invasive than the corresponding empty vector control cells (Figure 4A). However, inhibition of p38 kinase with SB203580 significantly suppressed the invasiveness of GDF15 stable cells (Figure 4B). These data indicate that p38 contributes to the pro-invasive phenotype of GDF15-overexpressing breast cancer cells.
Figure 3. GDF15 induces p38 phosphorylation in HER2-overexpressing breast cancer cells.
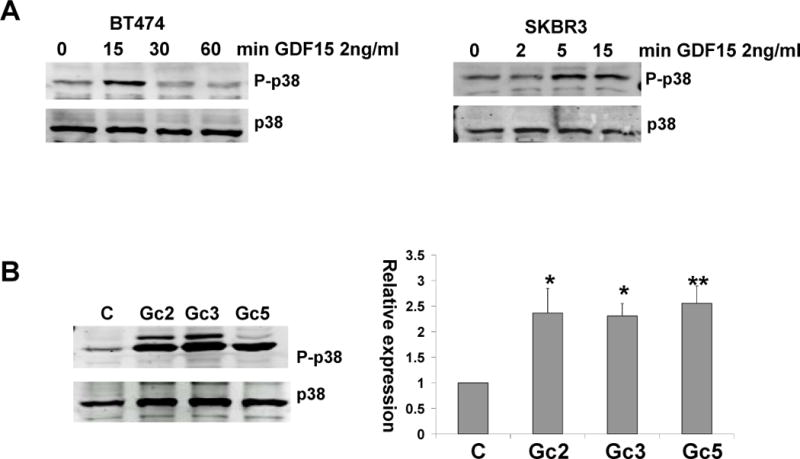
(A) BT474 (left) were serum-starved overnight and then stimulated with vehicle control (C) or 2 ng/mL rhGDF15 for 15, 30, or 60 min; SKBR3 (right) were serum-starved overnight and then stimulated with vehicle control (C) or 2 ng/mL rhGDF15 for 2, 5, or 15 min. Total protein lysates were Western blotted for phosphorylated and total p38. Blots were performed three times. (B) Total protein lysates from BT474 empty vector control clone (C) and GDF15 stable clones (Gc2, Gc3, Gc5) were Western blotted for phosphorylated and total p38. The graph shows quantification of phosphorylated p38 normalized to total p38 for three blots relative to control clone; error bars represent standard deviation between three replicates of the blots. compared to empty vector control clone, *p<0.05, **p<0.005, student’s t-test
Figure 4. GDF15 increases invasiveness of HER2-overexpressing breast cancer cells via p38.
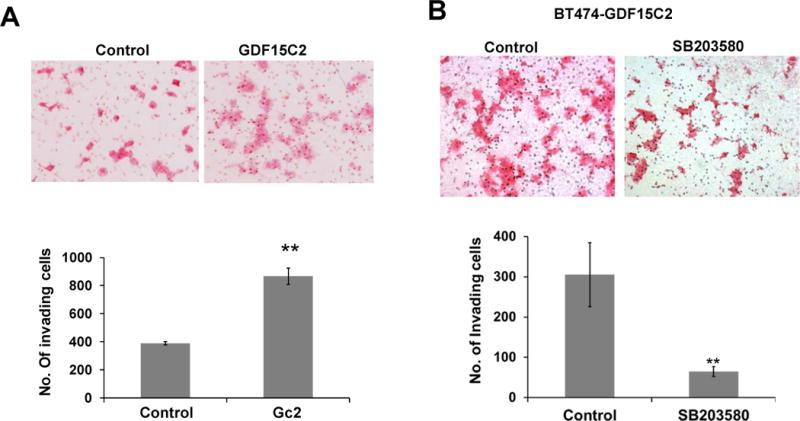
(A) BT474 stable empty vector control clone or BT474 stable GDF15 clone were plated in Trevigen Culture Coat R 24-well Low-BME Cell Invasion Optimization assay chambers. After 48 hours, cells were stained, and images were taken at 10× magnification. The total number of invading cells was counted in ten random fields, and the average from triplicate cultures is shown for each group. The experiment was performed twice; Gc2 versus control was compared by student’s t-test, **p<0.005. (B) BT474-GDF15 stable clone 2 (BT474-GDF15C2) cells (25,000) were plated in invasion chambers and treated with vehicle control (DMSO) or 10 μM p38 inhibitor SB203580 for 48 hours. Cells were then stained and imaged at 10× magnification. The total number of invading cells was counted in ten random fields, and the average is shown for triplicate cultures per group. The experiment was performed twice; **p<0.005, student’s t-test.
3.3 GDF15 overexpression significantly correlates with phosphorylation of p38 in human breast tumor tissues
Immunohistochemistry was performed for GDF15 and phosphorylated p38 on a breast tumor tissue array (Figure 5). Among 140 tumor tissues, 98 (70%) showed increased GDF15 staining, indicating that a high fraction of breast tumors express GDF15. Examining all tissues collectively, there was not a significant association between HER2 status alone and GDF15 (p=0.1432) and only a marginal association between HER2 status and p-p38 MAPK (p=0.0727) (Table 2). However, when GDF15-overexpressing tumors were examined independently from tissues that showed low or no GDF15 staining, HER2 was significantly associated with phosphorylation of p38 (p=0.0053). HER2-positive/GDF15-positive tumors were more likely to show phosphorylation of p38 in comparison to HER2-negative/GDF15-positive tumors. Thus, cancers with high expression of both HER2 and GDF15 showed increased phosphorylation of p38MAPK. These data support an association between GDF15 and p38 phosphorylation in HER2-positive breast tumors.
Figure 5. IHC for GDF15 and phosphorylated p38 in human breast tumor tissues.
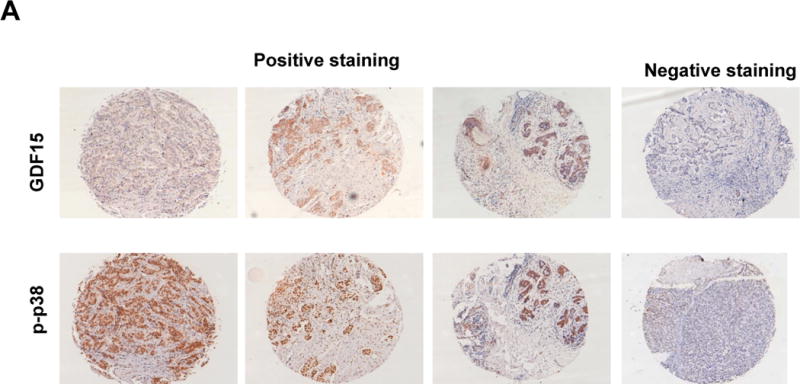
Representative IHC images of GDF15 and phosphorylated p38 on a commercially purchased breast tumor tissue microarray. Tissue microarray slides were deparaffinized, hydrated, and incubated with primary antibody overnight. Following secondary antibody incubation for one hour, the slides were developed with DAB. Slides were then dehydrated with xylene grades and mounted. Images were taken at 10× and scored based on intensity of staining.
Table 2.
IHC analysis of GDF15 and phosphorylated p38 on breast tumor array IHC (n=140 tumor tissues)
| One-to-one correlations: | ||||||||
|---|---|---|---|---|---|---|---|---|
| GDF15 | p-p38 | |||||||
| Negative | Positive | Negative | Positive | |||||
| HER2 status
|
N
|
%
|
N
|
%
|
N
|
%
|
N
|
%
|
| Negative (n=57) | 21 | 36.8 | 36 | 63.2 | 20 | 35.1 | 37 | 64.9 |
| Positive (n=83) | 21 | 25.3 | 62 | 74.7 | 17 | 20.5 | 66 | 79.5 |
| Correlations within GDF15-positive tumors: | ||||
|---|---|---|---|---|
| p-p38 | ||||
| Negative | Positive | |||
| N
|
%
|
N
|
%
|
|
| HER2−/GDF15+ (n=36) | 6 | 16.7 | 30 | 83.3 |
| HER2+/GDF15+ (n=62) | 1 | 1.6 | 61 | 98.4 |
GDF15 was not significantly associated with HER2 status alone (p=0.1432 by chi-square test).
HER2 and p-p38 showed a trend toward association (p=0.0727 by chi-square test).
Among 98 GDF15-positive tissues, HER2 was significantly associated with phosphorylation of p38 (p=0.0053; two-tailed Fisher’s exact test).
4. DISCUSSION
Trastuzumab is an important and fundamental treatment for HER2-positive breast cancer. However, some patients do not respond to trastuzumab, displaying so-called primary or intrinsic resistance. Among those who do respond, many will eventually demonstrate disease progression or acquired resistance to trastuzumab. Multiple mechanisms have been proposed to mediate the development of trastuzumab resistance, including hyper-activation of PI3K signaling through PIK3CA mutation or PTEN deletion. Activation of growth factor signaling cross-talk through various receptor kinases, such as IGF-IR and MET, has also been demonstrated in several models of trastuzumab resistance. We previously reported that trastuzumab-resistant cell lines significantly overexpress the transcript, endogenous protein, and secreted form of the cytokine GDF15 [3]. Elevated serum levels of GDF15 were previously reported in 6 of 10 (60%) metastatic breast carcinomas [12]. We [3] and others [7, 8] previously reported that GDF15 induces phosphorylation of HER2. However, the biological effects stimulated by GDF15 and the mechanistic basis for these effects in HER2-positive breast cancer cells remain unclear.
In the current study, we examined the role of p38MAPK in HER2-overexpressing breast cancer cells. We found that cells with intrinsic or acquired resistance to trastuzumab exhibited increased levels of phosphorylated p38. Resistance was overcome by genetic or pharmacological inhibition of p38MAPK, suggesting a direct contribution of p38 to the resistant phenotype (Figure 6). Thus, p38 activity may be elevated in trastuzumab-refractory HER2-positive disease and should be explored as a potential therapeutic target or predictive marker specifically within this setting. There are multiple isoforms of p38 expressed in breast epithelial cells, and we used reagents that affect or detect more than one isoform. SB203580 inhibits p38 alpha and beta isoforms [16], suggesting that one or both of these isoforms may be involved in the invasiveness and resistance mediated by p38 in our studies. Increased phosphorylation of p38 and p42/44 MAPKs has also been demonstrated in tamoxifen-resistant breast cancer [20]. Interestingly, the EGFR inhibitor gefitinib rescued tamoxifen resistance in association with reduced phosphorylation of p38. In a previous study, we performed IHC analysis for Ki-67, HER2, and phosphorylated p38 in 96 patients who received chemotherapy for node-positive breast cancer [21]. We found that progression-free survival was significantly reduced in patients whose tumors had increased staining for both Ki-67 and phosphorylated p38. However, this previous study failed to demonstrate a prognostic value for phosphorylated p38 specifically in HER2-positive tumors. These results may have been due to the relatively low sample size of 24 HER2-overexpressing tumors. Importantly, Antoon et al. [22] demonstrated that p38 kinase inhibition suppressed the in vivo tumor growth of the ER+/HER2+ tamoxifen-resistant, trastuzumab-resistant MDA361 breast cancer cell line. These published studies suggest that p38 kinase activity may contribute to cancer cell growth and resistance, providing pre-clinical rationale for studying p38 kinase inhibition as a potential therapeutic approach in breast cancer.
Figure 6. Schematic for GDF15-p38-mediated resistance and invasiveness.
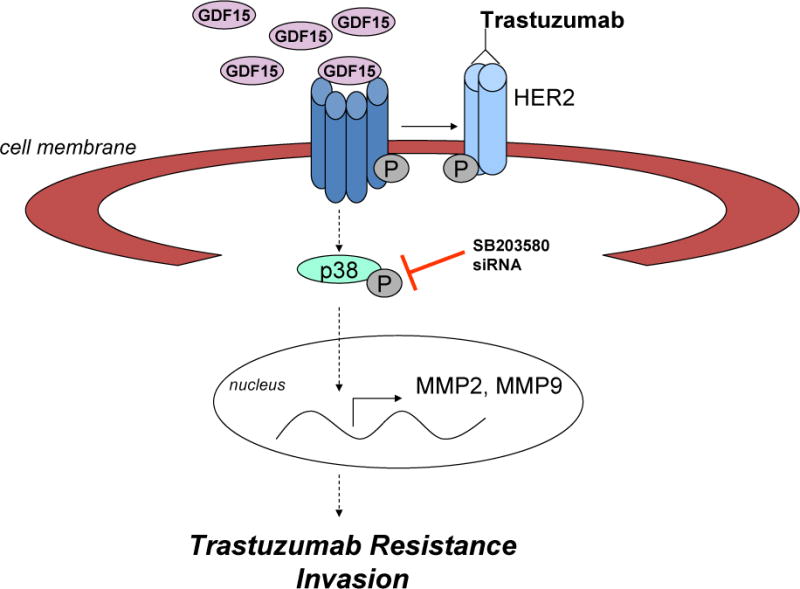
Multiple cell line models of intrinsic and acquired trastuzumab resistance exhibited increased phosphorylation of p38. Kinase inhibition or knockdown of p38 improved trastuzumab sensitivity. We previously reported that expression of growth differentiation factor 15 (GDF15) was increased in trastuzumab-resistant HER2-overexpressing breast cancer cells and induced phosphorylation of HER2. In this study, we found that exogenous GDF15 or stable overexpression of GDF15 stimulated p38 phosphorylation, suggesting a possible upstream mechanism by which p38 is activated in resistant cells. GDF15 stable clones showed significantly increased invasiveness, which was rescued by p38 kinase inhibition, suggesting that p38 plays a role in the pro-invasive phenotype conferred by GDF15. We also found a significant correlation between HER2 and phosphorylated p38 specifically in GDF15-positive breast tumor tissues. Our results suggest that p38 signaling drives trastuzumab resistance and invasiveness in HER2-overexpressing breast cancer. Upstream growth factor signals that have previously been implicated in trastuzumab resistance, such as GDF15, may contribute to the increased phosphorylation of p38 found in resistant cells.
Activation of MAPKs, including p38, occurs downstream of multiple receptor kinases. Thus, the increased levels of phosphorylated p38 in resistant cells may be due to activation of multiple upstream signaling pathways. For example, similar to GDF15, IGF-I and HGF signaling have been implicated in trastuzumab resistance, which may be related to the abilities of IGF-I and HGF to induce phosphorylation of p38. Indeed, IGF-IR phosphorylation has been associated with increased p38 phosphorylation in tamoxifen-resistant cells [20]. Thus, although we focus on the concept that GDF15-stimualted p38 phosphorylation drives resistance and invasion, additional upstream stimuli that induce p38 signaling may also contribute to the development of trastuzumab resistance.
An important finding from this study was that pharmacological inhibition or knockdown of p38 rescued GDF15-driven invasiveness in HER2-overexpressing breast cancer cells (Figure 6). These results provide a potential mechanism by which GDF15 promotes cellular invasion and suggest a possible targeted approach for inhibiting this process. The processes regulating invasiveness and resistance may differ; however, p38 appears to be essential for both processes, at least in the models examined in this study. Interestingly, we observed increased invasiveness in response to increased GDF15 not only in HER2-positive breast cancer cells and ovarian cells [6], but also in triple-negative MDA231 cells (Figure S1). Similar to our previous findings in ovarian tumors [6], we found significant correlations between MMP-2, a marker of invasiveness, and GDF15 positivity in HER2-positive cancers (Table S1). In addition, real-time PCR indicated that levels of MMPs and VEGF were elevated in trastuzumab-resistant cells, but that this was rescued by lentiviral knockdown of p38 (not shown). Although we did not over-express p38 in parental cells to determine if this increases invasiveness and leads directly to trastuzumab resistance, such studies would further strengthen the concept that p38 is a key mediator in these processes. In addition, in vivo analysis of the invasiveness and drug resistance of GDF15-overexpressing breast cancer cells will be important for validating GDF15 as a potential therapeutic target or mediator of breast cancer progression. Invasiveness and drug resistance both appear to be induced by GDF15 in a p38-mediated manner, which may ultimately result in disease progression. Overall, our results suggest roles for GDF15 and p38 signaling in the pro-invasive phenotype of HER2-positive breast cancer cells, providing rationale for further analysis of these signaling molecules in metastatic or drug-resistant breast cancer.
Our IHC studies established a striking correlation between HER2 and phospho-p38 specifically in GDF15-positive breast cancers. We found that GDF15 was expressed at high levels in 70% of breast tumors. In contrast, none of the “normal” breast fibroadenoma tissues stained positive for GDF15. Welsh et al. [15] previously reported that 60% (6 of 10) metastatic breast cancers showed increased staining for GDF15. Our findings together with those previously reported suggest that GDF15 is elevated in a majority of breast cancers. Approximately 74.7% of HER2-positive breast tumors showed increased expression of GDF15, which was slightly higher than all subsets considered together but not significantly different. We identified a strong and significant correlation between HER2 and phosphorylated p38 in 98 GDF15-positive tumors. Among the 98 GDF15-positive tumors identified on our tissue array, there was a trend (p=0.0727) correlating HER2 positivity and GDF15 positivity. Future studies will examine a larger set of tissues to determine whether this relationship reaches statistical significance. The breast tumor array used in this study is limited with respect to sample size and a lack of data regarding patient survival or response to treatment. Thus, further preclinical study of GDF15 in a larger set of breast tumor tissues with survival and response data is warranted to determine if GDF15 serves as a marker of disease progression. Based on our observations that GDF15 overexpression is found in resistant cell lines, a statistically significant correlation between GDF15 and HER2 may best be appreciated in advanced or treatment-refractory cases. Thus, future analysis of a larger set that includes early-stage and advanced treatment-refractory cases of HER2-positive breast cancer is being planned.
In summary, we present data suggesting that GDF15 and p38 mediate invasion and trastuzumab resistance in HER2-overexpressing breast cancer. Future studies will further delineate the mechanisms by which GDF15-p38 signaling promotes breast cancer progression and will examine the suitability of targeting this molecular pathway in vivo.
Conclusion
HER2 overexpression occurs in a subset of metastatic breast cancers. The first-line therapy, trastuzumab, is effective but limited by the development of drug resistance. We previously found that GDF15 expression is increased in trastuzumab-resistant cells. In the current study, we provide novel data implicating GDF15-mediated p38 phosphorylation in increased invasiveness and trastuzumab resistance of HER2-overexpressing breast cancer cells.
Supplementary Material
Figure S1. GDF15 stimulation increases the invasiveness of MDA231 triple-negative breast cancer cells. MDA231 cells were plated in matrigel-coated Boyden invasion chambers. Vehicle control or 20 ng/mL recombinant human GDF15 was placed in the lower chamber as a chemoattractant. After 48 h, cells were stained, and the numbers of invading cells were counted in ten random fields; the average of ten fields is shown from triplicate cultures per group. The experiment was performed twice; statistical analysis was performed using student’s t-test, **p<0.005
Acknowledgments
R. Nahta gratefully acknowledges funding from NIH R01CA157754, The Mary Kay Foundation, Georgia Cancer Coalition Distinguished Scholars Program, The Glenn Family Breast Program Pilot Grant, and support from NIH P30 CA138292 to Winship Cancer Institute. The authors acknowledge the Winship Cancer Institute Integrated Cellular Imaging Core Facility. Technical contributions by T. Ozbay and J. Joshi are acknowledged.
List of Abbreviations
- GDF15
growth differentiation factor 15
- HER2
human epidermal growth factor receptor 2
- MAPK
mitogen-activated protein kinase
- MMP
matrix metalloproteinase
Footnotes
Conflict of Interest
The authors acknowledge that they do not have any financial conflicts of interest. R. Nahta holds patent WO 2013/012648 “GDF15 in diagnostic and therapeutic applications”.
References
- 1.Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH, Allred DC, Bartlett JM, Bilous M, Fitzgibbons P, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: american society of clinical oncology/college of american pathologists clinical practice guideline update. J Clin Oncol. 2013;31(31):3997–4013. doi: 10.1200/JCO.2013.50.9984. [DOI] [PubMed] [Google Scholar]
- 2.Metro G, Mottolese M, Fabi A. HER-2-positive metastatic breast cancer: trastuzumab and beyond. Expert opinion on pharmacotherapy. 2008;9(15):2583–2601. doi: 10.1517/14656566.9.15.2583. [DOI] [PubMed] [Google Scholar]
- 3.Joshi JP, Brown NE, Griner SE, Nahta R. Growth differentiation factor 15 (GDF15)-mediated HER2 phosphorylation reduces trastuzumab sensitivity of HER2-overexpressing breast cancer cells. Biochem Pharmacol. 2011;82(9):1090–1099. doi: 10.1016/j.bcp.2011.07.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bootcov MR, Bauskin AR, Valenzuela SM, Moore AG, Bansal M, He XY, Zhang HP, Donnellan M, Mahler S, Pryor K, et al. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc Natl Acad Sci U S A. 1997;94(21):11514–11519. doi: 10.1073/pnas.94.21.11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu J, Kimball TR, Lorenz JN, Brown DA, Bauskin AR, Klevitsky R, Hewett TE, Breit SN, Molkentin JD. GDF15/MIC-1 functions as a protective and antihypertrophic factor released from the myocardium in association with SMAD protein activation. Circ Res. 2006;98(3):342–350. doi: 10.1161/01.RES.0000202804.84885.d0. [DOI] [PubMed] [Google Scholar]
- 6.Griner SE, Joshi JP, Nahta R. Growth differentiation factor 15 stimulates rapamycin-sensitive ovarian cancer cell growth and invasion. Biochem Pharmacol. 2013;85(1):46–58. doi: 10.1016/j.bcp.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim KK, Lee JJ, Yang Y, You KH, Lee JH. Macrophage inhibitory cytokine-1 activates AKT and ERK-1/2 via the transactivation of ErbB2 in human breast and gastric cancer cells. Carcinogenesis. 2008;29(4):704–712. doi: 10.1093/carcin/bgn031. [DOI] [PubMed] [Google Scholar]
- 8.Park YJ, Lee H, Lee JH. Macrophage inhibitory cytokine-1 transactivates ErbB family receptors via the activation of Src in SK-BR-3 human breast cancer cells. BMB Rep. 2010;43(2):91–96. doi: 10.5483/bmbrep.2010.43.2.091. [DOI] [PubMed] [Google Scholar]
- 9.Kim JS, Baek SJ, Sali T, Eling TE. The conventional nonsteroidal anti-inflammatory drug sulindac sulfide arrests ovarian cancer cell growth via the expression of NAG-1/MIC-1/GDF-15. Mol Cancer Ther. 2005;4(3):487–493. doi: 10.1158/1535-7163.MCT-04-0201. [DOI] [PubMed] [Google Scholar]
- 10.Yang H, Choi HJ, Park SH, Kim JS, Moon Y. Macrophage inhibitory cytokine-1 (MIC-1) and subsequent urokinase-type plasminogen activator mediate cell death responses by ribotoxic anisomycin in HCT-116 colon cancer cells. Biochem Pharmacol. 2009;78(9):1205–1213. doi: 10.1016/j.bcp.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 11.Brown DA, Ward RL, Buckhaults P, Liu T, Romans KE, Hawkins NJ, Bauskin AR, Kinzler KW, Vogelstein B, Breit SN. MIC-1 serum level and genotype: associations with progress and prognosis of colorectal carcinoma. Clin Cancer Res. 2003;9(7):2642–2650. [PubMed] [Google Scholar]
- 12.Mimeault M, Batra SK. Divergent molecular mechanisms underlying the pleiotropic functions of macrophage inhibitory cytokine-1 in cancer. J Cell Physiol. 2010;224(3):626–635. doi: 10.1002/jcp.22196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roth P, Junker M, Tritschler I, Mittelbronn M, Dombrowski Y, Breit SN, Tabatabai G, Wick W, Weller M, Wischhusen J. GDF-15 contributes to proliferation and immune escape of malignant gliomas. Clin Cancer Res. 2010;16(15):3851–3859. doi: 10.1158/1078-0432.CCR-10-0705. [DOI] [PubMed] [Google Scholar]
- 14.Staff AC, Bock AJ, Becker C, Kempf T, Wollert KC, Davidson B. Growth differentiation factor-15 as a prognostic biomarker in ovarian cancer. Gynecol Oncol. 2010;118(3):237–243. doi: 10.1016/j.ygyno.2010.05.032. [DOI] [PubMed] [Google Scholar]
- 15.Welsh JB, Sapinoso LM, Kern SG, Brown DA, Liu T, Bauskin AR, Ward RL, Hawkins NJ, Quinn DI, Russell PJ, et al. Large-scale delineation of secreted protein biomarkers overexpressed in cancer tissue and serum. Proc Natl Acad Sci U S A. 2003;100(6):3410–3415. doi: 10.1073/pnas.0530278100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao J, Semenova MM, Solovyan VT, Han J, Coffey ET, Courtney MJ. Distinct requirements for p38alpha and c-Jun N-terminal kinase stress-activated protein kinases in different forms of apoptotic neuronal death. J Biol Chem. 2004;279(34):35903–35913. doi: 10.1074/jbc.M402353200. [DOI] [PubMed] [Google Scholar]
- 17.Nahta R, Takahashi T, Ueno NT, Hung MC, Esteva FJ. P27(kip1) down-regulation is associated with trastuzumab resistance in breast cancer cells. Cancer Res. 2004;64(11):3981–3986. doi: 10.1158/0008-5472.CAN-03-3900. [DOI] [PubMed] [Google Scholar]
- 18.Rowe DL, Ozbay T, Bender LM, Nahta R. Nordihydroguaiaretic acid, a cytotoxic insulin-like growth factor-I receptor/HER2 inhibitor in trastuzumab-resistant breast cancer. Mol Cancer Ther. 2008;7(7):1900–1908. doi: 10.1158/1535-7163.MCT-08-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 20.Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, Weiss H, Rimawi M, Schiff R. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008;68(3):826–833. doi: 10.1158/0008-5472.CAN-07-2707. [DOI] [PubMed] [Google Scholar]
- 21.Esteva FJ, Sahin AA, Smith TL, Yang Y, Pusztai L, Nahta R, Buchholz TA, Buzdar AU, Hortobagyi GN, Bacus SS. Prognostic significance of phosphorylated P38 mitogen-activated protein kinase and HER-2 expression in lymph node-positive breast carcinoma. Cancer. 2004;100(3):499–506. doi: 10.1002/cncr.11940. [DOI] [PubMed] [Google Scholar]
- 22.Antoon JW, Bratton MR, Guillot LM, Wadsworth S, Salvo VA, Elliott S, McLachlan JA, Burow ME. Pharmacology and anti-tumor activity of RWJ67657, a novel inhibitor of p38 mitogen activated protein kinase. American journal of cancer research. 2012;2(4):446–458. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. GDF15 stimulation increases the invasiveness of MDA231 triple-negative breast cancer cells. MDA231 cells were plated in matrigel-coated Boyden invasion chambers. Vehicle control or 20 ng/mL recombinant human GDF15 was placed in the lower chamber as a chemoattractant. After 48 h, cells were stained, and the numbers of invading cells were counted in ten random fields; the average of ten fields is shown from triplicate cultures per group. The experiment was performed twice; statistical analysis was performed using student’s t-test, **p<0.005