

Abstract
Dual-emitting photonic nano-objects that can sense changes in the environmental pH are designed based on shell-crosslinked micelles assembled from amphiphilic block copolymers and crosslinked with pH-insensitive chromophores. The chromophoric crosslinkers are tetra-functionalized pyrazine molecules that bear a set of terminal aliphatic amine groups and a set of anilino amine groups, which demonstrate morphology-dependent reactivities towards the poly(acrylic acid) shell domain of the nano-objects. The extent to which the anilino amine groups react with the nano-object shell is shown to affect the hypsochromic shift (blue-shift). The ratio of fluorescence intensity at 496 nm over that of 560 nm is dependent upon the solution pH. We report, herein, observations on the pH-sensitive dual-emission photophysical properties of rod-shaped or spherical nano-objects, whose shell domains offer two distinct platforms for amidation reactions to occur—through formation of activated esters upon addition of carbodiimide or pre-installation of activated ester groups. We demonstrate that physical manipulations (changes in morphology or particle dimensions) or chemical manipulations of the crosslinking reaction (the order of installation of activated esters) lead to fine tuning of dual-emission over ca. 60 nm in a physiologically relevant pH range. Rod-shaped shell-crosslinked nanostructures with poly(p-hydroxystyrene) core show blue-shift as a function of increasing pH while spherical shell-crosslinked nanostructures with polystyrene core and poly(ethylene oxide) corona exhibit blue-shift as a function of decreasing pH.
1. Introduction
Stimuli-sensitive materials that respond to changes in various biologically relevant events with dual-emitting fluorescence as the output signals have been celebrated as a potential non-invasive diagnostic tool for various diseases. The types of stimuli of interest have naturally been associated with or caused by the characteristics of diseased cells, such as decreased pH,1-14 increased concentration of O2,15 presence of heavy-metal ions,16 or concentration of ATP17 or proteins such as avidin18 or RNase.19 Ratiometric sensing based on a dual-emission profile is superior to single-emission, as the output is independent of sensor concentration and absolute fluorescence emission intensity. Single-component materials whose dual-emissions span over ca. 70 nm have been fabricated: for instance, Fraser and co-workers recently reported a ratiometric O2 sensing film, prepared from iodide-substituted difluoroboron dibenzoylmethane-poly (lactic acid), which emitted fluorescence at 450 nm and 525 nm for tumour hypoxia imaging with a I450/I525 ratio ranging from ca. 0.22 to 0.41.15 For in vivo pH sensing, much of the recent developments have relied on the intrinsic pH-responsiveness of small molecule probes: the commercially available carboxy-seminaphthofluorescein (cSNARF®-1), for example, is a modified fluorescein molecule, whose emission spectrum undergoes a pH-dependent wavelength shift. The compound is usually excited between 488 nm and 530 nm while monitoring the fluorescence emission at two wavelengths, 580 nm and 640 nm, respectively, with I580/I640 ratios easily reaching values greater than thirty.20 Drastic improvements on quantum yield of the chromophore have been realized by Burgess and co-workers through synthesis of a pH probe equipped with two xanthene (the fluorescent core of fluorescein) donors and one boron-dipyrromethene (BODIPY) acceptor with I600/I525 between one and five.5 To minimize probe-protein interactions in vivo, small molecule chromophores have been encapsulated within the cavities of l-α-phosphatidylcholine-based liposomes, while maintaining pH responsiveness by providing minimal hindrance for the movement of protons across the liposome.21
While great advances have been achieved in the synthesis and utilization of small molecule probes for detecting pH, it is of wide interest to design dual-emitting macromolecular/supramolecular probes that are water soluble and able to sequester guest molecules for coincident imaging and treatment of diseases. Jin and co-workers recently reported fluorescein isothiocyanate-coated quantum dots, having a hydrodynamic diameter of 7 nm, with an I600/I515 dual emission ratio ranging from fourteen to four from pH 6 to 8.7 Peng and Wolfbeis et al. reported the preparation of a polyurethane-based nanogel loaded with the pH indicator bromothymol blue and the fluorophores Coumarin 6 and Nile Red as a two-component system that underwent Förster resonance energy transfer (FRET) in response to a pH change.10
In designing a single-component, dual-emitting, pH-responsive nanoscopic probe, which is based upon pH-insensitive small molecule chromophores, shell-crosslinked knedel-like nanoparticles (SCKs) have emerged as an interesting nanotechnology platform. SCKs are well-defined, discrete macromolecular assemblies with unique covalently stabilized core-shell morphology and serve as a robust template onto which orthogonal chemical reactions can take place. In preparation of SCKs, condensation reaction between the shell of amphiphilic block copolymer micelles and crosslinkers is the key step that ensures maintenance of the integrity of the final nanostructures under a wide variety of conditions (pH, ionic strength, dilution, etc.). We have previously utilized pH-insensitive pyrazine-based chromophoric crosslinkers in this critical step to impart pH-responsive enhancement of single-wavelength fluorescence emission intensities in the resulting fluorophore-SCKs for pH-sensing applications.22 Building upon our past advance, we envisioned a single-component, dual-emitting analogue as a powerful alternative to sense the pH, while utilizing the core/shell nature for loading of guest molecules23-25 and attaching targeting ligands for active-targeted delivery.26-29 Herein, we report preparation of pH-responsive, dual-emitting, single-component shell-crosslinked nano-objects and observation of their pH sensitive photophysical properties as a function of physical parameters (morphology or core/shell dimensions) or chemical parameters (stoichiometry of reagents added or pre-installation of reactive groups) using pH-insensitive chromophoric crosslinkers (Fig. 1). Two morphologies were utilized in this study: shell-crosslinked rod-shaped nanostructures (SCRs) and spherical nanoparticles (SCKs). SCRs were self-assembled from poly(acrylic acid)-b-poly(p-hydroxystyrene) (PAA140-b-PpHS50)30 and SCKs were self-assembled from either the same parent diblock copolymer, PAA140-b-PpHS50, or poly(ethylene oxide)-b-poly(N-acryloxysuccinimide)-b-polystyrene (PEO45-b-PNAS50-b-PS30) or PEO45-b-PNAS95-b-PS60.31 Surprisingly opposite behaviour for SCRs vs. SCKs was observed: SCRs exhibited increasing intensities of hypsochromic shifts (blueshift) as a function of increasing solution pH, whereas SCKs showed the same effect as a function of decreasing pH, over a solution pH range of 4.6 to 8.6. Both systems present themselves as promising dual-emitting ratiometric pH sensing materials. Much of the work reported here was, therefore, conducted to understand the different behaviours.
Fig. 1.
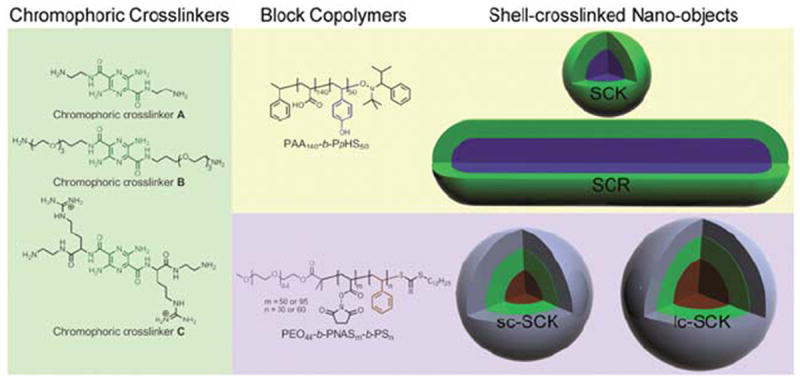
Chemical structures of chromophoric crosslinkers (A, B and C), block copolymers and schematic illustrations of the respective shell-crosslinked nano-objects.
2. Experimental
2.1. Materials
The universal alkoxyamine initiator 2,2,5-trimethyl-3-(1′-phenylethoxy)-4-phenyl-3-azahexane was graciously provided by Sigma-Aldrich. The corresponding nitroxide 2,2,5-trimethyl-4-phenyl-3-azahexane-3-nitroxide was synthesized according to the literature method.32 Prior to use, N-acryloxysuccinimide, purchased from Acros (99%), was recrystallized from dry ethyl acetate and stored under argon. The mono-methoxy terminated mono-hydroxy poly(ethylene glycol) (mPEG, Mw = 2000 Da, PDI = 1.06) was purchased from Intezyne Technologies and was used for the synthesis of macro-chain transfer agent (macro-CTA) without further purification. The mPEG2k macro-CTA and PEO45-b-PNAS95-b-PS60 were synthesized according to previous reports.31 All other chemicals and reagents were obtained from Aldrich and used as received, unless described otherwise. tert-Butyl acrylate (tBA) and 4-acetoxystyrene (AS) were filtered through a plug of aluminium oxide to remove the inhibitor. All reactions were performed under N2, unless noted otherwise.
2.2. Instrumental
1H NMR and 13C NMR spectra were recorded at 500 MHz and 125 MHz, respectively, as solutions with the solvent proton or carbon signal as a standard. UV-vis spectra were collected at ambient temperature in the region of 200–800 nm, using a Varian Cary 100 Bio UV-visible spectrophotometer. The fluorescence spectra were obtained at room temperature using a Varian Cary Eclipse fluorescence spectrophotometer. An excitation wave-length of the observed maximum absorption peak was used unless otherwise noted. Each fluorescence spectrum was normalized with respect to the absorbance at the excitation wavelength. The molar extinction coefficient (ε) of chromophoric crosslinkers (εA = 5163, εB = 5772, εC = 3463 M−1 cm−1 at 441 nm) was determined by a calibration curve in 5 mM PBS. The chromophoric crosslinker concentrations in the nano-objects were determined by UV-vis spectroscopy. IR spectra of neat films on NaCl plates were recorded using a Shimadzu Prestige21 IR spectrometer.
Gel permeation chromatography (GPC) was conducted on a Waters 1515 HPLC (Waters Chromatography, Inc.) equipped with a Waters 2414 differential refractometer, a PD2020 dual-angle (15° and 90°) light scattering detector (Precision Detectors, Inc.), and a three-column series PL gel 5 μm Mixed C, 500 Å, and 104 Å, 300 × 7.5 mm columns (Polymer Laboratories, Inc.). The system was equilibrated at 35 °C in anhydrous THF, which served as the polymer solvent and eluent with a flow rate of 1.0 mL min−1. Polymer solutions were prepared at a known concentration (ca. 4–5 mg mL−1) and an injection volume of 200 μL was used. Data collection and analysis were performed, respectively, with Precision Acquire software and Discovery 32 software (Precision Detectors, Inc.). The interdetector delay volume and the light scattering detector calibration constant were determined by calibration using a nearly monodispersed polystyrene standard (Pressure Chemical Co., Mp = 90 kDa, Mw/Mn < 1.04). The differential refractometer was calibrated with a standard polystyrene reference material (SRM 706 NIST) of known specific refractive index increment dn/dc (0.184 mL g−1). The dn/dc values of the analyzed polymers were then determined from the differential refractometer response.
The N,N-dimethylformamide (DMF) GPC was conducted on a Waters Chromatography Inc. (Milford, MA) system equipped with an isocratic pump model 1515, a differential refractometer model 2414, and a two-column set of Styragel HR 4 and HR 4E 5 μm DMF 7.8 × 300 mm columns. The system was equilibrated at 70 °C in pre-filtered DMF containing 0.05 M LiBr, which served as polymer solvent and eluent (flow rate set to 1.00 mL min−1). Polymer solutions were prepared at a concentration of ca. 3 mg mL−1 and an injection volume of 200 μL was used. Data collection and analysis were performed with Empower Pro software (Waters Inc.). The system was calibrated with poly (ethylene glycol) standards (Polymer Laboratories) ranging from 615 to 442 800 Da.
Dynamic light scattering measurements were conducted with a Brookhaven Instruments, Co. (Holtsville, NY) DLS system equipped with a model BI-200SM goniometer, BI-9000AT digital correlator, and a model EMI-9865 photomultiplier, and a model Innova 300 Ar ion laser operated at 514.5 nm (Coherent Inc., Santa Clara, CA). Measurements were made at 25 ± 1 °C. Prior to analysis, solutions were filtered through a 0.45 μm Millex®-GV PVDF membrane filter (Millipore Corp., Medford, MA) to remove dust particles. Scattered light was collected at a fixed angle of 90°. The digital correlator was operated with 522 ratio spaced channels, and an initial delay of 5 μs, a final delay of 50 ms, and a duration of 8 minutes. A photomultiplier aperture of 400 μm was used, and the incident laser intensity was adjusted to obtain a photon counting of between 200 and 300 kcps. The calculations of the particle size distributions and distribution averages were performed with the ISDA software package (Brookhaven Instruments Company), which employed single-exponential fitting, Cumulants analysis, and CONTIN particle size distribution analysis routines. All determinations were average values from ten measurements. Alternatively, DLS measurements were also conducted using a Delsa Nano C from Beckman Coulter, Inc. (Fullerton, CA) equipped with a laser diode operating at 658 nm. Size measurements were made in nanopure water. Scattered light was detected at 15° angle and analyzed using a log correlator over 70 accumulations for a 0.5 mL of sample in a glass size cell (0.9 mL capacity). The photomultiplier aperture and the attenuator were automatically adjusted to obtain a photon counting rate of ca. 10 kcps. The calculation of the particle size distribution and distribution averages was performed using CONTIN particle size distribution analysis routines using Delsa Nano 2.31 software. The peak average of histograms from intensity, volume and number distributions out of 70 accumulations were reported as the average diameter of the particles.
Transmission electron microscopy (TEM) bright-field imaging was conducted on a Hitachi H-7500 microscope, operating at 80 kV. The samples were prepared as follows: 4 μL of the dilute solution (with a polymer concentration of ca. 0.2–0.5 mg mL−1) was deposited onto a carbon-coated copper grid, which was pre-treated with absolute ethanol to increase the surface hydrophilicity. After 5 min, the excess of the solution was quickly wicked away by a piece of filter paper. The samples were then negatively stained with 4 μL of 1 wt% phosphotungstic acid (PTA) aqueous solution. After 1 min, the excess PTA solution was quickly wicked away by a piece of filter paper and the samples were left to dry under ambient conditions overnight.
2.3. Synthesis of chromophoric crosslinkers
2.3.1. Synthesis of chromophoric crosslinker A (3,6-diamino-N2,N5-bis(2-aminoethyl)pyrazine-2,5-dicarboxamide)
A mixture of sodium 3,6-diaminopyrazine-2,5-dicarboxylate (500 mg, 2.07 mmol), tert-butyl 2-aminoethylcarbamate (673 mg, 4.20 mmol), HOBt (836 mg, 5.46 mmol) and EDCI (1.05 g, 5.48 mmol) in DMF (25 mL) was allowed to stir for 16 h and was then concentrated. The residue was partitioned with 1 N NaHSO4 (200 mL) and EtOAc (200 mL). The organic layer was separated and washed with water (200 mL × 3), saturated NaHCO3 (200 mL × 3), and brine. It was then dried with MgSO4, filtered, and concentrated to afford the bisamide as an orange foam. 770 mg, 76% yield. 1H NMR (300 MHz, DMSO-d6, δ): major conformer, 8.44 (t, J = 5.7 Hz, 2H), 6.90 (t, J = 5.7 Hz, 2H), 6.48 (br, 4H), 2.93–3.16 (m, 8H), 1.37 (s, 9H), 1.36 (s, 9H). 13C NMR (75 MHz, DMSO-d6, δ): 165.1, 155.5, 155.4, 146.0, 126.2, 77.7, 77.5, 45.2, 44.5, 28.2. LC-MS (15–95% gradient acetonitrile in 0.1% TFA over 10 min), single peak retention time = 7.18 min on a 30 mm column, (M + H)+ = 483 amu. TFA (25 mL) was added to the product (770 mg, 1.60 mmol) in methylene chloride (100 mL), and the reaction was stirred at room temperature for 2 h. The mixture was concentrated and the residue was dissolved in methanol (15 mL). Diethyl ether (200 mL) was added and the orange solid precipitate was isolated by filtration and dried in high vacuum to afford an orange powder. 627 mg, 77% yield. IR (NaCl): 2951, 2928, 1811, 1759, 1233, 1090, 1067, 864, 831, 775 cm−1. 1H NMR (300 MHz, DMSO-d6, δ): 8.70 (t, J = 6 Hz, 2H), 7.86 (br, 6H), 6.50 (br, 4H), 3.46–3.58 (m, 4H), 3.26–3.40 (m, 4H); 13C NMR (75 MHz, DMSO-d6, δ): 166.4, 146.8, 127.0, 39.4, 37.4. LC-MS (15–95% gradient acetonitrile in 0.1% TFA over 10 min), single peak retention time = 2.60 min on a 30 mm column, (M + H)+ = 283 amu. UV-vis (100 mM in PBS): λabs = 435 nm. Fluorescence (100 nM): λex = 449 nm and λem = 562 nm. The product was converted to the HCl salt by co-evaporation (3 × 100 mL) with 1 N aqueous HCl.
2.3.2. Synthesis of chromophoric crosslinker B (3,6-diamino-N2,N5-bis(3-(2-(2-(3-aminopropoxy)ethoxy)ethoxy)propyl) pyrazine-2,5-dicarboxamide dihydrochloride)
Step 1
Synthesis of tert-butyl 1,1′-(3,6-diaminopyrazine-2, 5-diyl)bis(1-oxo-6,9,12-trioxa-2-azapentadecane-15,1-diyl)dicarbamate: a mixture of 3,6-diaminopyrazine-2,5-dicarboxylic acid (0.31g, 1.6 mmol), tert-butyl 3-(2-(2-(3-aminopropoxy)ethoxy) ethoxy) propyl carbamate (1.00 g, 3.12 mmol), EDC·HCl (0.72 g, 3.7 mmol) and HOBt (0.50, 3.7 mmol) was stirred in DMF (35 mL) for 16 h at room temperature. The residue was partitioned with EtOAc (100 mL) and saturated sodium bicarbonate (100 mL). The layers were separated and the EtOAc solution was washed with 5% aq. citric acid (100 mL) and brine (100 mL). The EtOAc layer was dried (MgSO4), filtered and concentrated to afford 1.2 g (48% yield) of the bisamide as an orange oil. The crude bis-amide was taken on to the next step with no further purification: HRMS calcd for C36H66N8O12Na, [M + Na]+ = 825.4692 g mol−1; observed, 825.4674 g mol−1.
Step 2
To the crude product mixture from Step 1 (~1.20 g, 1.50 mmol) was added 4 N HCl–dioxane (10 mL) and the resulting mixture was stirred for 1 h at room temperature. Concentration, in vacuo and pumping at high vacuum afforded 910 mg (90% yield) product as a viscous red oil: IR (NaCl): 2957, 2940, 1809, 1751, 1231, 1098, 1070, 866, 833, 775 cm−1. LCMS (5–95% gradient acetonitrile in 0.1% TFA over 10 min), single peak retention time = 5.70 min on a 30 mm column, HRMS calcd for C72H136N10O32 [M + H]+ = 603.3824 g mol−1. Observed M + H = 603.3823 g mol−1. UV/vis (100 μM in PBS) λabs = 435 nm. Fluorescence: (100 nM) λex = 449 nm and λem = 562 nm.
2.3.3. Synthesis of chromophoric crosslinker C (3,6-diamino-N2,N5-bis[N-(2-aminoethyl)-arginine amide]-pyrazine-2,5-dicarboxamide tetra TFA salt
Step 1
Synthesis of 3,6-diamino-N2,N5-bis(N-pbf-Arginine methyl ester)-pyrazine-2,5-dicarboxamide: a mixture of 3,6-diaminopyrazine-2,5-dicarboxylic acid (0.90 g, 4.5 mmol), H-Arg (pbf)-OMe·HCl (4.77 g, 9.99 mmol), EDC (1.53 g, 9.99 mmol), HOBt (1.34 g, 9.99 mmol) and TEA (726 μL, 9.99 mmol) was stirred in DMF (35 mL) for 6 h at room temperature. The reaction was concentrated in vacuo and partitioned between 125 mL EA and 100 mL saturated sodium bicarbonate. The organics were washed with 10% NaHSO4, brine, dried and concentrated to ½ volume and filtered through a plug of silica gel and the filtrate was concentrated to afford 2.4 g of a red oilglass. The crude bis-amide was taken on to the next step with no further purification.
Step 2
Synthesis of 3,6-diamino-N2,N5-bis(N-pbf-arginine)-pyrazine-2,5-dicarboxamide di-lithium salt: a solution of the product from Step 1 (2.40 g, 2.30 mmol) in THF (35 mL) was treated with a solution of lithium hydroxide (276 mg, 11.5 mmol) in water (5.0 mL). After stirring for 1 h at room temperature, HPLC analysis indicated reaction was complete. The reaction was quenched by the addition of dry ice and concentrated. This material was used in the next step without further purification.
Step 3
Synthesis of 3,6-diamino-N2,N5-bis[N-(2-boc-aminoethyl)-arginine amide]-pyrazine-2,5-di-carboxamide: a mixture of the product from Step 2 (1.00 g, 0.97 mmol), tert-butyl 2-aminoethyl-carbamate (350 mg, 2.19 mmol), EDC·HCl (420 mg, 2.19 mmol), HOBt (290 mg, 2.15 mmol) and TEA (~0.5 mL) in DMF (50 mL) was stirred at room temperature for 16 h. The reaction was concentrated and the residue was partitioned between 100 mL ethyl acetate and 100 mL saturated sodium bicarbonate. The organics were washed with 10 mL aqueous KHSO4, brine, and concentrated in vacuo and vacuum dried to afford 905 g (71% yield) of the product as a red semi-solid: MS (ESI) [M + H]+ = 1300 g mol−1; [M + Na]+ = 1323 g mol−1. This material was used in the next step without further purification.
Step 4
To the product from Step 3 (900 mg, 0.69 mmol) were added TFA (9.25 mL), water (25 μL), and triisopropyl silane (25 μL). The resulting mixture was stirred at room temperature for 72 h (convenience—over weekend). The reaction mixture was concentrated in vacuo. The residue was purified by preparative HPLC (C18, 30 × 150 mm column, 5% ACN in H2O to 95% over 12 min, 0.1% TFA) to afford 178 mg (26% yield) of the product as a red foam: IR (NaCl): 2957, 2934, 1811, 1749, 1233, 1094, 1067, 864, 831, 777 cm−1. HRMS calcd for C22H43N16O4, theoretical M+ H = 595.3648 g mol−1; observed M + H = 595.3654 g mol−1.
2.4. Preparation of shell-crosslinked nano-objects
2.4.1. Preparation of rod-shaped micelles (2)
To a 100 mL round bottom flask equipped with a magnetic stir bar were added PAA140-b-PpHS50 (93 mg, 5.7 μmol) and nanopure water (91 mL) to achieve a polymer concentration of ca. 1.0 mg mL−1. The mixture was allowed to stir at rt for 2 h. An aliquot of the solution (25 mL) was added to a 100 mL round bottom flask and diluted with nanopure water (60 mL) to achieve a final polymer concentration of ca. 0.3 mg mL−1. The solution was allowed to stir at rt overnight.
2.4.2. Preparation of spherical micelles (2)
To a 100 mL round bottom flask equipped with a magnetic stir bar was added 50 mL of PAA140-b-PpHS50 (15 mg, 0.9 μmol). The pH value was adjusted to ca. 12 by adding a pellet of NaOH to afford a clear solution. The micellization was initiated by decreasing the solution pH value to ca. 7 by adding dropwise HCl. The micelle solution was allowed to stir at rt for 12 h. Hav (average height) = 5 ± 2 nm (AFM); Dav (average diameter) = 16 ± 3 nm (TEM); Dh (hydrodynamic diameter) as measured by DLS was pH dependent—see ref. 33 for the data.
2.4.3. Preparation of SCR-As
To a 50 mL round bottom flask equipped with a magnetic stir bar was added a solution of 2 in nanopure H2O (28 mL or 21 mL, 72 μmol or 54 μmol of carboxylic acid residues). To this solution was added a solution of A (0.20 mg, 0.57 μmol (0.79 mol% relative to the acrylic acid residues) for 2% crosslinking extent; 1.0 mg, 2.8 μmol (3.9 mol% relative to the acrylic acid residues) for 6% crosslinking extent; or 2.0 mg, 5.6 μmol (7.9 mol% relative to the acrylic acid residues) for 10% crosslinking extent). The reaction mixture was allowed to stir at rt for 2 h. To this solution was added, dropwise via a syringe pump over 1 h, a solution of 1-[3′-(dimethylamino) propyl]-3-ethylcarbodiimide methiodide (EDCI): 0.40 mg, 1.4 μmol (stoichiometric) for 2% crosslinking extent; 2.1 mg, 7.2 μmol (stoichiometric) for 6% crosslinking extent; 4.3 mg, 14 μmol (stoichiometric) for 10% crosslinking extent; 0.52 mg, 1.8 μmol (2 molar excess) for 2% crosslinking extent; 2.6 mg, 8.8 μmol (2 molar excess) for 5% crosslinking extent; or 5.2 mg, 18 μmol (2 molar excess) for 9% crosslinking extent and the reaction mixture was further stirred at rt for 16 h. Finally, the reaction mixture was transferred to pre-soaked dialysis tubing (MWCO ca. 3500 Da) and dialyzed against 5 mM PBS (5 mM NaCl, pH 7.4) for a day then nanopure water for another day to remove the non-attached crosslinkers, excess small molecule starting materials and by-products, and afford aqueous solutions of shell-crosslinked cylinder, SCR-A2%, SCR-A6%, SCR-A10%, SCR-A2%, SCR-A5% or SCR-A9% (final polymer concentration: 0.30 mg mL−1, 0.30 mg mL−1 or 0.28 mg mL−1 for stoichiometric addition of EDCI and 0.22 mg mL−1, 0.23 mg mL−1 or 0.23 mg mL−1 for 2 molar excess of EDCI, respectively—where in each case, the % crosslinking was determined by UV-vis spectroscopic measurement of the amount of crosslinker remaining after purification). SCR solutions for UV-vis and fluorescence studies were further partitioned into four vials each containing 5 mM PBS (with 5 mM NaCl) at pH values of 4.6, 6.4, 7.4 and 8.4. SCRs measured 23 ± 2 nm in width and 100 nm to a micron length, by TEM.
2.4.4. Preparation of SCR-Bs
See ESI†.
2.4.5. Preparation of SCR-Cs
See ESI†.
2.4.6. Preparation of SCK-As
To a 50 mL round bottom flask equipped with a magnetic stir bar was added a solution of 2 in nanopure H2O (25 mL or 28 mL, 68 μmol or 72 μmol of carboxylic acid residues). To this solution was added a solution of A (0.19 mg, 0.54 μmol (0.79 mol% relative to the acrylic acid residues) for 2% crosslinking extent; 1.0 mg, 2.7 μmol (3.9 mol% relative to the acrylic acid residues) for 7% crosslinking extent; or 1.9 mg, 5.4 μmol (7.9 mol% relative to the acrylic acid residues) for 13% crosslinking extent). The reaction mixture was allowed to stir at rt for 2 h. To this solution was added, dropwise via a syringe pump over 1 h, a solution of 1-[3′-(dimethylamino) propyl]-3-ethylcarbodiimide methiodide (EDCI): 0.41 mg, 1.4 μmol (stoichiometric) for 2% crosslinking extent; 2.0 mg, 6.8 μmol (stoichiometric) for 7% crosslinking extent; 4.1 mg, 14 μmol (stoichiometric) for 13% crosslinking extent; 15 mg, 50 μmol (36 molar excess) for 2% crosslinking extent; 15 mg, 50 μmol (36 molar excess) for 8% crosslinking extent; 15 mg, 50 μmol (35 molar excess) for 14% crosslinking extent; 30 mg, 100 μmol (75 molar excess) for 2% crosslinking extent; 30 mg, 100 μmol (75 molar excess) for 7% crosslinking extent; or 30 mg, 100 μmol (75 molar excess) for 13% crosslinking extent and the reaction mixture was further stirred at rt for 16 h. Finally, the reaction mixture was transferred to pre-soaked dialysis tubing (MWCO ca. 3500 Da) and dialyzed against 5 mM PBS (5 mM NaCl, pH 7.4) for a day then nanopure water for another day to remove the non-attached crosslinker, excess small molecule starting materials and by-products, and afford aqueous solutions of shell-crosslinked spherical nanoparticles, SCK-A2%, SCK-A7%, SCK-A13%, SCK-A2%, SCK-A8%, SCK-A14%, SCK-A2%, SCK-A7% or SCK-A13% (final polymer concentration: 0.25 mg mL−1, 0.24 mg mL−1 or 0.24 mg mL−1 for stoichiometric addition of EDCI and 0.26 mg mL−1, 0.26 mg mL−1 or 0.26 mg mL−1 for 35 molar excess amount of EDCI and 0.27 mg mL−1, 0.27 mg mL−1 or 0.26 mg mL−1 for 75 molar excess amount of EDCI, respectively—where in each case, the % crosslinking was determined by UV-vis spectroscopic measurement of the amount of crosslinker remaining after purification). SCK solutions for UV-vis and fluorescence studies were further partitioned into four vials each containing 5 mM PBS (with 5 mM NaCl) at pH values of 4.6, 6.4, 7.4 and 8.4. SCKs measured 27 ± 3 nm by number-average distribution dynamic light scattering measurements and 23 ± 2 nm in diameter, by TEM.
2.4.7. Preparation of SCK-As with two sequential addition of stoichiometric amount of EDCI (SCK-A′)
To a 50 mL round bottom flask equipped with a magnetic stir bar was added a solution of SCK-A2%, SCK-A7% or SCK-A13% in nanopure H2O (28 mL, 64 μmol of combined carboxylic acid and amide residues). To this solution was added, dropwise via a syringe pump over 1 h, a solution of 1-[3′-(dimethylamino)propyl]-3-ethylcarbodiimide methiodide (EDCI): 0.38 mg, 1.3 μmol (stoichiometric) for 2% crosslinking extent; 1.9 mg, 6.4 μmol (stoichiometric) for 7% crosslinking extent or 3.8 mg, 13 μmol (stoichiometric) for 13% crosslinking extent. Finally, the reaction mixture was transferred to pre-soaked dialysis tubing (MWCO ca. 3500 Da) and dialyzed against 5 mM PBS (5 mM NaCl, pH 7.4) for a day then nanopure water for another day to remove excess small molecule starting materials and by-products, and afford aqueous solutions of shell-crosslinked spherical nanoparticles, SCK-A′2%, SCK-A′7% or SCK-A′13% (final polymer concentration: 0.26 mg mL−1, 0.26 mg mL−1 or 0.25 mg mL−1, respectively—where in each case, the % crosslinking was determined by UV-vis spectroscopic measurement of the amount of crosslinker remaining after purification). SCR solutions for UV-vis and fluorescence studies were further partitioned into four vials each containing 5 mM PBS (with 5 mM NaCl) at pH values of 4.6, 6.4, 7.4 and 8.4. SCKs measured 27 ± 3 nm by number-average distribution dynamic light scattering measurements and 23 ± 2 nm in diameter, by TEM.
2.4.8. Synthesis of PEO45-b-PNAS50
To a 25 mL Schlenk flask equipped with a magnetic stir bar dried with flame under N2 atmosphere were added the mPEG2k macro-CTA (0.19 g, 79 μmol) and 1,4-dioxane (5 mL). The reaction mixture was stirred for 0.5 h at rt to obtain a homogeneous solution. To this solution were added NAS (0.8 g, 4.7 mmol) and AIBN (0.76 mg, 4.7 μmol). The reaction flask was sealed and allowed to stir for 10 min at rt. The reaction mixture was degassed through several cycles of freeze–pump–thaw. After the last cycle, the reaction mixture was allowed to stir for 10 min at rt before being immersed into a pre-heated oil bath at 55 °C to start the polymerization. After 210 min, the monomer conversion reached ca. 75% by analyzing aliquots collected through 1H NMR spectroscopy. The polymerization was quenched by cooling the reaction flask with liquid N2. The polymer was purified by precipitation into 500 mL of cold diethyl ether at 0 °C three times. The precipitants were collected, washed with 100 mL of cold ether, and dried under vacuum overnight to afford the PEO45-b-PNAS50 block copolymer precursor as a yellow solid (0.68 g, 85% yield based upon monomer conversion). 1H NMR (500 MHz, DMSO-d6, ppm): δ 0.81 (t, J = 6 Hz, 3H, dodecyl CH3), 1.09 (br, 5H, CH3 and dodecyl CH2), 1.20 (br, 19H, CH3 and dodecyl CH2s), 1.30 (br, 2H, dodecyl CH2), 1.60 (t, J = 6 Hz, 2H, dodecyl CH2), 2.01 (br, PNAS backbone protons), 2.75 (NAS CH2CH2s), 3.09 (br, PNAS backbone protons), 3.20 (s, mPEG terminal OCH3), 3.47 (m, OCH2CH2O from the PEG backbone), 4.07 (br, 2H from the PEO backbone terminus connected to the ester linkage); 13C NMR (125 MHz, DMSO-d6, ppm): δ 25.2, 41.2, 69.8, 172.8. MnNMR = 10 800 Da, PDI = 1.2 (DMF GPC).
2.4.9. Synthesis of PEO45-b-PNAS50-b-PS30
To a 10 mL Schlenk flask equipped with a magnetic stir bar dried with flame under N2 atmosphere were added the PEO45-b-PNAS50 macro-CTA (0.5 g, 46 μmol), 1,4-dioxane (2.0 mL), and DMF (2.0 mL). The reaction mixture was allowed to stir for 0.5 h at rt to obtain a homogeneous solution. To this solution were added styrene (0.78 g, 7.5 mmol) and AIBN (0.41 mg, 2.5 μmol). The reaction flask was sealed and allowed to stir for 10 min at rt. The reaction mixture was degassed through several cycles of freeze–pump–thaw. After the last cycle, the reaction mixture was allowed to stir for 10 min at rt before being immersed into a pre-heated oil bath at 58 °C to start the polymerization. After 12.5 h, the monomer conversion reached ca. 18% by analyzing aliquots collected through 1H NMR spectroscopy. The polymerization was quenched by cooling the reaction flask with liquid N2. The polymer was purified by precipitation into 500 mL of cold diethyl ether at 0 °C three times. The precipitants were collected and dried under vacuum overnight to afford the block copolymer precursor as a yellow solid (0.55 g, 85% yield based upon monomer conversion). 1H NMR (500 MHz, DMSO-d6, ppm): δ 0.81 (br, dodecyl CH3), 1.10–2.40 (br, dodecyl Hs, PNAS, and PS backbone protons), 2.75 (NAS CH2CH2s), 3.15 (br, PNAS backbone protons), 3.28 (s, mPEG terminal OCH3), 3.60 (m, OCH2CH2O from the PEG backbone), 6.20–7.30 (br, Ar Hs); 13C NMR (125 MHz, DMSO-d6, ppm): δ 25.2, 41.6, 69.8, 125.7, 128.0, 145.2, 172.8. MnNMR = 13 900 Da, PDI = 1.2 (DMF GPC).
2.4.10. General procedure for self-assembly of PEO-b-PNAS-b-PS block copolymers
To a solution of PEO-b-PNAS-b-PS block copolymer in DMF (ca. 1.0 mg mL−1) was added dropwise an equal volume of nanopure H2O within 2 h via a syringe pump at a rate of 15.0 mL h−1. The mixture was further allowed to stir for 1 h at rt before used for cross-linking/functionalization reactions.
2.4.11. General procedure for cross-linking/functionalization of PEO-b-PNAS-b-PS micelles
To a solution of PEO-b-PNASb-PS micelles in DMF/H2O (v/v = 1 : 1) at rt was added dropwise over 10 min a solution of cross-linker A or B (0.1 eq., relative to the amounts of NAS residues, for nominal 20% of cross-linking) in nanopure water. The reaction mixture was allowed to stir for 48 h at rt in the absence of light. The reaction mixture was then divided into five portions (ca. 13 mL each) and transferred into pre-soaked dialysis tubing (MWCO 3500 Da) and dialyzed against 5.0 mM buffer solutions (with 5.0 mM NaCl) at pH 5.8, 6.5, 7.2, 7.9, and 8.6, respectively, for 7 days to remove DMF, unreacted crosslinkers, and the small molecule by-products to afford an aqueous solution of sc-SCK-A and B (from PEO45-b-PNAS50-b-PS30 block copolymer precursors) and lc-SCK-A and B (from PEO45-b-PNAS95-b-PS60 block copolymer precursors), respectively.
3. Results and discussion
3.1. Photophysical properties of SCRs
Block copolymers having two different compositions and three different block lengths were utilized to give rise to SCRs and SCKs with unique morphological and chemical properties. The pH-responsive diblock copolymer, PAA140-b-PpHS50, was used to create SCR precursors. Three chromophoric crosslinkers were then utilized in varying amounts to prepare SCR-A, SCR-B or SCR-C. Similarly, the spherical structural analog was created from the same block copolymer and subsequently shell cross-linked with the chromophoric crosslinker A to yield a set of SCKAs having different crosslinking extents. SCKs self-assembled from PEO45-b-PNAS50-b-PS30 and crosslinked with A or B gave rise to small core-SCK-A (sc-SCK-A) or sc-SCK-B. Likewise, PEO45-b-PNAS95-b-PS60 afforded large core-SCKs (lc-SCK-A or lc-SCK-B). These sets of nano-objects allowed for observation of pH-responsive photophysical properties due to the changes in morphology, spherical particle size or regioselective reactions within the shell region.
With the PAA140-b-PpHS50 block copolymer system, the shell crosslinking reactions involved condensation reactions between diamines of the chromophores and PAAs of the nanostructures in the presence of water-soluble carbodiimide, 1-[3′-(dimethylamino) propyl]-3-ethylcarbodiimide methiodide (EDCI). Typically, a 1 : 1 molar ratio or slight excess of carbodiimide to the aliphatic amines was added to the reaction mixture in order to form sufficient amounts of activated intermediates for intramicellar crosslinking reactions while avoiding intermicellar reactions. The chromophoric crosslinkers were based on a tetrasubstituted pyrazine ring structure that shows a strong yellowishgreen fluorescence in solution. While acylation of the terminal primary amines does not affect the emission wavelength, acylation of the anilino amine groups has been reported to cause blue-shifts in the fluorescence emission by ca. 50 nm, due to the decrease in the donor property of the amino groups.34 The chromophoric crosslinkers bear two terminal amine groups that are more reactive towards amidation of the PAAs than are the anilino amine groups on the pyrazine ring.Wehypothesized that the degree to which the amine groups would undergo reaction with the PAAs could be controlled by the amount of EDCI added to the reaction mixture during the shell crosslinking reaction. In order to assess the extent to which aromatic amines participated in the crosslinking reaction, the amounts of crosslinking chromophore loaded (2, 6 or 9% crosslinking density) as well as the EDCI loaded (stoichiometric or 2 molar excess, relative to the aliphatic amines of the crosslinker) were varied (Fig. 2). Physical mixtures ofAand rod-shaped block copolymer micelles resulted in a single fluorescence emission (Fig. 2, upper row); only when EDCI was added to the mixture did dual-emission arise from the resulting SCRs (Fig. 2, middle and lower rows).
Fig. 2.
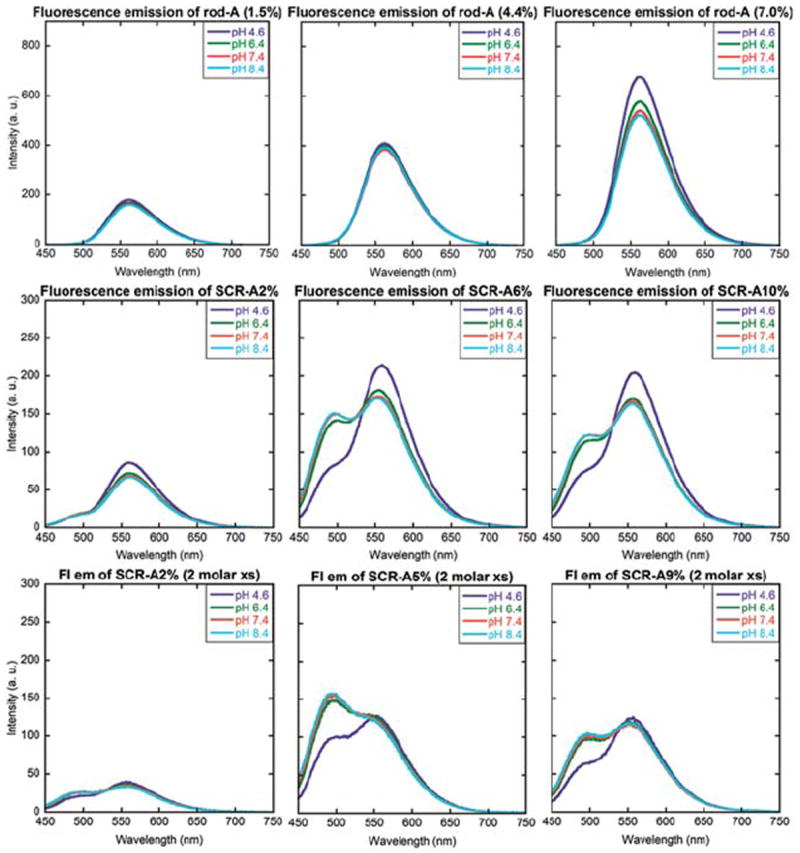
Normalized fluorescence emission spectra of SCR-A at 2%, 6% or 10% (left, middle, right) crosslinking density with the addition of 0, stoichiometric amount or 2 molar excess amount of EDCI (top, middle, bottom).
We were intrigued by the observation that the addition of stoichiometric amounts of EDCI to solutions of rod-shaped micelles with A displayed a significant amount of blue-shift. Such unique behavior (in comparison to their spherical structural analogs under identical reaction conditions, vide infra) is attributed to the linear section of the rods, which consists of densely packed polymer chains (Fig. 3). In contrast, the spherical assemblies and the rod end caps have higher curvature, which reduces the density of chain packing. From a previous literature report,34 acylation of both anilino amines (corresponding to ca. 100 nm blue-shift) of A is not very likely under such mild reaction conditions. Therefore, mono-anilino acylation (corresponding to ca. 53 nm blue-shift) is proposed to occur throughout the studies presented here. Addition of 2 molar excess amount of EDCI to the reaction mixture resulted in greater intensities of the blueshifted fluorescence emission for all SCR samples, further confirming the hypothesis that the extent to which anilino amine groups participated in the shell crosslinking reaction determined the degree of pyrazine units that experienced the blue-shift, exhibited by the resulting nanostructures. This finding represents a unique ability for the rod-shaped block copolymer micelles to create a local environment that facilitates enhanced crosscoupling reactions between the polymer chains and crosslinkers.
Fig. 3.
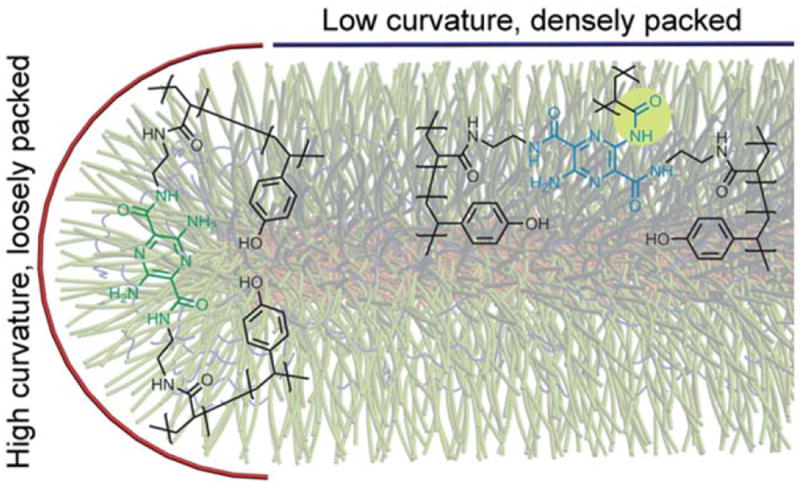
Schematic representation of SCR-A having two distinct local environments: end-caps mimic the environment found in the spherical polymer assemblies while the linear portion of the rods displays an opportunity to engage the anilino amine of the chromophoric crosslinker in acylation reactions to impart blue-shifted fluorescence emission.
We then performed a similar set of studies with chromophoric crosslinkers A, B and C, to observe that each exhibited increasing blue-shifted fluorescence emission intensity with increasing amounts of EDCI activator, and further extended the studies to allow for observation of their pH-responsive ratiometric dual-emission when incorporated into the SCR nanostructures (Fig. 4). With addition of a stoichiometric amount of EDCI, the SCR-A series displayed a moderate pH-responsiveness (I496/I560 ranging from 0.2 to 0.9). SCR-A series with an addition of 2 molar excess of EDCI exhibited an increase in absolute blue-shift and pH-sensitivity (I496/I560 ranging from 0.5 to 1.4). Likewise, the SCK-B series showed I496/I560 that ranged from 0 to 0.5, at a stoichiometric amount of EDCI, and increased from 0.1 to 1, at 2 molar excess of EDCI. Most interestingly, the SCR-C series demonstrated an absence of appreciable pH-sensitivity at a stoichiometric amount of EDCI. However, SCR-C6% exhibited a remarkable pH-sensitivity upon addition of a 2 molar excess amount of EDCI (I496/I560 ranging from 0.3 to 1), showing the most significant ratiometric response in the biologically relevant pH range of 5.0 to 7.4. In all cases, 5% to 7% crosslinked SCRs displayed the highest absolute blue-shift while 9% to 14% crosslinked SCRs suffered from self-quenching. The SCR series was also characterized by transmission electron microscopy (TEM), which revealed no apparent changes in morphology as a function of shell crosslinking density or solution pH value (Fig. 5).
Fig. 4.
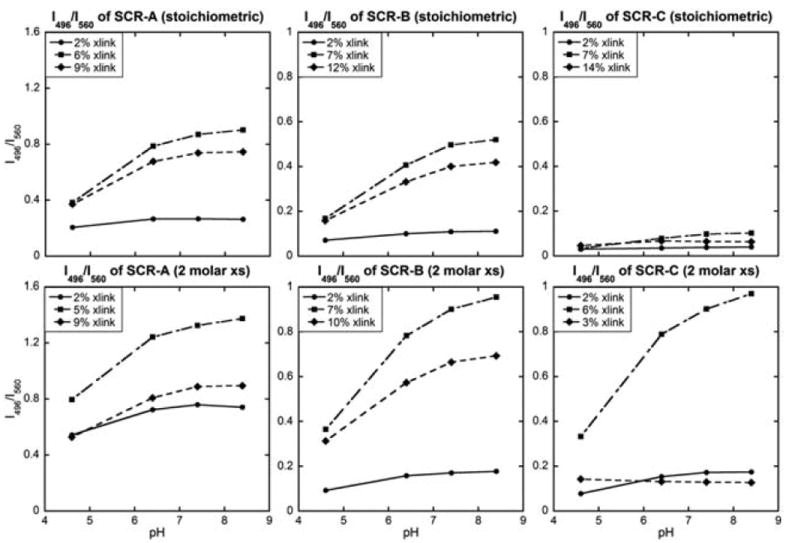
Relationship between solution pH and the fluorescence intensity ratio at two fixed wavelengths (496 nm and 560 nm) for SCR-A, SCR-B and SCR-C (left, middle, right, respectively) with stoichiometric (top) or 2 molar excess amounts of EDCI (bottom). The excitation wavelength was 433 nm.
Fig. 5.
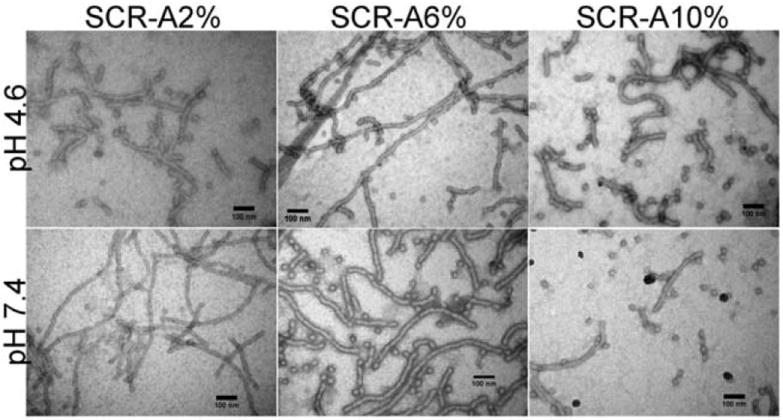
TEM images of SCR-A2%, 6% and 10% at pH 4.6 or 7.4.
3.2. Photophysical properties of SCKs
Similar experiments were conducted on SCKs, for which no blueshift was previously observed, to develop a better understanding of the chemistry involved and the influence of block copolymer morphology, by attempting to impart a blue-shift in the fluorescence emission. In this set of experiments, 35 or 70 molar excesses of EDCI were added to the spherical micelle solutions, where 70 molar excess of EDCI, relative to the amount of crosslinker aliphatic amines, was sufficient to activate essentially all carboxylic acid residues on the PAA chains. As the EDCI loading increased, the intensity of blue-shifted fluorescence emission became greater. The second highest crosslinker loading underwent the greatest relative amount of blue-shifted fluorescence (Fig. 6). In essence, we were able to achieve the photophysical consequences that were exclusive to shell-crosslinked rods within a spherical framework by manipulating the crosslinking reaction conditions with retention of morphology.
Fig. 6.
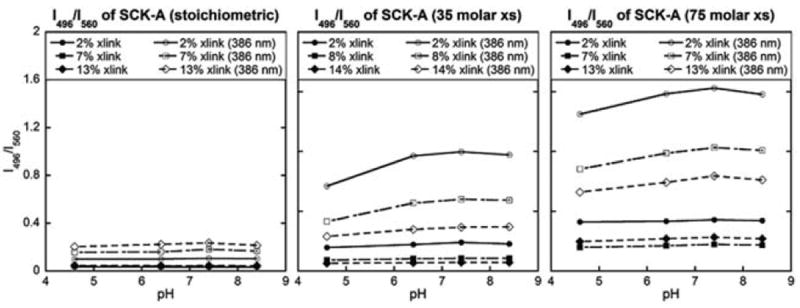
Relationship between the solution pH and the fluorescence intensity ratio at two fixed wavelengths (496 nm and 560 nm) for SCK-A with stoichiometric, 35 molar excess or 75 molar excess amount of EDCI (left, middle, right). The excitation wavelength was either 433 nm or 386 nm.
The above data indicate that the less reactive aromatic amines of the crosslinking chromophores were available for reactions with the acids after a shell crosslinking reaction with the aliphatic amines. Therefore, in this study, we applied sequential crosslinking reactions twice, each with a fixed amount of EDCI, with the intention of first crosslinking the structure and then imparting the blue-shifted fluorescence emission. We prepared a batch of SCK-A series with a stoichiometric amount of EDCI and purified the sample by dialysis to remove free crosslinking chromophores and urea by-products. To the purified batch was added an additional stoichiometric amount of EDCI to allow for reactions between unreacted amines and residual PAA units. The fluorescence emission spectra, collected as a function of pH, showed increased intensities for the blue-shifted emission after the second crosslinking reaction (Fig. 7).
Fig. 7.
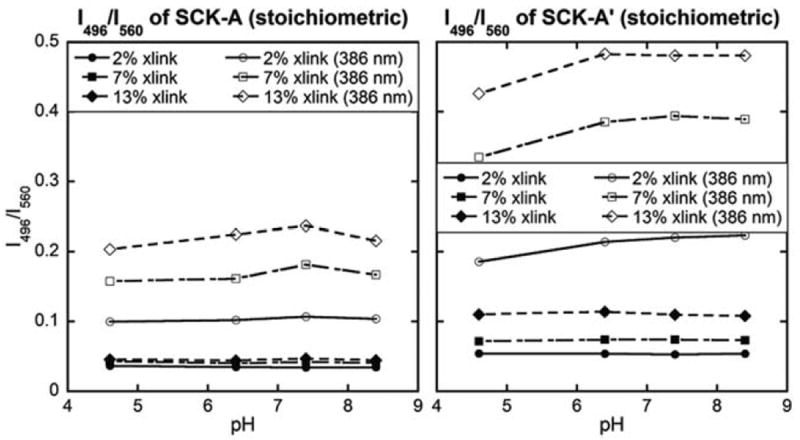
Relationship between the solution pH and the fluorescence intensity ratio at two fixed wavelengths (496 nm and 560 nm) for SCKs with one or two cycles of shell-crosslinking reactions by addition of stoichiometric amounts of EDCI during each cycle (left, right). The excitation wavelength was either 433 nm or 386 nm.
The pH-responsiveness of the ratiometric dual-emission of the SCKs, however, diminished in comparison to the SCRs. The degree to which the particles exhibited blue-shifted fluorescence emission was dependent upon the amount of EDCI added during the shell crosslinking reaction (whether at once (Fig. 6, left vs. middle vs. right plots) or consecutively in two batches (Fig. 7, left vs. right plots)), but unlike the rod-shaped isomers, these spherical analogs exhibited no pH-responsive behaviour, giving no appreciable 496 nm fluorescence emission intensity enhancement. We then utilized a unique triblock terpolymer system (PEO45-b-PNAS50-b-PS30 or PEO45-b-PNAS95-b-PS60) that was recently developed to give rise to SCKs with activated esters preinstalled within the shell domain.35 Addition of chromophoric crosslinkers to these SCK solutions resulted in direct formation of covalent bonds between the crosslinkers and the shell domain. The resulting photophysical properties revealed opposite pHresponsive dual-emission profiles (Fig. 8) than those observed for the EDCI activated rods (vide infra). In addition to having the pre-activated esters, these SCKs had a PEO corona and PS core, each differing compositionally from the EDCI-activated SCRs and SCKs.
Fig. 8.
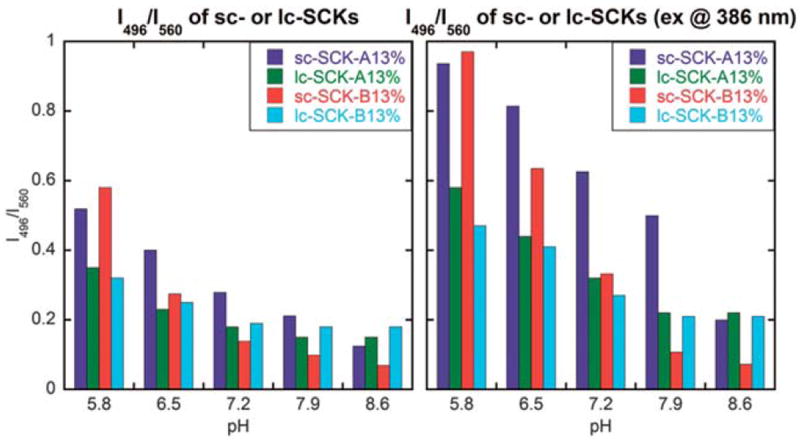
Relationship between the solution pH and the fluorescence intensity ratio at two fixed wavelengths (496 nm and 560 nm) for sc-SCK-A, sc-SCK-B, lc-SCK-A and lc-SCK-B (left, excited at 433 nm, the maximum absorbance wavelength of A and B; right, excited at 386 nm, the maximum absorbance wavelength of the nanostructures).
3.3. Photophysical properties of SCKs with pre-installation of activated esters
With the demonstration of the morphological effect (i.e., cylinders vs. spheres) on the photophysical properties of the photonic nanostructures, we continued to investigate other critical parameters of the nanoscale materials, namely, the shell composition and size of nanoparticles. As described above, the packing mode of the chromophoric crosslinkers throughout the hydrophilic domains of rod-shaped nanostructures played an important role in the fluorescence emission outputs. It can be speculated that, for spherical nanoparticles with a core–shell morphology, changes in the volumetric ratio between the hydrophilic shell (in which the chromophoric crosslinkers were accommodated) and the hydrophobic core domains could induce significant effects on tuning of their photophysical properties.
Two kinds of SCK nanoparticles, i.e., SCKs with relatively smaller and larger core domains (sc-SCK and lc-SCK, respectively), were prepared from aqueous self-assembly of PEO45-b-PNAS50-b-PS30 and PEO45-b-PNAS95-b-PS60 triblock terpolymer precursors, respectively, followed by crosslinking of the corresponding micelles with A or B at 13% of crosslinking extents, through amidation chemistry to afford sc-SCK-A13%, and sc-SCK-B13% or lc-SCK-A13%, and lc-SCK-B13%. Interestingly, although the repeat units of these two triblock terpolymers were different, we did not notice dramatic difference in the overall hydrodynamic diameter, as measured by DLS, for a majority of the constructed SCKs over the surveyed pH range (see ESI†). The structure analysis revealed that the sc-SCK had a relatively thicker PNAS domain, in comparison with lc-SCK, therefore, the chromophoric crosslinker was applied to a local environment that had more active esters during the amidation and the acylation of aromatic amines consequentially increased. Other factors, such as the steric packing modes of PNAS in the micelles with smaller core domains during crosslinking, and the resulting effects onto A and B ring moieties, after being incorporated into sc-SCKs, should also been taken into account. Ultimately, the residual NAS units underwent hydrolysis to afford spherical SCKs having PS cores and pyrazine-crosslinked PAA-based shells with PEO corona.
The increase in the fluorescence emission intensity at 496 nm relative to that at 560 nm with decreasing pH, opposite to the behavior observed for SCRs derived from EDCI-activated A or B crosslinking of PAA-b-PpHS micelles, is highly interesting. Because the sc-SCKs, lc-SCKs and SCRs give opposite pH responses, whereas the SCKs give no response, at the moment, we can only speculate that combinations of morphological differences and compositional variations between the core and corona chemistries may each play roles. Due to the low curvature and dense packing of polymer chains in the rod structures, it is expected that there would be regions near the interface that provide opportunities for significant interaction between the PpHS and PAA domains. Micellization conditions that favor formation of the rods may allow for intimate exposure of zones of shell carboxylates with zones of core phenols, possibly aided by hydrogen bonding interactions. When these types of micelles are exposed to EDCI and the chromophoric crosslinkers, standard shell crosslinking can proceed, but ester formation may also occur. Due to the close interfacial exposure of segments of the two domains in the rods, some reaction takes place on the aryl amines. Thus, both the normal 560 nm and the blue-shifted 496 nm fluorescence are observed. Also, as mentioned, there may be a significant presence of phenyl esters generated from the EDCI treatment. Closely spaced cross-linked pyrazines could then intercept the phenyl esters to form the arylamino amide derivative (Fig. 9), which could be promoted at elevated pH values, giving enhanced formation of the 496 nm emitting chromophore. Since the PEO-b-PNAS-b-PS preformed activated ester terpolymers have no phenolic groups, this type of morphology-driven arylamide formation pathway is not available. Finally, the fact that the I496/I560 ratio drops with increasing pH with the lc-SCKs and sc-SCKs (sc-SCK-A and sc-SCK-B) further supports the proposition that the phenyl esters are necessary for the increase in the blue-shifted fluorescence (Fig. 8). These SCKs have some 496 nm fluorescence due to non-selective cross-linking just as in the case of the rods. But, when the pH is increased in these systems there are no phenyl esters to further acylate the aminopyrazine groups. In this case, the existing pyrazine-arylamides that formed on EDCI treatment are probably hydrolyzed back to the desired difunctional pyrazine crosslinkers. Thus the 496 fluorescence is lost in favor of 560 nm and thus the I496/I560 ratio drops.
Fig. 9.
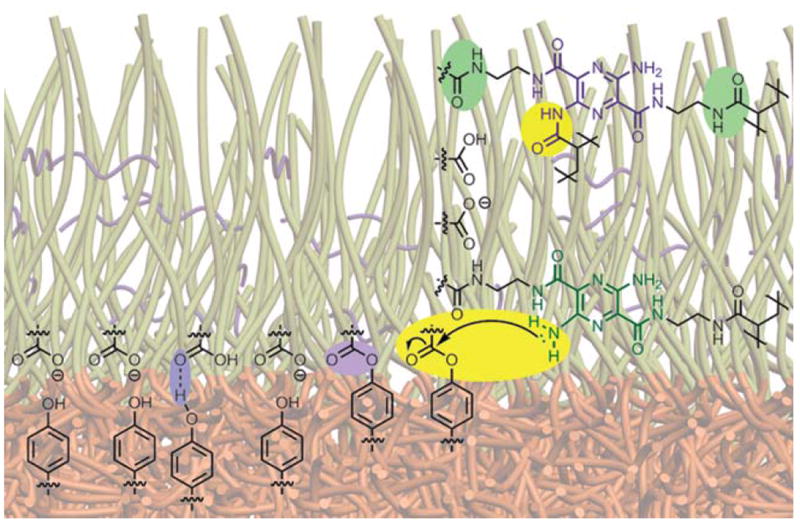
Schematic representation of the core–shell interface of SCR-A showing hydrogen bonding and phenyl esters between core and shell functionalities (highlighted in purple) to form arylamino amide derivatives at high pH (highlighted in yellow) as well as the desired crosslinking adduct (highlighted in green).
4. Conclusions
In the process of installing a chromophoric crosslinker into block copolymer nanostructures, we have made several important fundamental findings that may lead to the creation of responsive diagnostic nanomaterials. Utilization of a single parent diblock copolymer of acrylic acid and para-hydroxystyrene to create two structural isomers has allowed for studies of photophysical properties that were strictly due to the changes manifested by two different morphologies (rods vs. spheres). Rods, having more densely packed regions with a low interfacial curvature, provided unique shell domains, rich in high local concentrations of carboxylic acids for the crosslinkers to reside, while also having the possibility of formation of phenyl esters at the core–shell interface, both of which contributed towards formation of arylamide that was responsible for blue-shifted fluorescence emission. The extent to which the blue-shifting occurred was fine-tuned by the addition of varying amounts of activating carbodiimide during the shell crosslinking reaction. These nanorods underwent ratiometric pH-sensing, exhibiting increases in blue-shifted fluorescence emission with increasing pH over the range from 4.6 to 8.6, whereas the analogous spherical structures gave almost no pH response. Spherical nanoparticles derived from a different parent triblock terpolymer, having a terminal poly (ethylene oxide) chain segment, activated esters along the poly (acrylic acid) segment and polystyrene block, demonstrated opposite pH-responsiveness in photophysical properties than did the rods, presumably due to combined effects from the lack of reactive groups at the core/shell interface and differences in morphology. It is interesting that the rod-shaped nanostructures exhibited blue-shifted fluorescence emission in high pH solutions while the spherical nanoparticles showed similar behavior in low pH solutions. By having dual fluorescence emission, direct measurement of pH may be possible without the need for an internal standard or potential complications from fluorescence quenching. Given the exciting field of shape-dependent cell internalization research,28,36,37 these findings should provide further insight into designing future diagnostic tools, including diagnostic embedded therapeutics.
Supplementary Material
Acknowledgments
Financial support from Covidien is gratefully acknowledged. K. L. Wooley serves as a consultant to Covidien. N. S. Lee thanks GlaxoSmithKline for their financial support through an ACS Division of Organic Chemistry Graduate Fellowship. This work was also supported in part by the National Heart Lung and Blood Institute of the National Institutes of Health as a Program of Excellence in Nanotechnology (HHSN268201000046C) and by the Welch Foundation through the W. T. Doherty-Welch Chair in Chemistry, Grant No. A-0001. The authors thank Department of Otolaryngology, Washington University School of Medicine for access to the TEM.
Footnotes
Electronic supplementary information (ESI) available: Synthetic experimental details, UV-vis absorbance, fluorescence emission spectra, dynamic light scattering data and transmission electron microscopy images. See DOI: 10.1039/c1jm11854d
Notes and references
- 1.Dong S, Ma H, Li X, Sun M, Duan X. Anal Lett. 2004;37:2937–2948. [Google Scholar]
- 2.Niu C-G, Gui X-Q, Zeng G-M, Guan A-L, Gao P-F, Qin P-Z. Anal Bioanal Chem. 2005;383:349–357. doi: 10.1007/s00216-005-3422-y. [DOI] [PubMed] [Google Scholar]
- 3.Sun H, Scharff-Poulsen AM, Gu H, Almdal K. Chem Mater. 2006;18:3381–3384. [Google Scholar]
- 4.Allard E, Larpent C. J Polym Sci Part A: Polym Chem. 2008;46:6206–6213. [Google Scholar]
- 5.Han J, Loudet A, Barhoumi R, Burghardt RC, Burgess K. J Am Chem Soc. 2009;131:1642–1643. doi: 10.1021/ja8073374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiu Y-L, Chen S-A, Chen J-H, Chen K-J, Chen H-L, Sung H-W. ACS Nano. 2010;4:7467–7474. doi: 10.1021/nn102644u. [DOI] [PubMed] [Google Scholar]
- 7.Jin T, Sasaki A, Kinjo M, Miyazaki J. Chem Commun. 2010;46:2408–2410. doi: 10.1039/b921602b. [DOI] [PubMed] [Google Scholar]
- 8.Lee M, Gubernator NG, Sulzer D, Sames D. J Am Chem Soc. 2010;132:8828–8830. doi: 10.1021/ja101740k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lei J, Wang L, Zhang J. Chem Commun. 2010;46:8445–8447. doi: 10.1039/c0cc03310c. [DOI] [PubMed] [Google Scholar]
- 10.Peng H-S, Stolwijk JA, Sun L-N, Wegener J, Wolfbeis OS. Angew Chem Int Ed. 2010;49:4246–4249. doi: 10.1002/anie.200906926. [DOI] [PubMed] [Google Scholar]
- 11.Chan Y-H, Wu C, Ye F, Jin Y, Smith PB, Chiu DT. Anal Chem. 2011;83:1448–1455. doi: 10.1021/ac103140x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim S, Pudavar HE, Prasad PN. Chem Commun. 2006:2071–2073. doi: 10.1039/b600926c. [DOI] [PubMed] [Google Scholar]
- 13.Horvath TD, Kim G, Kopelman R, Ashkenazi S. Analyst. 2008;133:747–749. doi: 10.1039/b800116b. [DOI] [PubMed] [Google Scholar]
- 14.Doussineau T, Smaïhi M, Mohr GJ. Adv Funct Mater. 2009;19:117–122. [Google Scholar]
- 15.Zhang G, Palmer GM, Dewhirst MW, Fraser CL. Nat Mater. 2009;8:747–751. doi: 10.1038/nmat2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song Y, Cao X, Guo Y, Chen P, Zhao Q, Shen G. Chem Mater. 2009;21:68–77. [Google Scholar]
- 17.Kurishita Y, Kohira T, Ojida A, Hamachi I. J Am Chem Soc. 2010;132:13290–13299. doi: 10.1021/ja103615z. [DOI] [PubMed] [Google Scholar]
- 18.Kim H-J, Lee J, Kim T-H, Lee T-S, Kim J. Adv Mater. 2008;20:1117–1121. [Google Scholar]
- 19.Honda K, Nakata E, Ojida A, Hamachi I. Chem Commun. 2006:4024–4026. doi: 10.1039/b608684e. [DOI] [PubMed] [Google Scholar]
- 20.Whitaker JE, Haugland RP, Prendergast FG. Anal Biochem. 1991;194:330–344. doi: 10.1016/0003-2697(91)90237-n. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y-C, Ostafin A, Mizukami H. Nanotechnology. 2010;21:215501–215510. doi: 10.1088/0957-4484/21/21/215503. [DOI] [PubMed] [Google Scholar]
- 22.Lee NS, Sun G, Neumann WL, Freskos JN, Shieh JJ, Dorshow RB, Wooley KL. Adv Mater. 2009;21:1344–1348. doi: 10.1002/adma.200803053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nyström AM, Xu Z, Xu J, Taylor S, Nittis T, Stewart SA, Leonard J, Wooley KL. Chem Commun. 2008:3579–3581. doi: 10.1039/b805428b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin LY, Lee NS, Zhu J, Nyström AM, Pochan DJ, D RB, Wooley KL. J Controlled Release. 2011;152:37–48. doi: 10.1016/j.jconrel.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Hindi K, Watts KM, Taylor JB, Zhang K, Li Z, Hunstad DA, Cannon CL, Youngs WJ, Wooley KL. Chem Commun. 2010;46:121–123. doi: 10.1039/b916559b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qi K, Ma Q, Remsen EE, Clark CG, Jr, Wooley KL. J Am Chem Soc. 2004;126:6599–6607. doi: 10.1021/ja039647k. [DOI] [PubMed] [Google Scholar]
- 27.Joralemon MJ, Murthy KS, Remsen EE, Becker ML, Wooley KL. Biomacromolecules. 2004;5:903–913. doi: 10.1021/bm0344710. [DOI] [PubMed] [Google Scholar]
- 28.Zhang K, Rossin R, Hagooly A, Chen Z, Welch MJ, Wooley KL. J Polym Sci Part A: Polym Chem. 2008;46:7578–7583. doi: 10.1002/pola.23020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Becker ML, Liu J, Wooley KL. Biomacromolecules. 2005;6:220–228. doi: 10.1021/bm049551y. [DOI] [PubMed] [Google Scholar]
- 30.Lee NS, Wooley KL. Mater Matters. 2010;5:9–12. [Google Scholar]
- 31.Sun G, Lee NS, Neumann WL, Freskos JN, Shieh JJ, Dorshow RB, Wooley KL. Soft Matter. 2009;5:3422–3429. [Google Scholar]
- 32.Benoit D, Chaplinski V, Braslau R, Hawker CJ. J Am Chem Soc. 1999;121:3904–3920. [Google Scholar]
- 33.Lee NS, Li Y, Ruda CM, Wooley KL. Chem Commun. 2008:5339–5341. doi: 10.1039/b000000x/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shirai K, Yanagisawa A, Takahashi H, Fukunishi K, Matsuoka M. Dyes Pigm. 1998;39:49–68. [Google Scholar]
- 35.Sun G, Cui H, Lin LY, Lee NS, Yang C, Neumann WL, Freskos JN, Shieh JJ, Dorshow RB, Wooley KL. J Am Chem Soc. 2011;133:8534–3543. doi: 10.1021/ja200182t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Euliss LE, DuPont JA, Gratton S, DeSimone J. Chem Soc Rev. 2006;35:1095–1104. doi: 10.1039/b600913c. [DOI] [PubMed] [Google Scholar]
- 37.Zhang K, Fang H, Chen Z, Taylor J-SA, Wooley KL. Bioconjugate Chem. 2008;19:1880–1887. doi: 10.1021/bc800160b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.