

Highlights
-
•
We compared an enzyme cascade in vivo as well as in vitro.
-
•
Mechanistic insights were gained for the presented cascade.
-
•
Monomer production for biorenewable polyesters was investigated.
-
•
Pathway construction with non-related enzymes was performed.
Abbreviations: ADH, alcohol dehydrogenase; BVMO, Baeyer–Villiger monooxygenase; CFE, cell free extract; CHMO, cyclohexanone monooxygenase; ERED, enoate reductase; G6P, glucose 6-phosphate; G6P-DH, glucose 6-phosphate dehydrogenase; GC, gas chromatography; IPTG, isopropyl-β-d-thiogalactopyranoside; LB, Lysogeny broth; NAD+, nicotinamide adenine dinucleotide; NADH, nicotinamide adenine dinucleotide (reduced); NADP+, nicotinamide adenine dinucleotide phosphate; NADPH, nicotinamide adenine dinucleotide phosphate (reduced); OD, optical density; PMSF, phenylmethylsulfonyl fluoride; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; TB, terrific broth
Keywords: Multi-step biocatalysis, Baeyer–Villiger monooxygenase, Biorenewable polyesters, Enoate reductase, Alcohol dehydrogenase
Abstract
An artificial enzyme cascade composed of an alcohol dehydrogenase, an enoate reductase and a Baeyer–Villiger monooxygenase was investigated in vitro to gain deeper mechanistic insights and understand the assets and drawbacks of this multi-step biocatalysis. Several substrates composed of different structural motifs were examined and provided access to functionalized chiral compounds in high yields (up to >99%) and optical purities (up to >99%). Hence, the applicability of the presented enzymatic cascade was exploited for the synthesis of biorenewable polyesters.
1. Introduction
The use of enzymes for more efficient and cleaner chemical syntheses has increased during the past decades (Lopez-Gallego and Schmidt-Dannert, 2010, Ricca et al., 2011). Intense investigations in the field of biocatalysis led to a versatile biocatalytic toolbox composed of numerous enzymes which are capable of transforming a wide range of functional groups and concomitantly displaying a very promiscuous substrate profile. By exploiting the manifoldness of enzymes with their different catalytic entities it is possible to compose new synthetic pathways on the basis of the ‘retrosynthetic approach’ (Warren and Wyatt, 2008), which only very recently has been proposed as novel concept also for biocatalysis (Turner and O’Reilly, 2013). Since chemo-, regio- and enantioselective single step biotransformations have become indispensable for the production of fine chemicals (Meyer et al., 2007, Wohlgemuth, 2010) the next logical step in organic synthesis was to increase complexity and to introduce multi-enzymatic systems obtaining excess to more intricate synthetic schemes (Lopez-Gallego and Schmidt-Dannert, 2010). Take a leaf out of nature the combination of several (bio)catalysts in one-pot reactions without the need to isolate intermediates and to circumvent protection group strategies is also of environmental significance (Ricca et al., 2011) due to reduced waste production and increased atom-efficiency.
The use of whole cells (in vivo) as well as the application of isolated enzymes (in vitro) have been introduced as two major approaches first and were successfully implemented in the synthesis of complex molecules. Using an in vivo whole cell system has the major advantage of a fully functional living organism providing an efficient enzyme production machinery, increased enzyme stability due to their natural environment, and an automated cofactor regeneration system (Lopez-Gallego and Schmidt-Dannert, 2010). Additionally, no enzyme purification is required. However, in a multiple enzyme system usually preliminary genetic manipulations are required and therefore a different expertise is necessary.
On the contrary the in vitro system is less complex and reaction conditions are easier to control. Reaction parameters like substrate or enzyme concentrations, pH-value, temperature, and co-solvent additives can be varied and optimized in a more systematic way. Additionally, storage capabilities of cell free extracts, purified enzyme solutions or lyophilisates are often increased compared to living cells. Hence, all assets and drawbacks at hand, from a chemist's point of view an in vitro system with cell free extracts, although rather expensive cofactors have to be added, is often preferred. Furthermore, no special lab tools are required once the extracts are produced.
As previously reported we implemented an in vivo enzymatic toolbox for redox cascade reactions (Oberleitner et al., 2013) composed of three enzymes. Within this study we demonstrated the flexibility and applicability of the cascade in asymmetric synthesis. In particular we investigated a three step cascade (Scheme 1) whereas the first reaction from an unsaturated cyclic alcohol to the corresponding ketone is catalyzed by two different alcohol dehydrogenases either LK-ADH from Lactobacillus kefir (Weckbecker and Hummel, 2006) or RR-ADH from Rhodococcus ruber (Stampfer et al., 2003). For the subsequent reduction of the unsaturated double bond we chose the well-known OYE1 from Saccharomyces carlsbergensis (Padhi et al., 2009) or XenB from Pseudomonas sp. (Oberleitner et al., 2013). For the last step, a Baeyer–Villiger oxidation, we employed CHMO from Acinetobacter sp. (Donoghue et al., 1976).
Scheme 1.

Scheme of the interplay of enzymes, substrates and cofactors.
We emphasized to gain better understanding of each individual single reaction and the interaction of all enzymes in the cascade by in vitro experiments. Several single reactions as well as cascade reactions of all studied substrates were performed in assays with cell free extracts containing enzymes heterologously produced in Escherichia coli. In this paper we investigated the limits of the cascade by performing reactions at increased substrate concentrations and potential application in the production of biorenewable polyesters.
2. Materials and methods
2.1. Materials
All chemicals were purchased from Fluka (Buchs, Switzerland), Sigma (Steinheim, Germany), Merck (Darmstadt, Germany), VWR (Hannover, Germany) or Carl Roth (Karlsruhe, Germany) and were used without further purification unless otherwise specified.
2.2. Bacterial strains, plasmids and general culture conditions
E. coli BL21 (DE3) [fhuA2 [lon] ompT gal (λ DE3) [dcm] ΔhsdS] was purchased from New England Biolabs (Beverly, MA, USA). The plasmids pET22_LK-ADH (GenBank: AY267012.1) and pRR-ADH (GenBank: CAD36475.1) were provided by Prof. Hummel (University of Düsseldorf, Germany). For expression of the CHMO gene (GenBank: BAA86293.1) pMM04 was used (Chen et al., 1999). pGas_XenB (GenBank: KF055345) was obtained as described elsewhere (Oberleitner et al., 2013). Transformation of E. coli strains with the plasmids was carried out by the heat shock method as described by Chung et al. (1989).
E. coli strains were routinely cultured in LB medium (adapted from Bertani (1951): 10 g/l peptone, 5 g/l yeast extract, 10 g/l NaCl) and, if necessary, supplemented with ampicillin (100 μg/ml), chloramphenicol (25 μg/ml) or kanamycin (50 μg/ml), and incubated in baffled Erlenmeyer flasks in orbital shakers (InforsHT Multitron 2 Standard) at 200 rpm and 37 °C. Bacteria on agar plates were incubated in a Heraeus Instruments FunctionLine incubator under air. All materials and biotransformation media were sterilized by autoclaving at 121 °C for 20 min. Aqueous stock solutions were sterilized by filtration through 0.20 μm syringe filters. Agar plates were prepared with LB medium supplemented by 1.5% w/v agar.
2.3. Gene expression of different enzymes
In general, the cultivation for enzyme production was carried out by inoculation of TB (or LB) media (2006) supplied with the specified antibiotic with an overnight culture (pregrown in LB media) to an OD600 of 0.05. Cultivation was continued at 37 °C until OD600 = 0.6–0.7 was reached. Then, expression of genes coding for heterologous enzymes was induced either by addition of 0.1 mM IPTG or 0.2% Rhamnose. Addition of 1 mM ZnCl2 prior to induction (OD600 = 0.4) was necessary for RR-ADH (Stampfer et al., 2003). The expression was continued overnight at the specific temperatures (for further details see supplementary information Table S1). A value of 0.43 g/l cdw per OD600 unit was used to calculate biomass concentrations from optical density measurements.
2.3.1. CFE preparation
After the end of cultivation 200–400 ml of the culture were centrifuged at 6000 × g at 4 °C for 15 min. The supernatant was decanted and cells were resuspended in 10–20 ml of 50 mM TRIS–HCl at pH 7.5 and centrifuged again at 6000 × g at 4 °C for 15 min. After resuspension of cells in 5–10 ml 50 mM TRIS–HCl pH 7.5 supplemented with 0.1 mM PMSF sonication was performed with a Bandelin KE 76 sonotrode connected to a Bandelin Sonopuls HD 3200 wave generator. The cell suspension was repeatedly (9 cycles) sonicated for 5 s followed by 55 s pause on ice. Insoluble cell residues were removed by centrifugation at 14,000 × g at 4 °C for 40 min. The supernatant cell free extract was sterilized by 0.2 μm filtration. Protein concentration was determined with Bio-Rad Protein Assay.
2.3.2. Enzyme activity measurements
Activity measurements were performed spectrophotometrically on the Jasco V550 by observing NADPH consumption or NADPH formation for 120 s at 340 nm in 1 ml cuvettes. The activities of LK-ADH against 2-cyclohexen-1-ol (1a), 2-cyclohexen-1-one (1b), cyclohexanone (1c), cyclohexanol (1c-ol) were measured in Tris–HCl buffer (50 mM, pH 7.5) at RT with 4 mM of substrate and 0.3 mM NADPH or 0.3 mM NADP+ in triplicates. Crude cell extract solution (1–20 μl) with 2.75 mg/ml protein content was used, and the reaction mixture was adjusted to 1 ml with buffer.
2.4. GC-analysis
Samples (100 μl) were extracted with 500 μl ethyl acetate supplemented with 1 mM methyl benzoate serving as internal standard. Conversion and product purity were determined by GC using a Thermo Finnigan Focus GC/DSQ II equipped with a standard capillary column (BGB5, 30 m × 0.32 mm ID). Enantiomeric excess was determined by GC analysis using a BGB175 (30 m × 0.25 mm ID, 0.25 μm film) or BGB173 (30 m × 0.25 mm ID, 0.25 μm film) column on a ThermoQuest Trace GC 2000 and a Thermo Focus GC, both with FID detector. Amounts were validated using calibration curves. The GC methods used are listed in Table S2 in the supplementary information.
2.5. Substrates
2-cyclohexen-1-ol (1a), 4-methylcyclohex-2-en-1-ol (2a), 2-methylcyclohex-2-en-1-ol (3a), 3-methylcyclohex-2-en-1-ol (4a), (1R,5R)-carveol (5a), (1S,5R)-carveol (6a) and (1S,5S)-carveol (7a) were used as starting materials for the different cascades. The synthesis of all substrates used was described previously (Oberleitner et al., 2013).
2.6. Biocatalysis
Two different reaction setups were chosen for biotransformations: (i) a sequential cascade reaction where the three enzymes were added one after another, enabling a discrete initiation phase for each reaction step; (ii) a non-sequential approach with all enzymes, cofactors and recycling system mixed at once; this setup is more comparable with the in vivo approach previously reported.
All reactions were performed in closed glass vials at 24 °C and were shaken at 200 rpm.
2.6.1. Sequential cascade reaction
The first reaction of the cascade, the oxidation of the unsaturated alcohol to the ketone, was performed with CFE of LK-ADH with a total protein concentration of 5 mg/ml. Reaction volume was 1 ml in a 50 mM Tris–HCl buffer at pH 7.5. NADP+ (4.8 mM, 1.2 equiv.) was added as cofactor and the reaction started upon addition of 4 mM unsaturated alcohol (as a 1 M ethanolic stock solution). First sampling was conducted within a few minutes after mixing of the components (time point zero; ). After one hour another sample was taken and then the ERED was added to the reaction mixture to a final total protein concentration of 5 mg/ml. In this step there was no need to add additional cofactor as NADPH was produced in the LK-ADH reaction. After another hour reaction progress was sampled and the BVMO (CFE with 5 mg/ml total protein), 4.8 mM NADP+, 8 mM glucose-6-phosphate (G6P) and 1 U/ml glucose-6-phosphate dehydrogenase (G6P-DH) as cofactor recycling system were added. Sampling was performed after another hour (3 h) and continued up to 30 h.
2.6.2. Non-sequential addition cascade reaction
For all cascade reactions with RR-ADH the non-sequential addition of the enzymes was performed, due to NAD+ dependence of this ADH. CFEs of the ADH, the ERED and the BVMO were used at a total protein concentration of 5 mg/ml. 4.8 mM NAD+ (1.2 equiv.) and 4.8 mM NADP+ (1.2 equiv.) were added; 2 equiv. of G6P (8 mM) and 1 U/ml of G6P-DH served as cofactor recycling system. All CFEs, the cofactors, the recycling system and 4 mM substrate were mixed together at once in 50 mM Tris–HCl (pH 7.5).
The non-sequential cascade applying LK-ADH was performed accordingly just without addition of NAD+. Samples were taken at regular time intervals to be comparable to the sequential approach.
2.6.3. Substrate concentration study
Cascade reactions at increased substrate concentrations were performed according to the non-sequential addition method. To keep conditions as similar as possible, higher concentrated stocks of NADP+ and G6P were prepared in the reaction buffer (50 mM Tris–HCl pH 7.5). Substrate concentrations of 4 mM, 10 mM, 25 mM and 50 mM were chosen and amounts of NADP+, G6P and G6P-DH were scaled up, accordingly.
3. Results and discussion
We started our investigations to gain a better understanding of the presented redox cascade by characterizing each individual reaction and several different substrates (Scheme 2). In a first attempt we performed a sequential approach by adding all enzymes and their appropriate cofactors after defined time intervals. Our model reaction (Fig. 1) was the transformation of 2-cyclohexen-1-ol (1a) to the corresponding ɛ-caprolactone (1d).
Scheme 2.

Substrates investigated for the in vitro redox cascade.
Fig. 1.

Sequential in vitro cascade starting from cyclohexen-2-ol (1a).
After the addition of LK-ADH several interesting observations were made: The oxidation process to the corresponding α,β-unsaturated 2-cyclohexen-1-one was surprisingly fast. Even very rapid sample preparation after initialization of the reaction resulted in significant conversion of the starting material to the desired product. As a consequence t0 was defined as in this study. A slight background reaction caused by the native enoate reductase from E. coli (NemR (Mueller et al., 2010)) was noticed and traces of cyclohexanone (1c) were detected. Moreover, another by-product was found and could be identified as cyclohexanol (1c-ol) leading to the conclusion that the LK-ADH also accepts cyclohexanone as a substrate. In the second phase of the biotransformation the enoate reductase OYE1 was added and the formation of the undesired by-product cyclohexanol (1c-ol) was highly pronounced. Obviously, the second enzyme led to an equilibrium shift of the first oxidation step by converting the product (1b) to the saturated ketone (1c); this yielded in more than 95% conversion of the starting material. In the final stage of the cascade, formation of the desired end product was observed (1d) after addition of CHMOAcineto. Interestingly, the unfavored side product (1c-ol) was consumed as well and the enzyme cascade reached completion after already 6 h of reaction time (Table 1).
Table 1.
Relative conversion of substrates.
| Substrate | Product | Conversion (%) | ee/de (%) | Time (h) | |
|---|---|---|---|---|---|
| 2-Cyclohexen-1-ol |
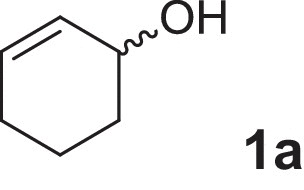 1a 1a
|
1d | >99 | n.a.a | 6 |
| 4-Methylcyclohex-2-en-1-ol | 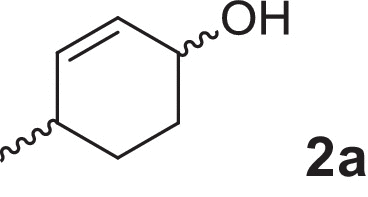 |
2d | >99 | >99 | 20 |
| 2-Methylcyclohex-2-en-1-ol | 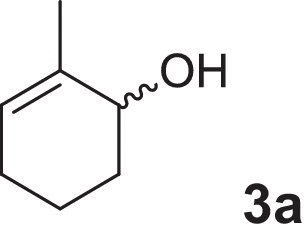 |
3d | 74 | 93 | 30 |
| 3-Methylcyclohex-2-en-1-ol | 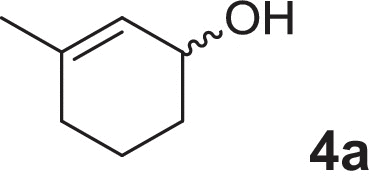 |
4d | 82 | >99 | 20 |
| 4eb | – | – | – | ||
| (1R,5R)-Carveol | 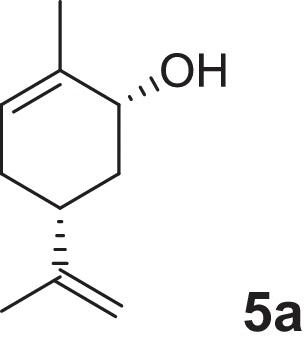 |
5d | 55 | >99 | 30 |
| (1S,5R)-Carveol | 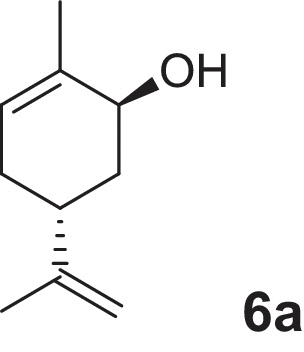 |
6d | Sequential: 99 | >99 | 6 |
| Non-sequential: 88 | >99 | 20 | |||
| (1S,5S)-Carveol | 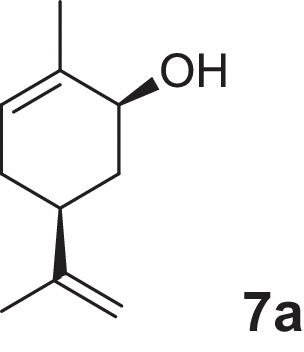 |
7d | 97 | >99 | 6 |
All substrates tested in 4 mM scale.
Not applicable.
No formation of the proximal lactone 4e was observed.
Consequently we investigated at the individual LK-ADH reactions in more detail. Based on simple activity measurements, supported by calculations of relative Gibbs free energy for the individual reactions at 25 °C and pH 7 in aqueous media we were able to describe and explain our observations (Table 2).
Table 2.
Relative Gibbs free energies refer to back and forward reactions compared to enzyme activities.
| Reaction | a (kcal/mol) | Enzyme activity (U/mg) |
|---|---|---|
| 2-Cyclohexen-1-ol + NADP+ → 2-Cyclohexen-1-one + NADPH | −0.0143 | 0.91 |
| 2-Cyclohexen-1-one + NADPH → 2-Cyclohexen-1-ol + NADP+ | +0.0143 | 0.29 |
| Cyclohexanol + NADP+ → Cyclohexanone + NADPH | +1.507 | 1.59 |
| Cyclohexanone + NADPH → Cyclohexanol + NADP+ | −1.507 | 30.08 |
ΔfG0 were estimated based on GCM software (http://lcsbweb.epfl.ch/GCMWebSite/Login.php) (Jankowski et al., 2008, Mavrovouniotis, 1990, Mavrovouniotis, 1991).
As depicted in Table 2 the oxidation of (1a) to the corresponding α,β unsaturated ketone was slightly favored in terms of thermodynamics (ΔrxnG0 < 0) as well as overall enzyme activity (threefold higher than for the reduction). In the following the first reaction step of the cascade proceeded smoothly and provided enough starting material for the subsequent (irreversible at the used conditions) enoate reduction. A slightly different picture was observed for the “side-reaction”. In this particular case the reduction from (1c) to (1c-ol) was preferred. The enzyme activity was almost 19 times higher for this reaction than for the desired oxidation and based on the ΔrxnG0 the oxidation is not spontaneous at all. Due to the fact, that the last step of our investigated reaction sequence includes an irreversible process, a continuous shift of equilibrium takes place and conversions up to 100% can be achieved.
We extended our investigations toward more complex substrates by introducing chirality examining the reaction starting from 4-methylcyclohex-2-en-1-ol (2a). Similar trends as in the model reaction were observed (see supplementary information). Again, the background reaction of NemR led to the formation of the saturated 4-methylcyclohexanone (2c) in the first phase of the enzyme cascade. Subsequent reduction of LK-ADH resulted in less than 10% 4-methylcyclohexanol (2c-ol). Finally, the reaction was finished within 18 h after the addition of CHMOAcineto (Table 1) and gave the desired lactone (2d) in perfect enantioselectivity (>99% ee). In this particular enzyme cascade the low substrate specificity of LK-ADH toward all isomers of the starting material (4a) was remarkable and beneficial for a successful and complete transformation. Inspired by this result we investigated the sequential biotransformation of 2-methylcyclohex-2-en-1-ol (3a). Interestingly LK-ADH showed a distinct selectivity and accepted preferably one enantiomer of the starting material, which was in contrast to the previous substrate. The transformation displayed a kinetic resolution fashion and slowed down significantly after 50% conversion (see supplementary information). On the contrary to the experiments described vide supra no side reaction of NemR was observed. Nonetheless, the back reaction of the intermediate (3c) to the corresponding saturated alcohol (3c-ol) was prominent and yielded in almost 25% of the undesired side product. Due to the addition of the two remaining enzymes the overall yield (Table 1) could be increased to 74% and lactone (3d) was obtained in high optical purity (of 92% ee). The bottleneck of this cascade was explicitly located in the first oxidation step of the LK-ADH. In the final phase of the biotransformation only remaining starting material and desired product could be detected.
A slightly different result was obtained by applying 3-methylcyclohex-2-en-1-ol (4a) to the sequential enzyme cascade. In this particular biotransformation each step turned out to be highly selective with respect to all different substrates and stereochemistry (Fig. 2). The alcohol oxidation of (4a) to the corresponding enone (4b) was rather slow, and intermediate (4b) was not accepted by the enoate reductase NemR at all. After addition of OYE1 saturated ketone (4c) was obtained in about 20% yield whereas no reduction to the corresponding saturated 3-methylcyclohexanol was observed. In the final step the Baeyer–Villiger monooxygenase CHMOAcineto converted (4c) in 82% overall yield toward (4d) with perfect regio- and enantioselectivity (>99% distal lactone, 99% ee).
Fig. 2.

Sequential in vitro cascade of 3-methyl-2-cyclohexen-1-ol (4a).
Finally we were interested in the synthetic application of our cascade concept for the synthesis of dihydrocarvides (5–7d) which were recently investigated as monomers useful for the production of functional biorenewable polyesters (Lowe et al., 2011). For a successful transformation of (1R,5R)-Carveol (5a), a second ADH from R. ruber (RR-ADH) was applied. Although the oxidation step was rather slow and limited the performance of the cascade, we were able to obtain the desired product (5d) after 30 h of reaction time in 55% yield and very high optical purity (>99% ee, Table 1). Neither the formation of (5c) due to NemR nor the reduction of dihydrocarvone (5c) mediated by RR-ADH was observed (see supplementary information). Within the above examples the ADH mediated oxidation often turned out to be the limiting step. In the present case, (1S,5R)-carveol (6a) was an excellent substrate (Fig. 3). After one hour all the starting material (6a) was converted to the first intermediate (6b) and almost no formation of side products (below 3%) was observed.
Fig. 3.

Sequential in vitro cascade starting from (1S,5R)-carveol (6a).
The addition of enoate reductase XenB yielded in 75% consumption of substrate (6b), the formation of dihydrocarvone (6c), and significant amounts of a by-product. This product was identified as an isomer of dihydrocarveol (6c-ol) and its formation confirms the fast reductive activity of LK-ADH toward dihydrocarvone (6c). Application of CHMOAcineto facilitated performance of the cascade by shifting the equilibrium of the ADH-reaction due to dissipation of (6c) up to an overall yield of 99% of (6d) with perfect regio- and stereoselectivity (Table 1).
For a better comparison of the in vitro with the in vivo cascades we further investigated a non-sequential reaction approach by mixing starting material, all enzymes and cofactors at once. By applying this one pot, single operation protocol we observed a significant increase in the production of the undesired by-product dihydrocarveol (6c-ol) in comparison to the sequential cascade of (1S,5R)-carveol (6a) (Fig. 4).
Fig. 4.

(1S,5R)-Carveol – in vitro non sequential cascade.
At first glance competition between LK-ADH and CHMOAcineto for the substrate (6c) took place. One explanation for this significant difference between the sequential and the non-sequential addition of the enzymes might be the cofactor dependency and availability. During the first step of the sequential addition LK-ADH converted carveol (6a) into carvone (6b) upon concomitant production of NADPH. The following XenB mediated reduction of (6b) toward dihydrocarvone (6c) consumed NADPH and yielded in a redox self-sustaining enzymatic cascade. The by-product formation of dihydrocarveol (6c-ol) performed by LK-ADH was limited due to the lack of the appropriate reducing equivalent NADPH. As soon as BVMO and cofactor recycling system were present the reaction was drawn to lactone (6d, Fig. 3). In the non-sequential approach (Fig. 4) where NADPH was readily available from the beginning, the high affinity of LK-ADH toward (2R,5R)-dihydrocarvone (6c) was in direct competition with the Baeyer–Villiger oxidation. The rate limiting step turned out to be the BVMO reaction and the formation of dihydrocarveol (6c-ol) was very prominent. Since the last step of the cascade was explicitly irreversible, the equilibrium was shifted by the presence of the BVMO and the final product lactone (6d) was obtained in 88% yield and >99% ee (Table 1).
Consequently, the non-sequential in vitro transformations were compared to our recently published in vivo approach (Oberleitner et al., 2013). Fig. 5 displays a comparative study between the in vitro and in vivo cascade of 3-methylcyclohex-2-en-1-ol (4a). The overall reaction performances were highly similar, but a faster transformation was observed in vitro.
Fig. 5.

Comparison of in vitro and in vivo results of 3-methyl-2-cyclohexen-1-ol cascade of LK-ADH, OYE1 and CHMOAcineto.
Finally, we investigated different substrate loads of our redox cascade. The substrate of choice was (1S,5S)-carveol (7a) due to its best performance with respect to reaction time and selectivity (Table 1). Standard substrate concentration at 4 mM was tested in comparison with 10, 25 and 50 mM. By increasing the substrate concentration from 4 to 10 mM a significant decrease of the reaction speed was observed. Nonetheless the reaction was nearly completed after 20 h. While the final lactone product (7d), could still be observed (up to 50%) when applying 25 mM concentration, a significantly decreased productivity was detected at 50 mM starting material. This result indicates either a product inhibition of the LK-ADH or a substrate inhibition for the following enoate reductase OYE1. Fig. 6 shows a comparison of all these experiments.
Fig. 6.

Comparison of the influence of different substrate concentrations on the cascade performance (7a–d).
4. Conclusions
In this follow up study we were able to investigate a modular enzymatic multi-step redox toolbox in vitro to gain deeper understanding of each individual reaction step and the overall influence on the cascade performance. Additionally we met the challenge of combining relatively unselective enzymes in the first reaction step, the oxidation of a secondary alcohol to an α,β-unsaturated ketone followed by the promiscuous reduction of these ketones by two EREDs (XenB, OYE1). Finally we exploited the oxygenation capabilities of BVMO CHMOAcineto.
By performing all enzymatic cascades in a sequential approach and depending on the individual substrate, the formation of several by-products was observed. Conclusively, a direct competition of the alcohol dehydrogenase and the Baeyer–Villiger monooxygenase became evident. A detailed picture of our observations can be seen in Scheme 3. The presence of NADPH promotes the reduction of the saturated ketone to the corresponding alcohol as well the formation to the lactone. Due to the fact, that the ADH reaction is reversible–in contrast to the BVMO oxidation–the overall performance of the cascade is slowed down, but not completely inhibited and the reaction equilibrium is shifted toward the favored product side. Our observations were corroborated by calculation of the relative Gibbs free energy for individual reactions and enzyme activities depicted in Scheme 3 and Table 2. (1a ⇆ 1b and 1c ⇆ 1c-ol).
Scheme 3.

Cascade and possible back reaction catalyzed by the ADH.
Additionally, we could demonstrate the applicability of our cascade for the production of monomers for the synthesis of functional biorenewable polyesters. Based on our results the limitations of the cascade are in the restricted substrate acceptance of the performed ADH.
Future work will focus on the optimization of reaction conditions, adjusting the protein levels according to the transformation rates and implementing an E. coli BL21(DE3) ΔnemA strain for protein production to minimize unwanted side reactions beside our efforts to identify more suitable alcohol dehydrogenases. Furthermore a simple kinetic model for future in vitro as well as in vivo studies for the investigated enzymatic cascade will be established to gain an even deeper understanding of the ongoing processes and identify potential bottlenecks.
Author contributions
M.D.M., U.T.B. and F.R. initiated the project and designed the overall strategy. C.P., cloned the enzymes into various vectors, expressed them to perform initial in vitro experiments on selected substrates and determined enzyme activities; N.O. performed all in vitro as well as in vivo studies, N.O. established the analytical methods; M.D.M., U.T.B. and F.R. cowrote the paper, and all authors read and edited the manuscript.
Conflict of interest statement
All authors declare no conflict of interest.
Acknowledgements
We thank (grant no. Bo1862/6-1), FWF (grant no. I723-N17 & P24483-B20) and Vienna University of Technology (grant no. GEV-TOP163) for financial support of this DACH-Program. We are also grateful to Prof. W. Hummel (University of Düsseldorf) for supplying the genes encoding LK-ADH and RR-ADH and Prof. J.D. Stewart (University of Florida) for the gene encoding OYE1.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.jbiotec.2014.04.008.
Appendix A. Supplementary data
The following are the supplementary data to this article:
References
- Bertani G. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 1951;62:293–300. doi: 10.1128/jb.62.3.293-300.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G., Kayser M.M., Mihovilovic M.D., Mrstik M.E., Martinez C.A., Stewart J.D. Asymmetric oxidations at sulfur catalyzed by engineered strains that overexpress cyclohexanone monooxygenase. New J. Chem. 1999;23:827–832. doi: 10.1039/a902283j. [DOI] [Google Scholar]
- Chung C.T., Niemela S.L., Miller R.H. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc. Natl. Acad. Sci. 1989;86:2172–2175. doi: 10.1073/pnas.86.7.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoghue N.A., Norris D.B., Trudgill P.W. The purification and properties of cyclohexanone oxygenase from nocardia globerula CL1 and acinetobacter NCIB 9871. Eur. J. Biochem. 1976;63:175–192. doi: 10.1111/j.1432-1033.1976.tb10220.x. [DOI] [PubMed] [Google Scholar]
- Jankowski M.D., Henry C.S., Broadbelt L.J., Hatzimanikatis V. Group contribution method for thermodynamic analysis of complex metabolic networks. Biophys. J. 2008;95:1487–1499. doi: 10.1529/biophysj.107.124784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Gallego F., Schmidt-Dannert C. Multi-enzymatic synthesis. Curr. Opin. Chem. Biol. 2010;14:174–183. doi: 10.1016/j.cbpa.2009.11.023. [DOI] [PubMed] [Google Scholar]
- Lowe J.R., Martello M.T., Tolman W.B., Hillmyer M.A. Functional biorenewable polyesters from carvone-derived lactones. Polym. Chem. 2011;2:702–708. doi: 10.1039/C0py00283f. [DOI] [Google Scholar]
- Mavrovouniotis M.L. Group contributions for estimating standard gibbs energies of formation of biochemical-compounds in aqueous-solution. Biotechnol. Bioeng. 1990;36:1070–1082. doi: 10.1002/bit.260361013. [DOI] [PubMed] [Google Scholar]
- Mavrovouniotis M.L. Estimation of standard gibbs energy changes of biotransformations. J. Biol. Chem. 1991;266:14440–14445. [PubMed] [Google Scholar]
- Meyer A., Pellaux R., Panke S. Bioengineering novel in vitro metabolic pathways using synthetic biology. Curr. Opin.Microbiol. 2007;10:246–253. doi: 10.1016/j.mib.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Mueller N.J., Stueckler C., Hauer B., Baudendistel N., Housden H., Bruce N.C., Faber K. The substrate spectra of pentaerythritol tetranitrate reductase, morphinone reductase, N-ethylmaleimide reductase and estrogen-binding protein in the asymmetric bioreduction of activated alkenes. Adv.Synth. Catal. 2010;352:387–394. doi: 10.1002/adsc.200900832. [DOI] [Google Scholar]
- Oberleitner N., Peters C., Muschiol J., Kadow M., Saß S., Bayer T., Schaaf P., Iqbal N., Rudroff F., Mihovilovic M.D., Bornscheuer U.T. An enzymatic toolbox for cascade reactions: a showcase for an in vivo redox sequence in asymmetric synthesis. ChemCatChem. 2013:3524–3528. doi: 10.1002/cctc.201300604. [DOI] [Google Scholar]
- Padhi S.K., Bougioukou D.J., Stewart J.D. Site-saturation mutagenesis of tryptophan 116 of saccharomyces pastorianus old yellow enzyme uncovers stereocomplementary variants. J. Am. Chem. Soc. 2009;131:3271–3280. doi: 10.1021/ja8081389. [DOI] [PubMed] [Google Scholar]
- Ricca E., Brucher B., Schrittwieser J.H. Multi-enzymatic cascade reactions: overview and perspectives. Adv. Synth. Catal. 2011;353:2239–2262. doi: 10.1002/adsc.201100256. [DOI] [Google Scholar]
- Stampfer W., Kosjek B., Kroutil W., Faber K. CIBA Specialty Chemicals Holding Inc. (Ed.); 2003. Purification and Characterization of a Secondary Alcohol Dehydrogenase ADH-A With High Solvent and Temperature Stability from Rhodococcus ruber; p. 56 pp. [Google Scholar]
- Terrific Broth . 2006. Cold Spring Harbor Protocols. pdb.rec8620. [DOI] [Google Scholar]
- Turner N.J., O’Reilly E. Biocatalytic retrosynthesis. Nat. Chem. Biol. 2013;9:285–288. doi: 10.1038/nchembio.1235. [DOI] [PubMed] [Google Scholar]
- Warren S., Wyatt P. Wiley-VCH; Weinheim: 2008. Organic Synthesis. The Disconnection Approach. [Google Scholar]
- Weckbecker A., Hummel W. Cloning, expression, and characterization of an (R)-specific alcohol dehydrogenase from Lactobacillus kefir. Biocatal. Biotransform. 2006;24:380–389. [Google Scholar]
- Wohlgemuth R. Asymmetric biocatalysis with microbial enzymes and cells. Curr. Opin. Microbiol. 2010;13:283–292. doi: 10.1016/j.mib.2010.04.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.