

Abstract
Resistance to chemotherapy, biological and targeted therapies is an important clinical problem. Resistance can arise and/or be selected for multiple mechanisms of action. Unfortunately, acquired resistance to antitumor agents or regimens is nearly inevitable in all patients with metastatic disease. Until recently, it was believed that this resistance was unalterable and irreversible, rendering retreatment with the same or similar drugs futile in most cases. However, the introduction of epigenetic therapies, including HDAC inhibitors and DNA methyltransferase inhibitors (DNMTIs), has provided oncologists with new strategies to potentially overcome this resistance. For example, if chemoresistance is the product of multiple non-genetic alterations, which develop and accumulate over time in response to treatment, then the ability to epigenetically modify the tumor to reconfigure it back to its baseline non-resistant state, holds tremendous promise for the treatment of advanced, metastatic cancer. This minireview aims (1) to explore the potential mechanisms by which a group of small molecule agents including HDACs (entinostat and vorinostat), DNA hypomethylating agents such as the DNMTIs (decitabine (DEC), 5-azacytidine (5-AZA)) and redox modulators (RRx-001) may reprogram the tumors from a refractory to non-refractory state, (2) highlight some recent findings in this area, and (3) discuss the therapeutic potential of resensitization approaches with formerly failed chemotherapies.
Keywords: Epigenetics, HDAC inhibitors, reactive oxygen species, resensitization
INTRODUCTION
In contrast to the permanence of DNA mutations, the reversibility of epigenetic aberrations constitutes an attractive therapeutic target. Therefore, an innovative treatment approach to the widespread problem of chemoresistance is to reprogram the tumor to restore sensitivity to therapies that were initially effective, but subsequently failed secondary to the development of resistance. While conceptually challenging, it is becoming increasingly evident that chemoresistance may be reversible in some situations. Here we discuss some promising approaches to this problem and the putative mechanisms of action of the agents that may be useful for this purpose.
DNA METHYLTRANSFERASE INHIBITORS (DNMTIs)
Methyltransferases (MTases) transfer a methyl group to the C5 position of cytosine guanine dinucleotides (CpG). Of the three active DNA MTases identified (DNMT1, DNMT3A and DNMT3B), DNMT1 is the most abundant enzyme of this family found in mammals. DNA Methyl transferases require S- adenosylmethionine as the methyl group donor and result in the formation S-adenosylhomocysteine as a product of the reaction (Fig 1). Overexpressed MTases lead to CpG hypermethylation around transcriptional start sites which are associated with gene silencing and cancer [1]. MTases are important players in many processes and thus their inhibition disrupts multiple signaling pathway nodes [2]. The prototype DNMTIs are nucleoside inhibitors 5-azacytidine (5-azaCdR) and 2'-deoxy-5-azacytidine (decitabine). These cytidine analogues are incorporated into DNA and RNA during replication and transcription, inhibiting the action of MTases; this results in hypomethylation and activation of silenced genes that are important for control of pathways critical to proliferation and differentiation.
Fig. (1).

DNA cytosine-5 methylation, catalyzed by DNMT. SAM: S-adenosylmethionine, SAH: S-adenosylhomocysteine.
These agents, known as antimetabolites, (Fig. 2) were initially used as conventional cytotoxic therapies, but now, particularly at lower doses, are also recognized as DNA demethylators. FDA approval for the treatment of myelodysplasia and acute myelogenous leukemia (AML), was granted for these epigenetic modifiers in 2004 and 2006, respectively [3].
Fig. (2).

Nucleoside DNMT inhibitors.
Resensitization to previously failed therapies has been directly demonstrated with these agents, most notably in ovarian cancer, to restore sensitivity in patients with platinum-resistant disease. Matei et al., [4] administered low-dose decitabine prior to carboplatin in 17 patients with heavily-pretreated and platinum-resistant ovarian cancer in a Phase 2 clinical trial, resulting in a 35% objective response rate (RR) and progression-free survival (PFS) of 10.2 months, with nine patients (53%) free of progression at 6 months. This was encouraging, given the relatively low rate of objective responses (<10%), usually of short duration, observed in this patient population. In addition, Li et al., [5] reported a phase I/II study of 5-azacitidine and carboplatin that demonstrated durable responses (median duration of therapy 7.5 months) with an overall response rate of 13.8% and a disease control rate (partial response plus stable disease) in 45% (13/29 evaluable patients) with platinum-resistant or refractory ovarian cancer. Further confirmatory studies in this patient population are anticipated.
Juergens et al., conducted a combination phase I/II trial in extensively pretreated patients with recurrent metastatic non-small cell lung cancer with azacytidine and entinostat (see HDACi below), inhibitors of DNA methylation and histone deacetylation, respectively. Objective responses were observed, [6] and survival benefits (>1 year in approximately 20% of the patients and a median OS of 6.4 months) exceeded historical controls [3]. Interestingly, the authors did not attribute the long survival to prolonged stable disease, but rather to an “unusually robust response to subsequent cytotoxic therapies, with which the majority of patients were treated” [3]. A similar observation was also made in the Phase 1 trial of RRx-001, discussed below. Subsequent therapies in the NSCLC trial included permetrexed, docetaxel, erlotinib, an anti PD-1 monoclonal antibody, gemcitabine, irinotecan/bevacizumab, and cisplatin, suggesting that this combination of epigenetic inhibitors reverted the tumor to a less resistant phenotype, making it more widely susceptible to a variety of subsequent therapeutic agents.
The non-nucleoside DNMTIs constitute a distinct class of inhibitors that act predominately by binding the active site of MTases, without the requirement of incorporation into DNA or RNA (Fig. 3). This class of inhibitors is currently an area of active research, with researchers anticipating similar epigenetic activity to the nucleosides [7, 8], though with the promise of lower toxicity. Although preclinical studies have shown reactivation of tumor suppressor genes [9], none of these compounds have been investigated clinically. While the exact mechanisms of resensitization—and sensitization—is very poorly understood, the promiscuous demethylation activity of these DNA Methyltransferase Inhibitors, which hit multiple targets including the tumor suppressor gene p53 [10], and can affect metabolic pathways (e.g. shift the tumor cells away from glycolysis) [11, 12], may alter or reset the tumor and thereby affect the sensitivity of the tumor to a variety of subsequent or concomitant therapies.
Fig. (3).

Non-nucleoside DNMT inhibitors.
HISTONE DEACTYLATING INHIBITORS (HDACi)
The histone deactylating inhibitors (HDACs) inhibit the enzyme histone deacetylase responsible for gene silencing through hypoacetylation of histones. Histone deacetylation increases the electrostatic attraction between the positive charges of the histones and negative charges of the DNA, which ensures tight binding and renders promoter regions inaccessible to polymerases for gene transcription (Fig. 4).
Fig. (4).

Histone acetylation and repression or activation of transcription.
Cancer is linked to histone hypoacetylation, due to over-expression of HDACs, and the anti-cancer effects of HDAC inhibitors have been attributed to the restoration of the histone acetylation balance [13, 14]. However, this process is complex, involving at least six human HDAC enzymes, a broad spectrum of protein classes, multiple mechanisms that include induction of reactive oxygen species (ROS) and pleiotropic biologic effects for which the putative target is unknown or uncertain [15]. Acetylation has broad regulatory functions on histones and non-histone proteins. Substrates of nonhistone acetylation are multiple and include important cellular factors involved in cellular homeostasis such as p53, nuclear factor-κB (NF-κB), and hypoxia-inducible factor (HIF-1α) [16] which overlap with the DNA methylation inhibitors described above and RRx-001 described below. In particular the effect on p53 is highlighted for this review: the p53 tumor suppressor protein and glycolytic regulator is activated directly through deactylation [17] and indirectly through ROS-induced DNA damage [18]. HDAC inhibitors fall into a number of broad classes. These include the short chain fatty acids and the hydroxamic acids (Fig. 5) and the benzamides and cyclic tetrapeptides (Fig. 6).
Fig. (5).

HDAC Inhibitors: Short chain fatty acids and hydroxamic acids.
Fig. (6).

HDAC Inhibitors: benzamides and cyclic tetrapeptides.
Two HDAC inhibitors, vorinostat and entinostat, have been approved by the FDA for the treatment of leukemia. They have also been studied in solid tumors, as a component of combination therapy, with results suggesting the potential for a resensitization effect. While there was no significant activity in two non-small lung cancer trials, [19, 20] perhaps due, in part, to dosing considerations, a randomized phase II breast cancer trial of the aromatase inhibitor exemestane with the HDACi entinostat versus exemestane alone demonstrated significantly improved overall survival. [21] These encouraging results merit additional testing to better characterize this survival benefit, and elucidate the underlying mechanism of the improved efficacy in the combination therapy arm.
RRx-001
RRx-001 is a novel therapeutic, originating from the aerospace industry, with a chemical structure and mechanism of action that is clearly differentiated from classic cytotoxic and epigenetic agents (Fig. 7). As a redox active agent, RRx-001 inhibits tumor promotion and leads to apoptosis through the induction of oxidative and nitrosative stress in tumors that are already overburdened with free radicals leading to modulation and/or inhibition of multiple targets including HDACs and DNMTs. RRx-001 has shown promise as a novel cancer therapeutic agent in a number of cell lines and tumor models, in the absence of unacceptable off-target effects and systemic toxicity.
Fig. (7).
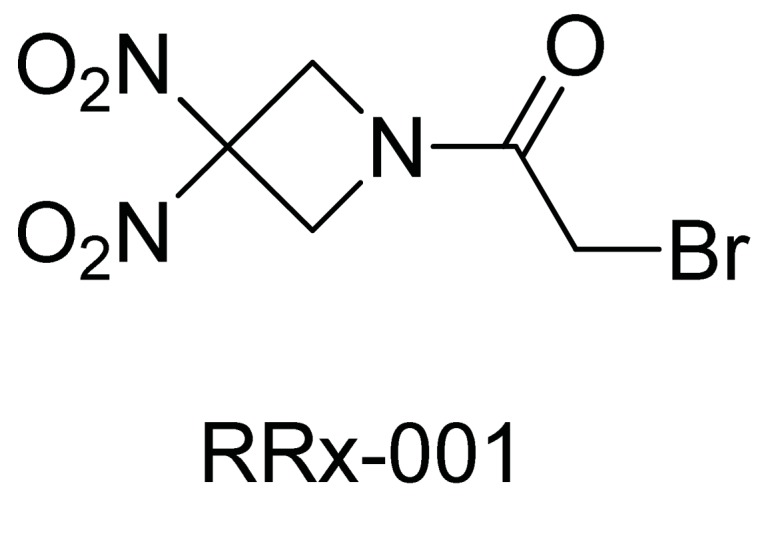
RRx-001.
In a multi-center Phase 1 dose escalation study, RRx-001 demonstrated an acceptable safety profile at the maximal dose of 83 mg/m2 and evidence of anti-cancer activity, including one partial response and disease stabilization in nine patients lasting at least 16 weeks. At 16.8 months, 50% of patients were still alive. Similar to the observation in the azacytidine and entinostat combination NSCLC trial, there was prolonged overall survival that greatly exceeded that which was expected on the basis of the regorafenib CORRECT trial [22], in which the median OS was 6.4 months. This increased survival is attributed, in part, to robust clinical responses with subsequent post-progression therapies, suggesting that tumors and/or the tumor microenvironment was modified leading to resensitization of tumors to previously failed therapies. It is believed that RRx-001- mediated epigenetic modification of the tumors may have played a role in this apparent resensitization.
RRx-001- enhanced susceptibility to chemotherapy was observed in at least 5 patients, 4 with colorectal cancer and 1 with NSCLC, with drugs to which the patients were previously resistant. A case report was recently published describing the resensitization of two colorectal cancer patients to FOLFIRI, which both patients had previously failed [23]. RRx-001 has multiple mechanisms of action, including induction of redox and metabolic stress on the tumor, which can disrupt multiple cellular pathways via sulfhydryl oxidation, Nrf2 activation and p53 upregulation, PARP cleavage, HIF-1 alpha and inhibition of G6PD, a central substrate of the Pentose Phosphate Pathway (RadioRx, unpublished data). Based on the potential for episensitization a number of Phase 2 studies in multiple tumor types including colorectal, breast, brain and liver will be initiated.
EPIGENETIC EFFECTS AND OXIDATIVE STRESS
The influence of ROS on epigenetic events has been studied extensively, particularly in the field of aging [24] and age-related illnesses, supporting a direct and indirect causal relationship between oxidative stress and epigenetic changes. Indirectly, oxidative stress leads to glutathione depletion and impairment of the one carbon cycle: DNA methylation and histone methylation depend upon the availability of methyl groups from S-adenosylmethionine (SAM). An increase in cysteine synthesis for GSH regeneration leads to a depletion of methionine, resulting in decreased synthesis of SAM, and an overall defect in methylation reactions. Oxidative stress also directly inactivates HDACs [25]. HDAC inhibition results in histone acetylation and a more open or permissive chromatin conformation, rendering DNA more susceptible to ROS damage [26]. Indeed, HDAC inhibitors are associated with ROS generation [27, 28], resulting in DNA damage induction and repair, as measured through increased gamma-H2AX expression [29], thereby fueling a self-propelling loop of ROS-mediated cytotoxicity followed by repair and resolution processes (Fig. 8).
Fig. (8).
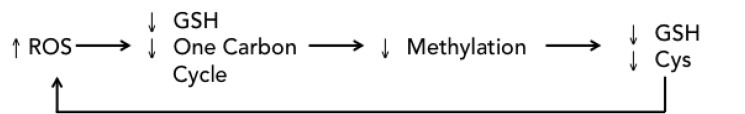
ROS epigenetic cycle.
HDAC inhibitors increase the activity of DNA synthesis inhibitors such as fludarabine. The widely used anti epileptic and mood stabilizer, valproic acid (VPA), a small, branched fatty acid, has been investigated as an HDAC inhibitor both preclinically and clinically [30]. Preclinically, it enhanced fludarabine-induced apoptosis via mitochondria-mediated apoptosis, which in turn was associated with ROS generation, intracellular ATP depletion and reduced levels of ATM, an important protein in DNA damage response [31]. In addition, VPA increased the cytotoxicity of the aurora kinase inhibitor, VE465, as evidenced by higher levels of cleaved PARP. Ovarian cells were particularly sensitive [32].
Clinically, in two studies in patients with high-risk acute myeloid leukemia or myelodysplastic syndrome, the combination of VPA and a demethylating agent demonstrated higher OS and CR rates compared to the combination of VPA and chemotherapy, such as all-trans retinoic acid ATRA [33, 34]. Other studies in have similarly reported improvements in survival associated with epigenetic changes [35-37]. As an inhibitor of HDACs, which collectively deacetylate a variety of non-histone proteins, VPA has pleiotropic effects that are linked to increased levels of reactive oxygen species [38]. Leukemia is particularly sensitive to oxidative damage [39]. These results suggest that the chemosensitizing effect of VPA is associated with oxidative stress and induction of p53, with a resulting inhibition of glycolysis and improved therapeutic efficacy.
We hypothesize that RRx-001 has epigenetic modifying effects because it significantly affects the intracellular redox status of tumor cells [40]; the enzymes responsible for histone methylation and DNA methylation rely on critical redox intermediates derived from metabolic pathways, potentially leading to changes in gene expression. Furthermore, RRx-001 induces double-stranded DNA breaks secondary to the generation of ROS and RNS, and affects a variety of signaling pathways, which can impact the activity of p53 and p21, and deregulate tumor metabolism and cellular energetics. In this way, oxidative stress, through the alteration of DNA-methylation and histone modifications, may contribute to epigenetic changes. This is currently an area of active laboratory and clinical investigation, with several phase 2 trials planned with RRx-001 both as a single agent and in combination with chemotherapy and radiation therapy.
CONCLUSIONS AND FUTURE PERSPECTIVES
The ability to modulate gene expression of tumors by either epigenetic modification or other mechanisms of action in order to resensitize them to previously failed therapies presents an entirely new paradigm for the treatment of cancer. As discussed here, data, in some cases preliminary, suggest that drugs such as HDACs, DNMTIs and RRx-001 may have the ability to resensitize or ‘epi-sensitize’ tumors to formerly failed chemotherapy drugs/regimens and that this episensitization is dependent upon the disruption of the expression or activity of an array of cellular targets, some of which may affect cellular energetics. These targets include p53, PARP, NFκB, and HIF-1α. While the specific molecular mechanisms involved in episensitization are admittedly unclear, multiplicity may be more important than specificity i.e. the simultaneous inhibition of multiple pharmacologic targets that are crucial to cellular metabolism and energy status. In particular, the anti-cancer agents described in this review all activate and upregulate p53, which can have numerous effects, including its function as a gatekeeper at the crossroads of metabolism in cancer cells, with potential secondary effects on multiple other targets as well as the tumor glycolytic rate [12].
Since, RRx-001, HDAC inhibitors and DNA methyltransferase inhibitors all disrupt multiple signaling pathways, perhaps this lack of specificity (i.e. affecting multiple pathways and cellular functions) contributes to their ability to resensitize tumor cells to treatments [3] to which they have become resistant. This is an active area of investigation, the results of which may provide promising new therapeutic strategies for enhancing the responsiveness of patients’ tumors to subsequent therapies following progression of disease. This new paradigm has near term translational applicability, with the potential to impact the natural history of a variety of refractory and metastatic cancers.
ACKNOWLEDGEMENTS
Declared none.
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflict of interest.
REFERENCES
- 1.Huang TH, Perry MR, Laux DE. Methylation profiling of CpG islands in human breast cancer cells. Hum. Mol. Genet. 1999;8(3 ):459–470. doi: 10.1093/hmg/8.3.459. [DOI] [PubMed] [Google Scholar]
- 2.Robertson KD. DNA methylation, methyltransferases, and cancer. Oncogene. 2001;20(24 ): 3139–3155. doi: 10.1038/sj.onc.1204341. [DOI] [PubMed] [Google Scholar]
- 3.Azad N, Zahnow CA, Rudin CM, Baylin SB. The future of epigenetic therapy in solid tumours-lessons from the past. Nat. Rev. Clin. Oncol. 2013;10(5 ): 256–266. doi: 10.1038/nrclinonc.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matei D, Fang F, Shen C, Schilder J, Arnold A, Zeng Y, Berry WA, Huang T, Nephew KP. Epigenetic resensitization to platinum in ovarian cancer. Cancer Res. 2012;72(9 ):2197–205. doi: 10.1158/0008-5472.CAN-11-3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Y, Hu W, Shen DY, Kavanagh JJ, Fu S. Azacitidine enhances sensitivity of platinum-resistant ovarian cancer cells to carboplatin through induction of apoptosis. Am. J. Obstet. Gynecol. 2009;200(2 ): 177. e1–9. doi: 10.1016/j.ajog.2008.08.030. [DOI] [PubMed] [Google Scholar]
- 6.Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, Sebree R, Rodgers K, Hooker CM, Franco N, Lee B, Tsai S, Delgado IE, Rudek MA, Belinsky SA, Herman JG, Baylin SB, Brock MV, Rudin CM. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2012;1(7 ): 598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asgatay S, Champion C, Marloie G, Drujon T, Senamaud-Beaufort C, Ceccaldi A, Erdmann A, Rajavelu A, Schambel P, Jeltsch A, Lequin O, Karoyan P, Arimondo PB, Guianvarc'h D. Synthesis and evaluation of analogues of N-phthaloyl-l-tryptophan (RG108):as inhibitors of DNA methyltransferase 1. J. Med. Chem. 2014;57(2 ):421–434. doi: 10.1021/jm401419p. [DOI] [PubMed] [Google Scholar]
- 8.Valente S, Liu Y, Schnekenburger M, Zwergel C, Cosconati S, Gros C, Tardugno M, Labella D, Florean C, Minden S, Hashimoto H, Chang Y, Zhang X, Kirsch G, Novellino E, Arimondo PB, Miele E, Ferretti E, Gulino A, Diederich M, Cheng X, Mai A. Selective non-nucleoside inhibitors of human DNA methyltransferases active in cancer including in cancer stem cells. J. Med. Chem. 2014;57(3 ): 701–713. doi: 10.1021/jm4012627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brueckner B, Garcia Boy R, Siedlecki P, Musch T, Kliem HC, Zielenkiewicz P, Suhai S, Wiessler M, Lyko F. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 2005;65: 6305–6311. doi: 10.1158/0008-5472.CAN-04-2957. [DOI] [PubMed] [Google Scholar]
- 10.Karpf AR, Moore BC, Ririe TO, Jones DA. Activation of the p53 DNA damage response pathway after inhibition of DNA methyltransferase by 5-aza-2'-deoxycytidine. Mol. Pharmacol. 2001;59: 751–757. [PubMed] [Google Scholar]
- 11.Karahoca M, Momparler RL. Pharmacokinetic and pharmacodynamic analysis of 5-aza-2'-deoxycytidine (decitabine) in the design of its dose-schedule for cancer therapy. Clin. Epigenetics. 2013;5: 3. doi: 10.1186/1868-7083-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puzio-Kuter AM. The Role of p53 in Metabolic Regulation. Genes Cancer. 2011;2: 385–391. doi: 10.1177/1947601911409738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang J, Plass C, Gerhauser C. Cancer chemoprevention by targeting the epigenome. Curr. Drug Targets. 2010;12: 1925–1956. doi: 10.2174/138945011798184155. [DOI] [PubMed] [Google Scholar]
- 14.Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors overview and perspectives. Mol. Cancer Res. 2007;5: 981–989. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- 15.Pediconi N, Guerrieri F, Vossio S, Bruno T, Belloni L, Schinzari V, Scisciani C, Fanciulli M, Levrero M. HSirT1-dependent regulation of the PCAF-E2F1-p73 apoptotic pathway in response to DNA damage. Mol. Cell Biol. 2009;29:1989–1998. doi: 10.1128/MCB.00552-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh BN, Zhang G, Hwa YL, Li J, Dowdy SC, Jiang SW. Nonhistone protein acetylation as cancer therapy targets. Expert Rev. Anticancer Ther. 2010;10: 935–954. doi: 10.1586/era.10.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90: 95–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 18.Liu B, Chen Y, St Clair DK. ROS and p53: A versatile partnership. Free Radic. Biol. Med. 2008;44: 1529–1535. doi: 10.1016/j.freeradbiomed.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reguart N, Rosell R, Cardenal F, Cardona AF, Isla D, Palmero R, Moran T, Rolfo C, Pallares MC, Insa A, Carcereny E, Majem M, De Castro J, Queralt C, Molina MA, Taron M. Phase I/II trial of vorinostat (SAHA) and erlotinib for non-small cell lung cancer (NSCLC) patients with epidermal growth factor receptor (EGFR) mutations after erlotinib progression. Lung Cancer. 2014;84(2 ):161–167. doi: 10.1016/j.lungcan.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 20.Witta SE, Jotte RM, Konduri K, Neubauer MA, Spira AI, Ruxer RL, Varella-Garcia M, Bunn PA. Randomized phase II trial of erlotinib with and without entinostat in patients with advanced non-small-cell lung cancer who progressed on prior chemotherapy. J. Clin. Oncol. 2012;30:2248–2255. doi: 10.1200/JCO.2011.38.9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yardley DA, Ismail-Khan RR, Melichar B, Lichinitser M, Munster PN, Klein PM, Cruickshank S, Miller KD, Lee MJ, Trepel JB. Randomized phase II, double-blind, placebocontrolled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J. Clin. Oncol. 2013;31: 2128–2135. doi: 10.1200/JCO.2012.43.7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, Humblet Y, Bouche O, Mineur L, Barone C, Adenis A, Tabernero J, Yoshino T, Lenz H. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2012;381:303–312. doi: 10.1016/S0140-6736(12)61900-X. [DOI] [PubMed] [Google Scholar]
- 23.Reid T, Dad S, Korn R, Oronsky B, Knox S, Scicinski J. Two case reports of resensitization to previous chemotherapy with the novel hypoxia-activated hypomethylating anticancer agent RRx-001 in metastatic colorectal cancer patients. Case Rep. Oncol. 2014;7: 79–85. doi: 10.1159/000358382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cencioni C, Spallotta F, Martelli F, Valente S, Mai A, Zeiher AM, Gaetano C. Oxidative stress and epigenetic regulation in ageing and age-related diseases. Int. J. Mol. Sci. 2013;14: 17643–17663. doi: 10.3390/ijms140917643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Afanas' ev I. Nucleophilic mechanism of ROS/RNS signaling in cancer epigenetic modifications. Am. J. Biomed. Sci. 2012;4: 285–306. [Google Scholar]
- 26.Rajendran P, Ho E, Williams DE, Dashwood RH. Dietary phytochemicals, HDAC inhibition, and DNA damage/repair defects in cancer cells. Clin. Epigenetics. 2012;3(1 ):1–4. doi: 10.1186/1868-7083-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug. Discov. 2006;5: 769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 28.Rosato RR, Grant S. deacetylase inhibitors: Insights into mechanisms of lethality. Exp. Opin. Ther. Targets. 2005;9: 809–824. doi: 10.1517/14728222.9.4.809. [DOI] [PubMed] [Google Scholar]
- 29.Rosato RR, Almenara JA, Maggio SC, Coe S, Atadja P, Dent P, Grant S. Role of histone deacetylase inhibitor-induced reactive oxygen species and DNA damage in LAQ-824/fludarabine antileukemic interactions. Mol. Cancer Ther. 2008;7: 3285–3297. doi: 10.1158/1535-7163.MCT-08-0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duenas-Gonzalez A, Candelaria M, Perez-Plascencia C, Perez-Cardenas E, De la Cruz-Hernandez E, Herrera LA. Valproic acid as epigenetic cancer drug Preclinical, clinical and transcriptional effects on solid tumors. Cancer Treat. Rev. 2008;34: 206–222. doi: 10.1016/j.ctrv.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 31.Yoon J, Ishdorj G, Graham B, Johnston J, Gibson S. Valproic acid enhances fludarabine-induced apoptosis mediated by ROS and involving decreased AKT and ATM activation in B-cell-lymphoid neoplastic cells. Apoptosis. 2014;19: 191–200. doi: 10.1007/s10495-013-0906-7. [DOI] [PubMed] [Google Scholar]
- 32.Li Y, Liu T, Ivan C, Huang J, Shen DY, Kavanagh JJ, Bast RC Jr, Fu S, Hu W, Sood AK. Enhanced cytotoxic effects of combined valproic acid and the aurora kinase inhibitor VE465 on gynecologic cancer cells. Front. Oncol. 2013;3: 1–58. doi: 10.3389/fonc.2013.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raffoux E, Cras A, Recher C, Boelle PY, De Labarthe A, Turlure P, Marolleau JP, Reman O, Gardin C, Victor M, Maury S, Rousselot P, Malfuson JV, Maarek O, Daniel M. Phase 2 clinical trial of 5-azacitidine, valproic acid, and all-trans retinoic acid in patients with high-risk acute myeloid leukemia or myelodysplastic syndrome. Oncotarget. 2011: 34–42. doi: 10.18632/oncotarget.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soriano AO, Yang H, Faderl S, Estrov Z, Giles F, Ravandi F, Cortes J, Wierda WG, Ouzounian S, Quezada A, Pierce S, Estey EH, Issa JP, Kantarjian HM, Garcia-Manero G. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood. 2007;110: 2302–2308. doi: 10.1182/blood-2007-03-078576. [DOI] [PubMed] [Google Scholar]
- 35.Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, Yang H, Rosner G, Verstovsek S, Rytting M, Wierda WG, Ravandi F, Koller C, Xiao L, Faderl S, Estrov Z, Cortes J, O'Brien S, Estey E, Bueso-Ramos C, Fiorentino J, Jabbour E, Issa JP. Phase 1/2 study of the combination of 5-aza-2'-deoxycytidine with valproic acid in patients with leukemia. Blood. 2006;108: 3271–3279. doi: 10.1182/blood-2006-03-009142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maslak P, Chanel S, Camacho LH, Soignet S, Pandolfi PP, Guernah I, Warrell R, Nimer S. Pilot study of combination transcriptional modulation therapy with sodium phenylbutyrate and 5-azacytidine in patients with acute myeloid leukemia or myelodysplastic syndrome. Leukemia. 2006;20: 212–217. doi: 10.1038/sj.leu.2404050. [DOI] [PubMed] [Google Scholar]
- 37.Blum W, Klisovic RB, Hackanson B, Liu Z, Liu S, Devine H, Vukosavljevic T, Huynh L, Lozanski G, Kefauver C, Plass C, Devine SM, Heerema NA, Murgo A, Chan KK, Grever MR, Byrd JC, Marcucci G. Phase I study of decitabine alone or in combination with valproic acid in acute myeloid leukemia. J. Clin. Oncol. 2007;25:3884–3891. doi: 10.1200/JCO.2006.09.4169. [DOI] [PubMed] [Google Scholar]
- 38.Boudadi E, Stower H, Halsall JA, Rutledge CE, Leeb M, Wutz A, O'Neill LP, Nightingale KP, Turner BM. The histone deacetylase inhibitor sodium valproate causes limited transcriptional change in mouse embryonic stem cells but selectively overrides Polycomb-mediated Hoxb silencing. Epigenetics Chromatin. 2013;6:11. doi: 10.1186/1756-8935-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pei S, Minhajuddin M, Callahan KP, Balys M, Ashton JM, Neering SJ, Lagadinou E. Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J. Biol. Chem. 2013;288:33542–33558. doi: 10.1074/jbc.M113.511170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ning S, Bednarski M, Oronsky B, Scicinski J, Saul G, Knox SJ. Dinitroazetidines are a novel class of anticancer agents and hypoxia-activated radiation sensitizers developed from highly energetic materials. Cancer Res. 2012;72: 2600–2608. doi: 10.1158/0008-5472.CAN-11-2303. [DOI] [PubMed] [Google Scholar]