

Introduction
Pulmonary fibrosis describes the process of progressive scarring and ultimately structural changes to the basement membranes of the alveolar interstitium, leading to impaired gas exchange and respiratory insufficiency. This process occurs in the interstitial lung diseases (ILDs), of which there are greater than 150 disease types. ILDs are also known as diffuse parenchymal lung diseases (DPLDs) which include not only the alveolar interstitium, but adjacent structures such as capillaries, terminal and respiratory bronchioles, and lymphatics of the bronchovascular bundle and interlobular septae [1] The ILDs are a clinically heterogeneous group with both known and unknown causes of fibrosis. The known causes of pulmonary fibrosis include occupational and environmental exposures (both inorganic and organic), drug toxicities, and collagen vascular diseases [2]. Pulmonary fibrosis can also occur in the absence of these causes as an isolated finding limited to the lungs. This latter category is known as the idiopathic interstitial pneumonias (IIPs) which were defined by the American Thoracic Society in 2002 into distinct classifications [3]. The IIPs are composed of several subtypes, identified by their clinico-radiological- pathological classification. The IIPs consist of idiopathic pulmonary fibrosis (IPF), nonspecific interstitial pneumonia (NSIP), respiratory bronchiolitis-associated interstitial lung disease (RB-ILD), desquamative interstitial pneumonia (DIP), cryptogenic organizing pneumonia (COP), acute interstitial pneumonia (AIP), and lymphoid interstitial pneumonia (LIP). Of these, IPF is the most common and the most severe [4]. IPF is associated with fibrosing interstitial pneumonia of unknown cause, a usual interstitial pneumonia pattern (UIP) on surgical lung biopsy and/or high-resolution CT scan (HRCT), an older age of onset, (usually >50 years), and exclusion of other known causes of ILD. IPF affects men greater than women and has been associated with cigarette smoking. IPF has a median survival of 3–5 years [2] and currently there is no approved treatment other than lung transplantation [4]. While IPF is thought to be a disease of unknown origin, risk factors such as cigarette smoking, certain environmental exposures (e.g. metal dusts, wood dusts, farming, bird-raising, hair-dressing), microbial agents (viruses-EBV, CMV, HHV-7,8), gastroesophageal reflux and genetic factors are risk factors for this disease [4].
Several lines of evidence exist that pulmonary fibrosis has a genetic basis. First, it is known that pulmonary fibrosis is a consistent finding in other rare pleiotropic hereditary conditions. Pulmonary fibrosis is a reported finding in such heritable disorders such as Hermansky-Pudlak syndrome (HPS) [5], neurofibromatosis (NF) [6], Gaucher’s disease [7], familial hypocalciuric hypercalcemia [8], tuberous sclerosis (TS) [9] and dyskeratosis congenita (DC) [10]. Second, it has been shown that there is variability in the developmental outcome of pulmonary fibrosis amongst workers exposed to similar exposures, whether it was fibrogenic dusts or organic antigens [11, 12]. Third, mouse models have shown a difference in the development of pulmonary fibrosis when exposed to bleomycin or asbestos depending upon whether they were wild-type mice or inbred strains [13, 14]. The most compelling epidemiological evidence for a genetic basis is the observation of familial clustering of pulmonary fibrosis. This clustering has been reported in monozygotic twins raised apart [15–17], in consecutive generations of families [18], and in family members separated at an early age [19]. The majority of pedigrees exhibit an autosomal dominant with reduced penetrance transmission pattern [18, 20] although one report proposed an autosomal recessive pattern [20]. The recognition of familial clustering has since led to a designation of a familial form of pulmonary fibrosis, familial interstitial pneumonia (FIP).
Familial Interstitial Pneumonia (FIP)
While no formal definition has been proposed for familial forms of pulmonary fibrosis, multiple attempts to describe it have been made. Previous descriptions were termed “familial IPF” in which at least two or more primary biological family members (parent, sibling or child) had a clinical classification of IPF [2]. Past studies [21, 22] have reported sporadic IPF and familial IPF to be indistinguishable from a clinical, radiological and pathological standpoint and to have similar male: female (males>females) affection ratios, yet differences in age of onset such as younger age of onset (≈ 55 years in familial IPF vs. ≈ 67 years in sporadic IPF) have been reported [22]. However, we reported up to 45% of pedigrees demonstrated heterogeneous diagnoses of greater than one type of IIP within families with a median age of onset of 68 yrs similar to sporadic IPF [18]. The various forms of IIP were thought to have clinical, radiological and pathological distinction (Figure 1). In addition, this report [18] showed an independent association with cigarette smoking within families, significant enough to be considered a risk factor for individuals with a family history of IPF. The findings suggested a possible common underlying pathogenic mechanism amongst the different forms of IIPs within families. The authors suggested that a gene in families may predispose individuals to develop pulmonary fibrosis, but it would take another event such as another gene, environmental exposure or a medical condition to result in a specific type of IIP, a possible “two-hit” model. Due to these findings, a more accurate description of these families would conceivably be familial interstitial pneumonia (FIP). In our studies, we have defined FIP as the presence of two or more cases of definite or probable idiopathic interstitial pneumonia within three generations of a family, and at least one of those cases having the phenotype of IPF [23]. Familial IPF is best used to refer to a pedigree where all affected members have an IPF phenotype. In contrast, familial interstitial pneumonia (FIP) refers to a pedigree with two or more cases of IIP (i.e. NSIP and IPF in a pedigree, or 2 or more cases of IPF). Consequently, studies comparing sporadic IPF to familial IPF must be interpreted with these distinctions in mind. However, these studies do not clearly distinguish multiple types of IIP within a pedigree and therefore, FIP would be a better term to encompass the familial forms of pulmonary fibrosis.
Figure 1.
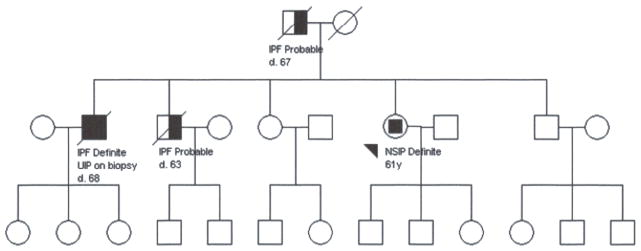
The pedigree demonstrates multiple affected individuals in the second generation and male to male transmission consistent with autosomal dominant inheritance. The third generation is unaffected at the time of ascertainment reflecting late age of disease onset. Phenotypic heterogeneity is reflected by both NSIP and UIP in two siblings.
Epidemiology
No studies on incidence of FIP have been performed, but an early report from Marshall et al [22] in 2000 estimated familial cases to be 0.5–2.2% of IIP cases and the prevalence to be 1.34 cases per 106 in the United Kingdom in the largest cohort of families reported to date at the time. Hodgson et al [24] reported a prevalence of 3.3–3.7% in Finland. Subsequent reports have since showed a higher prevalence, showing a possible underestimation of FIP. Vanderbilt University Hospital lung transplant center described 19% of patients undergoing lung transplant reported a positive family history of ILD [25]. In a Netherlands ILD clinic, 10% of IIP patients reported a positive family history [26]. The National Institute of Respiratory Diseases in Mexico has published that 20% of IPF patients reported a family history [27]. From these latter studies it appears that FIP is more prevalent than originally reported. But until a formal definition is established and a better way of tracking or confirming the diagnoses within families occurs, the true prevalence and incidence of FIP will remain an estimate.
Genetic Associations
Many studies have been performed searching for genetic associations in IPF. These studies have focused on candidate genes involved with such areas as inflammation, matrix turnover, oxidative stress, profibrotic molecules, the surfactant pathways, coagulation pathway genes and matrix metalloproteinase amongst others[4]. Outcomes in association with IPF vary depending on the gene studied, and further investigations should be pursued in larger cohorts.
The familial studies have been the most successful in providing insight into genetic associations, although for the most part the rare genetic mutations reported represent a very low frequency amongst families with FIP, indicating a need for further research. Those genes with rare variants identified to date from studies on FIP include surfactant genes SFTPC, SFTPA2, and telomerase genes TERT and TERC. A candidate gene, ELMOD2 has been linked to FIP in a Finnish cohort. More recently, a common variant in the promoter polymorphism of mucin gene MUC5B has been identified with a higher proportion of FIP and IPF (Table 1).
Table 1.
Genes associated with Familial Interstitial Fibrosis
| Gene Symbol (OMIM#)1 ,2 | Name | Protein | Normal Function | Chromosome Position | Variant Type(s) | Inheritance Patterns in FIP |
|---|---|---|---|---|---|---|
| SFTPC (#178620) | Surfactant, Pulmonary-Associated Protein C | SPC | Essential for lung function after birth, prevent alveolar collapse | 8p21.3 | Intronic (splice site), Exonic (missense) | AD with reduced penetrance |
| SFTPA2 (#178642) | Surfactant, Pulmonary-Associated Protein A2 | Isoform of Surfactant Protein A | Host defense of lung | 10q22.3 | Missense | AD |
| TERT (#187270) | Telomerase Reverse Transcriptase | Telomerase | Telomere integrity during cell division | 5p15.33 | Missense, frameshift, intronic (splice-site) | AD with reduced penetrance |
| TERC (#602322) | Telomerase RNA Component | Telomerase | Telomere integrity during cell division | 3q26.2 | Missense | AD with reduced penetrance |
| ELMOD2 (#610196) | Elmo Domain-Containing Protein 2 | Elmo Domain-Containing Protein 2 | Gene expression in all tissues, including lung | 4q31.1 | None detected | unknown |
| MUC5B (#60070) | MUCIN 5, Subtype B | Mucin protein | Major gel-forming mucin in mucus, contributes to normal saliva, lung, and cervical mucus | 11p15.5 | Common promoter SNP rs35705950, G>T | Common variant |
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim/
National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov/
Surfactant Proteins
Pulmonary surfactant is a mixture of proteins and phospholipids secreted by type II alveolar cells and are responsible for lowering surface tension and preventing collapse at the air-liquid interface of the alveoli. Disorders of surfactant proteins are more commonly seen in the pediatric forms of interstitial lung diseases (PILD). PILD is a heterogeneous group of disorders with some overlap of their adult counterparts, but are generally thought to be separate entities due to differences in frequency, clinical presentation and spectrum of disease. Overlapping histological patterns such as DIP, NSIP and COP in adult and pediatric ILD exist, but do not seem to carry out a similar clinical course [28, 29]. Pulmonary surfactant deficiency was first described in newborns with severe respiratory distress syndrome (RDS) as early as 1959 [30]and since then mutations in surfactant protein A (SFTPA), surfactant protein B (SFTPB), ATP-binding cassette A3 (ABCA3) and surfactant protein C (SFTPC) have been identified in inherited forms of ILD [31]. SFTPB and ABCA3 deficiency are considered recessive disorders and most often present with lethality in the newborn period with RDS. ABCA3 can contain milder mutations with a clinical course that extends into later childhood. SFTPC mutations are dominantly inherited if not de novo, with reduced penetrance. SFTPC mutations have been reported in families with both PILD and FIP, and have been reported as a rare cause of sporadic cases of IPF. SFTPA mutations have been described in adult onset FIP linked with cancer and sporadic cases of IPF. Compound heterozygosity for a SFTPC mutation and ABCA3 mutation has been reported and shown to be a possible modifier of SFTPC associated lung disease [32].
Surfactant Protein C (SFTPC)
The surfactant protein C gene (SFTPC) encodes a hydrophobic, pulmonary protein known as SPC which is essential for lung function and produced exclusively by type II alveolar cells. SPC is one of multiple surfactant proteins that make up pulmonary surfactant which prevents lung collapse by lowering surface tension in the alveoli. SFTPC mutations are not considered to be strongly represented in FIP, but can be considered in earlier onset cases of FIP such as family members affected under the age of 40, or families with both pediatric and adult-onset of ILD. In 2001, Nogee et al [33] reported a kindred involving a female infant with biopsy proven cellular NSIP, whose mother had been diagnosed with DIP, and the maternal grandfather had died from lung disease of unknown cause. Tissue samples were obtained from both infant and mother and genetic and protein analyses were performed. The results showed a heterozygous SFTPC mutation (c.460 +1 G>A) in the infant and mother, which was consistent with the autosomal dominant transmission inheritance pattern in the family. In 2002 Thomas et al [34]reported a large kindred spanning adults with UIP and children with cellular NSIP to have heterogyzosity for a SFTPC mutation. The mutation found in this family was exon 5 + 128 T>A/L188Q, a transversion mutation associated with both cellular NSIP in children and adults with UIP which segregated with disease. It is estimated that based on these studies, SFTPC mutations only account for approximately 3% of FIP [35]. Recently in a Dutch study, van Moorsel reported 5 out of 20 unrelated patients with FIP had SFTPC mutations, (25%, CI 10–49%) [26], the highest frequency of SFTPC mutations yet reported in FIP. Due to the above findings, SFTPC mutations have been explored in the sporadic forms of IIP in at least two studies, in which only sequence variations were found and of those, only one had been previously reported in an infant with lung disease [36, 37]. Therefore, SFTPC mutations are not considered to be a significant cause of sporadic types of IPF, perhaps as low as 1% or less, but may vary based on ethnicity.
Surfactant Protein A2 (SFTPA2)
Wang et al [38] reported a large kindred with adult early-onset pulmonary fibrosis and lung cancer cosegregating in an autosomal dominant pattern. The lung cancer was described as adenocarcinoma with features of broncholalveolar cell carcinoma (BAC). Other related family members without fibrosis had either pulmonary adenocarcinoma or BAC. Whole genome linkage analysis was performed on 29 family members of this kindred and affected family members (affected = pulmonary fibrosis and/or lung cancer) shared an identical-by-descent region on chromosome 10. Candidate genes in the region were narrowed to genes encoding surfactant proteins A and D, which are both highly expressed in the lungs and whose function is to maintain the alveoli of the lung. There are two isoforms of surfactant protein A, SP-A1 and SP-A2 and each are encoded by genes SFTPA1 and SFTPA2 respectively. These genes resided in close proximity within the described linked region and are >90% identical in their coding regions. Further sequencing of these two genes in the proband and other affected family members yielded a misense mutation (c.692G>T, G231V) in a heterozygous state in SFTPA2. Interrogation of 58 other probands in their familial cohort for SFTPA1 and SFTPA2 mutations revealed a heterozygous mutation (c.593T>C, F198S) in the gene SFTPA2 in a proband with IPF and lung cancer. No rare variants were reported in SP-D and the rare and common variants in SP-A were found not to impact protein function. While an important finding, mutations in surfactant proteins are not well represented in the FIP population.
Telomerase Mutations (TERT/TERC)
As previously mentioned, pulmonary fibrosis is a pleiotropic finding in several other known hereditary syndromes. In one of those syndromes, Dyskeratosis Congenita (DC), pulmonary disease described as reduced diffusion capacity and/or restrictive defect was reported to occur in 20% of patients [10]. DC is a rare inherited syndrome classically known for a triad of mucocutaneous findings of oral leukoplakia, skin hyperpigmentation and nail dystrophy. Other multisystem disorders in addition to the pulmonary disease were recognized in patients with DC such as bone marrow failures, liver disease, osteoporosis, premature graying/hair loss and malignancy. DC may manifest as X-linked, autosomal recessive or autosomal dominant forms. Mortality in DC results from bone marrow failure disorders (60–70%), pulmonary disease (10–15%), and malignancy (10%), and it is now recognized that bone marrow disorders may be the presenting features of DC rather than the classical mucocutaneous features [39]. The genes involved in the development of DC have been discovered and thus far are made up of seven genes (DKC1, TERC, TERT, NOP10, NHP2, TINF2, and TCAB1) in what is known as the telomerase pathway. DKC1 is responsible for the X-linked forms of DC while TERC, TERT and TINF2 are implicated in the autosomal dominant forms of DC.
Telomerase is an enzyme that serves in the end-replication repair of telomeres during cell division. Telomeres are composed of DNA sequences of TTAGGG tandem repeats located at chromosomal ends. During cell division, asymmetric replication of DNA leaves newly synthesized DNA strands shorter than the original template. Telomerase solves this end-replication problem by providing the addition of repetitive sequences to the telomeres to maintain chromosomal integrity. Telomerase has several critical subunits, two of which are telomerase reverse transcriptase (hTERT) and a specialized RNA component hTR, or hTERC (telomerase RNA component) [40, 41]. These two components are encoded by genes TERT and TERC respectively. Of these two critical components, hTERC is expressed in all tissues, but hTERT is usually repressed in somatic cells causing those telomeres to shorten with each cell cycle. However, germ-line cells, stem cells and immortal cancer cells continue to express hTERT for longevity [42]. Absence or dysfunction of telomerase causes the telomeres to shorten with each cell division, until the cell eventually undergoes apoptosis. This process has been implicated in age-related diseases [43].
Telomerase mutations in TERT and TERC associated with FIP were first reported by two independent groups in 2007 using different approaches. In the first study, Armanios et al [44] performed a candidate gene study of TERT and TERC in 73 probands of families with FIP in their cohort and discovered that 8% had heterozygous mutations in TERT or TERC. None of these mutations were found in healthy controls. By studying the families, they found the pattern of inheritance to be consistent with autosomal dominance with reduced penetrance, and that the onset of disease may be age-dependent. Mutant telomerase was further shown to be associated with shortened telomeres in the probands and asymptomatic mutation carriers compared to healthy controls and noncarrier relatives. Telomere length in peripheral blood mononuclear cells for mutation carriers fell below the 10th percentile of the controls. In the second study, Tsakiri et al [45] used genome-wide linkage analysis in two large FIP families and discovered several mutations within a linked region to chromosome 5, in which lies the TERT gene. Further sequencing of TERT in probands of 44 additional FIP families discovered additional mutations, and applying the same techniques to 44 individuals with sporadic ILD revealed a missense mutation in a similar codon found in one of the TERT mutation FIP families. Knowing that telomerase is made up of the two essential components of TERT and TERC and that TERC mutations have been reported in patients with DC, they sequenced TERC in their 46 FIP probands and their cases of sporadic pulmonary fibrosis and found one heterozygous mutation in an FIP proband. Overall, functional analyses of the combined mutations showed variable activity of the telomerase enzyme, ranging from zero to 100% of wild-type activity. Telomere measurements in mutation carriers (symptomatic or asymptomatic) indicated a significant shorter telomere length over family members without mutations, and all mutations resulted in shorter telomeres. Collectively, these two studies showed that heterozygous telomerase mutations in TERT and TERC were associated with pulmonary fibrosis in 8–15% of families with FIP without the classical mucocutaneous features of DC. Interestingly, a few of the families between the two studies did exhibit extrapulmonary disease in the spectrum of DC including mild to moderate anemia, axial osteoporosis, liver cirrhosis of unknown etiology and aplastic anemia. Further studies of a large familial cohort found 18% of FIP was associated with TERT mutations [46].
As discussed previously, mutations in TERT and TERC have been linked to shortened telomeres in mutation carriers with FIP, DC and bone marrow failure [40]. Further studies of both sporadic and familial cases of pulmonary fibrosis have revealed heterozygous mutations in TERT and TERC in 1–3% of sporadic IPF/IIP cases and shortened telomeres in a significant fraction of affected individuals from FIP, sporadic IPF and IIP in the absence of TERT and TERC mutations [43, 47]. This may indicate that other genetic loci or phenotypic characteristics (older age or smoking) may well be involved in the process of telomere shortening in patients with FIP or sporadic IIP. In one report, family members who did not inherit a TERT mutation had shorter telomere lengths than controls, demonstrating that factors other than mutations in TERT (such as mutations in other genes, epigenetic inheritance, or a shared environment) can affect telomere length [46]. A large GWAS, primarily consisting of patients with sporadic IIP, identified common variants in both TERT and TERC to be associated with increased risk of developing pulmonary fibrosis demonstrates that common variants in TERT or TERC with smaller effect-size also predispose to pulmonary fibrosis [48]. Telomere length has been reported to be a heritable trait with parental affects [47]. Along with FIP, rare variants and mutations in the genes TERT and TERC have also been reported in aplastic anemia (along with shortened telomeres) [49, 50], myelodysplastic syndrome [51], liver disease [52] and acute leukemia [41], all disorders previously described in families with DC. Families with FIP as described previously may have one or more of these diagnoses as pleiotropic manifestations of telomerase mutations or shortened telomeres. The spectrum of disease phenotypes associated with mutations in TERT and TERC has been put forth as a “telomere syndrome” or a “telomeropathy” recognizing telomere-mediated disease to have an underlying common etiology [40, 41] (Figure 2). In more than one report, IPF was the most common manifestations of telomerase syndromes [40, 53]. Shortened telomeres in the presence or absence of telomerase mutations may be the reason FIP and the sporadic IIPs occur at a later age of onset. Furthermore, mutations in TERT and TERC, and heritability of a shorter telomere set-point in families may explain earlier diagnoses of family members with FIP.
Figure 2.

The pedigree demonstrates the pleiotropic manifestations of telomere-mediated disease in FIP. Family members may be affected with one or more phenotypes, with IPF being the most common manifestation. A thorough family history should include inquiries regarding telomere-mediated diseases as seen in Dyskeratosis Congenita (DC).
ELMOD2
ELMOD2 was first described to be associated with FIP after Hodgson et al [54] performed a genome-wide linkage analysis in six families with FIP from Finland. Five of those families originated from the southeastern area of Finland, where there is a higher frequency of both sporadic and familial pulmonary fibrosis than in the rest of the country, and the prevalence of FIP was 50-fold higher than the rest of the country. The sixth family was not from this region. Three loci were of interest on chromosomes 3, 4, and 13, and further investigation led to an additional two loci on chromosomes 9 and 12 of interest due to a shared haplotype. Fine mapping, interrogation of these regions and of shared haplotypes amongst the six pedigrees and the addition of remaining multiplex families found that 8 of 24 families shared a haplotype on chromosome 4. A candidate gene, ELMOD2 was found to be located within the shared haplotype; however no mutations in ELMOD2 were discovered. ELMOD2 is expressed in the lung, but not exclusively, and the authors performed expression studies of ELMOD2 in both healthy lung and in IPF lung samples. They observed significantly lower expression levels of ELMOD2 among patients with IPF than controls. These findings provided some evidence for ELMOD2 to be a susceptibility gene for FIP. However, these findings have not been validated in other populations. Follow-up studies in Finland set out to determine the function of ELMOD2 [55]. By thoroughly interrogating the gene-signaling pathways of ELMOD2 in human alveolar epithelial cells, they determined that ELMOD2 regulated interferon-related antiviral responses and there was a decrease in the expression of ELMOD2 in response to viral infection. Furthermore, they were able to show that ELMOD2 was expressed in lung epithelial cells as well as alveolar macrophages, both of which are cell types infected by respiratory viruses. These results supported their hypothesis that viral infections may be involved in the development of IPF.
MUC5B
A genome-wide linkage study in 82 multiplex families with FIP led to the discovery of a linked region on chromosome 11 [23]. Once the linkage to chromosome 11 was established, further interrogation of the region by fine mapping was performed in an independent cohort of FIP, sporadic IPF and controls. This narrowed the region to 11p15 in which there were several SNPs in the region of a mucin gene cluster inclusive of MUC2, MUC5AC and MUC5B genes. Further evaluation in a third independent cohort of FIP (n=85), IPF (n=494) and controls (n=332) revealed a MUC5B promoter-region SNP (rs35705950, G>T) located three kilobases (3kb) upstream of the transcription start site of MUC5B that was the most significantly associated with both sporadic IPF and FIP. The minor allele (T) was present at a frequency of 34% in FIP cases, 38% in IPF cases, and 9% in controls. Odds ratios for disease in FIP heterozygotes and homozygotes were 6.8 and 20.8 respectively and for sporadic IPF heterozygotes and homozygotes the odds ratios were 9.0 and 21.8 respectively. To show the effect of the rs35705950 SNP on MUC5B gene expression, the authors utilized lung tissue from 33 subjects with IPF and 47 unaffected subjects and showed MUC5B expression to be 14.1 times higher in IPF subjects as controls. There was also a 37.4-fold increase in MUC5B expression in unaffected subjects who carried at least one copy of the SNP in contrast to those subjects with two copies of the wild-type allele (G). This work was further validated in four independent cohorts [48, 56–58]. While it is unclear what role the MUC5B promoter SNP is playing in the development of IPF and FIP, Seibold et al [23] hypothesize that the MUC5B SNP could be impairing mucosal defense due to excess production of MUC5B protein, interfering in alveolar repair, or directly causing lung injury; either one or all three working together. The common variant rs35705950 in the putative promoter of the MUC5B gene has been the most strongly associated genetic variant to IPF and FIP to date. Furthermore, this finding was the first to implicate mucin genes in the development of IPF and FIP, presenting an opportunity of future targeted research and therapeutics. In a follow-up study, association between the MUC5B promoter polymorphism and IPF showed improved survival amongst patients who were heterozygous or homozygous carriers of the MUC5B SNP (either GT or TT) when compared to those with the wild type genotype (GG). This study utilized retrospective data on two independent cohorts of sporadic IPF from an international clinical trial in IPF and from an ILD clinic at the University of Chicago [58]. A genetic test developed for the detection of the promoter SNP could be used to identify persons at risk for IPF/FIP, in particular family members, and aiding clinicians in potentially assessing survival of patients with IPF once the clinical implications of the survival data is determined.
Genome-wide association studies (GWAS) in IPF
In a report by a Japanese group who performed a genome-wide association study (GWAS) on 159 patients with sporadic IPF and 934 controls, they found nine single nucleotide polymorphisms (SNPs) that were significant in relation to IPF [59]. To verify their findings, they further interrogated the nine SNPs in an independent case-control study from additional samples of IPF and controls from a Japanese biobank. They found evidence of association between a common SNP found in the TERT gene (rs2736100) and development of IPF. The authors suggested the common SNP in TERT contributed significantly to susceptibility of sporadic IPF, thereby validating TERT’s presence in IPF and FIP. A GWAS with 542 IPF cases and 542 controls identified three SNPs in TOLLIP (rs111521887, rs5743894, rs5743890) and one SNP MUC5B (rs35705950) in the adjacent mucin gene cluster on chromosome 11p15, one SPPL2C SNP (rs17690703), and one MDGA2 SNP (rs7144383) that reached genome wide significance (P, 5 × 10 −8). All SNPs except rs7044383 were reproduced in validation in an independent population[57]. The authors interpreted the TOLLIP associations to be independent of MUC5B rs35705950 based on low linkage disequilibrium. Having the TOLLIP minor allele rs5743890 was associated with decreased risk of IPF and increased mortality. Recently, a large GWAS study of individuals with fibrotic IIPs inclusive of FIP (N=1,616) and controls (N= 4,683) and then subsequently validated with a replication population of cases (N= 876) and controls (N=1,890) confirmed association with genes TERT, MUC5B and a region near TERC as well as identifying seven newly identified loci [48]. The most highly associated SNP in this study was in the promoter region of MUC5B in 11p15 in which that region has been previously reported to be associated with IPF and FIP [23]. In addition, the SNP previously reported in TERT by the Japanese study previously mentioned [59], rs2736100 was validated in this study, and another SNP in TERT, rs2853676 was discovered. In addition, the TERC gene was again implicated in its presence in IIPs and FIP in this study as a SNP near the TERC gene, rs1881984. These findings show that MUC5B, TERT and TERC maintain a consistent representation in FIP and IPF and must be important in the development of these diseases, whether acting as rare or common variants. The study identified seven novel loci (FAM13A at 4q22, DSP at 6p24, OBFC1 at 10q24, ATP11A at 13q34, DPP9 at 19p13 and regions 7q22 and 15q14–15) that in addition to the confirmed genes above appear to be involved in host defense, cell-cell adhesion and DNA repair.
Genetic Testing
Clinical genetic testing is available for genes TERT, TERC, additional genes in the telomerase pathway, SFTPC, and MUC5B sequencing. Patients with FIP are appropriate to test as 8–18% of these patients are expected to have rare variants in the SFTPC, TERT or TERC genes, and 50–60% of individuals with FIP have variants in the promoter of MUC5B. Patients presenting with IIP who have extrapulmonary features associated with a telomeropathy, such as hematological failures, liver cirrhosis, osteoporosis, and premature graying (the latter with variable expressivity) with or without a family history of lung fibrosis are also candidates for sequencing in TERT and TERC. As stated previously, family members may be pleiotropically affected with some members manifesting IPF, liver cirrhosis or another manifestation of Dyskeratosis Congenita. IPF is the most common finding in these telomeropathies. In sporadic cases of IPF without a personal or family history of clinical manifestations of a telomeropathy, TERT and TERC sequencing would not necessarily be appropriate as there is a 1–3% chance that a rare variant or mutation would be found. Patients presenting with IPF under the age of 40 with or without a family history of lung fibrosis may be considered for SFTPC sequencing, especially if pediatric ILD is reported in the family. However, the representation of SFTPC mutations in FIP remains lower than that of the telomerase genes at approximately 3%. It is most appropriate to test a patient with FIP over unaffected family members for TERT/TERC and SFTPC initially. Genetic testing for SFTPA2 and the MUC5B SNP are not readily available clinically, but this may change in the near future.
Genetic counseling is highly recommended prior to and after genetic testing. Risk to other family members to develop FIP is a major concern in the families who have multiple members affected. Since FIP appears to follow an autosomal dominant with reduced penetrance transmission in families, it represents a challenge to provide specific risk numbers to family members, especially in the absence of a known mutation in the family. The exact penetrance of FIP is unknown. Therefore, risk to offspring of patients with FIP is up to 50%, but with reduced penetrance is most likely less. Second and third degree relatives of patients diagnosed with FIP are presumed to have lower risk, but increased over the general population. Based on the MUC5B SNP, unaffected family members of FIP patients with one or two copies of the SNP could have anywhere from a 6.8 to 20.8 fold risk, respectively, to develop pulmonary fibrosis.
Clinicians, patients and family members may be concerned for the potential of genetic discrimination in genetic testing for FIP. Genetic discrimination is a common fear in the general population when unaffected family members are at risk for a familial disease and genetic testing is available. The Genetic Information and Nondiscrimination Act (GINA) passed in 2008 [60] is a federal law that prevents genetic test results of an individual or a family member, family health history, or receipt of genetic services (e.g. genetic counseling, genetic testing, genetic research) from being used to discriminate against a person for health care coverage and employment. GINA prevents health insurance companies from raising premiums, setting eligibility or coverage, premium and contribution amounts based on genetic information as described above. It is important to note that this law does not extend to elective types of insurance such as life insurance. Therefore, if a specific mutation is known in the family, unaffected family members could be tested for the same mutation to find out if they are carriers without full risk of it impacting their health insurance based on the coverage GINA provides. Genetic counseling surrounding the issues of genetic testing and questions on genetic discrimination is recommended prior to and after any genetic testing is performed.
Conclusion
Based upon the vast amount of evidence presented, there leaves little doubt that FIP is a recognized entity. FIP may present clinically similar to sporadic cases of IIP with the family history being the only distinguishing feature, or can present with younger age of onset. The younger age of onset may be a misrepresentation due to heightened awareness of the disease amongst family members who may seek early screening and are diagnosed early. The genetics of the two appear to be telling us something different, as the majority of genes discovered have some overlap between FIP and the sporadic forms of IIP. It is very possible that patients with FIP are more heavily weighted with genetic risk factors than their sporadic counterparts. For example, rare variants in TERT or TERC that have large deleterious effects are more penetrant, and have a higher representation in FIP than in sporadic IPF (8–18% vs. 1–3% respectively), whereas common variants with lower penetrance and small effect may be more prevalent in sporadic IPF. Overall, the genes discovered through familial studies and large GWA studies have confirmed at least three genes, TERT, TERC and MUC5B as key genes, as well as several other loci in the development of pulmonary fibrosis. This alone shows that FIP and IIPs in general are multigenic and cannot be labeled as a single-gene disorder. These, along with evidence of environmental components support a multifactorial cause for lung fibrosis within FIP as well as the sporadic forms of IIP.
Acknowledgments
Sources of Funding
Funding Sources include National Institute of Health, Veterans Administration, and National Heart, Lung and Blood Institute.
Footnotes
Conflicts of Interest
M. Steele and J. Talbert report no conflicts of interests or disclosures. D. Schwartz has a patent pending on utilizing MUC5B for the diagnosis and prognosis of pulmonary fibrosis.
Contributor Information
Janet L. Talbert, Email: talbertj@njhealth.org, Certified Genetic Counselor, National Jewish Health, 1400 Jackson Street, F107, Denver, CO 80206, (P) 303-398-1022, (F) 303-270-2240.
David A. Schwartz, Email: david.schwartz@ucdenver.edu, Professor and Chair of Medicine, University of Colorado, School of Medicine, Director, Center for Genes, Environment, and Health, National Jewish Health.
Mark P. Steele, Email: mark.p.steele@vanderbilt.edu, Professor of Medicine, Division of Allergy, Pulmonary, and Critical Care Medicine, Medical Director, Lung Transplant Program, Vanderbilt University School of Medicine.
Bibliography
- 1.Steele MP, Brown KK. Genetic predisposition to respiratory diseases: infiltrative lung diseases. Respiration. 2007;74:601–8. doi: 10.1159/000110204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Society AT. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS) Am J Respir Crit Care Med. 2000;161:646–64. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- 3.American Thoracic S and European Respiratory S. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165:277–304. doi: 10.1164/ajrccm.165.2.ats01. [DOI] [PubMed] [Google Scholar]
- 4.Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DePinho RA, Kaplan KL. The Hermansky-Pudlak syndrome. Report of three cases and review of pathophysiology and management considerations. Medicine (Baltimore) 1985;64:192–202. [PubMed] [Google Scholar]
- 6.Riccardi VM. Von Recklinghausen neurofibromatosis. N Engl J Med. 1981;305:1617–27. doi: 10.1056/NEJM198112313052704. [DOI] [PubMed] [Google Scholar]
- 7.Schneider EL, Epstein CJ, Kaback MJ, et al. Severe pulmonary involvement in adult Gaucher’s disease. Report of three cases and review of the literature. Am J Med. 1977;63:475–80. doi: 10.1016/0002-9343(77)90288-1. [DOI] [PubMed] [Google Scholar]
- 8.Auwerx J, Boogaerts M, Ceuppens JL, et al. Defective host defence mechanisms in a family with hypocalciuric hypercalcaemia and coexisting interstitial lung disease. Clin Exp Immunol. 1985;62:57–64. [PMC free article] [PubMed] [Google Scholar]
- 9.Harris JO, Waltuck BL, Swenson EW. The pathophysiology of the lungs in tuberous sclerosis. A case report and literature review. Am Rev Respir Dis. 1969;100:379–87. doi: 10.1164/arrd.1969.100.3.379. [DOI] [PubMed] [Google Scholar]
- 10.Dokal I. Dyskeratosis congenita in all its forms. British Journal of Haematology. 2000;110:768–79. doi: 10.1046/j.1365-2141.2000.02109.x. [DOI] [PubMed] [Google Scholar]
- 11.Polakoff PL, Horn BR, Scherer OR. Prevalence of radiographic abnormalities among Northern California shipyard workers. Ann N Y Acad Sci. 1979;330:333–9. doi: 10.1111/j.1749-6632.1979.tb18736.x. [DOI] [PubMed] [Google Scholar]
- 12.Selikoff IJ, Lilis R, Nicholson WJ. Asbestos disease in United States shipyards. Ann N Y Acad Sci. 1979;330:295–311. doi: 10.1111/j.1749-6632.1979.tb18732.x. [DOI] [PubMed] [Google Scholar]
- 13.Ortiz LA, Lasky J, Hamilton RF, Jr, et al. Expression of TNF and the necessity of TNF receptors in bleomycin-induced lung injury in mice. Exp Lung Res. 1998;24:721–43. doi: 10.3109/01902149809099592. [DOI] [PubMed] [Google Scholar]
- 14.Warshamana GS, Pociask DA, Sime P, et al. Susceptibility to asbestos-induced and transforming growth factor-beta1-induced fibroproliferative lung disease in two strains of mice. Am J Respir Cell Mol Biol. 2002;27:705–13. doi: 10.1165/rcmb.2002-0096OC. [DOI] [PubMed] [Google Scholar]
- 15.Javaheri S, Lederer DH, Pella JA, et al. Idiopathic pulmonary fibrosis in monozygotic twins. The importance of genetic predisposition. Chest. 1980;78:591–4. doi: 10.1378/chest.78.4.591. [DOI] [PubMed] [Google Scholar]
- 16.Bonanni PP, Frymoyer JW, Jacox RF. A Family Study of Idiopathic Pulmonary Fibrosis. A Possible Dysproteinemic and Genetically Determined Disease. Am J Med. 1965;39:411–21. doi: 10.1016/0002-9343(65)90208-1. [DOI] [PubMed] [Google Scholar]
- 17.Solliday NH, Williams JA, Gaensler EA, et al. Familial chronic interstitial pneumonia. Am Rev Respir Dis. 1973;108:193–204. doi: 10.1164/arrd.1973.108.2.193. [DOI] [PubMed] [Google Scholar]
- 18.Steele MP, Speer MC, Loyd JE, et al. Clinical and pathologic features of familial interstitial pneumonia. Am J Respir Crit Care Med. 2005;172:1146–52. doi: 10.1164/rccm.200408-1104OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swaye P, Van Ordstrand HS, McCormack LJ, et al. Familial Hamman-Rich syndrome. Report of eight cases. Dis Chest. 1969;55:7–12. doi: 10.1378/chest.55.1.7. [DOI] [PubMed] [Google Scholar]
- 20.Tsukahara M, Kajii T. Interstitial pulmonary fibrosis in two sisters. Possible autosomal recessive inheritance. Jinrui Idengaku Zasshi. 1983;28:263–7. doi: 10.1007/BF01876789. [DOI] [PubMed] [Google Scholar]
- 21.Lee HL, Ryu JH, Wittmer MH, et al. Familial idiopathic pulmonary fibrosis: clinical features and outcome. Chest. 2005;127:2034–41. doi: 10.1378/chest.127.6.2034. [DOI] [PubMed] [Google Scholar]
- 22.Marshall RP, Puddicombe A, Cookson WO, et al. Adult familial cryptogenic fibrosing alveolitis in the United Kingdom. Thorax. 2000;55:143–6. doi: 10.1136/thorax.55.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seibold MA, Wise AL, Speer MC, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364:1503–12. doi: 10.1056/NEJMoa1013660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hodgson U, Laitinen T, Tukiainen P. Nationwide prevalence of sporadic and familial idiopathic pulmonary fibrosis: evidence of founder effect among multiplex families in Finland. Thorax. 2002;57:338–42. doi: 10.1136/thorax.57.4.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loyd JE. Pulmonary fibrosis in families. American Journal of Respiratory Cell and Molecular Biology. 2003;29:S47–50. [PubMed] [Google Scholar]
- 26.van Moorsel CH, van Oosterhout MF, Barlo NP, et al. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am J Respir Crit Care Med. 2010;182:1419–25. doi: 10.1164/rccm.200906-0953OC. [DOI] [PubMed] [Google Scholar]
- 27.Garcia-Sancho C, Buendia-Roldan I, Fernandez-Plata MR, et al. Familial pulmonary fibrosis is the strongest risk factor for idiopathic pulmonary fibrosis. Respir Med. 2011;105:1902–7. doi: 10.1016/j.rmed.2011.08.022. [DOI] [PubMed] [Google Scholar]
- 28.Fan LL, Deterding RR, Langston C. Pediatric interstitial lung disease revisited. Pediatr Pulmonol. 2004;38:369–78. doi: 10.1002/ppul.20114. [DOI] [PubMed] [Google Scholar]
- 29.Deutsch GH, Young LR, Deterding RR, et al. Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med. 2007;176:1120–8. doi: 10.1164/rccm.200703-393OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamvas A, Cole FS, Nogee LM. Genetic disorders of surfactant proteins. Neonatology. 2007;91:311–7. doi: 10.1159/000101347. [DOI] [PubMed] [Google Scholar]
- 31.Whitsett JA, Wert SE, Weaver TE. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annual Review of Medicine. 2010;61:105–19. doi: 10.1146/annurev.med.60.041807.123500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bullard JE, Nogee LM. Heterozygosity for ABCA3 mutations modifies the severity of lung disease associated with a surfactant protein C gene (SFTPC) mutation. Pediatr Res. 2007;62:176–9. doi: 10.1203/PDR.0b013e3180a72588. [DOI] [PubMed] [Google Scholar]
- 33.Nogee LM, Dunbar AE, 3rd, Wert SE, et al. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. 2001;344:573–9. doi: 10.1056/NEJM200102223440805. [DOI] [PubMed] [Google Scholar]
- 34.Thomas AQ, Lane K, Phillips J, 3rd, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med. 2002;165:1322–8. doi: 10.1164/rccm.200112-123OC. [DOI] [PubMed] [Google Scholar]
- 35.Fernandez BA, Fox G, Bhatia R, et al. A Newfoundland cohort of familial and sporadic idiopathic pulmonary fibrosis patients: clinical and genetic features. Respir Res. 2012;13:64. doi: 10.1186/1465-9921-13-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Markart P, Ruppert C, Wygrecka M, et al. Surfactant protein C mutations in sporadic forms of idiopathic interstitial pneumonias. Eur Respir J. 2007;29:134–7. doi: 10.1183/09031936.00034406. [DOI] [PubMed] [Google Scholar]
- 37.Lawson WE, Grant SW, Ambrosini V, et al. Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax. 2004;59:977–80. doi: 10.1136/thx.2004.026336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Kuan PJ, Xing C, et al. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet. 2009;84:52–9. doi: 10.1016/j.ajhg.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dokal I. Dyskeratosis congenita. Hematology Am Soc Hematol Educ Program. 2011;2011:480–6. doi: 10.1182/asheducation-2011.1.480. [DOI] [PubMed] [Google Scholar]
- 40.Armanios M. Syndromes of telomere shortening. Annu Rev Genomics Hum Genet. 2009;10:45–61. doi: 10.1146/annurev-genom-082908-150046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Calado RT, Young NS. Telomere diseases. N Engl J Med. 2009;361:2353–65. doi: 10.1056/NEJMra0903373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garcia CK, Wright WE, Shay JW. Human diseases of telomerase dysfunction: insights into tissue aging. Nucleic Acids Res. 2007;35:7406–16. doi: 10.1093/nar/gkm644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alder JK, Chen JJ, Lancaster L, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 2008;105:13051–6. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Armanios MY, Chen JJ, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356:1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 45.Tsakiri KD, Cronkhite JT, Kuan PJ, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci USA. 2007;104:7552–7557. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diaz de Leon A, Cronkhite JT, Katzenstein AL, et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS One. 2010;5:e10680. doi: 10.1371/journal.pone.0010680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cronkhite JT, Xing C, Raghu G, et al. Telomere shortening in familial and sporadic pulmonary fibrosis. American Journal of Respiratory and Critical Care Medicine. 2008;178:729–37. doi: 10.1164/rccm.200804-550OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fingerlin TE, Murphy E, Zhang W, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013 doi: 10.1038/ng.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vulliamy T, Marrone A, Dokal I, et al. Association between aplastic anaemia and mutations in telomerase RNA. Lancet. 2002;359:2168–70. doi: 10.1016/S0140-6736(02)09087-6. [DOI] [PubMed] [Google Scholar]
- 50.Yamaguchi H, Calado RT, Ly H, et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med. 2005;352:1413–24. doi: 10.1056/NEJMoa042980. [DOI] [PubMed] [Google Scholar]
- 51.Yamaguchi H, Baerlocher GM, Lansdorp PM, et al. Mutations of the human telomerase RNA gene (TERC) in aplastic anemia and myelodysplastic syndrome. Blood. 2003;102:916–8. doi: 10.1182/blood-2003-01-0335. [DOI] [PubMed] [Google Scholar]
- 52.Calado RT, Brudno J, Mehta P, et al. Constitutional telomerase mutations are genetic risk factors for cirrhosis. Hepatology. 2011;53:1600–7. doi: 10.1002/hep.24173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Armanios M. Telomerase and idiopathic pulmonary fibrosis. Mutation Research. 2012;730:52–8. doi: 10.1016/j.mrfmmm.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hodgson U, Pulkkinen V, Dixon M, et al. ELMOD2 is a candidate gene for familial idiopathic pulmonary fibrosis. Am J Hum Genet. 2006;79:149–54. doi: 10.1086/504639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pulkkinen V, Bruce S, Rintahaka J, et al. ELMOD2, a candidate gene for idiopathic pulmonary fibrosis, regulates antiviral responses. FASEB Journal. 2010;24:1167–77. doi: 10.1096/fj.09-138545. [DOI] [PubMed] [Google Scholar]
- 56.Zhang Y, Noth I, Garcia JG, et al. A variant in the promoter of MUC5B and idiopathic pulmonary fibrosis. N Engl J Med. 2011;364:1576–7. doi: 10.1056/NEJMc1013504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Noth Iea. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Resp Med. 2013;1:309–17. doi: 10.1016/S2213-2600(13)70045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peljto AL, Zhang Y, Fingerlin TE, et al. Association Between the MUC5B Promoter Polymorphism and Survival in Patients With Idiopathic Pulmonary Fibrosis. JAMA. 2013:1–8. doi: 10.1001/jama.2013.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mushiroda T, Wattanapokayakit S, Takahashi A, et al. A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J Med Genet. 2008;45:654–6. doi: 10.1136/jmg.2008.057356. [DOI] [PubMed] [Google Scholar]
- 60.Genetic Information Nondiscrimination Act of 2008, Public Law 110–233, 42 USC, 122 STAT 181. 2008.