

Abstract
To provide potential new leads for the treatment of orthopoxvirus infections, the 5-position of the pyrimidine nucleosides have been modified with a gem diether moiety to yield the following new nucleosides: 5-(dimethoxymethyl)-2′-deoxyuridine (2b), 5-(diethoxymethyl)-2′-deoxyuridine (3b), 5-formyl-2′-deoxyuridine ethylene acetal (4b), and 5-formyl-2′-deoxyuridine propylene acetal (5b). These were evaluated in human foreskin fibroblast cells challenged with the vaccinia virus or cowpox virus. Of the four gem diether nucleosides, only the dimethyl gem diether congener showed significant antiviral activity against both viruses. This antiviral activity did not appear to be related to the decomposition to the 5-formyl-2′-deoxyuridine, which was itself devoid of anti-orthopoxvirus activity in these assays. Moreover, at the pH of the in vitro assays, 2b was very stable with a decomposition (to aldehyde) half-life of >15 d. The anti-orthopoxvirus activity of pyrimidine may be favored by the introduction of hydrophilic moieties to the 5-position side chain.
Introduction
The events of September 11, 2001 forced terrorism to the forefront of national consciousness.1-12 Contamination of government, press, and mail facilities, numerous infections, and several deaths from anthrax vividly demonstrated the potential13-23 of bioterrorism.12,24-32 However, smallpox represents an even more serious bioterrorist threat than anthrax 10,33-45 to the civilian and military populations46-50 because of its high fatality rates and transmissibility. The lethality of the disease and its ease of transmissibility place the variola virus at the top of the CDC's list of high-threat (category A) agents. One drug, cidofovir (Vistide), is licensed to treat cytomegalovirus (CMV) retinitis in HIV-infected patients; however, it is available through an investigational new drug (IND) protocol to treat smallpox vaccine reactions (http://www.bt.cdc.gov/agent/smallpox/vaccination/cidofovir.asp) if the vaccine immune globulin (VIG) fails. Cidofovir might be used to treat generalized vaccinia, eczema vaccinatum, or progressive vaccinia.51-60 Cidofovir, when intravenously administered, produces nephrotoxicity; however, it remains the only drug available (IND) to treat vaccination complications or, on a compassionate basis, to treat smallpox itself. Progress has been made on the development of oral dosage forms of Cidofovir9,61-63,82, but these are not yet available in the clinic. Even though other candidates such as ST-246 that targets the smallpox virus core protein cysteine proteinase,64 are in development, to date, there is no drug approved by the FDA to treat smallpox (variola) itself. It is, therefore, the stated role of the U. S. government to have available two anti-smallpox drugs possessing different mechanisms of action and to have two more such drugs in the pipeline.65
Herein we report on the first of our inquiries using the nucleoside scaffold as a point of departure in the search for antiviral drugs targeting orthopoxviruses.
Results
Strategy and Chemistry
Our cornerstone for the exploration of 5-substituted pyrimidine nucleoside chemical space has been the known 5-formyl-2′-deoxyuridine (1b), which recruits the rich and extensive chemistry of the aldehyde carbonyl to this undertaking and permits the introduction of electronegative hydrophilic substituents to the pyrimidine side chain.
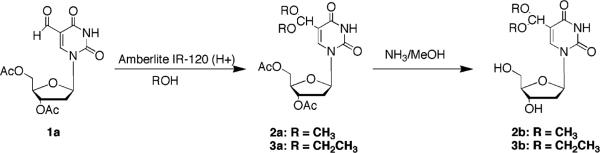
The preparation of the pyrimidine nucleoside 5-substituted gem diether side chains began with the known 3′,5′-di-O-acetyl-5-formyl-2′-deoxyuridine (1a).66 3′,5′-Di-O-acetyl-5-formyl-2′-deoxyuridine dimethylacetal (2a) and 3′,5′-di-O-acetyl-5-formyl-2′-deoxyuridine diethylacetal (3a) were prepared by refluxing a methanol or ethanol solution of 1a in the presence of an acidic resin used as a catalyst (Scheme 1). Ammonia/CH3OH deprotection afforded 5-formyl-2′-deoxyuridine dimethylacetal (2b) (Scheme 1) and 5-formyl- 2′-deoxyuridine diethylacetal (3b).
Scheme 1.
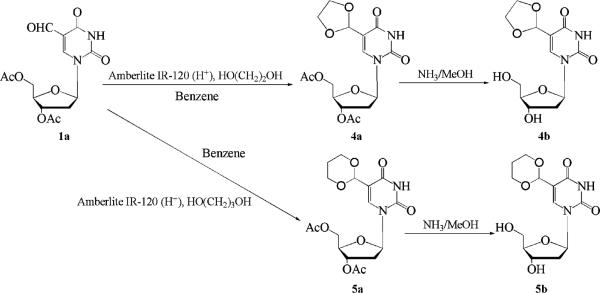
The two cyclic acetal acetates, 3′,5′-di-O-acetyl-5-formyl-2′-deoxyuridine ethylene acetal (4a) (Scheme 2) and 3′,5′-di-O-acetyl-5-formyl-2′-deoxyuridine propylene acetal, (5a) (Scheme 2) were obtained by refluxing a benzene solution of 1a and either ethylene glycol or propylene glycol in the presence of an acidic catalyst. These two acetal products were deacetylated by treating them with NH3/MeOH to give 4b and 5b.
Scheme 2.
Finally, 5-formyluracil dimethylacetal (7) was prepared from 5-formyluracil (6).
Biological Activities. Anti-Orthopoxvirus Activity of Novel 5-Substituted Pyrimidine Nucleosides
The antiviral activities of compounds (Table 1) were determined in human foreskin fibroblast cells, and the challenge orthopoxviruses were either the vaccinia virus (VV) or the cowpox virus (CV). An initial evaluation was performed using a viral cytopathogenic effect as the endpoint. A second confirmatory assay involved plaque reduction. The concentration of the agent that inhibited viral CPE or plaque formation by 50% was defined as the EC50 value. The effect of the potential antiviral agent on uninfected host cell viability was ascertained by neutral red uptake as a measure of toxicity. The concentration of the applied agent that reduced neutral red uptake by 50% was defined as the CC50 value. Neutral red toxicity assays were performed with confluent monolayers. None of the compounds described here had any significant cytopathic effect on uninfected cells under these conditions. All of the CC50 values were in excess of 300 μM.
Table 1.
Anti-Orthopoxvirus Activities of Nucleosidesa
| efficacy EC50b (μM) |
toxicity CC50c (μM) |
||||
|---|---|---|---|---|---|
| compd | VVd CPE | VVd PR | CVd CPE | CVd PR | neutral red uptake |
| Cidofovir | 6.9–8.2 | 9–9.8 | 8.3–9 | 9–16 | >300 |
| 1b | >300 | NT | >300 | NT | >300 |
| 1a | >300 | NT | >300 | NT | >300 |
| 2a | 296 | NT | >300 | NT | >300 |
| 2b | 8.4 | 9.0 ± 1.3e | 11.7 | 7.4 ± 3.0e | ±300 ± 0e |
| 3a | 38 | 79 | 52.4 | 25.8 | >300 |
| 3b | >60 | NT | >60 | NT | >300 |
| 4a | >300 | NT | >300 | NT | >300 |
| 4b | >300 | NT | >300 | NT | >300 |
| 5a | >300 | NT | >300 | NT | >300 |
| 5b | >300 | NT | >300 | NT | >300 |
| 7 | >300 | NT | >300 | NT | >300 |
Assays were performed according to the procedures described previously for activity against the vaccinia virus (VV) and cowpox virus (CV) and for cytotoxicity (neutral red uptake assay) in human foreskin fibroblast (HFF) cells. Briefly, to determine efficacy, initial cytopathogenic effect (CPE) assays were performed in 96-well plates seeded with HFF cells. Varying concentrations of the drug were challenged with VV or CV at 1000 PFU/well (incubation at 37 °C for 7 days). Confirmatory assays involving plaque reduction (PR) were performed using HFF cells seeded in 6-well plates, 2 days prior to use and infected with either VV or CV by the addition of 20–30 PFU/well. Plates were incubated for 1 h, and various concentrations of the drug were then added to triplicate wells, and the plates were incubated at 37 °C for 3 days. Toxicity was evaluated using HFF cells seeded in 96-well plates incubated with various concentrations of the drug for 7 days at 37 °C. Neutral red toxicity assays were performed with confluent monolayers.
EC50: effective concentration to reduce viral cytopathogenicity or plaque formation by 50%; NT, not tested.
CC50: the concentration that causes a cytotoxic effect (as ascertained by neutral red uptake) on 50% of uninfected cells.
Virus used for challenge: VV (Copenhagen) or CV (Brighton).
Values are the mean (standard deviation of two or more assays. In instances where the means and standard deviations are provided, at least two or more assays have been done. If there is no such indication, then only one assay was performed.
Of all the compounds bioassayed, by far, the most active one was the gem diether (2b), which displayed EC50 values of 8.4 and 9.0 μM against VV as determined by the cytopathogenic effect and plaque reduction methods, respectively. Compound 2b also was highly active against CV showing EC50 values of 11.7 and 7.4μM as determined by CPE and plaque reduction, respectively. Its 3′,5′-diacetate was devoid of significant activity. The other compound with significant, albeit much reduced, antiviral activity was the 3′,5′-diacetate ester of diethylacetal (3a); however, this activity was not increased when free nucleoside 3b was evaluated. The extremely modest antiviral activity of the diethylacetal (3a) coupled with the lack of activity shown by its free nucleoside may imply a different mode of action from that of the dimethylacetal or some difference by which the infected cells process 3a and b relative to 2a and b, or their activities may not be related at all.
For the remaining congeners, there was a dramatic drop-off of antiviral activity when the alcoholic moieties of the nucleoside aldehyde acetal were combined in the form of either ethylene or propylene glycols. These five- and six-membered dioxolanes possessed no anti-orthopoxvirus activity either as free nucleo-sides or as their corresponding diacetates. In addition, the aglycone, 5-formyluracil dimethylacetal (7), was completely devoid of antiviral activity.
Chemical Stability of 5-(Dimethoxymethyl)-2′-Deoxyuri-dine (2b) Under Aqueous Conditions
Proton NMR was used to follow the rate of hydrolysis of the gem diether (2b) at (1) pH 7.5 in a phosphate buffer and (2) pH 6.0. From Figure 1a, it is clear that 2b had a half-life for hydrolysis of approximately 16 days in the buffer at pH 7.5. As expected, the half-life significantly diminished (to approximately 18 h) in water at pH 6.0 (Figure 1b). In both instances, no other transformation product other than 5-formyl-2′-deoxyuridine was noted.
Figure 1.

Decomposition of Compound 2b at pH 7.5 (a) and pH 6.0 (b).
Stability of 5-(Dimethoxymethyl)-2′-deoxyuridine (2b) to Esterase
Shown in Figure 2 are experiments conducted to determine the efficiency of the conversion of 2a to free unesterified nucleoside 2b in the presence of purified porcine liver esterase to simulate the various protease-esterases that would be present in the cell-virus assay. From Figure 2, it is apparent that under the conditions employed, tdiacetate 2a was rapidly degraded. However, an immediate product was produced, most probably the 3′-monoacetate (although we have not pursued this identification), and this was de-esterified relatively slowly to provide free nucleoside 2b. Under these conditions, the half-life for the loss of the presumed 3′-monoacetate was 1.5 h, which was the same as the half-life for the formation of free nucleoside 2b.
Figure 2.
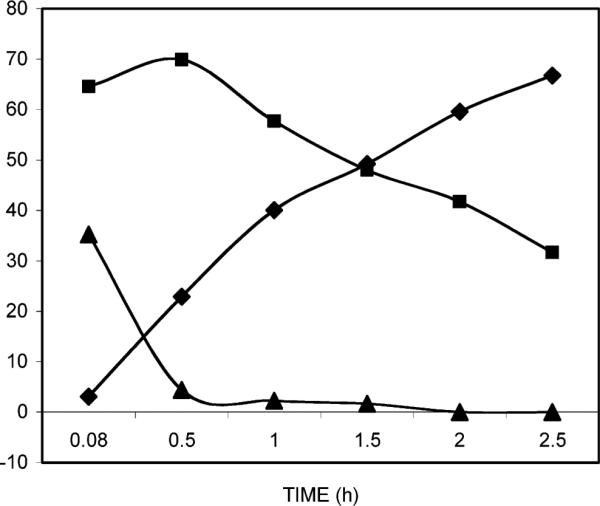
Action of porcine liver esterase on 5-(dimethoxymethyl)-2′-deoxyuridine 3′,5′-diacetate (2a, ■) resulting in the production of the intermediate presumed to be 5-(dimethoxymethyl)-2′-deoxyuridine 3′-acetate (▲) and the ultimate fully deacetylated product 5-(dimethoxymethyl)-2′-deoxyuridine (2b, ◆).
Discussion
Acetals have been employed as prodrug candidates;67,68 therefore, the possibility exists that the acetals prepared in this study may owe their mode of action to the hydrolysis to 5-formyl-2′-deoxyuridine which, in contrast to the present study, has been reported to have some anti-vaccinia virus activity.69 In the present situation, It seems reasonably certain that the antiviral potency of the gem diether 2b cannot be due to its action as a prodrug of 5-formyl-2′-deoxyuridine (1b) because the latter is completely devoid of biological activity in these assay systems (Table 1) and because 2b was stable at pH of 7.5, which is the pH employed for these assays. It might be argued that perhaps the pH within the regions of the virus-infected cells could provide a more acidic milieu. Indeed, in the case of the orthopoxviruses, cell-associated enveloped virions may enter cells through endocytosis followed by a low pH disruption of their outer members and subsequent fusion with endosomal membranes. Instead, in the absence of definitive mechanistic studies, it would be reasonable to argue that the gem diether or dimethylacetal (2b) may possess inherent antiviral activity by virtue of its particular structure. There was no activity associated with the aglycone (7); therefore, antiviral activity was not attributable to cleavage to the heterocyclic base. 5-Isopropyl-2′-deoxyuridine has been reported to possess anti-herpes virus activity,70-72 and 5-cyclopropyl-4′-thio-2′-deoxyuridine possesses significant activity against both herpes simplex-1 and -2 as well VV.73 The distantly related pyrimidine 5-dimethoxymethyl substituent of compound 2b would be significantly less hydrophobic than 5-alkylpryimidine nucleo-sides of comparable chain length and configuration because it contains the electronegative hydrogen-bond-acceptor oxygen atoms capable of interacting with a more hydrophilic protein domain.
In contrast to results with the free nucleoside, dimethylacetal 2b in the diacetate form (2a) does not provide antiviral activity (Table 1). The inactivity of the diacetate derivative becomes relevant in the context of potential prodrug formulations that may be envisioned in the future. Therefore, we sought to develop further information regarding this inactivity. We examined the conversion of 2a to free unesterified nucleoside 2b in the presence of purified porcine liver esterase to model the various protease-esterases present in the cell culture milieu. Although diester 2a was rapidly transformed by esterase, free nucleoside 2b was slow to form, most probably because of the intermediate formation of the esterase-resistant 3′-acetate. The facile cleavage of the primary alcoholic ester would be expected, as would the more difficult cleavage of the secondary alcohol ester. The hypothesis may be forwarded that the inactivity of diester 2a may be related to the kinetics of this slower cleavage and the resultant failure to provide sufficient inhibitory concentrations of nucleoside 2b. However, the long duration of the antiviral assays in the cell culture might have been expected to provide sufficient time for the esterase cleavage of even the secondary ester. Thus, the cellular milieu may not provide sufficient esterase activity to effect the required de-esterification.
Conclusions
The nucleoside acetal, 5-(dimethoxymethyl)-2′-deoxyuridine (2b) can be readily synthesized from 3′,5′-diacetate of 5-formyl-2′-deoxyuridine and displays potent in vitro antiviral activity against two representative orthopoxviruses, namely, VV and CV. Its activity against both viruses is comparable to the activity of cidofovir. This activity is strongly dependent on the nature of the pyrimidine 5-substituent. The replacement of the 5-dimethoxymethyl group by the diethoxymethyl group, the 1,3-dioxolan-2-yl moiety, or the 1,3-dioxan-2-yl substituent all resulted in the complete loss of anti-orthopoxvirus activity. Present evidence is consistent with the inherent activity of 5-(dimethoxymethyl)-2′-deoxyuridine itself as opposed to its action as an acetal prodrug form of 5-formyl-2′-deoxuridine. The potent anti-orthopoxvirus activity of compound 2b is likely to be related to the pyrimidine 5-substituent oxygens, which enable polar and hydrogen bonding interactions, thereby permitting binding to more hydrophilic protein target domains than many previously described 5-substiuted pyrimidine nucleosides that bear hydro-phobic alkyl or alkenyl groups. At the least, the antiviral activity of 5-(dimethoxmethyl)-2′-deoxyuridine provides important clues to the design of anti-orthopoxvirus agents.
Experimental Section
Melting points were recorded with a Barnstead 1201D electro-thermal melting point apparatus and are uncorrected. 1H NMR and 13C NMR spectra were recorded on a Varian 400 MHz spectrometer. CDCl3, CD3OD, or DMF-d7 is used as the solvent for different compounds. The chemical shifts of the deuterated solvent served as the internal standard. The mass spectra were performed on an HP1100 MSD spectrometer at the HT Laboratories, San Diego, CA. The HRMS (High-Resolution Mass Spectra) were performed on a JEOL HX 110A spectrometer at the Department of Chemistry, University of Arizona. Silica gel column chromatography was conducted with Sigma-Aldrich silica gel (70-230 mesh). 5-Formyl-2′-deoxyuridine (1b) was prepared essentially as described by protecting the carbonyl group in 1a as the dimethylacetal (2a), which was then sequentially treated with NaOMe/MeOH and AcOH/H2O to give 1b.
Chemical Stability Studies
1. Compound 2b (11.8 mg, 39.1 μmol) was dissolved in D2O (1 mL). The pH of this final solution was 6.0. The solution was then monitored by 1H NMR. 2. A second 1H NMR study of 5-formyl-2′-deoxyuridine dimethylacetal (2b) was carried out in KD2PO4-D2O buffer (pH ) 7.5) at the same final concentration as that above (11.8 mg, 39.1 μmol in 1 mL buffer). The resulting solution was monitored by 1H NMR.
Procedure for Porcine Liver Esterase Assay
Enzyme or control assay mixtures were prepared by adding 40 μL of a DMSO solution (1 mM) of the compound to be assayed to 1750 μL of H2O plus 200 μL of KH2PO4 buffer (100 mM, pH 7.5). Reaction was initiated by the addition of 10 μL of enzyme, or for the control mix, 10 μL of H2O. Enzyme reaction mixtures (or control) were incubated at 37 °C, and aliquots of 300 μL were removed at 0.08, 0.5, 1, 1.5, 2, and 2.5 h. These were flash-frozen in a dry ice/acetone bath and stored at −80 °C until HPLC analysis at which time, individual time-point samples were rapidly thawed and injected into the HPLC together with a thymine solution (0.1 mM) as an internal standard. HPLC elution was with a stepwise elution of solvent A (50mM NH4Oac, pH 7) into solvent B (1:1 CH3CN/H2O).
1. Preparation of 3′,5′-Di-O-acetyl-5-formyl-2′-deoxyuridine (1a)
To a solution of K2S2O8 (16.6 g, 61.4 mmol) and CuSO4’ 5H2O (3.0 g) in 110 mL H2O was added a CH3CN solution (100 mL) containing 3′,5′-di-O-acetyl-thymidine (10.0 g, 30.6 mmol) and 2,6-lutidine (12.2 mL). The mixture was stirred at 65 °C for 2 h. Upon completion, the mixture was concentrated to half of the initial volume, and the remaining solution was extracted with EtOAc. The organic layer was washed with H2O. The aqueous layers were combined and back-extracted with CHCl3. Then the organic layers were combined, dried over Na2SO4, and then concentrated. The residue was purified through silica gel column chromatography with a mixture of EtOAc and hexane (2:1, v/v) as eluant. The fractions were collected and concentrated. The solid product was crystallized from EtOAc to give 1a as white crystals (3.68 g, 35.4%); mp 148-150 °C; 1H NMR (CDCl3): δ 2.12 (s, 3H, CH3), 2.17 (s, 3H, CH3), 2.23-2.26 (m, 1H, H2′-1), 2.58-2.64 (m, 1H, H2′-2), 4.31-4.42 (m, 3H, H4′, H5′), 5.25-5.27 (m, 1H, H3′), 6.30-6.34 (m, 1H, H1′), 8.45 (s, 1H, H6), 9.05 (br s, 1H, NH), 10.01 (s, 1H, CHO). 13 C NMR (CDCl3) δ: 20.84, 21.04, 38.81, 63.79, 74.22, 83.25, 86.38, 111.81, 144.84, 149.67, 162.39, 170.60, 170.82, 186.16. FAB MS m/e: 341 (MH+), 363 (MNa+).
2. Preparation of 3′,5′-Di-O-acetyl-5-formyl-2′-deoxyuridine Dimethylacetal (2a)
In the presence of Amberlite IR-120 (100 mg), 3′,5′-di-O-acetyl-5-formyl-2′-deoxyuridine (1a, 340 mg, 1.0 mmol) in 20 mL of anhydrous methanol was refluxed with stirring for 2 h. The mixture was filtered to remove the solid acid, and the filtrate was concentrated. The residue was purified through silica gel column chromatography with a mixture of EtOAc and hexane (2:1, v/v) as eluant to give a colorless solid (2a, 327 mg, 84.7%). Compound 3a was prepared in a similar manner from 1a and anhydrous ethanol.
Compound 2a
Mp 120-122 °C; 1H NMR (CDCl3) δ: 2.04 (s, 3H, CH3), 2.08 (s, 3H, CH3), 2.10-2.18 (m, 1H, H2′-1), 2.38-2.43 (m, 1H, H2′-2), 3.26 (s, 3H, CH3), 3.33 (s, 3H, CH3), 4.18-4.26 (m, 3H, H4′, H5′), 5.17-5.19 (m, 1H, H3′), 5.24 (s, 1H, CH), 6.27-6.30 (m, 1H, H1′), 7.66 (s, 1H, H6).13 C NMR (CDCl3) δ: 20.71, 21.06, 37.91 54.16, 54.94, 64.29, 74.86, 82.57, 85.29, 98.45, 112.76, 137.61, 150.49, 162.32, 170.68. FAB MS m/e: 387 (MH+), 409 (MNa+). HRMS (ESI): calcd for C16H22N2NaO9, 409.1223 (MNa)+; found, 409.1217.
Compound 3a
Mp 100-102 °C; 1H NMR (CDCl3) δ: 1.11-1.17 (m, 6H, 2CH3), 2.05 (s, 3H, CH3), 2.10 (s, 3H, CH3), 2.10-2.19 (m, 1H, H2′-1), 2.39-2.44 (m, 1H, H2′-2), 3.42-3.72 (m, 4H, 2 × CH2), 4.19-4.28 (m, 3H, H4′, H5′), 5.17-5.19 (m, 1H, H3′), 5.33 (s, 1H, CH), 6.25-6.29 (m, 1H, H1′), 7.73 (s, 1H, H6), 8.27 (br s, 1H, NH).13 C NMR (CDCl3) δ: 15.36, 15.41, 20.98, 21.12, 38.02, 63.39, 63.86, 64.34, 74.86, 82.68, 85.60, 96.71, 114.13, 137.46, 150.24, 161.96, 170.61, 170.66. FAB MS m/e: 415 (MH+), 437 (MNa+). HRMS (ESI): calcd for C18H26N2O9, 437.1536 (MNa)+; found, 437.1518.
3. Preparation of 3′,5′-Di-O-acetyl-5-formyl-2′-deoxyuridine Ethylene Acetal (4a)
To a flask containing 3′,5′-di-O-acetyl-5-formyl-2′-deoxyuridine (1a, 340 mg, 1.0 mmol) and Amberlite IR-120 (100 mg) were added 0.3 mL ethylene glycol and 20 mL anhydrous benzene. The mixture was then refluxed with stirring for 1 h. Upon completion, the solid acid was removed by filtration. The filtrate was washed with water (20 mL × 2). The aqueous phase was extracted with chloroform (10 mL × 2), and the combined organic phases were then dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified through silica gel column chromatography with a mixture of EtOAc and Hexane (2:1, v/v) as eluant to give a colorless solid (4a, 334 mg, 86.9%). Compound 5a was prepared in a similar manner from 1a and propylene glycol.
Compound 4a
Mp 176-178 °C; 1H NMR (CDCl3) δ: 2.11-2.21 (m, 7H, 2CH3, H2′-1), 2.50-2.55 (m, 1H, H2′-2), 3.96-4.07 (m, 4H, -CH2CH2-), 4.28-4.36 (m, 3H, H4′, H5′), 5.24-5.26 (m, 1H, H3′), 5.75 (s, 1H, CH), 6.28-6.32 (m, 1H, H1′), 7.76 (s, 1H, H6), 8.33 (br s, 1H, NH). 13 C NMR (CDCl3) δ: 20.88, 21.12, 38.27, 64.22, 65.32, 65.46, 74.75, 82.85, 85.75, 98.10, 112.05, 137.58, 149.88, 161.45, 170.55. FAB MS m/e: 385 (MH+), 407 (MNa+). HRMS (FAB): calcd for C16H20N2O9, 385.1247 (MH)+; found, 385.1261.
5a
Mp 172-173 °C; 1H NMR (CDCl3) δ: 1.39-1.44 (m, 1H, CH2-1), 2.05-2.23 (m, 8H, CH2-2, 2xCH3, H2′-1), 2.46-2.51 (m, 1H, H2′-2), 3.91-3.98 (m, 2H, CH2), 4.13-4.19 (m, 2H, CH2), 4.28-4.38 (m, 3H, H4′, H5′), 5.24-5.26 (m, 1H, H3′), 5.52 (s, 1H, CH), 6.31-6.35 (m, 1H, H1′), 7.80 (s, 1H, H6), 8.42 (br s, 1H, NH). 13 C NMR (CDCl3) δ: 20.88, 21.11, 38.16, 64.36, 67.67, 74.96, 82.77, 85.61, 95.46, 113.36, 138.13, 149.88, 161.22, 170.63. FAB MS m/e: 399 (MH+), 421 (MNa+). HRMS (FAB): calcd for C17H22N2O9, 399.1404 (MH)+; found, 399.1390.
4. Preparation of 5-Formyl-2′-deoxyuridine Dimethylacetal (2b)
3′,5′-Di-O-acetyl-5-formyl-2′-deoxyuridine dimethylacetal (2a, 193 mg, 0.5 mmol) was dissolved in 2 mL of anhydrous methanol. To this solution was added 2 mL of 7 N NH3/MeOH solution. The mixture was stirred at 0 °C for 3 h and then at ambient temperature for 5 h. Upon completion, the solvent was removed under high vacuum. The residue was purified through column chromatography (chloroform/methanol, 8:1, v/v) to give the corresponding product (2b, 137 mg, 91%). Compounds 3b, 4b, and 5b were prepared in a similar manner.
1-((2R,4R,5R)-Tetrahydro-4-hydroxy-5-(hydroxymethyl)furan-2-yl)-5-(dimethoxymethyl)pyrimidine-2,4(1H,3H)-dione (2b)
Mp 144-145 °C; 1H NMR (CD3OD) δ: 2.21-2.30 (m, 2H, H2′-1, 2), 3.34 (s, 6H, 2CH3), 3.69-3.79 (m, 2H, H5′), 3.92-3.94 (m, 1H, H4′), 4.37-4.40 (m, 1H, H3′), 5.26 (s, 1H, CH), 6.26-6.29 (m, 1H, H1′), 8.11 (s, 1H, H6).13 C NMR (CD3OD) δ: 40.41, 53.81, 54.14, 61.94, 71.17, 85.18, 88.15, 98.56, 111.02, 138.970, 150.75, 162.53, 170.7. FAB MS m/e: 303 (MH+), 325 (MNa+). HRMS (ESI): calcd for C12H18N2O7, (MNa)+ 325.1012; found, 325.1006.
1-((2R,4R,5R)-Tetrahydro-4-hydroxy-5-(hydroxymethyl)furan-2-yl)-5-(diethoxymethyl)pyrimidine-2,4(1H,3H)-dione (3b)
Mp 112-114 °C, 1H NMR (CDCl3) δ: 1.21-1.24 (m, 6H, 2 × CH3), 1.77 (br s, 1H, OH), 2.32-2.46 (m, 2H, H2′), 2.88 (br s, 1H, OH), 3.56-3.91 (m, 6H, 2 CH2, H5′), 4.01-4.03 (m, 1H, H4′), 4.56-4.59 (m, 1H, H3′), 5.39 (s, 1H, CH), 6.13-6.17 (m, 1H, H1′), 7.84 (s, 1H, H6). 13 C NMR (CDCl3) δ: 15.40, 40.32, 62.61, 63.57, 64.60, 71.81, 87.48, 87.85, 96.82, 113.28, 139.65, 150.42, 162.10. FAB MS m/e: 331 (MH+), 353 (MNa+). HRMS (ESI): calcd for C14H22N2NaO7, 353.1325 (MNa)+; found, 353.1313.
5-(1,3-Dioxolan-2-yl)-1-((2R,4R,5R)-tetrahydro-4-hydroxy-5-(hydroxymethyl)-furan-2-yl)pyrimidine-2,4(1H,3H)-dione (4b)
Mp 192-193 °C; 1H NMR (CD3OD) δ: 2.19-2.33 (m, 2H, H2′), 3.70-3.81 (m, 2H, H5′), 3.91-3.96 (m, 3H, CH2, H4′), 4.03-4.07 (m, 2H, CH2), 4.38-4.41 (m, 1H, H3′), 5.74 (s, 1H, CH), 6.25-6.27 (m, 1H, H1′), 8.24 (s, 1H, H6). 13 C NMR (CD3OD) δ: 40.43, 61.47, 64.92, 64.96, 70.93, 85.67, 87.86, 98.50, 111.31, 139.83, 150.79, 163.20. FAB MS m/e: 301 (MH+), 323 (MNa+). HRMS (ESI): calcd for C12H17N2O7, 301.1037 (MH)+; found, 301.1038.
5-(1,3-Dioxan-2-yl)-1-((2R,4R,5R)-tetrahydro-4-hydroxy-5-(hydroxymethyl)furan-2-yl)pyrimidine-2,4(1H,3H)-dione (5b)
Mp 186-188 °C; 1H NMR (CD3OD) δ: 1.39-1.44 (m, 1H, CH2-1), 2.04-2.32 (m, 3H, H2′, CH2-2), 3.69-3.79 (m, 2H, H5′), 3.90-3.95 (m, 3H, H4′, CH2), 4.11-4.15 (m, 2H, CH2), 4.36-4.39 (m, 1H, H3′), 5.45 (s, 1H, CH), 6.23-6.27 (m, 1H, H1′), 8.14 (s, 1H, H6). 13 C NMR (CD3OD) δ: 25.54, 40.18, 61.63, 67.32, 67.36, 70.96, 85.66, 87.72, 95.70, 112.28, 139.92, 150.64, 162.85. FAB MS m/e: 315 (MH+), 337 (MNa+). HRMS (FAB): calcd for C13H18N2O7, 315.1193 (MH)+; found, 315.1189.
5. Preparation of 5-Formyluracil Dimethylacetal (7)
5-Formyluracil (6, 280 mg, 2.0 mmol) was added to a flask containing 20 mL of anhydrous methanol and Amberlite IR-120 (200 mg). The mixture was then heated to 80 °C with stirring for 1.5 h. Upon completion (monitored by TLC), the solid acid was removed through filtration. The filtrate was allowed to cool to room temperature, whereupon the product precipitated. The solid so formed was collected by filtration to give 7 (338 mg, 91%).
Compound 7
1H NMR (DMF-d6) δ: 3.47 (s, 6H, 2 × CH3), 5.41 (s, 1H, CH), 7.64 (s, 1H, H6), 11.08 (br s, 1H, NH), 11.29 (br s, 1H, NH).13 C NMR (DMF-d7) δ: 53.35, 98.70, 110.08, 140.33, 151.71, 163.55. FAB MS m/e: 187 (MH+), 209 (MNa+).
Acknowledgment
We acknowledge the US Army Medical Research Material Command contract USAMRIID DAMD 17-03-C-0081 and the State of Arizona Proposition 301 Funds for financial support and Robert Smith and Shalisa Sanders for excellent technical assistance. The in vitro evaluation for antiviral activity was supported by Public Health Service Contract No. NO1-AI-30049 from NIAID, NIH, Bethesda, MD.
Footnotes
Supporting Information Available: The HPLC purity of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Alberts B, Fineberg HV. Harnessing new science is vital for biodefense and global health. Proc. Natl. Acad. Sci. U.S.A. 2004;101:11177. doi: 10.1073/pnas.0404433101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alibek K. Smallpox: a disease and a weapon. Int. J. Infect. Dis. 2004;8:S3–8. doi: 10.1016/j.ijid.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 3.Arnon SS, Schechter R, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Hauer J, Layton M, Lillibridge S, Osterholm MT, O'Toole T, Parker G, Perl TM, Russell PK, Swerdlow DL, Tonat K. Botulinum toxin as a biological weapon: medical and public health management. JAMA, J. Am. Med. Assoc. 2001;285:1059–1070. doi: 10.1001/jama.285.8.1059. [DOI] [PubMed] [Google Scholar]
- 4.Artenstein AW. Biodefense: medicine in the time of bioterrorism. Med. Health R. I. 2003;86:201–203. [PubMed] [Google Scholar]
- 5.Atlas RM, Reppy J. Globalizing biosecurity. Biosecur. Bioterror. 2005;3:51–60. doi: 10.1089/bsp.2005.3.51. [DOI] [PubMed] [Google Scholar]
- 6.Borio L, Frank D, Mani V, Chiriboga C, Pollanen M, Ripple M, Ali S, DiAngelo C, Lee J, Arden J, Titus J, Fowler D, O'Toole T, Masur H, Bartlett J, Inglesby T. Death due to bioterrorism-related inhalational anthrax: report of 2 patients. JAMA, J. Am. Med. Assoc. 2001;286:2554–2559. doi: 10.1001/jama.286.20.2554. [DOI] [PubMed] [Google Scholar]
- 7.Borio L, Inglesby T, Peters CJ, Schmaljohn AL, Hughes JM, Jahrling PB, Ksiazek T, Johnson KM, Meyerhoff A, O'Toole T, Ascher MS, Bartlett J, Breman JG, Eitzen EM, Jr., Hamburg M, Hauer J, Henderson DA, Johnson RT, Kwik G, Layton M, Lillibridge S, Nabel GJ, Osterholm MT, Perl TM, Russell P, Tonat K. Hemorrhagic fever viruses as biological weapons: medical and public health management. JAMA, J. Am. Med. Assoc. 2002;287:2391–2405. doi: 10.1001/jama.287.18.2391. [DOI] [PubMed] [Google Scholar]
- 8.Bray M. Defense against filoviruses used as biological weapons. AntiViral Res. 2003;57:53–60. doi: 10.1016/s0166-3542(02)00200-0. [DOI] [PubMed] [Google Scholar]
- 9.Bray M, Buller M. Looking back at smallpox. Clin. Infect. Dis. 2004;38:882–889. doi: 10.1086/381976. [DOI] [PubMed] [Google Scholar]
- 10.Bray M, Roy CJ. Antiviral prophylaxis of smallpox. J. Antimicrob. Chemother. 2004;54:1–5. doi: 10.1093/jac/dkh286. [DOI] [PubMed] [Google Scholar]
- 11.Bray M, Wright ME. Progressive vaccinia. Clin. Infect. Dis. 2003;36:766–774. doi: 10.1086/374244. [DOI] [PubMed] [Google Scholar]
- 12.Dennis DT, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Friedlander AM, Hauer J, Layton M, Lillibridge SR, McDade JE, Osterholm MT, O'Toole T, Parker G, Perl TM, Russell PK, Tonat K. Tularemia as a biological weapon: medical and public health management. JAMA, J. Am. Med. Assoc. 2001;285:2763–2773. doi: 10.1001/jama.285.21.2763. [DOI] [PubMed] [Google Scholar]
- 13.Goodrich A, Parveen Z, Dornburg R, Schnell MJ, Pomerantz RJ. Spliced spleen necrosis virus vector RNA is not encapsidated: implications for retroviral replication and vector design. Mol. Ther. 2004;9:557–565. doi: 10.1016/j.ymthe.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 14.Henderson DA. The looming threat of bioterrorism. Science. 1999;283:1279–1282. doi: 10.1126/science.283.5406.1279. [DOI] [PubMed] [Google Scholar]
- 15.Henderson DA. Countering the posteradication threat of smallpox and polio. Clin. Infect. Dis. 2002;34:79–83. doi: 10.1086/323897. [DOI] [PubMed] [Google Scholar]
- 16.Henderson DA, Inglesby TV, Bartlett JG, Ascher MS, Eitzen E, Jahrling PB, Hauer J, Layton M, McDade J, Osterholm MT, O'Toole T, Parker G, Perl T, Russell PK, Tonat K. Smallpox as a biological weapon: medical and public health management. Working Group on Civilian Biodefense. JAMA, J. Am. Med. Assoc. 1999;281:2127–2137. doi: 10.1001/jama.281.22.2127. [DOI] [PubMed] [Google Scholar]
- 17.Kaiser J. Biodefense. Report faults smallpox vaccination. Science. 2005;307:1540. doi: 10.1126/science.307.5715.1540b. [DOI] [PubMed] [Google Scholar]
- 18.O'Toole T. The problem of biological weapons: next steps for the nation. Public Health Rep. 2001;116:108–111. doi: 10.1016/S0033-3549(04)50152-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Toole T. Emerging illness and bioterrorism: implications for public health. J. Urban Health. 2001;78:396–402. doi: 10.1093/jurban/78.2.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O'Toole T, Inglesby TV. Facing the biological weapons threat. Lancet. 2000;356:1128–1129. doi: 10.1016/S0140-6736(00)02751-3. [DOI] [PubMed] [Google Scholar]
- 21.O'Toole T, Inglesby TV. Epidemic response scenario: decision making in a time of plague. Public Health Rep. 2001;116:92–103. doi: 10.1016/S0033-3549(04)50150-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Toole T, Mair M, Inglesby TV. Shining light on “Dark Winter”. Clin Infect. Dis. 2002;34:972–983. doi: 10.1086/339909. [DOI] [PubMed] [Google Scholar]
- 23.Smith BT, Inglesby TV, O'Toole T. Biodefense R&D: anticipating future threats, establishing a strategic environment. Biosecur. Bioterror. 2003;1:193–202. doi: 10.1089/153871303769201842. [DOI] [PubMed] [Google Scholar]
- 24.Bryant JE, Barrett AD. Comparative phylogenies of yellow fever isolates from Peru and Brazil. FEMS Immunol. Med. Microbiol. 2003;39:103–118. doi: 10.1016/S0928-8244(03)00238-4. [DOI] [PubMed] [Google Scholar]
- 25.Cohen J, Marshall E. Bioterrorism. Vaccines for biodefense: a system in distress. Science. 2001;294:498–501. doi: 10.1126/science.294.5542.498. [DOI] [PubMed] [Google Scholar]
- 26.Couzin J. Biodefense. U. S. agencies unveil plan for biosecurity peer review. Science. 2004;303:1595. doi: 10.1126/science.303.5664.1595a. [DOI] [PubMed] [Google Scholar]
- 27.Davis CJ. Nuclear blindness: An overview of the biological weapons programs of the former Soviet Union and Iraq. Emerging Infect. Dis. 1999;5:509–512. doi: 10.3201/eid0504.990408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Enserink M. Infectious diseases. New biodefense splurge creates hotbeds, shatters dreams. Science. 2003;302:206–207. doi: 10.1126/science.302.5643.206a. [DOI] [PubMed] [Google Scholar]
- 29.Enserink M. Biodefense. Smallpox vaccines: looking beyond the next generation. Science. 2004;304:809. doi: 10.1126/science.304.5672.809a. [DOI] [PubMed] [Google Scholar]
- 30.Giangrego E. Biodefense: it's more than duct tape. CDS Rev. 2003;96:10–16. [PubMed] [Google Scholar]
- 31.Gilfillan L, Smith BT, Inglesby TV, Kodukula K, Schuler A, Lister M, O'Toole T. Taking the measure of countermeasures: leaders' views on the nation's capacity to develop biodefense countermeasures. Biosecur. Bioterror. 2004;2:320–327. doi: 10.1089/bsp.2004.2.320. [DOI] [PubMed] [Google Scholar]
- 32.Goodman L. Biodefense cost and consequence. J. Clin. Invest. 2004;114:2–3. doi: 10.1172/JCI22418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bray M. Viral bioterrorism and antiviral countermeasures. In: Torrence PF, editor. AntiViral Drug DiscoVery for Emerging Diseases and Bioterrorism Threats. John Wiley & Sons; New York: 2005. pp. 17–30. [Google Scholar]
- 34.Torrence PF. Introduction: pestilence, plague, bioterrorism. In: Torrence PF, editor. Antiviral Drug DiscoVery for Emerging Diseases and Bioterrorism Threats. John Wiley & Sons; New York: 2005. pp. 3–16. [Google Scholar]
- 35.Arita I, Wickett J, Nakane M. Eradication of infectious diseases: its concept, then and now. Jpn. J. Infect. Dis. 2004;57:1–6. [PubMed] [Google Scholar]
- 36.Bhalla DK, Warheit DB. Biological agents with potential for misuse: a historical perspective and defensive measures. Toxicol. Appl. Pharmacol. 2004;199:71–84. doi: 10.1016/j.taap.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 37.Debord T. Smallpox and bioterrorism. Med. Mal. Infect. 2004;34:6–11. doi: 10.1016/j.medmal.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 38.Eichner M, Schwehm M. Smallpox: a vulnerable specter. Epidemiology. 2004;15:258–260. doi: 10.1097/01.ede.0000121548.84183.04. Discussion 260-251. [DOI] [PubMed] [Google Scholar]
- 39.Guharoy R, Panzik R, Noviasky JA, Krenzelok EP, Blair DC. Smallpox: clinical features, prevention, and management. Ann. Pharmacother. 2004;38:440–447. doi: 10.1345/aph.1D272. [DOI] [PubMed] [Google Scholar]
- 40.Jacobs LM, Emanuelsen K, McKay C, Burns K. Bioterrorism preparedness-Part II. Smallpox vaccination in a hospital setting. Conn. Med. 2004;68:27–35. [PubMed] [Google Scholar]
- 41.Kaplan EH. Preventing second-generation infections in a smallpox bioterror attack. Epidemiology. 2004;15:264–270. doi: 10.1097/01.ede.0000121821.02642.a4. [DOI] [PubMed] [Google Scholar]
- 42.Kuljic-Kapulica N. Smallpoxsin the past or not? Srp. Arh. Celok. Lek. 2004;132:272–276. doi: 10.2298/sarh0408272k. [DOI] [PubMed] [Google Scholar]
- 43.Leissner KB, Holzman RS, McCann ME. Bioterrorism and children: unique concerns with infection control and vaccination. Anesthesiol. Clin. North Am. 2004;22:563–577. doi: 10.1016/j.atc.2004.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lupatkin H, Lupatkin JF, Rosenberg AD. Smallpox in the 21st century. Anesthesiol. Clin. North Am. 2004;22:541–561. doi: 10.1016/j.atc.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 45.Mahalingam S, Damon IK, Lidbury BA. 25 years since the eradication of smallpox: why poxvirus research is still relevant. Trends Immunol. 2004;25:636–639. doi: 10.1016/j.it.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 46.Mayr A. Taking advantage of the positive side-effects of smallpox vaccination. J. Vet. Med., Ser. B. 2004;51:199–201. doi: 10.1111/j.1439-0450.2004.00763.x. [DOI] [PubMed] [Google Scholar]
- 47.Slifka MK, Hanifin JM. Smallpox: the basics. Dermatol. Clin. 2004;22:263–274. doi: 10.1016/j.det.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 48.Spuls PI, Bos JD, Rudikoff D. Smallpox: what the dermatolo-gist should know. Skinmed. 2004;3:197–206. doi: 10.1111/j.1540-9740.2004.02294.x. quiz 207-198. [DOI] [PubMed] [Google Scholar]
- 49.Tennyson HC, Mair EA. Smallpox: what every otolaryngologist should know. Otolaryngol. Head Neck Surg. 2004;130:323–333. doi: 10.1016/j.otohns.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 50.Thornton R, Court B, Meara J, Murray V, Palmer I, Scott R, Wale M, Wright D. Chemical, biological, radiological and nuclear terrorism: an introduction for occupational physicians. Occup. Med. (London) 2004;54:101–109. doi: 10.1093/occmed/kqh025. [DOI] [PubMed] [Google Scholar]
- 51.Bray M, Martinez M, Kefauver D, West M, Roy C. Treatment of aerosolized cowpox virus infection in mice with aerosolized cidofovir. Antiviral Res. 2002;54:129–142. doi: 10.1016/S0166-3542(01)00220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bray M, Martinez M, Smee DF, Kefauver D, Thompson E, Huggins JW. Cidofovir protects mice against lethal aerosol or intranasal cowpox virus challenge. J. Infect. Dis. 2000;181:10–19. doi: 10.1086/315190. [DOI] [PubMed] [Google Scholar]
- 53.De Clercq E. Cidofovir in the therapy and short-term prophylaxis of poxvirus infections. Trends Pharmacol. Sci. 2002;23:456–458. doi: 10.1016/S0165-6147(02)02091-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Clercq E. Cidofovir in the treatment of poxvirus infections. Antiviral Res. 2002;55:1–13. doi: 10.1016/S0166-3542(02)00008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neyts J, Leyssen P, Verbeken E, De Clercq E. Efficacy of cidofovir in a murine model of disseminated progressive vaccinia. Antimicrob. Agents Chemother. 2004;48:2267–2273. doi: 10.1128/AAC.48.6.2267-2273.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quenelle DC, Collins DJ, Kern ER. Efficacy of multiple- or single-dose cidofovir against vaccinia and cowpox virus infections in mice. Antimicrob. Agents Chemother. 2003;47:3275–3280. doi: 10.1128/AAC.47.10.3275-3280.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roy CJ, Baker R, Washburn K, Bray M. Aerosolized cidofovir is retained in the respiratory tract and protects mice against intranasal cowpox virus challenge. Antimicrob. Agents Chemother. 2003;47:2933–2937. doi: 10.1128/AAC.47.9.2933-2937.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smee DF, Bailey KW, Sidwell RW. Comparative effects of cidofovir and cyclic HPMPC on lethal cowpox and vaccinia virus respiratory infections in mice. Chemotherapy. 2003;49:126–131. doi: 10.1159/000070618. [DOI] [PubMed] [Google Scholar]
- 59.Smee DF, Bailey KW, Wong MH, Wandersee MK, Sidwell RW. Topical cidofovir is more effective than is parenteral therapy for treatment of progressive vaccinia in immunocompromised mice. J. Infect. Dis. 2004;190:1132–1139. doi: 10.1086/422696. [DOI] [PubMed] [Google Scholar]
- 60.Toro JR, Sanchez S, Turiansky G, Blauvelt A. Topical cidofovir for the treatment of dermatologic conditions: verruca, condyloma, intraepithelial neoplasia, herpes simplex and its potential use in smallpox. Dermatol. Clin. 2003;21:301–309. doi: 10.1016/s0733-8635(02)00116-x. [DOI] [PubMed] [Google Scholar]
- 61.Aldern KA, Ciesla SL, Winegarden KL, Hostetler KY. Increased antiviral activity of 1-O-hexadecyloxypropyl-[2-(14)C]-cidofovir in MRC-5 human lung fibroblasts is explained by unique cellular uptake and metabolism. Mol. Pharmacol. 2003;63:678–681. doi: 10.1124/mol.63.3.678. [DOI] [PubMed] [Google Scholar]
- 62.Bradbury J. Orally available cidofovir derivative active against smallpox. Lancet. 2002;359:1041. doi: 10.1016/S0140-6736(02)08115-1. [DOI] [PubMed] [Google Scholar]
- 63.Ciesla SL, Trahan J, Wan WB, Beadle JR, Aldern KA, Painter GR, Hostetler KY. Esterification of cidofovir with alkoxyalkanols increases oral bioavailability and diminishes drug accumulation in kidney. Antiviral Res. 2003;59:163–171. doi: 10.1016/s0166-3542(03)00110-4. [DOI] [PubMed] [Google Scholar]
- 64.Byrd CM, Bolken TC, Mjalli AM, Arimilli MN, Andrews RC, Rothlein R, Andrea T, Rao M, Owens KL, Hruby DE. New class of orthopoxvirus antiviral drugs that block viral maturation. J. Virol. 2004;78:12147–12156. doi: 10.1128/JVI.78.22.12147-12156.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tseng CK. Overview of antiviral drug discovery and development. In: Torrence PF, editor. AntiViral Drug DiscoVery for Emerging Diseases and Bioterrorism Threats. John Wiley & Sons; New York: 2005. pp. 31–82. [Google Scholar]
- 66.Ono A, Okamoto T, Inada M, Nara H, Matsuda A. Nucleosides and nucleotides. 131. Synthesis and properties of oligonucleotides containing 5-formyl-2′-deoxyuridine. Chem. Pharm. Bull. (Tokyo) 1994;42:2231–2237. doi: 10.1248/cpb.42.2231. [DOI] [PubMed] [Google Scholar]
- 67.Nomura M, Shuto S, Matsuda A. Synthesis of the cyclic and acyclic acetal derivatives of 1-(3-C-ethynyl-beta-D-ribo-pentofuranosyl)cytosine, a potent antitumor nucleoside. Design of prodrugs to be selectively activated in tumor tissues via the bio-reduction-hydrolysis mechanism. Bioorg. Med. Chem. 2003;11:2453–2461. doi: 10.1016/s0968-0896(03)00104-4. [DOI] [PubMed] [Google Scholar]
- 68.Mahmud NP, Garrett SW, Threadgill MD. The 5-nitrofuran-2-ylmethylidene group as a potential bioreductively activated prodrug system for diol-containing drugs. Anticancer Drug Des. 1998;13:655–662. [PubMed] [Google Scholar]
- 69.Park JS, Chang CT, Schmidt CL, Golander Y, De Clercq E, Descamps J, Mertes MP. Oxime and dithiolane derivatives of 5-formyl-2′-deoxyuridine and their 5′-phosphates: antiviral effects and thymidylate synthetase inhibition. J. Med. Chem. 1980;23(3):661–665. doi: 10.1021/jm00180a016. [DOI] [PubMed] [Google Scholar]
- 70.Otvos L, Tudos FH. Hevizos ointment efficacy in view of molecular pharmacology. Acta Pharm. Hung. 1993;63:237–242. [PubMed] [Google Scholar]
- 71.Sagi J, Szabolcs A, Szemzo A, Michaela V, Jaroslav K, Jitka S, Alena S, Otvos L. Incorporation of the antiherpetic 5-isopropyl-2′-deoxyuridine into a synthetic DNA and the consequence of incorporation on structure and functions of the DNA. Acta Pharm. Hung. 1993;63:204–214. [PubMed] [Google Scholar]
- 72.Sagi J, Szabolcs A, Szemzo A, Otvos L. Modified polynucleotides. VII. Impaired integrity of a synthetic DNA containing the antiherpetic agent 5-isopropyl-2′-deoxyuridine. Nucleic Acids Res. 1986;14:3449–3462. doi: 10.1093/nar/14.8.3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Verri A, Focher F, Duncombe RJ, Basnak I, Walker RT, Coe PL, de Clercq E, Andrei G, Snoeck R, Balzarini J, Spadari S. Anti-(herpes simplex virus) activity of 4′-thio-2′-deoxyuridines: a biochemical investigation for viral and cellular target enzymes. Biochem. J. 2000;351:319–326. [PMC free article] [PubMed] [Google Scholar]