

Abstract
MC4-NN2-0453 is a novel, long-acting, selective, melanocortin-4-receptor agonist developed for treatment of obesity. This first-human-dose, randomized, double-blind, placebo-controlled trial investigated the safety, tolerability, pharmacokinetics, and pharmacodynamics of single and multiple doses of MC4-NN2-0453 in overweight to obese but otherwise healthy subjects. The trial included a single-dose part of ascending subcutaneous 0.03–1.50 mg/kg doses in overweight to obese but otherwise healthy men, and a multiple-dose part of ascending subcutaneous 0.75–3.0 mg/day doses in obese but otherwise healthy men/women. The single-dose part included 7 cohorts of 8 subjects, randomized 6:2 to active drug/placebo; the multiple-dose part included 4 cohorts of 20 subjects, randomized 16:4 to active drug/placebo. MC4-NN2-0453 was well tolerated and raised no safety concerns except for nonserious skin-related adverse events, this along with lack of weight loss effect led to premature termination of the trial. Headache, sexual–arousal disturbance, and penile erection were also reported. Single-dose pharmacokinetics showed dose-linearity and dose-proportionality. Maximum plasma concentration was observed after 50–100 hours, which then declined with a 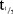 of approximately 250 hours. Plasma concentration reached steady state after 4 weeks for 0.75 and 1.5 mg/day multiple-dose cohorts, and the
of approximately 250 hours. Plasma concentration reached steady state after 4 weeks for 0.75 and 1.5 mg/day multiple-dose cohorts, and the 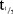 was similar to single dose. There were no significant pharmacodynamic effects, including effect on body weight.
was similar to single dose. There were no significant pharmacodynamic effects, including effect on body weight.
Keywords: alpha-melanocyte-stimulating hormone, obesity, pharmacokinetics, pharmacodynamics
Obesity is associated with multiple co-morbidities, predisposing people to cardiovascular diseases and type 2 diabetes.1,2 Weight reduction reduces the risk of developing type 2 diabetes in obese people3 and is therefore recommended by the American Diabetes Association (ADA) as a preventive treatment for type 2 diabetes4; weight reduction has also been shown to mitigate the risk of cardiovascular disease.5
Few treatment options for obesity are available today that conform to the US Food and Drug Administration (FDA) draft guidance, which states that an anti-obesity drug should be safe and cause significant weight loss that is sustained for at least 12 months (loss of >5% of initial body weight). Currently marketed anti-obesity drugs have limited efficacy or a high frequency of adverse effects at the most effective doses.6,7 However, recently approved obesity treatments, Qsymia® (combination of phentaramine and topiramate, Vivus, Inc., Mountain View, CA, USA) and Belviq® (lorcaserin, serotonin 2C receptor agonist, Arena Pharmaceuticals, Inc., San Diego, CA, USA) have shown clinically meaningful results in terms of safety and efficacy.
The melanocortin system has been explored as a target for treatment of obesity due to its dual role in feeding behavior and energy expenditure contributing to body weight control.8 Five types of melanocortin receptors (MCR) have been identified, and evidence confirms an association between MC4R and energy regulation.9 MC4R knock-out mouse studies have demonstrated the involvement of MC4R in feeding behavior and energy expenditure; the nonselective melanocortin agonist melanotan-II (MT-II) was shown to have no effect on food intake or energy expenditure in MC4R knock-out mice, but it did exert these effects in wild-type mice.10 Genetic studies in humans link obesity with lack of or mutations in MC4R.11,12 This evidence indicates MC4R as a possible target for the treatment of obesity.
Alpha-melanocyte-stimulating hormone (α-MSH) analog MC4-NN2-0453 is a long-acting, selective MC4R agonist investigated for the treatment of obesity. Several MC4R peptide agonists have been identified and characterized using in vitro and acute in vivo assays.13 Peptide agonist MC4-NN1-0182, referred to as peptide 11 and its derivative peptide 19 (analog MC4-NN2-0453), were found to have the desired potency, selectivity, solubility, and stability; however, MC4-NN2-0453 was comparatively more soluble at physiologic pH and more stable in solution than MC4-NN1-0182. Proof-of-principle for MC4-NN2-0453 was first established with the close analog MC4-NN1-0182. MC4-NN1-0182 significantly reduced body weight in diet-induced obese (DIO) rats when administered subcutaneously (s.c.) in a dose of 0.1 and 0.3 mg/kg once daily for 3 weeks, and in obese minipigs when given as a s.c. bolus dose of 30 mg on Day 1 followed by 0.1 mg/kg every other day for 8 weeks.14 An unpublished study conducted with MC4-NN2-0453 in DIO rats with s.c. doses of 0.1 and 0.3 mg/kg produced similar results. These results, along with those of nonclinical pharmacokinetic (PK) analysis and toxicology studies, indicated that MC4-NN2-0453 was generally well tolerated and was a candidate for clinical development as a weight-reducing agent.
Based on the above findings, this first-human-dose (FHD) trial was conducted to evaluate the safety, tolerability, PK, and pharmacodynamic (PD) effects of single doses (SD) of MC4-NN2-0453 followed by a multiple-dose (MD) part in overweight or obese but otherwise healthy subjects.
Methods
Subjects
The SD part enrolled overweight or obese but otherwise healthy men, aged 18–64 years, with body mass index (BMI) of ≥27.0 to ≤39.0 kg/m2, and the MD part enrolled obese but otherwise healthy men and women aged 18–64 years, with BMI of ≥30.0 to ≤39.0 kg/m2. Female subjects of childbearing potential were to follow a double-barrier method of contraception. Subjects with the same inclusion criteria were used for controls. Main exclusion criteria included obesity due to other endocrinological disorders, thyroid stimulating hormone (TSH) values outside the 0.4–6.0 mIU/L range, aggressive diet attempts, binge eating, use of weight-management drugs or medications that may cause significant weight gain in the 3 months before screening, weight change of ≥5 kg in the last 3 months, any surgical weight-reduction procedures such as liposuction, abdominoplasty, or similar procedure within 1 year of randomization, history of mood depressive disorder or severe psychiatric disease in the last 2 years, any clinically significant disease, laboratory values outside normal limits unless deemed not clinically significant, and lifetime history of suicide attempt. Subjects in both parts provided written informed consent prior to screening for trial participation. The trial was conducted at Covance Clinical Research Unit, Inc. (Evansville, IN) in accordance with the Declaration of Helsinki15 and International Conference on Harmonisation Good Clinical Practice,16 and was approved by local ethics committee.
Trial Design
This was a randomized, double-blind, placebo-controlled, two-part trial (a sequential s.c. SD ascending part and a semi-sequential s.c. MD ascending part) conducted to investigate the safety, tolerability, PK, and PD of MC4-NN2-0453 in overweight or obese but otherwise healthy male and female subjects.
Single-Dose Trial Design (Part 1)
The SD part consisted of seven dose cohorts (0.03, 0.06, 0.15, 0.30, 0.60, 1.00, and 1.50 mg/kg body weight), in each of which 8 subjects were randomized 6:2 to receive either active drug or placebo, respectively, except for the 0.60 mg/kg cohort, for which the ratio 7:2 was due to a dropout, resulting in one additional subject randomization. In this sequential SD part, dosing of subjects in a successive cohort would commence only after safety evaluation was performed, considering safety data, PK, laboratory results, and adverse events (AEs) reported 10–14 days after dosing of subjects from the previous cohort. All doses were administered by s.c. injection into the abdomen after an overnight fast. All subjects attended an information visit, a screening visit (Days −28 to −2), an extended in-house treatment visit (Days −1 to 6), ambulatory safety visits (Days 7–18), and an ambulatory follow-up visit (Day 22).
Multiple-Dose Trial Design (Part 2)
The MD part consisted of four dose cohorts. In each cohort, 20 subjects were to be randomized 16:4 to receive active drug (0.75, 1.5, 3.0, and 5.0 mg/day) or placebo. Approximately 30 days after the start of each MD cohort, an internal trial safety group evaluated the cumulative safety, tolerability, and PK before agreeing upon the next dose level.
All subjects attended an information visit, a screening visit (Days −28 to −2), two extended in-house treatment visits (Days −1 to 5 and Days 69 to 74), nine ambulatory treatment visits (Days 8, 15, 22, 29, 36, 43, 50, 57, and 64), five ambulatory safety visits (Days 77, 84, 91, 98, and 105), an ambulatory follow-up visit (Day 112), and an ambulatory visit for sampling anti-MC4-NN2-0453 antibodies (Day 210). All dose administrations except for those on Days 1 and 4 were done by the subjects themselves after instruction. The drug was to be administered once daily after an overnight fast on Days 1–70.
Dermatological Follow-Up
Because of a high incidence of nonserious skin-related AEs in the active treatment subjects, all subjects on active treatment from both the SD and the MD parts were called to a dermatological safety follow-up visit, approximately 1 year after last dosing; during this follow-up visit only skin-related AEs were assessed.
Blood Collection for PK Analysis
For the SD part, blood was drawn for PK analysis just before dosing (0 hour) and at time points 2, 6, 12, 18, 24, 32, 40, 48, 56, 64, 72, 80, 88, 96, 104, 112, 120, 128, 144, 168, 216, 264, 312, 360, 408, and 504 hours after dosing (Days 1–22).
For the MD part, blood was drawn for PK analysis just before dosing on Days 1, 2, 3, 4, 5, 8, 15, 22, 29, 36, 43, 50, 57, 64, and 70 (trough samples), and at time points 2, 6, 12, and 18 hours post-dosing at Days 1 and 4. Following the last dose (Day 70) for cohort 1, blood was drawn at 2, 6, 12, 18, 24, 32, 40, 48, 72, 96, 168, 336, 504, 672, 840, and 1,008 hours (42 days later) post-final-dose. For cohort 2, the last pre-dose sample was drawn on Day 50 (n = 12) or Day 36 (n = 2), 22 or 15 (n = 1 per day) and for cohort 3, the last pre-dose sample was drawn at Day 15 (n = 14) or Day 8 (n = 2). Dosing was terminated within days hereafter for cohort 2 and cohort 3 subjects. Blood was thereafter drawn at 168, 336, 504, 672, 840, and 1,008 hours post-last-dose.
The blood samples were collected by venipuncture or cannulation in EDTA tubes and frozen at −20°C after centrifugation at 4°C.
Injection Site and Methodology
All dose administrations were given before breakfast as s.c. injections into the abdomen while seated. Subjects had the injection site marked with a surgical marker (circle of at least 3 in. in diameter), the area appropriately treated with alcohol, and the s.c. injection made according to customary s.c. abdominal injection practices. Volume of the injection did not exceed 2 mL per injection and at higher “2-0453 doses,” in the SD part, multiple injections were required and injected in the same area.
In the MD part dose, administration on odd days (1st, 3rd, etc.) were done in the right site of the abdomen; dose administration on even days (2nd, 4th, etc.) were done in the left site of the abdomen.
2-0453 solution was used for injection at concentration 5 mg/mL (only to be used in Part 2), 10 mg/mL (to be used in Part 1 and 2), 20 mg/mL (only to be used in Part 1).
Safety Evaluation
The primary objective of the trial was safety and tolerability of ascending SD and MD of MC4-NN2-0453 assessed by AEs including sexual disturbance (examination for priapism by an urologist) and hypoglycemic episodes, clinical laboratory tests (anti-MC4-NN2-0453 antibodies for the MD part), Holter monitoring (for the SD part), 12-lead ECG, physical examination, vital signs including blood pressure (BP) monitoring, pulse and temperature, injection-site assessment, and determination of SD and MD maximum tolerated dose (MTD). Mental health was assessed using the PHQ-9 (patient health questionnaires): PoMS (The Profile of Mood States Questionnaire) and C-SSRS (The Columbia Suicidality Severity Rating Scale Questionnaire) questionnaires.
The trial was put on hold because of unexpected skin-related AEs, and all subjects were called in for a dermatologic assessment by a dermatologist. All subjects were offered biopsy and standard care by the dermatologist. Twenty-two (22) of the 27 subjects with clinically atypical nevi underwent biopsies including a histology evaluation. The dermatological report showed seven subjects diagnosed with mild histologic atypia. Three (3) of the 27 subjects did not consent to the biopsy, and 2 subjects did not attend the dermatologic assessment visit.
Given the dermatological findings, the sponsor decided to terminate the trial prematurely. All subjects in the MD-part were transferred to Visit 13 (ambulatory safety visit) and were followed as per the protocol for the next 10 weeks, and as described earlier all drug-exposed subjects were invited for an extra dermatological follow-up visit 1 year later.
Pharmacokinetic Evaluation
MC4-NN2-0453 was assayed in plasma by a validated LC-MS/MS assay including Plasma Protein Precipitation, (electrospray ionization, positive MRM mode, m/z: 698.5 → 279.5). The method was validated according to current guidelines.17,18 A stable isotope labeled analog of MC4-NN2-0453 labeled in 16 positions was used as an assay internal standard (electrospray ionization, positive MRM mode, m/z: 703.9 → 279.5). The concentration of MC4-NN2-0453 in plasma samples was calculated using the peak area ratios analyte/IS. Calibration graphs based on nine plasma samples spiked with MC4-NN2-0453 in the concentration range 10.0–2,500 ng/mL were constructed by weighted linear regression (1/x).
The SD and the MD PK parameters at steady state were estimated using a standard noncompartmental analysis (Table1).19
Table 1.
Definition of Pharmacokinetic Parameters
| Pharmacokinetic Parameter | Definition |
|---|---|
| tmax | Time of observed maximum concentration |
| Cmax | Observed maximum concentration |
| Ctrough | Concentration just prior to dosing measured once weekly in multiple dose studies |
| AUC | Area under the curve, from dosing to infinity. Calculated by the linear trapezoidal rule time 0 hour extrapolated to infinity |
| AUC0–24h | Area under the concentration–time curve from 0 to 24 hours post-dose, calculated on Days 1 and 4 for multiple dose studies |
| AUC0–96h | Area under the concentration–time curve from 0 to 96 hours post-dose |
| AUClast | Area under the concentration–time curve calculated by the linear trapezoidal rule time 0 hour to last time “t” hours after dosing, where t is the time point of last observable drug concentration |
| t1/2 | Terminal half life estimated as 0.693/λz, λz is the terminal rate constant determined by log-linear regression on the terminal log-linear part of the concentration–time curve |
| Vz/F | Volume of distribution after extravascular administration, based on terminal phase Vz/F = Dose/(AUC × λz) |
| CL/F | Apparent total plasma clearance of drug after extravascular administration CL/F = Dose/AUC |
| RAC | Ratio of accumulation defined as AUC0–24h from multiple dose data/AUC0–24h from single dose data |
Pharmacodynamic Evaluation
The PD evaluation for the SD part included change in body weight and lipid profile, and the PD evaluation for the MD part included change in body weight, lipid profile, waist and hip circumference, waist–hip ratio, subjective self-reported appetite variables [appetite, taste, hunger, thirst, nausea, cravings, palatability, and fullness using visual analog scale (VAS) scores], caloric intake, and homeostatic model for insulin resistance (HOMA IR).
Statistical Analysis
The mean AUC (SD only), AUC0–24h, and Cmax were plotted for all doses used in this trial to observe and test for dose proportionality using log-transformed data. Dose proportionality was tested for the mean AUC (SD only), AUC0–24h, and Cmax for both single and MDs using the linear model:
where log AUC (similarly for log AUC0–24h and log Cmax) is the log-transformed PK parameter and log Dose is the log-transformed dose. It was tested whether the coefficient β equals 1. Deviation from dose proportionality was only detected if the hypothesis was rejected.
Results
All dose cohorts in the SD part completed the trial, whereas only cohort 1 (0.75 mg/day) in the MD part completed the trial. Because of unexpected incidents of nonserious skin-related AEs, cohorts 2 (1.5 mg/day) and 3 (3.0 mg/day) were prematurely terminated after approximately 49 and 14 days of treatment, respectively; cohort 4 (5.0 mg/day) was not initiated. The results for all initiated cohorts are presented, including the 1-year dermatological follow-up results for subjects who were on active treatment.
In the SD part, 53 of 57 randomized subjects completed the trial. Four subjects in the SD part discontinued the trial due to withdrawal of consent for reasons unrelated to safety. The demographic features and baseline characteristics of the trial subjects are shown in Table2 for the SD part. The mean age of the SD trial population was 34.9 ± 9.4 years, and mean BMI was 31.7 ± 3.2 kg/m2.
2a.
Baseline Characteristics: Single Dose (Part 1): All Randomized Subjects
| Placebo (n =14) | 0.03 mg/kg (n = 6) | 0.06 mg/kg (n = 6) | 0.15 mg/kg (n = 6) | 0.30 mg/kg (n = 6) | 0.60 mg/kg (n = 7) | 1.00 mg/kg (n = 6) | 1.50 mg/kg (n = 6) | |
|---|---|---|---|---|---|---|---|---|
| Age (years) | 32.9 (6.7) | 37.3 (11.2) | 32.3 (12.3) | 39.3 (11.3) | 35.2 (11.8) | 37.6 (4.5) | 35.0 (7.1) | 31.8 (13.6) |
| Race, n (%) | ||||||||
| White | 12 (86) | 6 (100) | 5 (83) | 5 (83) | 4 (67) | 2 (29) | 4 (67) | 5 (83) |
| Black or African American | 2 (14) | 0 | 1 (17) | 1 (17) | 2 (33) | 4 (57) | 2 (33) | 0 |
| Asian | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Other | 0 | 0 | 0 | 0 | 0 | 1 (14) | 0 | 1 (17) |
| Body weight (kg) | 105.0 (17.3) | 96.6 (13.7) | 93.2 (8.9) | 95.1 (13.1) | 89.4 (10.6) | 97.0 (8.8) | 101.5 (15.1) | 97.8 (12.3) |
| BMI (kg/m2) | 32.1 (3.1) | 32.2 (4.8) | 30.8 (2.5) | 31.0 (3.4) | 30.4 (2.0) | 32.3 (2.1) | 33.0 (4.2) | 31.2 (3.5) |
Data are mean (standard deviation) unless otherwise specified.
BMI, body mass index.
Sixty randomized subjects were assigned to one of the three cohorts in the MD part. While the MD part was ongoing, two subjects withdrew consent (not safety-related), one subject was lost to follow-up, and one subject was withdrawn due to nonadherence to scheduled visits. The demographic features and baseline characteristics of the trial subjects are shown in Table3. In the MD part, there were 21 males and 39 females. The mean age of the MD part population was 40.1 ± 11.2 years, and mean BMI was 34.4 ± 2.3 kg/m2 across treatment cohorts.
2b.
Baseline Characteristics: Multiple Dose (Part 2): All Randomized Subjects
| Placebo | 0.75 mg/day | 1.5 mg/day | 3.0 mg/day | |
|---|---|---|---|---|
| Age (years) | 35.4 (11.1) | 43.8 (11.9) | 38.6 (8.2) | 41.4 (12.6) |
| Sex, M:F [n (%)] | 5:7 (42:58) | 4:12 (25:75) | 8:8 (50:50) | 4:12 (25:75) |
| Race, n (%) | ||||
| White | 10 (83) | 13 (81) | 8 (50) | 14 (88) |
| Black or African American | 2 (17) | 3 (19) | 6 (38) | 2 (13) |
| Asian | 0 | 0 | 1 (6) | 0 |
| Other | 0 | 0 | 1 (6) | 0 |
| BMI (kg/m2) | 34.5 (2.3) | 34.9 (2.4) | 34.5 (2.2) | 33.8 (2.3) |
Data are mean (standard deviation) unless otherwise specified.
M, male; F, female, BMI, body mass index.
Safety
No serious AEs, deaths, or discontinuations due to AEs were reported. In the SD part, 41 (71.9%) subjects reported a total of 94 treatment-emergent adverse events (TEAEs); headache (21.1%) followed by increase in frequency and duration of penile erection (14%), and decreased appetite (12%) were the most common TEAEs. All AEs were mild to moderate in severity.
In the MD part, 54 (90%) subjects reported a total of 264 TEAEs; melanocytic nevus (45%), skin hyperpigmentation (33.3%), headache (30%), and sexual disturbance (increase in frequency and duration of penile erection or sexual arousals) (27%) were the most common TEAEs. All but three subjects in the MD part recovered; one subject experienced AEs of seborrheic dermatitis and blepharospasm; one subject experienced sciatica and depression; and one subject reported hypothyroidism. All the AEs reported by these three subjects were assessed to be unlikely related to the trial drug.
Skin-related AEs
In the SD part, clinically atypical melanocytic nevus was reported by one subject (16.7%) in the 1.0 mg/kg cohort and by one subject in the 1.5 mg/kg dose cohort, both revealed mild histological atypia after biopsy. Two subjects (28.6%) in the 0.60 mg/kg dose cohort and one subject (16.7%) in the 1.5 mg/kg dose cohort reported skin hyperpigmentation on sun-exposed areas. None receiving placebo reported either melanocytic nevus or skin hyperpigmentation. No increase in these events with increasing doses was observed. All skin-related AEs were mild in severity. All subjects recovered except for one subject in the 1.5 mg/kg active treatment cohort who reported with typical melanocytic nevus and who was recovering; the subject withdrew from follow-up.
In the MD part, clinically atypical melanocytic nevus including the palmar and the plantar location was reported for 6 (37.5%), 12 (75%), and 9 (56.3%) subjects on active treatment in the 0.75, 1.5, and 3.0 mg/day dose cohorts, respectively. All these subjects with clinically atypical nevus were called for a dermatological evaluation during which biopsy performed revealed mild histological atypia for five subjects. There were no reports of atypical nevus with placebo. Skin pigmentation was observed in 6 (37.5%), 10 (62.5%), and 3 (18.8%) subjects on active treatment in the 0.75, 1.5, and 3.0 mg/day dose cohorts, respectively; 1 subject (8.3%) receiving placebo also had skin hyperpigmentation. The skin changes occurred typically 3–6 weeks after treatment initiation. All skin-related AEs were mild. All subjects recovered except one subject with skin hyperpigmentation who was lost to follow-up.
All results from the dermatological visit including clinical assessment and biopsy results were presented to three external dermatology experts, who concluded that all findings were benign but recommended 1-year follow-up visits for subjects treated with MC4-NN2-0453.
One-year dermatologic follow-up
At the 1-year dermatologic follow-up visit, 29 of the 53 subjects in the SD part and 42 of the 56 subjects in the MD part consented to participate. Three subjects (10%) in the SD part of the trial reported four skin-related AEs; all events were benign melanocytic nevus confirmed to be without atypia on biopsy. In the MD part of the trial, six subjects (14%) reported eight AEs, of which six events were skin-related; all skin-related events were benign melanocytic nevus confirmed to be without atypia on biopsy except one with mild cellular atypia. None of these events were serious, all were judged by the investigator to be possibly related to the drug, and all the events showed complete recovery. No melanoma was diagnosed in any of the examined subjects. The remaining two AEs in the MD part reported by one subject included wound infection at the biopsy site and vaginal candidiasis (due to the treatment of the infection), which were unlikely to be related to the drug.
Urogenital AEs
Seven males on active treatment and one male on placebo in the SD part experienced an increase in frequency as well as duration of penile erection. During the MD trial, six males (all on active treatment) were noted to have increased frequency as well as duration of penile erection, and seven females (one on placebo) had disturbance in sexual arousal; however, tolerance to this effect was seen with repeated dosing. None of these events were dependent on exposure, and there was no priapism reported.
Vital signs
No clinically relevant change in BP was observed. Mean change from baseline in systolic blood pressure (SBP) and diastolic blood pressure (DBP) over time with intensive measuring are presented in Figure 1a,b for the SD part, and with a less intensive assessment in Figure 1c,d for the MD part. Similarly, no clinically relevant changes were observed in other vital signs such as pulse, respiratory rate and temperature.
Figure 1.
(a) Change (mean ± SEM) in systolic blood pressure over time—single-dose part (n = 6). For simplicity only data for three doses are presented (the lowest, a medium, and the highest dose). Refer to supplementary figures for the data on the other doses not presented here. (b) Change (mean ± SEM) in diastolic blood pressure over time—in single-dose part (n = 6). For simplicity only data for three doses are presented (the lowest, a medium, and the highest dose). Refer to supplementary figures for the data on the other doses not presented here. (c) Change (mean ± SEM) in systolic blood pressure over time—multiple-dose part. (d) Change (mean ± SEM) in diastolic blood pressure over time—multiple-dose part.
There were no clinically relevant changes in clinical lab tests, ECG (2 subjects in the SD part experienced incidences of asymptomatic nonsustained ventricular tachycardia, but with normal ECG), and physical examination (except for skin-related AEs). No subjects experienced symptoms of hypoglycemia.
Other assessments
Three subjects in the MD part tested positive for low-titer, non-neutralizing, non-cross-reacting antibodies.
No injection-site reactions were reported. There were no clinically relevant psychological changes as assessed by PHQ-9 questionnaires.
MTD was not established for the SD part. However, the maximum feasible dose reached in the SD part was 1.5 mg/kg. All the doses were nontolerable in the MD part because of the onset of skin-related AEs in all dose cohorts, leading to premature termination of the trial.
Pharmacokinetic Analysis
Forty-three subjects on active treatment in the SD part and 48 subjects on active treatment in the MD part were included in the PK analysis.
Single-dose part
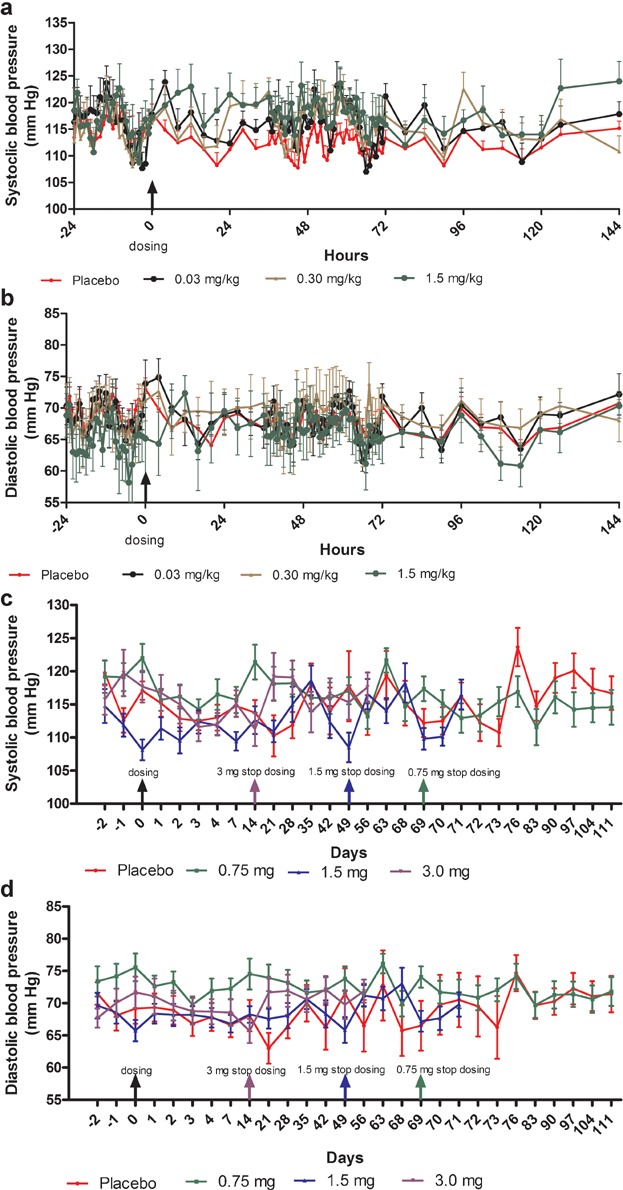
Mean plasma concentration–time profiles following single s.c. doses of MC4-NN2-0453 are presented in Figure 2a. There was a dose-proportional increase in the mean exposure to MC4-NN2-0453, with tmax ranging from 50 to 100 hours over the dose range 0.03–1.5 mg/kg. Dose proportionality was observed during systemic exposure to MC4-NN2-0453 with respect to AUC0–24h, AUClast, and AUC0–∞ (Figure 3a) over the dose range 0.03–1.5 mg/kg, with the value of the constant β estimated to be 0.98 [95% confidence interval (CI); 0.9105–1.0587] for AUC0–24h, 0.95 [95% CI; 0.9110–0.9906] for AUClast, and 0.95[95% CI; 0.9073–0.9998] for AUC0–∞. Dose proportionality was thus statistically significant only for AUC0–24h. Cmax increased with the dose (Figure 3b), but dose proportionality could not be confirmed (0.93 [95% CI; 0.8932–0.9797]. However, dose proportionality was significant for all the above PK parameters in the dose range 0.15–1.5 mg/kg, excluding doses 0.03 and 0.06 mg/kg (95% CI for slope of AUC0–24h, AUClast, AUC0–∞, and Cmax included unity).
Figure 2.
MC4-NN2-0453 plasma concentration versus time by dose group (mean ± SD) for single-dose cohorts (a) and multiple-dose (MD) cohorts (b), including the 42 days post-last-dosing for all three MD cohorts with last pre-dose sampling at Day 70 (cohort 1; n = 14), Day 50 (cohort 2, n = 12), and Day 15 (cohort 3, n = 14).
Figure 3.
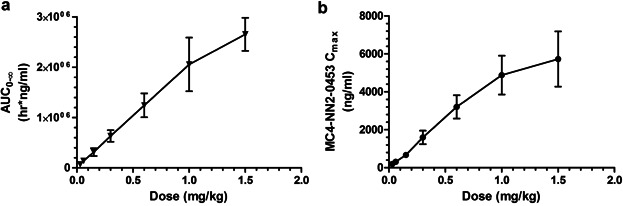
AUC0–∞ (a) and Cmax in plasma (b) (mean ± SD) versus single dose of MC4-NN2-0453 (0.03–1.5 mg/kg; n = 6 per dose group).
The plasma PK parameters following administration of SDs of MC4-NN2-0453 are presented in Table4. The mean tmax ranged between 50 and 100 hours (approx. 2–4 days) post-dose. Thereafter, the plasma MC4-NN2-0453 concentration declined, with mean 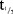 values of 234–274 hours, which was fairly consistent across all dose cohorts. With each dose escalation from 0.03 to 1.5 mg/kg, a 1.5–2-fold increase was observed in both mean Cmax, which increased from 195 to 6,540 ng/mL, and mean AUC0–∞ increased from 67,600 to 2,650,000 h × ng/mL. There was also a dose-dependent increase in mean AUC0–24h and mean AUClast. The total plasma clearance (CL/F) was noted to be almost constant at around 0.0005 L/h/kg, while the volume of distribution (Vz/F) ranged between 0.15 and 0.23 L/kg and was dose-independent.
values of 234–274 hours, which was fairly consistent across all dose cohorts. With each dose escalation from 0.03 to 1.5 mg/kg, a 1.5–2-fold increase was observed in both mean Cmax, which increased from 195 to 6,540 ng/mL, and mean AUC0–∞ increased from 67,600 to 2,650,000 h × ng/mL. There was also a dose-dependent increase in mean AUC0–24h and mean AUClast. The total plasma clearance (CL/F) was noted to be almost constant at around 0.0005 L/h/kg, while the volume of distribution (Vz/F) ranged between 0.15 and 0.23 L/kg and was dose-independent.
3a.
Single Dose (Part 1): Pharmacokinetic Parameters for MC4-NN2-0453
| 0.03 mg/kg (n = 6) | 0.06 mg/kg (n = 5)a | 0.15 mg/kg (n = 6) | 0.30 mg/kg (n = 6) | 0.60 mg/kg (n = 6) | 1.00 mg/kg (n = 6) | 1.50 mg/kg (n = 5)b | |
|---|---|---|---|---|---|---|---|
| Cmax (ng/mL) | 195 (23.1) | 309 (39.1) | 672 (108) | 1,590 (358) | 3,200 (616) | 4,880 (1,020) | 6,540 (909) |
| tmax (hour) | 61.3 (14.9) | 60.8 (31.3) | 100 (27.1) | 61.3 (56.0) | 50.7 (6.5) | 53.3 (9.7) | 70.7 (26.5) |
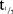 (hour) (hour) |
234 (48.8) | 268 (46.9) | 270 (35.8) | 268 (28.6) | 259 (30.2) | 260 (77.6) | 274 (61.1) |
| AUC0–24h (h ng/mL) | 2,080 (431) | 2,990 (960) | 6,730 (1,280) | 15,500 (5,220.2) | 44,500 (11,200) | 59,300 (12,700) | 68,500 (14,100) |
| AUClast (h ng/mL) | 51,400 (6,640) | 98,200 (16,700) | 212,000 (36,500) | 454,000 (70,683.8) | 912,000 (164,000) | 1,470,000 (299,000) | 1,980,000 (385,000) |
| AUC0–∞ (h ng/mL) | 67,600 (11,300) | 138,000 (30,100) | 313,000 (77,800) | 633,000 (113,315.6) | 1,240,000 (237,000) | 2,050,000 (534,000) | 2,650,000 (329,000) |
| CL/F (L/h/kg) | 0.0005 (0.0001) | 0.0005 (0.0001) | 0.0005 (0.0001) | 0.0005 (0.0001) | 0.0005 (0.0001) | 0.0005 (0.0001) | 0.0006 (0.0001) |
| Vz/F (L/kg) | 0.15 (0.02) | 0.17 (0.03) | 0.19 (0.02) | 0.19 (0.02) | 0.18 (0.03) | 0.18 (0.04) | 0.23 (0.07) |
Data are mean (standard deviation).
Except for AUC0–24h which was calculated based on n = 6.
Except for Cmax, tmax, and AUC0–24h which were calculated based on n = 6.
Multiple-dose part
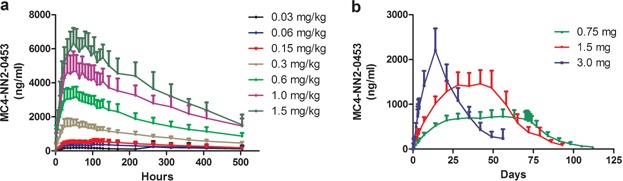
Mean plasma concentration–time profiles for MC4-NN2-0453 at steady state following MDs of MC4-NN2-0453 are presented in Figure 2b. The mean exposure to MC4-NN2-0453 increased with increasing dose after multiple dosing with 0.75, 1.5, and 3.0 mg/day. The plasma PK parameters at steady state are presented in Table5. The mean Cmax and AUC0–24h on days 1 and 4 increased approximately dose-proportionally after multiple dosing of 0.75, 1.5, and 3.0 mg/day. However, the dose proportionality could not be determined, as only one dose cohort was finalized. Steady state was achieved after Day 29 for the 0.75 and 1.5 mg/day cohorts. From data available for all three cohorts, the mean tmax on days 1 and 4 ranged between 19.9 and 23.9 hours. Similar to the SD part, 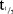 (232–253 hours) was fairly long and consistent across dose cohorts. Accumulation was 25 times in the 0.75 mg/day cohort. AUC0–∞ was 275,000 h × ng/mL for the 0.75 mg/day dose cohort; AUC0–∞ was not estimated for the 1.5 or 3.0 mg/day dose cohorts as the trial was prematurely terminated.
(232–253 hours) was fairly long and consistent across dose cohorts. Accumulation was 25 times in the 0.75 mg/day cohort. AUC0–∞ was 275,000 h × ng/mL for the 0.75 mg/day dose cohort; AUC0–∞ was not estimated for the 1.5 or 3.0 mg/day dose cohorts as the trial was prematurely terminated.
3b.
Multiple Dose (Part 2): Pharmacokinetic Parameters for MC4-NN2-0453
| 0.75 mg/day | 1.5 mg/day | 3.0 mg/day | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Day 1 (n = 16) | Day 4 (n = 16) | Day 70 (n = 14) | Day 1 (n = 16) | Day 4 (n = 16) | Day 70 (n = 16) | Day 1 (n = 16) | Day 4 (n = 16) | Day 70 (n = 15) | |
| Cmax (ng/mL) | 49.7 (12.4) | 199 (41.7) | 723 (155) | 99.4 (21.9) | 387 (63.4) | NA | 229 (47.4) | 947 (184) | NA |
| tmax (hour) | 23.6 (1.5) | 19.9 (5.2) | 24.9 (15.4) | 23.9 (0.0) | 22.1 (3.6) | NA | 23.9 (0.0) | 21.3 (4.3) | NA |
| t½ (hour) | NA | NA | 232 (31.0) | NA | NA | 234 (40.4) | NA | NA | 253 (31.5) |
| AUC0–24h (h ng/mL) | 698a (168) | 4,320a (862) | 16,500a (3,580) | 1,140a (397) | 8,330a (1,420) | NA | 3,320a (954) | 20,800a (4,170) | NA |
Data are mean (standard deviation).
NA, not applicable.
Cohort 2 (1.5 mg/day) terminated around Visit 9 and were transferred to Visit 13 within 1 week.
Cohort 3 (3.0 mg/day) terminated around Visit 4 and were transferred to Visit 13 within 1 week.
Steady state has not been reached.
Pharmacodynamic Analysis
In both the SD and the MD parts, no change in body weight could be demonstrated.
Furthermore, no change was observed in other PD variables including waist circumference, hip circumference, waist–hip ratio, HOMA, and lipid profile, and no definitive conclusions could be drawn for any of the cohorts because of inconsistent data for subjective self-reported appetite variables and caloric intake.
Discussion
This was the first human-dose trial of the long-acting MC4R agonist MC4-NN2-0453, developed for weight loss and weight maintenance in overweight to obese individuals. There were no serious or severe AEs reported in this trial. The trial was prematurely terminated because of unexpected incidences of nonserious skin-related AEs in the three highest dose cohorts in the SD part and in all dose cohorts in the MD part. Though, these skin related AEs of mild severity were well tolerated (these AEs showed recovery) and did not lead to any discontinuations, no possible weight loss benefits were seen at doses where these nonserious skin related AEs started appearing and thus leading to trial termination. All the skin-related AEs were considered likely to be related to the trial drug (all events except one occurred in active treatment groups). Other common AEs reported were headache, sexual disturbance, nausea, and injection-site hemorrhage observed with both MC4-NN2-0453 and placebo.
MC1 receptors are located in the skin on human melanocytes mediating the melanogenic and proliferative effects of melanotropic peptides.20,21 The skin hyperpigmentation was therefore unexpected given the low affinity of MC4-NN2-0453 for MC1 receptors and high affinity for the MC4R (MC4:MC1R selectivity 1,000:1).13 Melanocortin analogs such as melanotan-I and -II are potent, nonselective melanocortin agonists. They have been tested as skin-tanning agents in clinical trials with the therapeutic potential to prevent skin cancer (and intolerance to sun exposure),22–25 suggesting that regulated use of these agents at clinical doses could be safe. Unlicensed use of these nonselective α-MSH analogs has resulted in skin hyperpigmentation and darkening of existing nevus.26,27 This could be explained by their action through MC1R in the skin by increased signaling and proliferation of well-differentiated melanocytes.21
Skin hyperpigmentation occurring in the SD part could possibly be due to the constant and very high exposure to MC4-NN2-0453; exposure levels of 2.3–3.1 µM were observed, which is close to the Ki value of 2.7 µM of MC4-NN2-0453 on the MC1R.13 Even in the presence of albumin binding, this may be enough to stimulate the melanocytes. In the MD part as well, steady-state exposure levels were 0.34 and 0.67 µM with doses of 0.75 and 1.5 mg/day, respectively; Ctrough reached with the 3.0 mg dose was 1.05 µM. It is known that exposure to α-MSH analogs leads to up-regulation of MC1 receptors in the skin and in congenital nevi expression of MC1 receptors are increased28; this, along with exposure to UV radiation (which indirectly stimulates α-MSH secretion), leads to increased expression of MC1 receptors.29,30 Furthermore, a synergistic effect on tanning of melanotan-I and UV-B light was reported by Dorr et al.25 Although MC4-NN2-0453 is an analog with very high receptor specificity for the MC4R versus the MC1R, the high-exposure level and the eventual increase in MC1R expression could explain the increase in number and pigmentation of nevi both in sun-exposed and acral area as well as the hyperpigmentation induced by MC4-NN2-0435 in this trial. Acral nevi have been shown to be associated with ethnicity, pigmentation, age, and cutaneous melanoma risk factors and are not uncommon phenomena.31
Change in penile erection observed during the trial was reported more frequently in the active treatment group than in the placebo group. The changes can be explained by the role of melanocortin in erectile function. Human trials with melanotan-II and its derivative PT141 have demonstrated erectogenic activity in males with erectile dysfunction32,33 and arousal in females with sexual behavior disorders,34 indicating a potential for the treatment of sexual dysfunction. In contrast, a clinical trial conducted with the MC4R agonist MK-0493 failed to demonstrate the same effect35; it also failed to demonstrate effect on body weight and energy intake,36 which perhaps indicates that exposure was too low to affect penile erection and body weight.
In the current trial, MC4-NN2-0453 exhibited a neutral effect on BP despite being an MC4R agonist. This is in contrast to the MC4R agonist LY2112688, which significantly (P < .001) increased both SBP and DBP.37 The melanocortin system through MC4R regulates BP by modulating central sympathetic flow.37 The lack of effect on BP in the current trial could have been due to the limited capacity of MC4-NN2-0453 to cross the blood–brain barrier. Though, there was an initial effect on penile erection, indicating some availability to the central nervous system, as penile erection is thought to be centrally mediated,33 apparently the central exposure may not have been high enough to mediate an effect on body weight.
In the SD part, the PK of MC4-NN2-0453 was dose-proportional and linear, but dose proportionality was statistically significant only for AUC0–24h. AUC0–24h, however, most likely represented only a partial drug exposure during the absorption phase, as tmax occurred after 50 hours. After multiple dosing with 0.75 and 1.5 mg, steady state was reached after 29 days of treatment. 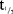 was 232–274 hours and was dose-independent both in the SD and the MD parts. The accumulation was 25 times with 0.75 mg/day dose, which is consistent with the dosing interval and the long elimination
was 232–274 hours and was dose-independent both in the SD and the MD parts. The accumulation was 25 times with 0.75 mg/day dose, which is consistent with the dosing interval and the long elimination 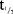 .
.
MC4-NN2-0453 did not show any noticeable PD effects in the SD part. In the MD part, no definitive conclusions could be drawn on the effect of MC4-NN2-0453 on body weight, subjective self-reported appetite variables, or caloric intake because of inconsistent data and premature closure of the MD part. Proof-of-principle was obtained with the nonclinical studies conducted with MC4-NN1-0182 and MC4-NN2-0453, demonstrating a significant reduction in body weight, which was attributed to reduction in food intake and increased energy expenditure.14 A similar effect with the peptide agonist MC4-NN2-0453 in humans could not be observed with the doses studied in this trial. Phase 1 and 2 clinical trials conducted by Krishna et al with the MC4R agonist MK-0493 also demonstrated a similar lack of efficacy,36 indicating either that exposure was too low or that differential mechanisms or pathways were involved in regulation of human body weight; this would bring the viability of targeting MC4R in body weight regulation into some question.
The site of anorectic action of melanocortins has been demonstrated to be the paraventricular nucleus (PVN), suggesting accessibility to peripherally administered melanocortins. However, the effect on energy expenditure is mediated through MC4Rs located on neurons in regions of the brain other than the PVN.38 Thus an MC4R agonist like MC4-NN2-0453 must access these regions to regulate food intake and energy expenditure and we could possibly attribute the lack of efficacy of the molecule in humans to accessibility.
Conclusions
Overall, MC4-NN2-0453 was generally well tolerated with no safety concerns except for the skin-related AEs (skin hyperpigmentation and melanocytic nevus).
PK of MC4-NN2-0453 was dose-proportional and linear, and the 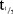 was long, indicating a potential for once-weekly dosing.
was long, indicating a potential for once-weekly dosing.
MC4-NN2-0453 treatment did not appear to affect body weight in humans in the doses or treatment exposure tested.
Our data suggest the need for further research and insight into whether α-MSH-mediated weight control is centrally or peripherally mediated especially in the context of obesity, also whether a constant high exposure to the MC4R agonist downregulates the receptor response. Finally, the predictivity of the used animal models should be further investigated in general within obesity.
Acknowledgments
We thank the trial participants for their participation. We also thank Dr. Vathsala Jayanth, employee of Novo Nordisk, Bangalore, India, for providing medical writing and editorial assistance and Michael Pilgård Andersen for providing assistance in assaying MC4-NN2-0453.
Declaration of Conflicting Interests
Gitte Konradsen, Ole Eskerod, Birgit S. Hansen, and Birgitte S. Wulff are employees of Novo Nordisk A/S.
Funding
The study was sponsored by Novo Nordisk (Bagsvaerd, Denmark).
References
- 1.Wilson PWF, D'Agostino RB, Sullivan L, Parise H, Kannel WB. Overweight and obesity as determinants of cardiovascular risk. The Framingham experience. Arch Intern Med. 2002;162:1867–1872. doi: 10.1001/archinte.162.16.1867. [DOI] [PubMed] [Google Scholar]
- 2.Mhurchu CN, Parag V, Nakamura M, Patel A, Rodgers A, Lam HT. Body mass index and the risk of diabetes mellitus in the Asia-Pacific region. Asia Pac J Clin Nutr. 2006;15(2):127–133. [PubMed] [Google Scholar]
- 3.Sjostrom CD, Peltonen M, Wedel H, Sjostrom L. Differentiated long-term effects of intentional weight loss on diabetes and hypertension. Hypertension. 2000;36(1):20–25. doi: 10.1161/01.hyp.36.1.20. [DOI] [PubMed] [Google Scholar]
- 4.American Diabetes Association. Standards of medical care in diabetes—2011. Diabetes Care. 2010;34:S11–S61. doi: 10.2337/dc11-S011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fleming RM. The effect of high-, moderate-, and low-fat diets on weight loss and cardiovascular disease risk factors. Prev Cardiol. 2007;5(3):110–203. doi: 10.1111/j.1520-037x.2002.01231.x. [DOI] [PubMed] [Google Scholar]
- 6.Guidance for industry developing products for weight management. Draft guidance. Food and Drug Administration website http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM071612.pdf. Accessed December 28, 2012.
- 7.Cooke D, Bloom S. The obesity pipeline: current strategies in the development of anti-obesity drugs. Nat Rev Drug Discov. 2006;5(11):919–931. doi: 10.1038/nrd2136. [DOI] [PubMed] [Google Scholar]
- 8.Vergoni AV, Bertolini A. Role of melanocortins in the central control of feeding. Eur J Pharmacol. 2000;405:25–32. doi: 10.1016/s0014-2999(00)00538-0. [DOI] [PubMed] [Google Scholar]
- 9.Vergoni AV, Bertolini A, Mutulis F, Wikberg JES, Schiöth HB. Differential influence of a selective melanocortin MC4 receptor antagonist (HS014) on melanocortin-induced behavioral effects in rats. Eur J Pharmacol. 1998;362(2):95–101. doi: 10.1016/s0014-2999(98)00753-5. [DOI] [PubMed] [Google Scholar]
- 10.Chen AS, Metzger JM, Trumbauer ME, et al. Role of the melanocortin-4 receptor in metabolic rate and food intake in mice. Transgenic Res. 2000;9(2):145–154. doi: 10.1023/a:1008983615045. [DOI] [PubMed] [Google Scholar]
- 11.Vaisse C, Clement K, Durand E, Hercberg S, Guy-Grand B, Froguel P. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest. 2000;106(2):253–262. doi: 10.1172/JCI9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farooqi IS, Yeo GSH, Keogh JM, et al. Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J Clin Invest. 2000;106(2):271–279. doi: 10.1172/JCI9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conde-Frieboes K, Thøgersen H, Lau JF, et al. Identification and in vivo and in vitro characterization of long acting and melanocortin 4 Receptor (MC4-R) selective α-melanocyte-stimulating hormone (α-MSH) analogues. J Med Chem. 2012;55(5):1969–1977. doi: 10.1021/jm201489a. [DOI] [PubMed] [Google Scholar]
- 14.Raun K, Dahl K, Nilsson C, et al. Novel MC4 agonist peptide effectively reduces body weight in diet induced obese rats and minipigs. Obesity. 2011;19:S153–S235. [Google Scholar]
- 15.World Medical Association. Declaration of Helsinki. Ethical Principles for Medical Research Involving Human Subjects. 52nd WMA General Assembly, Edinburgh, Scotland, October 2000. Last amended with Note of Clarification on Paragraph 29 by the WMA General Assembly, Washington. 2002, and Note of Clarification on Paragraph 30 by the WMA General assembly, Tokyo. 2004.
- 16.ICH guideline E9. Vol. 63. Published in the Federal Register; 1998. statistical principles for clinical trials. [PubMed] [Google Scholar]
- 17.Guidance for industry, bioanalytical method validation, May 2001. Food and Drug Administration website http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf. Accessed August 23, 2013.
- 18.Viswanathan CT, Bansal S, Booth B, et al. Workshop/Conference report—Quantitative bioanalytical methods validation and implementation: best practices for chromatographic and ligand binding assay. AAPS J. 2007;9(1):E30–E42. doi: 10.1007/s11095-007-9291-7. [DOI] [PubMed] [Google Scholar]
- 19.Collection of terms, symbols, equations, and explanations of common pharmacokinetic and pharmacodynamic parameters and some statistical functions. 2004. February. AGAH website http://www.agah.eu/fileadmin/_migrated/content_uploads/PK-glossary_PK_working_group_2004.pdf. Accessed August 23, 2013.
- 20.Suzaki I, Cone RD, Sungbin IM, Nordlund J, Abdel-Malek Z. Binding of melanotropic hormones to the melanocortin receptor MClR on human melanocytes stimulates proliferation and melanogenesis. Endocrinology. 1996;137(5):1627–1633. doi: 10.1210/endo.137.5.8612494. [DOI] [PubMed] [Google Scholar]
- 21.Abdel-Malek Z, Swope VB, Suzuki I, et al. Mitogenic and melanogenic stimulation of normal human melanocytes by melanotropic peptides. Proc Natl Acad Sci. 1995;92(5):1789–1793. doi: 10.1073/pnas.92.5.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barnetson RSC, Ooi TKT, Zhuang L, et al. [Nle4-D-Phe7]-α-melanocyte-stimulating hormone significantly increased pigmentation and decreased UV damage in fair-skinned Caucasian volunteers. J Invest Dermatol. 2006;126(8):1869–1878. doi: 10.1038/sj.jid.5700317. [DOI] [PubMed] [Google Scholar]
- 23.Levine N, Sheftel SN, Eytan T, et al. Induction of skin tanning by subcutaneous administration of a potent synthetic melanotropin. JAMA. 1991;266(19):2730–2736. [PubMed] [Google Scholar]
- 24.Dorr RT, Lines R, Levine N, et al. Evaluation of melanotan-II, a superpotent cyclic melanotropic peptide in a pilot phase-I clinical study. Life Sci. 1996;58(20):1777–1784. doi: 10.1016/0024-3205(96)00160-9. [DOI] [PubMed] [Google Scholar]
- 25.Dorr RT, Ertl G, Levine N, et al. Effects of a superpotent melanotropic peptide in combination with solar UV radiation on tanning of the skin in human volunteers. Arch Dermatol. 2004;140(7):827. doi: 10.1001/archderm.140.7.827. [DOI] [PubMed] [Google Scholar]
- 26.Thestrup-Pedersen K, Søndergaard K. Melatonin used for tanning induces and augments lentigines and naevi. Acta Derm Venereol. 2010;90(6):643–644. doi: 10.2340/00015555-0928. [DOI] [PubMed] [Google Scholar]
- 27.Ferrándiz-Pulido C, Fernández-Figueras MT, Quer A, Ferrándiz C. An eruptive pigmented lesion after melanotan injection. Clin Exp Dermatol. 2011;36(7):801–802. doi: 10.1111/j.1365-2230.2011.04045.x. [DOI] [PubMed] [Google Scholar]
- 28.Loir B, Pérez Sánchez C, Ghanem G, Lozano J, García-Borrón J, Jiménez-Cervantes C. Expression of the MC1 receptor gene in normal and malignant human melanocytes. A semiquantitative RT-PCR study. Cell Mol Biol (Noisy-le-grand) 1999;45(7):1083–1092. [PubMed] [Google Scholar]
- 29.Schiller M, Brzoska T, Böhm M, et al. Solar-simulated ultraviolet radiation-induced upregulation of the melanocortin-1 receptor, proopiomelanocortin, and α-melanocyte-stimulating hormone in human epidermis in vivo. J Invest Dermatol. 2004;122(2):468–476. doi: 10.1046/j.0022-202X.2004.22239.x. [DOI] [PubMed] [Google Scholar]
- 30.Scott MC, Suzuki I, Abdel-Malek Z. Regulation of the human melanocortin 1 receptor expression in epidermal melanocytes by paracrine and endocrine factors and by ultraviolet radiation. Pigment Cell Res. 2002;15(6):433–439. doi: 10.1034/j.1600-0749.2002.02051.x. [DOI] [PubMed] [Google Scholar]
- 31.Palicka GA, Rhodes AR. Acral melanocytic nevi: prevalence and distribution of gross morphologic features in white and black adults. Arch Dermatol. 2010;146(10):1085–1094. doi: 10.1001/archdermatol.2010.299. [DOI] [PubMed] [Google Scholar]
- 32.Diamond LE, Earle DC, Rosen RC, Willett MS, Molinoff PB. Double-blind, placebo-controlled evaluation of the safety, pharmacokinetic properties and pharmacodynamic effects of intranasal PT-141, a melanocortin receptor agonist, in healthy males and patients with mild-to-moderate erectile dysfunction. Int J Impot Res. 2004;16(1):51–59. doi: 10.1038/sj.ijir.3901139. [DOI] [PubMed] [Google Scholar]
- 33.Wessells H, Gralnek D, Dorr R, Hruby VJ, Hadley ME, Levine N. Effect of an alpha-melanocyte stimulating hormone analog on penile erection and sexual desire in men with organic erectile dysfunction. Urology. 2000;56(4):641–646. doi: 10.1016/s0090-4295(00)00680-4. [DOI] [PubMed] [Google Scholar]
- 34.Safarinejad MR. Evaluation of the safety and efficacy of bremelanotide, a melanocortin receptor agonist, in female subjects with arousal disorder: a double-blind placebo-controlled, fixed dose, randomized study. J Sex Med. 2008;5(4):887–897. doi: 10.1111/j.1743-6109.2007.00698.x. [DOI] [PubMed] [Google Scholar]
- 35.Krishna R, Wong P, Stevens C, et al. Lack of erectogenic activity of a novel selective melanocortin-4 receptor agonist in a clinical experimental model. J Clin Pharmacol. 2008;48(10):1237–1241. doi: 10.1177/0091270008320925. [DOI] [PubMed] [Google Scholar]
- 36.Krishna R, Gumbiner B, Stevens C, et al. Potent and selective agonism of the melanocortin receptor 4 With MK-0493 does not induce weight loss in obese human subjects: energy intake predicts lack of weight loss efficacy. Clin Pharmacol Ther. 2009;86(6):659–666. doi: 10.1038/clpt.2009.167. [DOI] [PubMed] [Google Scholar]
- 37.Greenfield JR, Miller JW, Keogh JM, et al. Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med. 2009;360(1):44–52. doi: 10.1056/NEJMoa0803085. [DOI] [PubMed] [Google Scholar]
- 38.Balthasar N, Dalgaard LT, Lee CE, et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123(3):493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]