

Abstract
Chromosomal translocations can promote cancers by eliciting the expression of fusion genes with oncogenic activity. The identification of translocations affecting RAF genes in prostate and gastric cancers and melanoma provides compelling evidence for the key role of RAF signaling in a subset of these cancers and suggests possible new avenues for personalized cancer therapy.
The identification of molecular changes that occur specifically in cancer cells is an essential step toward development of targeted cancer therapies. Unlike traditional cancer therapy, which typically kills rapidly dividing cells, targeted cancer therapies interfere with specific proteins involved in tumor maintenance. The archetype for such therapies is the drug imatinib, targeted to the BCR-ABL fusion protein, which is synthesized in chronic myelogenous leukemia as a result of a translocation between chromosomes 9 and 22—the so-called Philadelphia chromosome.
In this issue of Nature Medicine, Palanisamy et al.1 identify translocations of either BRAF or RAF1—two members of the family of RAF genes—in a small percentage of prostate and gastric cancers and melanoma1. This work expands the portfolio of diseases in which oncogenic RAF signaling is implicated in cancer initiation or progression. Furthermore, the data may have implications for personalized therapy with agents that target the RAF signaling pathway2 (Fig. 1) in a subset of patients with cancer.
Figure 1.
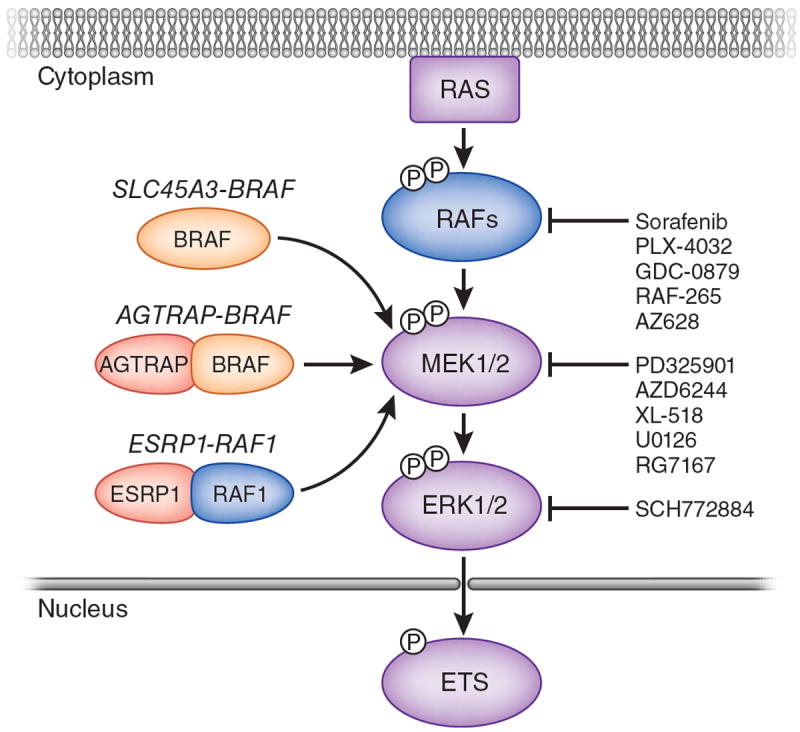
RAF fusion genes in prostate and gastric cancer and melanoma. The RAF family of protein kinases are activated by RAS GTPases. In turn, RAF kinases activate the protein kinases MEK1 and MEK2 by serine phosphorylation at two sites within the activation segment of the kinase domain. MEK1 and MEK2 activate the ERK MAP kinases by phosphorylation of a threonine and a tyrosine residue in the activation segment of the kinase domain. Among the plethora of ERK1/2 MAP kinase targets are a number of ETS family transcription factors, which are also implicated in prostate carcinogenesis, as ETS genes are frequent targets of chromosome translocations12,13. Palanisamy et al.1 identified fusion transcripts resulting from chromosome translocations that lead to expression of three fusion genes: SLC45A3-BRAF or ESRP1-RAF2 in prostate cancer or type 1 angiotensin II receptor–associated protein (AGTRAP) BRAF in gastric cancer2. The SCL45A3-BRAF fusion mRNA encodes a 329–amino acid protein that comprises only a C-terminal fragment of BRAF. By contrast, the ESRP1-RAF2 and AGTRAP-BRAF fusions encode proteins with substantial contribution of N-terminal sequences from the RAF fusion partner. The common feature of the proteins encoded by these fusion mRNAs is the presence of a RAF kinase domain that is likely to be constitutively activated. A large number of pharmacological inhibitors of the RAF→MEK→ERK MAP kinase pathway are being developed, a subset of which are listed in the figure2.
RAF genes first came to light as retroviral oncogenes with the potential to transform a broad range of cell types3. Subsequent research identified three mammalian RAF genes (ARAF, BRAF and RAF1) encoding serine-specific protein kinases that are involved in a signaling pathway that controls many cellular functions. Specifically, RAF proteins transduce signals from activated RAS GTPases to the extracellular signal–regulated kinase-1/2 (ERK1/2) mitogen-activated protein kinase (MAP) kinases by phosphorylating the dual-specificity protein kinases ERK kinase-1 (MEK1) and MEK2 (Fig. 1). Although the role of mutated RAS in human cancer was first identified in 1982, definitive evidence that RAF genes could be mutated to become human oncogenes was only reported in 2002 through the ‘smoking gun’ of somatic gene mutation in BRAF in a wide range of human malignancies4.
The most common mechanism of oncogenic activation of BRAF is point mutation. Although more than 60 point mutations in BRAF have been identified to date, a single T→A transversion at position 1799 in exon 15, leading to expression of the constitutively active BRAFV600E protein, accounts for about 90% of all BRAF point mutations found in human cancers4. The generation of mouse models allowing for conditional expression of oncogenic BRAFV600E recapitulate key aspects of the genetics and pathophysiology of human cancers5,6, provided direct evidence for the role of oncogenic BRAFV600E in cancer initiation. Moreover, early-stage clinical trials of pharmacological inhibitors of RAF kinases have reported striking clinical responses in advanced metastatic melanoma, a disease that has defied the best efforts of medical oncology for many years7,8, suggesting that BRAFV600E is also important in cancer maintenance.
Although point mutations are most common, RAF genes can be mutationally activated by chromosome translocations, as observed in thyroid cancer and pilocytic astrocytoma9-11. Chromosome translocations are also a common feature of prostate cancers, but in these tumors translocations typically affect members of the ETS family of transcription factors, which are downstream effectors of RAF signaling (Fig. 1)12,13. However, because transcription factors are hard to target with pharmacological agents, Palanisamy et al.1 searched for chromosome translocations involving potentially ‘druggable’ targets in prostate cancer.
The strategy they used was to sequence fragments of cDNA generated from prostate cancer–derived mRNAs—a method referred to as paired-end transcriptome sequencing—to identify fusion mRNAs resulting from chromosome translocations. To discriminate authentic fusion mRNAs from the false positives that such strategies generate, they developed a prioritization scoring system and validated the system with mRNA from cells known to carry ETS gene chromosome translocations. With this approach, Palanisamy et al.1 identified fusion transcripts involving either the BRAF gene or RAF1 gene in two prostate cancer specimens.
The BRAF fusion mRNA resulted from a translocation involving the SLC45A3 gene (encoding solute carrier family 45, member 3) on chromosome 1 and BRAF on chromosome 7. SLC45A3 is a known androgen-regulated, prostate-specific gene that probably confers androgen regulation and prostate-specific expression on the SLC45A3-BRAF fusion gene. The SLC45A3-BRAF fusion mRNA initiates translation at codon 441 of the normal BRAF mRNA and encodes a 329–amino acid C-terminal fragment of BRAF that has constitutive kinase activity. The fusion mRNA does not encode any portion of the SLC45A3 protein.
For the CRAF translocation, gene sequences encoding epithelial splicing regulatory factor-1 (ESRP1) located on chromosome 8 are fused to CRAF on chromosome 3, resulting in the expression of two different fusion mRNAs. The first transcript contains exons 1–13 of the ESRP1 gene fused to exons 6–17 of CRAF and is predicted to encode a 1,060–amino acid protein, of which 606 amino acids are derived from ESRP1 and the remainder from the C terminus of CRAF. Unlike SLC45A3, ESRP1 expression is neither androgen regulated nor prostate specific. It is also not known whether ESRP1 coding sequences have a role in any of the oncogenic properties of the ESRP1-CRAF fusion protein. In contrast, the authors’ data suggest that the kinase activity of CRAF is essential for any oncogenic effects of the ESRP1-CRAF fusion protein1.
Notably, the reciprocal fusion transcript is predicted to encode a 269–amino acid protein in which amino acids 1–194, encompassing the Ras-binding domain and conserved region-1 of RAF1, are fused to the C-terminal 75 amino acids of ESRP1. It has previously been shown that expression of an N-terminal fragment of CRAF encompassing the Ras-binding domain and conserved region-1 has a dominant inhibitory activity on RAS-regulated signaling, owing to the ability of these fragments to bind and sequester active GTP-bound RAS proteins14. However, the authors did not test whether this fusion protein has such effects in cells1.
The authors independently validated the presence of these fusion genes, identified by paired-end transcriptome sequencing strategy, by using fluorescence in situ hybridization and immunoblotting techniques. They then applied these two technologies to a larger panel of human prostate cancer specimens, detecting translocations in BRAF in six of 349 specimens and in RAF1 in four of 450 specimens—a frequency of RAF translocations of about 1% (ref. 1). Interestingly, prostate tumors carrying RAF gene aberrations tended not to have ETS gene translocations and also had more aggressive characteristics than tumors with ETS gene translocations.
By using either fluorescence in situ hybridization or paired-end sequencing technologies, the authors also detected BRAF translocations in two of 105 gastric cancers and singleton BRAF or RAF1 rearrangements in melanoma1. In the gastric cancer case, the fusion transcript comprised exons 1–5 of AGTRAP, a gene that encodes a type 1 angiotensin II receptor–associated protein, fused to exons 8–18 of BRAF. The fusion mRNA encodes a 597–amino acid protein encompassing the BRAF kinase domain.
No RAF gene alterations were detected in breast, endometrial or liver tumors. Moreover, no BRAF exon 15 point mutations were detected in 274 prostate cancers, a result consistent with a previous study15.
Whereas the SLC45A3-BRAF fusion elicited tumorigenic conversion of NIH 3T3 mouse fibroblast cells, surprisingly, the ESRP1-CRAF fusion failed to do so. However, in a benign immortalized prostate epithelial cell line (RWPE cells), both SLC45A3-BRAF and ESRP1-CRAF fusions promoted cell proliferation, migration and modest tumor growth, which were sensitive to inhibition by pharmacological inhibitors of RAF (sorafenib) or MEK (U0126). The oncogenic potential of the AGTRAP-BRAF fusion was not tested.
The finding of mutated RAF genes in prostate cancer is consistent with a previous observation that oncogenic BRAFV600E can initiate prostate cancer in mouse models16 and may have major implications for therapy. Although RAF gene translocations occur in only 1% to 2% of prostate cancers, this frequency may reflect up to 5,000 individuals who are identified with this disease every year in the US. Given reports that BRAFV600E-driven cancers are exquisitely sensitive to the antitumor effects of pharmacologic RAF or MEK inhibition5,6,17, it will probably become important to screen individuals with prostate cancer to identify those whose cancers are maintained by RAF signaling, so that their therapy can be appropriately tailored to the underlying genetic damage that maintains the disease.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Palanisamy N, et al. Nat Med. 2010;16:793–798. doi: 10.1038/nm.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khazak V, Astsaturov I, Serebriiskii IG, Golemis EA. Expert Opin Ther Targets. 2007;11:1587–1609. doi: 10.1517/14728222.11.12.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wellbrock C, Karasarides M, Marais R. Nat Rev Mol Cell Biol. 2004;5:875–885. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- 4.Davies H, et al. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 5.Dankort D, et al. Nat Genet. 2009;41:544–552. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dankort D, et al. Genes Dev. 2007;21:379–384. doi: 10.1101/gad.1516407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chapman P, et al. Eur J Cancer Suppl. 2009;7:5. [Google Scholar]
- 8.Flaherty K, et al. J Clin Oncol. 2009;27:15s. [Google Scholar]
- 9.Ciampi R, et al. J Clin Invest. 2005;115:94–101. doi: 10.1172/JCI23237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones DT, et al. Cancer Res. 2008;68:8673–8677. doi: 10.1158/0008-5472.CAN-08-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones DT, et al. Oncogene. 2009;28:2119–2123. doi: 10.1038/onc.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomlins SA, et al. Nature. 2007;448:595–599. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 13.Tomlins SA, et al. Science. 2005;310:644–688. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 14.Kolch W, Heidecker G, Lloyd P, Rapp UR. Nature. 1991;349:426–428. doi: 10.1038/349426a0. [DOI] [PubMed] [Google Scholar]
- 15.MacConaill LE, et al. PLoS One. 2009;4:e7887. doi: 10.1371/journal.pone.0007887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeong JH, et al. PLoS One. 2008;3:e3949. doi: 10.1371/journal.pone.0003949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Solit DB, et al. Nature. 2006;439:358–362. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]