

Abstract
Grain amaranths, edible C4 dicots, produce pseudo-cereals high in lysine. Lysine being one of the most limiting essential amino acids in cereals and C4 photosynthesis being one of the most sought-after phenotypes in protein-rich legume crops, the genome of one of the grain amaranths is likely to play a critical role in crop research. We have sequenced the genome and transcriptome of Amaranthus hypochondriacus, a diploid (2n = 32) belonging to the order Caryophyllales with an estimated genome size of 466 Mb. Of the 411 linkage single-nucleotide polymorphisms (SNPs) reported for grain amaranths, 355 SNPs (86%) are represented in the scaffolds and 74% of the 8.6 billion bases of the sequenced transcriptome map to the genomic scaffolds. The genome of A. hypochondriacus, codes for at least 24,829 proteins, shares the paleohexaploidy event with species under the superorders Rosids and Asterids, harbours 1 SNP in 1,000 bases, and contains 13.76% of repeat elements. Annotation of all the genes in the lysine biosynthetic pathway using comparative genomics and expression analysis offers insights into the high-lysine phenotype. As the first grain species under Caryophyllales and the first C4 dicot genome reported, the work presented here will be beneficial in improving crops and in expanding our understanding of angiosperm evolution.
Keywords: Caryophyllales, grain amaranth, Amaranthus hypochondriacus, lysine biosynthesis, C4 photosynthesis
1. Introduction
Grain amaranths, collectively referring to all the three grain-producing species of Amaranthus under the family Amaranthaceae, are among the most neglected crops in human history. Grain amaranths, A. caudatus, A. cruentus, and A. hypochondriacus, were domesticated for human consumption some 8,000 years ago and were the staple food for the Inca and the Aztec civilizations for thousands of years.1 However, cultivation of grain amaranths remains obscure during modern times following the ban on cultivation. In 1980s, the USA declared grain amaranths ‘the crop of the future’ based on its unique nutritional profile.1–3 The excitement over grain amaranths grew in 1972 when an Australian scientist John Downton reported that the edible cereal-like grains of amaranths display lysine content as high as in milk.1 The importance of this finding is even more crucial considering decades of failed attempts, with existing engineering strategies, to improve the lysine content in rice and wheat because of the inherent inverse correlation between lysine content and yield.4
Grain amaranths are among the rare edible dicots that have evolved to use C4 photosynthesis. It is well established that the species with C4 photosynthesis are more efficient during drought and other environmental stress conditions. Considering that all species producing protein-rich edible grains, such as legumes and nuts, use C3 photosynthesis, C4 photosynthesis must be one of the most sought-after phenotypes in edible dicot crops. Now that the genomes of many C3 legumes have been sequenced,5,6 the genome of one of the grain amaranths is likely to aid in the improvement of protein-rich dicot plants using comparative genomics. Besides, it has been shown that even under carbon dioxide-rich conditions, C4 dicot species under the Amaranthaceae family perform better than respective C3 dicots in terms of dry mass,7 making this species a contestant among other plant species as bioenergy resources.
Grain amaranths are also important contestants for expanding our narrow food base. Decorating the phylogenetic tree of angiosperms based on the Angiosperm Phylogeny Group (APG) III system of classification with edible plants (Supplementary Fig. S1) reveals that all the major clades across the plant kingdom, except Caryophyllales, are loaded with edible crops. The order Caryophyllales (http://www.cs.man.ac.uk/~david/flora/flora.html) comprises 70 genera and 800 species8 and is relatively void of edible crops. All the cereals are classified under monocots, the legumes, fruits, and nuts under Rosids, and the staple root crops such as potato and sweet potato under Asterids. Hence, grain amaranths, classified under the order Caryophyllales, offer an attractive alternative for expanding our food base via supplementing protein-rich cereals in the plant-based diet.
The species under the order Caryophyllales, including grain amaranths, are also unique from other angiosperms with regard to the synthesis of important secondary metabolites.9 Unlike the use of phenylalanine-derived anthocyanins for use as colouring pigments by most angiosperms, species under Caryophyllales use betalains for the same purpose, which are derived from tyrosine.10,11 Since tyrosine is also the precursor for melanin, the colouring pigment in mammals, the genome of a grain amaranth can, perhaps, also be exploited to develop cosmetics and cure for various skin disorders.12 For example, based on the ethnopharmaceutical survey, one of the plant species used by Rwandese to cure skin discolouration is a species under Caryophyllales, Chenopodium ugandae.12 To this end, more recently, the genome of the first Caryophyllales, Beta vulgaris, has been reported and will aid in expanding our understanding of betalains.13 The genome of one of the grain amaranths, whose flowers are already used as food colouration, will add to the study of enzymes involved in betalain synthesis.
Despite the huge potential of grain amaranths with regard to human health and disease, the genome of grain amaranths is yet to be deciphered. A partial transcriptome of A. hypochondriacus, one of the grain amaranths, has been reported, which was generated to understand how species under the Amaranthaceae family respond to environmental stress.14 In another application, limited sequencing of the genome of Amaranthus tuberculatus, a weed variety, has been generated to decipher the mechanism by which this species develops resistance to triple herbicides.15 A more systematic genomic effort on grain amaranths includes creation of BAC libraries for all the three grain species,16 developing microsatellite markers,17 and creating a single-nucleotide polymorphism (SNP)-based linkage map.18
Genomic studies of grain amaranths from the point of view of their unique nutritional profiles and C4 photosynthesis among edible dicots are limited. In various parts of South Asia including Nepal, grain amaranths have been cultivated as staple crops for consumption as an alternative to cereals by the locals.19 Here, we present the draft genome and transcriptome of a landrace that belongs to A. hypochondriacus (Rajgira) obtained from stable lines from the farmers in northern Karnataka, India, to aid in deciphering the genotype behind these important and unique traits from the edible variety.
2. Materials and methods
2.1. Sample collection for genome and transcriptome
We obtained the domesticated grains of A. hypochondriacus from farmers in northern Karnataka growing this as a crop for consumption in the name of ‘Rajgira’ or ‘Rajeera’ throughout southern India. We purchased the seeds of other grain species, A. cruentus and A. caudatus, from Park Seeds sold in the names of ‘Autumn's Touch’ and ‘Love-Lies-Bleeding’, respectively. These three types of seeds were grown in campus grounds in three separate lots for taxonomic purposes (Fig. 1). Large numbers of seeds from the first round from each grain species were whitish or pinkish for A. hypochondriacus, whitish for A. cruentus, and reddish for A. caudatus (shown inset in Fig. 1). Amaranthus hypochondriacus produced two types of plants—one with white and the other with red inflorescences. Separating the seeds of white and red varieties of A. hypochondriacus and growing for over two generations retained the inflorescence of the parent plants in the successive generations, thus suggesting purity of lines obtained from farmers. Also, repeated generations produced not only 100% white seeds for the white plants of A. hypochondriacus, but also the plants looked very similar in size, inflorescence, and yield. We have used the tissues from the plants of A. hypochondriacus with white inflorescence after two generations for sequencing both the genome and the transcriptomes reported here.
Figure 1.

Photographs of the three grain species of Amaranthus grown on campus grounds. The plants from left to right are: A. hypochondriacus, A. cruentus, and A. caudatus, as identified by their taxonomic features including those of the inflorescence and the leaves. Amaranthus hypochondriacus contains both a red variety and a white variety (the sequenced one). Insets are the corresponding seeds obtained after two generations of growing these species on campus grounds. For A. hypochondriacus (left), the seeds from red plants are at the bottom left and those from white plants are shown at the top right.
2.2. Taxonomy of Amaranthus
Amaranthus is a monoecious plant and the inflorescence is a thyrse with a racemose (catkin) main axis and cymose clusters (cymules) of one male flower and two or more female flowers. Amaranthus hypochondriacus is characterized by its apical erect inflorescence, which is heavily loaded with small edible seeds at maturity. The inflorescence can be bright red or whitish green depending on the presence or absence of betalain pigments in the plant variety (left, Fig. 1). The inflorescence of A. cruentus is also apical but with a Christmas-tree like topology that may gradually turn orange at maturity (middle, Fig. 1). The seeds are comparatively larger in size and the stem more robust than that of the other two species. Amaranthus caudatus produces apical but drooping inflorescences that are red in colour and produce small reddish seeds (right, Fig. 1). The plant is also comparatively fragile unlike the other two grain species.
2.3. Karyotyping
The seeds of the plants selected for sequencing were allowed to germinate in water for 24 h. The root tips were incubated at 4°C for 4 h and then fixed using Carnoy's solution. The processed root tips were excised and treated with 1 N HCl for 1 min, followed by treatment with 45% acetic acid for 10 min, staining with 0.2% aceto-orcein along with gentle heat fixing over a spirit lamp, squashing with the thumb, and mounting using 50% glycerol on a microscopic glass slide. The image was captured at 100× magnification under a compound light microscope. As shown in Fig. 2, the number of chromosomes in the selected diploid plant was confirmed to be 32 (n = 16) for sequencing purposes.
Figure 2.
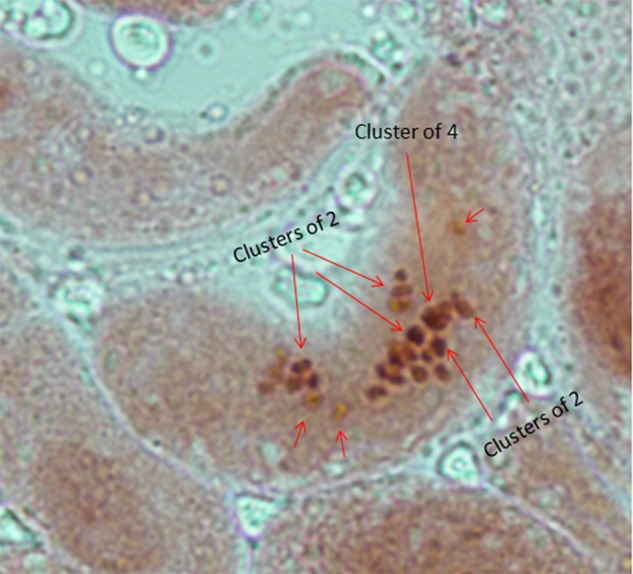
Karyotype of A. hypochondriacus, the species selected for sequencing, showing diploidy with 32 chromosomes in mitotic root tips. The karyotype shows 32 chromosomes under 100× magnification stained with 0.2% aceto-orcein, as would be expected from the diploid root tips of A. hypochondriacus. This figure appears in colour in the online version of DNA Research.
2.4. Extraction of genomic DNA
DNA for sequencing was extracted from fresh leaves of 45- to 50-day-old plants using the DNEasy Mini Plant DNA Extraction kit (Qiagen). The quality of the extracted DNA was checked using fluorometry (Qubit, Invitrogen) and by agarose gel electrophoresis.
2.5. Extraction of total RNA
For RNA extraction, leaf and stem tissues were collected from a pool of more than 10 individuals, from the plants grown from the same seed collection from which the genome was sequenced, at (i) 15 days, (ii) 25 and (iii) 30 days of age, and from (iv) mature seeds. The leaf and stem tissues, on excision, were immediately cleaned with DEPC-treated water, flash-frozen in liquid nitrogen, and stored at −80°C until RNA extraction was done. The mature seeds were collected from the inflorescence which was sun-dried, threshed, cleaned, and then stored at room temperature until RNA extraction was done. Total RNA was extracted using the conventional phenol–chloroform extraction method20 as standardized in the laboratory. Quality assessment of the total RNA was done on 1% agarose gel and the Agilent 2100 Bioanalyzer using high-sensitivity RNA chips.
2.6. Library preparation
For genome sequencing, one paired-end (PE; with an insert size of 300 bp) and four mate-pair (MP; with insert sizes of 1.75, 3, 5 and 10 kb) DNA libraries were made using the TruSeq DNA Sample Preparation Kit (Illumina) by following the manufacturer's low-throughput (LT) protocol. One microgram and 10 µg of the genomic DNA were used for the preparation of the PE and the MP libraries, respectively. DNA molecules were sheared by Covaris S2 according to the manufacturer's instructions and processed as follows.
2.6.1. PE genomic libraries
The fragmented DNA molecules were made blunt by performing end repair in End-Repair Mix (containing T4 polynucleotide kinase for 5′ phosphorylation, T4 DNA polymerase to fill in the 5′ overhangs, and Klenow to remove the 3′ overhangs) for 30 min at 30°C, and purified using Agencourt AMPure XP beads (Beckman Coulter) according to the manufacturer's recommendations. Furthermore, 3′ adenylation of the blunt-end DNA was carried out by incubating with A-Tailing Mix (containing a Klenow fragment) at 37°C for 30 min. Ligation of TruSeq adapters was done using DNA Ligase Mix (containing T4 DNA ligase) by incubating at 30°C for 10 min, and the reaction was stopped by adding Stop Ligase Mix. The adenylated DNA was purified using the Agencourt AMPure XP beads as done previously. Library size selection (for an insert size of 300 bp) was done using agarose gel electrophoresis, and the adapter-ligated purified DNA was further enriched by PCR for 10 cycles using adapter complementary primers (P5 and P7) followed by a clean-up using the Agencourt AMPure XP beads. The quality and quantity of the library were estimated by spectrophotometry (NanoDrop, Thermo Scientific) and fluorometry (Qubit, Invitrogen), and the size distribution was analysed on the Agilent 2100 Bioanalyzer using high-sensitivity DNA chips.
2.6.2. MP genomic libraries
The fragmented DNA molecules were made blunt by performing end repair in End-Repair Mix for 15 min at 20°C, followed by end labelling for 15 min at 20°C using biotin-dNTP mix and purification using the Agencourt AMPure XP beads. DNA of appropriate size (1.75, 3, 5, and 10 kb) was selected using agarose gel electrophoresis and circularized using Circularization Ligase. Any remaining linear DNA in the circularization step was digested using Exonuclease, and the circular DNA was again sheared using Covaris S2 as done previously. The biotinylated DNA fragments were purified using Dynal magnetic M-280 streptavidin beads as per the manufacturer's guidelines. Again, end repair was performed by incubating the biotinylated DNA fragments (immobilized on the streptavidin beads) in End-Repair Mix for 30 min at 20°C and purified; 3′ adenylation was done by incubating the immobilized DNA molecules with A-Tailing Mix at 37°C for 30 min. Ligation of TruSeq adapters was done using DNA Ligase Mix by incubating at 20°C for 15 min and the reaction was stopped by adding Stop Ligase Mix. The biotinylated adapter-ligated immobilized DNA was further enriched by PCR for 18 cycles using adapter complementary primers followed by a clean-up using the Agencourt AMPure XP beads. The quality and quantity of the library were estimated by spectrophotometry (NanoDrop, Thermo Scientific) and fluorometry (Qubit, Invitrogen), and the size distribution was analysed on the Agilent 2100 Bioanalyzer using high-sensitivity DNA chips.
Transcriptome libraries of 155 bp were prepared using the TruSeq RNA Sample Preparation Kit (Illumina) following the manufacturer's LT protocol. Two micrograms of total RNA were subjected to mRNA selection using poly-T oligo-attached magnetic beads using two rounds of purification. The selected poly-A RNA was subjected to fragmentation to sizes between 100 and 300 nt by incubating in fragmentation mix for 8 min at 94°C. First-strand cDNA was synthesized by adding 1 µl of Superscript II reverse transcriptase (Invitrogen) to solution containing primed RNA and first-strand master mix followed by incubation at 25°C for 10 min, 42°C for 50 min, and 70°C for 15 min. The complementary second-strand cDNA was synthesized by incubating first-strand cDNA in second-strand master mix at 16°C for 60 min. End repair was performed to remove the 3′ overhangs and fill the 5′ overhangs by incubating the DNA in End-Repair Mix containing T4 polynucleotide kinase, T4 DNA polymerase, and a large (Klenow) fragment of DNA polymerase I for 30 min at 30°C, and purified using Agencourt AMPure XP beads (Beckman Coulter) according to the manufacturer's recommendations. A-tailing of DNA was performed at 37°C for 30 min with the Klenow fragment followed by ligation of TruSeq adaptors using T4 DNA ligase by incubating at 30°C for 10 min. The reaction was stopped by adding stop-ligase mix and purified by using Agencourt AMPure XP beads. Adaptor-ligated DNA was then subjected to PCR enrichment with adaptor complementary primers for 15 cycles followed by clean-up using Agencourt AMPure XP beads. The quality and quantity of the library were estimated by NanoDrop and fluorometry (Qubit, Invitrogen), and the size distribution was analysed on the Agilent Bioanalyzer using high-sensitivity DNA chips.
2.7 Quantification of prepared library and sequencing
The libraries were quantified using SYBR-Green-based qPCR reagents (Kapa Biosystems). The qPCR results were compared with the predetermined concentration of the PhiX library. Eight picomolar of all the DNA libraries were used for cluster generation in cBot (Illumina). Also, 8 pM of the four RNA libraries prepared were pooled in equimolar ratio and seeded on four lanes of the flow cell for cluster generation. All the libraries were sequenced on Illumina Genome Analyzer IIx following the manufacturer's standard protocols. PE genomic reads of length 75 bases, MP genomic reads of length 36 bases, and PE transcriptomic reads of length 72 bases were generated.
2.8. Assembly
2.8.1. QC of sequence reads
The reads were analysed for their quality and all those with >75% of bases with phred scores of ≥20 and <15 Ns were considered to be of high quality.
2.8.2. Assembly of genomic reads from Illumina GAIIx platform
After testing for quality, the reads were assembled using SOAPdenovo (SOAPdenovo31mer version 1.05).21 Assembly was performed in two stages in a high configuration system with 96 GB RAM. First, 465,246,312 PE reads of 72-mers were assembled using default parameters and a k-mer size of 31. In the other set, using the same parameters, all the reads totalling 869,113,408 both from PE and MP libraries were assembled. The resulting scaffolds were subject to various analyses such as synteny, heterozygosity, Gene Ontology (GO) annotation, repeat analysis, duplication study, and proteome prediction, using available tools and scripts developed in-house.
2.8.3. Assembly of transcriptomic reads from 454 platform
Transcriptomic reads with the seed accession ID of IC 38040 (PI 480569) were downloaded from NCBI-SRA (SRX055331) and assembled into contigs using MIRA.22
2.8.4. Assembly of transcriptomic reads from Illumina platform
The high-quality reads from various tissues were pooled together and used as a set with Trinity (Release 16 February 2013) for assembly.23 Trinity, a de novo transcriptome assembler, uses a fixed k-mer length of 25 and it was run on a high configuration system with 48 GB RAM to generate trinity components.
2.9. Filtering criteria
The coverage of the scaffolds was measured by aligning the genomic scaffolds of A. hypochondriacus with various public sequencing data. All comparisons with the same species were done at the DNA level (blastn) and with other species at the amino acid levels (tblastn or tblastx).24 A filtering criterion of >99% over a contiguous stretch of >100 bases is used to establish identity with sequences from the same species, and a filtering criterion of 60% match over a contiguous stretch of 35 or more amino acids is used to establish the orthology of genes with other plant species such as Arabidopsis thaliana, Vitis vinifera, and Solanum lycopersicum.
2.10. Synteny analyses
All the synteny analyses were performed using SyMap25 and in-house perl scripts.
2.11. Heterozygosity analysis
Frequency of mutation in the genomic scaffolds was estimated by piling up the genomic reads to the scaffolds using Bowtie26 and SAMtools27 and counting the number of reads with an alternate base (minor allele) from that present in the reference. For the study, positions with depth between 10X and 315X (three times the read coverage) and minor allele frequency of 0.3 or more were considered. Also, the k-mer spectra of the sequenced genome and transcriptome were generated from the high-quality sequence reads using SOAPdenovo. The k-mer size was selected as 31 for the purpose as well as for the assembly of the genomic reads. The k-mer count with a very low frequency of 1 and 2 was removed.
2.12. GO annotation
The GO annotation for all the A. thaliana genes with orthology to other species was compared using the ‘GO annotation search, functional categorization and download’ tool available at TAIR.28 The percentage of A. thaliana genes within each GO category that are orthologous to scaffolds in the draft genomes of V. vinifera, S. lycopersicum, and A. hypochondriacus is computed as a percentage of the total number of A. thaliana proteins within the respective category for that species. The percentage of genes within each category reported by GO analysis for each species is normalized with respect to that of A. thaliana to estimate the number of genes deciphered within each GO category for the three species compared.
2.13. Proteome prediction
Ab initio gene predictions were done on the genomic scaffolds using GENSCAN,29 AUGUSTUS,30 and GeneMark,31 with the smallest predicted sequence being 100 nucleotides long. These predicted sequences were annotated using BLAST against plant protein databases. Extrinsic prediction from genomic scaffolds and transcripts was also done using BLAST against plant proteomes downloaded from PlantGDB32 including UniProt, mRNA, and HTC databases.
2.14. Repeat analysis
We have used both extrinsic and ab initio methods to assess the extent of repeat elements in A. hypochondriacus, compared with selected plant species. For extrinsic analysis, we have used Repbase33 using A. thaliana repeats as templates. For the de novo method, we have used RepeatScout34 and RepeatModeler35 to predict novel repeats from the genomic scaffolds and masked the genome with the respective repeat element using RepeatMasker.36,37
2.15. Duplication event
Protein-level homology was studied between paralogous pairs in several species belonging to different plant orders including A. thaliana, Glycine max, and V. vinifera from Rosids, S. lycopersicum from Asterids, A. hypochondriacus and B. vulgaris (USDA version) from Caryophyllales, and Oryza sativa, Sorghum bicolor, and Zea mays from the monocots. The proteomes for A. thaliana, G. max, V. vinifera, S. lycopersicum, O. sativa, S. bicolor, and Z. mays were downloaded from public repositories (TAIR28 and PGDD38), whereas those for A. hypochondriacus and B. vulgaris were predicted ab initio using Genscan from genomic scaffolds. Also, the GENSCAN-predicted proteome of S. lycopersicum was used as control to negate any possibility of bias in the result due to the gene prediction tool used for A. hypochondriacus and B. vulgaris. Furthermore, frequency polygons and kernel density plots of percentage identities of significant hits (those that were not self-hits or isoforms with 100 percent identity, and where the alignment covered at least 80% of the query or the subject sequence, whichever was longer) were plotted using R.39
2.16. Phylogenetic tree
Amino acid sequences of aspartate kinase (AK) and phosphoenolpyruvate carboxylase (PEPC) genes available at GenBank for plants of interest were collected. Sequences for the remaining species for which genomic scaffolds were available were extracted from the results of BLAST24 against the sequence from A. thaliana or from the closest taxon for which the sequence was available.
Multiple sequence alignment was carried out using MUSCLE,40 present as an application in MEGA5, using the default parameters. The evolutionary history was inferred using the Neighbor-Joining method.41 The bootstrap consensus tree inferred from 1,000 replicates is taken to represent the evolutionary history of the taxa analysed.42 Branches corresponding to partitions reproduced in <50% bootstrap replicates are collapsed. The evolutionary distances were computed using the Poisson correction method in the units of the number of amino acid substitutions per site.43 The analyses involved 25 AK and 75 PEPC sequences. Positions in the alignment containing gaps and missing data were eliminated to get a contiguous stretch of aligned sequences. There were a total of 229 and 278 positions in the final dataset for AK and PEPC proteins, respectively. Evolutionary analyses were conducted in MEGA5.44 The phylogenetic trees were visualized using iTOL.45,46
2.17. Gene expression profiling
RNA-seq reads from each sample were mapped using bowtie to the full-length cDNA obtained for all the enzymes in the lysine biosynthesis pathway by virtual splicing from genomic scaffolds and transcriptome. The read counts were converted to reads per kilo base of exons per million reads mapped (RPKM) to normalize the measurement across samples.
3. Results and discussion
3.1. Coverage by sequencing, assembly, and mapping
The species A. hypochondriacus (Rajgira), grown in northern Karnataka, India, was selected for sequencing. Following plant selection and genomic DNA extraction, five PE libraries of 300 bp and four MP libraries with insert sizes ranging from 1.5 to 10 kb were created. A total of 869,113,408 high-quality reads including 465,246,312 75-mers from PE and 403,867,096 36-mers from MP libraries were sequenced to produce a total of 49.4 billion bases, thus providing a coverage of 106-fold over the estimated genome size of 466 million bases.16 Also, we have generated 8.6 billion bases of transcriptome from three stages of shoot development and from mature seeds.
Table 1 includes the statistics from the assembly of genomic reads into scaffolds. The total ATGC content, without Ns, within the assembled scaffolds is 273 million bases. Addition of MP reads improved the size of the largest scaffold by 20 folds to 485,353 from 24,878 bases and scaffold N50 to 35,089 from contig N50 of 1,884. The genome of A. hypochondriacus was found to be AT-rich with 66% AT content (Supplementary Fig. S2), which compares with the genomes of other plants.47 Figure 3 shows the uniform coverage of the 16 chromosomes by genomic scaffolds, which harbour 355 (86%) SNPs of the 411 linkage SNPs reported for grain amaranths.18
Table 1.
Summary of statistics of genome and transcriptome assembly
| Assembled ‘ome | Genome | Genome | Transcriptome |
|---|---|---|---|
| Types of reads assembled | Paired end (PE) | PE and mate pair (MP) | PE |
| Read length (bases) | 75 | PE: 75 MP: 36 |
72 |
| Total number of high-quality reads sequenced | 465,246,312 | PE: 465,246,312 MP: 403,867,096 |
119,875,998 |
| Total number of bases sequenced (in high-quality reads) | 34,893,473,400 | 49,432,688,856 | 8,631,071,856 |
| Total number of bases in the assembly (% of genome) | 315,828,977 (63.2%) | 645,211,960 (>100%) | 136,933,803 (29.26%) |
| Number of A, T, G, and C in the assembly (% of genome) | 276,891,658 (58.9%) | 273,809,695 (58.3%) | 136,933,803 (29.26%) |
| G+C base content (% of the ‘ome) | 91,893,918 (19.63%) | 90,943,537 (19.43%) | 52,037,597 (38%) |
| Total number of assembled sequences | 491,569 | 367,441 | 57,658 |
| Longest sequence assembled (bases) | 24,878 | 485,353 | 17,471 |
| N50/NG50 (bases) | 1,884/648 | 35,089/50,869 | 3,279/– |
| Number of sequences above N50/NG50 | 42,507/111,228 | 4,897/2,826 | 15,134/– |
Figure 3.

Sixteen supercontigs of A. hypochondriacus corresponding to 16 haploid chromosomes drawn in centiMorgan scale showing the location of respective scaffolds anchored using 355 linkage SNPs out of the 411 reported. The red labels represent scaffolds containing the 355 SNPs and those in blue show the scaffolds that only map to one side of the SNPs with high confidence.
The assembly statistics for transcriptome reads are also included in Table 1. The transcriptome is also AT-rich like that of other plant transcriptomes.48 The total number of non-redundant bases resulting from assembly is 47 million bases without any intervening Ns, which would suggest that 10% of the A. hypochondriacus genome is transcribed in the tissues sequenced. Also, 74% of raw transcript reads map onto the genomic scaffolds, which is much more than what can be expected based on the ATGC content of the assembled genomic scaffolds which comprise 58% of the genome.
The genomic scaffolds reported here were also compared with a publicly available transcriptome sequence (SRX055331) with the seed accession of IC 38040/PI 480569 (India 38040) generated using 454 platform by another independent effort.14 Despite the fact that India 38040 has a reddish stem and drooping inflorescence (personal communication), we find significant homology between the edible white variety sequenced here and India 38040. For example, of the total 6,222,321 non-redundant bases in the assembled transcriptome from India 38040, 5,185,660 bases (83.34%) aligned to the genomic scaffolds with identities greater than 99% over a contiguous stretch greater than 100 bases.
3.2. Quality assessment and synteny of genomic scaffolds with B. vulgaris
The recently reported genome of B. vulgaris is the only reported genome of a member of the Caryophyllales plant order,13 which is used here to both assess the level of synteny between the two species and to assess the quality of the scaffolds reported here. All the 4,897 scaffolds above the N50 of 35,089 bases, constituting 322.6 Mb (69% including Ns in the scaffolds) of the A. hypochondriacus genome, find synteny for collinear blocks adding up to 85 Mb with the genome of B. vulgaris. As shown in Fig. 4, among the top 100 scaffolds (greater than 161,660 bases), 60% finds unique synteny with the genome of B. vulgaris compared with only 4% with A. thaliana. Sixty-nine of the top 100 scaffolds have collinear blocks syntenic to unique loci on B. vulgaris chromosomes, whereas 7 scaffolds show synteny, which is split over two different chromosomes of B. vulgaris. Twenty-four scaffolds that find no synteny with B. vulgaris are not necessarily from a gene-less region, but contain predicted genes most represented in GO category under ‘other cellular processes’ and ‘other metabolic processes’. These predicted genes may be present in B. vulgaris, but may have diverged significantly beyond the filtering criteria used for synteny analysis.
Figure 4.

Comparative analyses of the longest 100 scaffolds (in blocks of 20) of A. hypochondriacus with the nine chromosomes of B. vulgaris showing a high level of synteny between the two species. It is also evident that the collinear blocks in A. hypochondriacus cover individual scaffolds across their compete lengths in most cases indicating a good quality of the de novo assembly. This figure appears in colour in the online version of DNA Research.
3.3. Comparative coverage by homology to A. thaliana proteome
An amino acid level comparison of A. thaliana proteome49 against genomic scaffolds of A. hypochondriacus shows that 60.11% of A. thaliana proteins find orthology within the genomic scaffolds using the filter mentioned in the Materials and Methods section. Similar efforts with the scaffolds of two other species, V. vinifera50 and S. lycopersicum,51 finds comparable, 17,311 (63%) and 16,538 (60.32%), respectively, orthology to the A. thaliana proteome. Also, as shown in Fig. 5, the orthology of 14,590 A. thaliana genes common to the genomic scaffolds of V. vinifera, S. lycopersicum, and A. hypochondriacus suggests that functions of these genes are common to all dicots. A GO annotation of the A. thaliana orthologs in V. vinifera, S. lycopersicum, and A. hypochondriacus scaffolds shows that more than 80% of the genes annotated under each GO category in A. hypochondriacus has been deciphered (Fig. 6), and that the coverage of A. hypochondriacus genes within each category is comparable to the other two reported genomes of V. vinifera and S. lycopersicum.
Figure 5.

Comparative proteome analysis of A. hypochondriacus, V. vinifera and S. lycopersicum. The pie chart shows the source of evidence for proteins from the assembled transcriptome and genome. 17,770 proteins find evidence in both homology and presence in the transcriptome, 3,880 are ab initio predicted with presence in the transcriptome, and 3,179 are those with only evidence from homology to known plant proteins. The Venn diagram compares the total numbers of A. thaliana genes that are orthologous to the respective genomic scaffolds of A. hypochondriacus, V. vinifera and S. lycopersicum with 14,590 proteins common to all the three species representing three major clades under dicots. This figure appears in colour in the online version of DNA Research.
Figure 6.

Comparative GO annotation of three species including A. hypochondriacus, V. vinifera and S. lycopersicum. The percentage of genes deciphered for A. hypochondriacus, S. lycopersicum and V. vinifera within each category of GO as annotated by comparison with A. thaliana based on orthology and further normalized for the percentage of genes in A. thaliana shows that annotation of the draft genome is comparable to that of the published genomes. This figure appears in colour in the online version of DNA Research.
3.4. Genomic features of A. hypochondriacus
The mRNA reads from transcriptome sequencing efforts were assembled into transcripts with an N50 of 3,279 containing 15,179 transcripts above the median. Both extrinsic and ab initio methods were chosen for predicting the number of genes coded by A. hypochondriacus from both 57,658 assembled transcripts and 367,441 genomic scaffolds. For extrinsic prediction, we have used plant proteome databases including UniProt, HTC, and plant mRNA at plantGDB. For ab initio prediction of genes from the genomic scaffolds, we have used gene prediction tools including GENSCAN with 36,487, Augustus with 28,662, and GeneMark with 63,044 predicted genes. From the assembled transcriptome of A. hypochondriacus, the total number of potential proteins is estimated at 21,650 genes with either evidence of homology in the plant proteome database and/or evidence of gene prediction from the genomic scaffolds. An additional 3,179 potential proteins were added from genomic scaffolds, which were missed by gene prediction methods and not represented in the transcriptome, but show homology to the plant proteome database, thus making the total number of estimated proteins coded by A. hypochondriacus genome 24,829. This compares with the 27,421 proteins predicted for B. vulgaris, the other sequenced member of Caryophyllales.13
Mapping of raw genomic reads onto the assembled scaffolds revealed ∼1 SNP per 1,000 bases based on 282,692 SNPs out of 260 Mb base positions scanned. This indicates that the genome of A. hypochondriacus is relatively more homozygous than other sequenced plants. For example, the frequency of SNPs in Malus domestica, O. sativa, Z. mays, V. vinifera, and Fragaria vesca is 1 in 288 bases, 1 in 268 bases, 1 in 124 bases, 1 in 100 bases, and 1 in 15 bases, respectively.52–56 The k-mer spectrum of the genome (not shown) also gives a unimodal curve, supporting the high level of homozygosity within the assembled part of the genome.57
Characterizing the repeat elements in the A. hypochondriacus genome is challenged by the fact that repeats from species under the plant order Caryophyllales is missing in major repeat databases such as RepBase. Masking the draft genomes of a number of species including an unpublished draft of B. vulgaris using A. thaliana repeats as templates reveals that the A. hypochondriacus genome contains a relatively low percentage of repeat elements (Table 2). Also, masking the draft genomes of several species using the predicted repeats from the respective genomes using RepeatScout reveals 7.47% of repeats in the A. hypochondriacus genome, compared with 69.86% in B. vulgaris (USDA version, unpublished), 68.34% in S. lycopersicum, 55.02% in V. vinifera, 18.43% in A. thaliana, 68.67% in S. bicolor, and 47.69% in O. sativa (Table 2). A more rigorous prediction of repeats using RepeatModeler masked 13.76% of the A. hypochondriacus genome, still far below the 63% for B. vulgaris using RepeatModeler reported recently.13 Based on the number of proteins predicted for the genome and the success achieved in splicing full-length cDNA for a majority of genes from select biochemical pathways, we rule out the possibility that the low percentage of repeats in the A. hypochondriacus genome could stem from a fragmented assembly.
Table 2.
Comparative repeat element analysis using both A. thaliana repeats and predicted repeats
| Ah | Bv | Sla | Sl | Vv | At | Sb | Os | |
|---|---|---|---|---|---|---|---|---|
| Genome size (Mb) | 466 | 760 | 2,800 | 950 | 475 | 157 | 698 | 430 |
| LTR | 0.69% | 4.60% | 4.41% | 6.68% | 4.87% | 6.66% | 4.49% | 2.08% |
| Gypsy | 0.23% | 2.07% | 3.50% | 4.28% | 3.38% | 5.15% | 3.36% | 1.35% |
| Copia | 0.45% | 2.51% | 0.88% | 2.40% | 1.49% | 1.44% | 1.11% | 0.71% |
| LINE | 0.07% | 0.50% | 0.00% | 0.03% | 0.11% | 1.01% | 0.04% | 0.03% |
| L1 | 0.07% | 0.50% | 0.00% | 0.03% | 0.11% | 1.00% | 0.04% | 0.03% |
| Total genome masked using Repbase with A. thaliana as the template | 1.70% | 6.25% | 7.18% | 7.14% | 5.30% | 14.90% | 4.95% | 2.74% |
| Total genome masked using RepeatScout predicted repeats from respective genomes | 7.47 | 69.86 | NA | 68.34 | 55.02 | 18.43 | 68.67 | 47.69 |
The species used for comparison are—Ah: A. hypochondriacus; Bv: B. vulgaris (USDA version); Sla: Silene latifolia;58 Sl: S. lycopersicum; Vv: V. vinifera; At: A. thaliana; Sb: S. bicolor; Os: O. sativa.
Estimation of 24,829 proteins provided an opportunity to look for any sign of recent whole genome duplication using a method reported by Axelsen et al.59 Figure 7 shows the comparison of proteomes of various species to find levels of divergence among paralogs. The peak near 90% for Z. mays, A. thaliana, and G. max confirms the recent whole genome duplication events reported for these species.60 Amaranthus hypochondriacus and B. vulgaris, both species classified under the order Caryophyllales, lack the peak near 90% similar to V. vinifera, suggesting no recent whole genome duplication event in A. hypochondriacus and B. vulgaris. The peak near 30 for all dicot species represent a paleohexaploidy event common to all dicots,50 including the two species of Caryophyllales studied here. Based on this analysis, we have redrawn the place for A. hypochondriacus in Fig. 8, which is reproduced with permission from the authors.61
Figure 7.
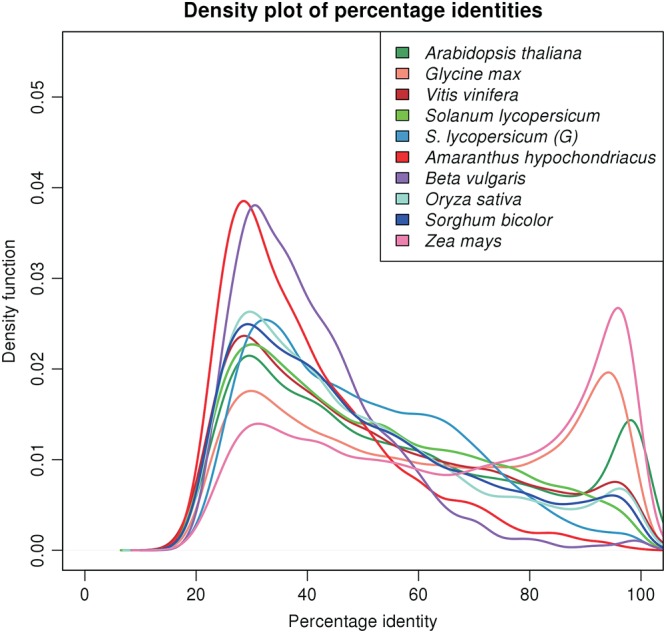
A genome duplication study in A. thaliana, G. max, V. vinifera, S. lycopersicum, A. hypochondriacus, B. vulgaris, O. sativa, S. bicolor, and Z. mays by comparison of the distribution of percentage identities among paralogous pairs from the respective proteomes. The GENSCAN-predicted proteome of S. lycopersicum is labelled as S. lycopersicum (G) in the figure. The Kernel density plot of the density function of the number of paralogous pairs against the percentage identities for the diverse sequenced plant species shows the first peak at 20–40% identity corresponding to the paleaohexaploidy event and the sharp peak at 90–100% identity representing a recent whole genome duplication event. From the plot, it is evident that A. hypochondriacus, like B. vulgaris, did not undergo any recent whole genome duplication.
Figure 8.

Rooted tree comparing the relative whole genome duplication events in many plant species including A. hypochondriacus. The tree is taken from PNAS journal with permission from the authorand modified to include A. hypochondriacus, creating a branch for Caryophyllales, using the phylogenetic tree generated in-house using rbcL gene (not shown) from 75 species across all plant orders. The predicted and reported ages of the duplication events are retained from the original paper. This figure appears in colour in the online version of DNA Research.
We find that there is 15% overestimation of the genome size of A. hypochondriacus based solely on assembly, even when only scaffolds greater than 1,000 are considered for the estimation. Similar overestimation has been reported for the V. vinifera genome, which is attributed to 11% hemizygosity.55 Since, in genomes with a significant percentage of hemizygosity, the coverage of the homozygous regions can be expected to be two times compared with the hemizygous regions, assembly tools are likely to be more biased towards the homozygous regions, thus providing the basis for high coverage of coding regions.
3.5. C4 evolution
It is now well established that PEPC (EC 4.1.1.31) is the key enzyme in the C4 pathway.62 Thus, comparative analysis of PEPC genes across plant kingdom, including C3 and C4 plants, has been attempted by many groups.63,64 There are multiple PEPC isoenzymes in all plant species including C3 plants. Work by various groups has shown that the C4-specific PEPC gene harbours a serine in place of alanine at position 780 of maize PEPC gene with accession CAA33317 (A780S) among both dicots and monocots.65 For example, one of the four sorghum PEPC genes harbours this mutation.
We have identified full-length cDNA sequences for all the four PEPC isoenzymes from the genome of A. hypochondriacus with only one harbouring the A780S mutation. In order to check if all PEPC genes across various species harbouring A780S mutation result from divergent or convergent evolution, we have created a phylogenetic tree using the multiple sequence alignment of all the PEPC isoenzymes from representative species under various major plant orders including many C3 and C4 from both dicots and monocots (Fig. 9). All PEPC isoenzymes harbouring the A780S mutation, among monocot species, cluster together, suggesting C4 evolution predating speciation in monocots. However, the PEPC gene harbouring the A780S mutation in C4 dicots (represented by A. hypochondriacus and Mollugo cerviana from Caryophyllales and Flaveria trinervia from Asterids), clusters in distal clades. Based on this observation, there can be two hypotheses: (i) the C4-specific mutation occurred independently under Asterid and Caryophyllales; (ii) all C3 dicot plants have selectively lost an ancestral C4-specific isoform during the course of evolution. The first hypothesis has been reported in the literature65 and our observation supports the same. In other words, C4 switch in dicots is a convergent evolution.65 For the second hypothesis to be true, one would need to show that at least one C3 dicot plant continues to retain a C4-specific PEPC gene.
Figure 9.

Rooted phylogram of PEPC 1, 2, 3, and 4. Branches labelled A780S are C4-specific PEPC isoforms containing alanine to serine mutation at residue 780 of Z. mays C4-specific gene with accession CAA33317. All the bacterial-type PEPC isoforms including A. hypochondriacus 4 and B. vulgaris 4 cluster in one clade. All the C4-specific PEPC genes from monocots cluster together. Among dicots, C4-specific PEPC genes are split into two clades representing Asterids and Caryophyllales. There is only one C4-specific PEPC gene in A. hypochondriacus as is also found in other C4 plants. In the figure, the protein sequence of PEPC taken from GenBank (gb ADO15315.1) has been labelled as A. hypochondriacus gb and the one extracted from the genomic scaffolds is labelled as A. hypochondriacus 1. This figure appears in colour in the online version of DNA Research.
3.6. Functional characterization and profiling of genes in the lysine biosynthetic pathway
In plants, the aspartate family pathway is responsible for the biosynthesis of four amino acids—threonine, isoleucine, methionine, and lysine.66 Based on the lysine biosynthetic pathway in KEGG,67,68 here are a total of seven enzymatic steps in the synthesis of l-lysine starting from l-aspartate, of which the first two steps are shared for all four amino acids before the lysine biosynthesis pathway diverges. The number of isoenzymes encoded by A. hypochondriacus for all the seven enzymatic steps in the aspartate pathway of lysine biosynthesis has been identified and is listed in Table 3 for comparison with those in other plant species.
Table 3.
Isozyme number polymorphism in A. hypochondriacus compared with other species
| EC number | EC 2.7.2.4 | EC 1.2.1.11 | EC 4.3.3.7 | EC 1.17.1.8 | EC 2.6.1.83 | EC 5.1.1.7 | EC 4.1.1.20 |
|---|---|---|---|---|---|---|---|
| Enzyme name | AK | ASD | DHDPS | DHDPR | DAPAT | DAPE | DAPDC |
| A. thaliana | 3 | 1 | 2 | 4 | 2 | 1 | 2 |
| O. sativa | 3 | 1 | 2 | 4 | 2 | Short | 1 |
| Z. mays | 1 | 1 | 2 | 3 | 1 | 2 | 1 |
| S. bicolor | 3 | 1 | 2 | 2 | 2 | 1 | 1 |
| G. max | 4 | 2 | 3 | 3 | 3 | 2 | 2 |
| V. vinifera | 2 | 2 | 3 | 2 | 3 | 2 | 2 |
| Ricinus communis | 1 | 1 | 1 | 2 | 3 | 1 | 1 |
| A. hypochondriacus | 1 | 1 | 2 | 2 | 2 | 1 | 2 |
The enzymes listed in the table are: AK: monofunctional aspartate kinase; ASD: aspartate semialdehyde dehydrogenase; DHDPS: dihydrodipicolinate synthase; DHDPR: dihydrodipicolinate reductase; DAPAT: diaminopimelate aminotransferase; DAPE: diaminopimelate epimerase; DAPDC: diaminopimelate decarboxylase.
It is known that both in prokaryotes and photosynthetic eukaryotes, lysine biosynthesis is mainly regulated by two allosteric enzymes.69 These are (i) monofunctional AK (EC 2.7.2.4), the first and the critical enzyme in the aspartate family pathway, and (ii) dihydrodipicolinate synthase (DHDPS, EC 4.3.3.7), the first enzyme towards lysine biosynthesis within the aspartate pathway. Given this, polymorphisms of any kind within these two gene loci can be expected to have a high likelihood of correlating with the high-lysine phenotype in grain amaranth.
There are varying numbers of monofunctional AK isoenzymes with varying lysine sensitivity in plants. For example, AK1, AK2, and AK3 genes of A. thaliana vary significantly in their affinity to lysine, with AK1 being the least sensitive.69 In this context, it is of interest to know if the lysine sensitivity of the only monofunctional AK gene of A. hypochondriacus compares with AK1 or the other two enzymes of A. thaliana. Phylogenetic analysis (Fig. 10) using the multiple sequence alignment of protein sequences of AK gene paralogs from diverse species, clusters the AK gene of A. hypochondriacus in the same clade as the AK1 gene of A. thaliana, suggesting that the AK gene of A. hypochondriacus is similar in lysine sensitivity to the AK1 gene. Loss of orthologs of the two lysine-sensitive AK2 and AK3 genes in A. hypochondriacus may be one of the reasons for the high-lysine phenotype in A. hypochondriacus, thus providing a testable hypothesis.
Figure 10.

Unrooted phylogram of monofunctional aspartate kinases, the first enzyme in the asparate family pathway. The tree shows that A. hypochondriacus AK gene is in the same clade as AK1 (A. thaliana 1) of A. thaliana, B. vulgaris, and V. vinifera. The AK2 and AK3 (A. thaliana 2 and 3) cluster with V. vinifera 3. The sequences that could not be annotated to any of AK 1/2/3 are labelled as x1, x2, and so on. This figure appears in colour in the online version of DNA Research.
DHDPS, the other most important set of enzymes in the lysine biosynthesis, is known to be orders of magnitude more sensitive to lysine.70 Similar to many other plant species from various plant orders, there are two DHDPS isoenzymes in A. hypochondriacus. Profiling gene expression levels from transcriptome sequencing for three stages of shoot development and mature seeds, we observe that the expression levels of both DHDPS isoenzymes decrease with developmental stages of the shoot and significantly spike up in the seeds (Table 4). The high levels of expression of DHDPS isoenzymes in seeds suggest yet another hypothesis for producing high-lysine grains in grain amaranths.
Table 4.
Expression level of genes implicated in the lysine biosynthetic pathway in the shoot at three stages of development and in mature seeds
| Enzyme | EC # AT ID |
RPKM |
|||
|---|---|---|---|---|---|
| Shoot 15 days |
Shoot 25 days |
Shoot 30 days |
Mature seeds | ||
| AK | 2.7.2.4 AT5G13280 |
12.21 | 11.13 | 12.82 | 4.17 |
| AK-homoserine dehydrogenase | 2.7.2.4–1.1.1.3 | 23.80 | 20.43 | 16.28 | 6.98 |
| Aspartate semialdehyde dehydrogenase | 1.2.1.11 AT1G14810 |
8.36 | 6.91 | 7.73 | 3.67 |
| Dihydrodipicolinate synthase | 4.3.3.7 AT2G45440 |
11.51 | 11.88 | 7.43 | 30.77 |
| 4.3.3.7 AT3G60880 |
5.09 | 5.00 | 5.16 | 18.57 | |
| Dihydrodipicolinate reductase | 1.17.1.8 AT3G59890 |
2.37 | 1.16 | 1.35 | 0.26 |
| 1.17.1.8 AT2G44040 |
2.77 | 2.45 | 2.29 | 0.61 | |
| 1.17.1.8 AT5G52100 |
27.58 | 19.22 | 8.36 | 0.00 | |
| Diaminopimelate aminotransferase | 2.6.1.83 AT4G33680 |
0.00 | 0.05 | 0.00 | 0.00 |
| 2.6.1.83 AT2G13810 |
7.06 | 6.36 | 6.18 | 1.64 | |
| Diaminopimelate epimerase | 5.1.1.7 AT3G53580 |
42.28 | 37.48 | 34.32 | 4.91 |
| Diaminopimelate decarboxylase | 4.1.1.20 AT3G14390 |
10.67 | 10.19 | 12.64 | 3.37 |
| 4.1.1.20 AT5G11880 |
25.53 | 20.79 | 18.06 | 3.06 | |
To correlate the observed gene number polymorphism for AK genes and eQTL of the DHDPS gene, we have generated horizontal and vertical profiles of free lysine across the different developmental stages of all three grain species using ultra-fast liquid chromatography. The study reveals that the free lysine content in seeds of the genus is relatively higher than that in the shoot (0.152 µmol/100 mg in 25-day-old shoot and 0.189 µmol/100 mg in mature seeds). The free lysine content analysis correlates with the expression levels of the DHDPS gene, suggesting eQTL for the high-lysine phenotype. However, the expression profiles of all the other enzymes including AK shows no correlation with high-lysine content in seeds compared with the shoot. Perhaps, the absence of orthologs of the two lysine-sensitive AK genes in A. hypochondriacus may be sufficient for high-lysine phenotype.
4. Conclusion
We have reported the draft genome of the first grain species under the plant order Caryophyllales and the first C4 dicot to be sequenced. Using the reported SNPs derived from homozygous regions of grain amaranths,18 we conclude that as high as 86% of the homozygous gene-rich region has been deciphered. Similarly, annotation of the genome using various criteria, including transcriptome mapping, gene prediction, and GO annotation, vindicates the conclusion. Also, based on the high level of synteny to the genome of B. vulgaris, we authenticate the quality of the reported scaffolds. We also confirm that the paleohexaploidy event reported to be common to all sequenced dicot species under Rosids and Asterids is also common to Caryophyllales. We have corroborated that the C4 evolution under Caryophyllales has occurred independently of other C4 dicots, for example, F. trinervia under Asterid. Also, we have proposed two testable hypotheses for the high-lysine phenotype including gene number polymorphism for the AK gene and eQTL for DHDPS. We have demonstrated that the draft genome reported here can be useful in advancing our understanding of the diverse phenotypes that are unique to grain amaranths including the unique nutritional profile, aggressive growth, drought resistance and adaptability to environmental stress and characterize genes involved in the biosynthesis of betalains.
4.1. Availability
The short genomic reads used in the study are deposited at NCBI-SRA under the accession ID of SRP031880. The scaffolds for the A. hypochondriacus genome and transcriptome are available at GenBank under the BioProject IDs of PRJNA214803 and PRJNA214804, respectively, and also at http://resource.ibab.ac.in/Plant_Genomics for download. An instance of NCBI's wwwBLAST71 has also been set up at the same url where users can BLAST a given gene against both the genomic and transcriptomic scaffolds sequenced here and other plant genomes compared here. The seeds of the sequenced plant are in the germplasm currently maintained at IBAB under the accession ID of IAh0001. We are in the process of depositing to the germplasm repository maintained by the National Bureau of Plant Genetic Resources in India. Also, the multiple sequence alignment files in .meg (MEGA5) format used in figs 9 and 10, and the rs IDs for the anchored scaffolds in fig. 3 are available from the authors on request.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
The sequencing work reported here is covered by grants to GANIT Labs from the Department of Information Technology, Government of India (Ref no: 18(4)/2010-E-Infra., 31-03-2010) and the Department of Information Technology, Biotechnology and Science & Technology, Government of Karnataka, India (Ref no: 3451-00-090-2-22). IBAB is recognized as a ‘Centre of Excellence for Research and Training in Bioinformatics’ by the Department of Electronics and Information Technology, Government of India which also covers the computational resources utilized for the work. Department of Biotechnology (DBT), New Delhi, India has provided support to Dr S.S. and the Experimental work via the Ramalingaswamy fellowship (Ref no: BT/HRD/35/02/17/2009) and to Mr S.R.N. via BINC fellowship. Support from DST-FIST was obtained as infrastructural grant to IBAB. Funding to pay the Open Access publication charges for this article was also provided by the Department of Biotechnology to Dr S.S. (Ref no:BT/HRD/35/02/17/2009).
Supplementary Material
Acknowledgements
The authors wish to extend special thanks to Dr S.S. Shivakumara of IBAB for detailed comments on the initial manuscript but for which the manuscript will not be in the current form. The authors would like to acknowledge Dr Imad Eujayl, NWISRL-ARS-USDA and Kimberly, Idaho for sharing information on the BvvSeq-1 assembly, the only other sequenced genome under the plant order Caryophyllales. The authors would also like to acknowledge Yves Van de Peer for allowing them to both use and modify the phylogenetic tree from his publication that depicts the age of recent whole genome duplications across several sequenced plant species.61 Last but not the least, the authors also wish to acknowledge IBAB for providing the facilities for field and laboratory work.
References
- 1.Innovation, N.R.C. (U.S.) A.C. Amaranth: Modern Prospects for an Ancient Crop. Washington, DC, USA: National Academies; 1984. [Google Scholar]
- 2.Marx J.L. Amaranth: a comeback for the food of the Aztecs? Science. 1977;198:40. doi: 10.1126/science.198.4312.40. [DOI] [PubMed] [Google Scholar]
- 3.Caselato-Sousa V.M., Amaya-Farfán J. State of knowledge on amaranth grain: a comprehensive review. J. Food Sci. 2012;77:R93–104. doi: 10.1111/j.1750-3841.2012.02645.x. [DOI] [PubMed] [Google Scholar]
- 4.Ferreira R.R., Varisi V.A., Meinhardt L.W., Lea P.J., Azevedo R.A. Are high-lysine cereal crops still a challenge? Braz. J. Med. Biol. Res. Rev. Bras. Pesqui. Médicas E Biológicas Soc. Bras. Biofísica Al. 2005;38:985–94. doi: 10.1590/s0100-879x2005000700002. [DOI] [PubMed] [Google Scholar]
- 5.Schmutz J., Cannon S.B., Schlueter J., et al. Genome sequence of the palaeopolyploid soybean. Nature. 2010;463:178–83. doi: 10.1038/nature08670. [DOI] [PubMed] [Google Scholar]
- 6.Varshney R.K., Chen W., Li Y., et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 2012;30:83–9. doi: 10.1038/nbt.2022. [DOI] [PubMed] [Google Scholar]
- 7.Ward J., Tissue D., Thomas R., Strain B. Comparative responses of model C3 and C4 plants to drought in low and elevated CO2. Glob. Change Biol. 1999;5:857–67. [Google Scholar]
- 8.Kadereit G., Borch T., Weising K., Freitag H. Phylogeny of Amranthaceae and Chenopodiaceae and the evolution of C4 photosynthesis. Int. J. Plant Sci. 2003;164:959–86. [Google Scholar]
- 9.Brockington S.F., Walker R.H., Glover B.J., Soltis P.S., Soltis D.E. Complex pigment evolution in the Caryophyllales. New Phytol. 2011;190:854–64. doi: 10.1111/j.1469-8137.2011.03687.x. [DOI] [PubMed] [Google Scholar]
- 10.Stafford H.A. Anthocyanins and betalains: evolution of the mutually exclusive pathways. Plant Sci. 1994;101:91–8. [Google Scholar]
- 11.Gandía-Herrero F., García-Carmona F. Biosynthesis of betalains: yellow and violet plant pigments. Trends Plant Sci. 2013;18:334–43. doi: 10.1016/j.tplants.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 12.Kamagaju L., Morandini R., Bizuru E., et al. Tyrosinase modulation by five Rwandese herbal medicines traditionally used for skin treatment. J. Ethnopharmacol. 2013;146:824–34. doi: 10.1016/j.jep.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 13.Dohm J.C., Minoche A.E., Holtgräwe D., et al. The genome of the recently domesticated crop plant sugar beet (Beta vulgaris) Nature. 2014;505:546–9. doi: 10.1038/nature12817. [DOI] [PubMed] [Google Scholar]
- 14.Délano-Frier J.P., Avilés-Arnaut H., Casarrubias-Castillo K., et al. Transcriptomic analysis of grain amaranth (Amaranthus hypochondriacus) using 454 pyrosequencing: comparison with A. tuberculatus, expression profiling in stems and in response to biotic and abiotic stress. BMC Genomics. 2011;12:363. doi: 10.1186/1471-2164-12-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Riggins C.W., Peng Y., Stewart C.N., Jr, Tranel P.J. Characterization of de novo transcriptome for waterhemp (Amaranthus tuberculatus) using GS-FLX 454 pyrosequencing and its application for studies of herbicide target-site genes. Pest Manag. Sci. 2010;66:1042–52. doi: 10.1002/ps.2006. [DOI] [PubMed] [Google Scholar]
- 16.Maughan P.J., Sisneros N., Luo M., Kudrna D., Ammiraju J.S.S., Wing R.A. Construction of an bacterial artificial chromosome library and genomic sequencing of herbicide target genes. Crop. Sci. 2008;48:S85. [Google Scholar]
- 17.Mallory M.A., Hall R.V., McNabb A.R., Pratt D.B., Jellen E.N., Maughan P.J. Development and characterization of microsatellite markers for the grain amaranths. Crop. Sci. 2008;48:1098. [Google Scholar]
- 18.Maughan P., Smith S., Fairbanks D., Jellen E. Development, characterization, and linkage mapping of single nucleotide polymorphisms in the grain amaranths (Amaranthus sp.) Plant Genome. 2011;4:92–101. [Google Scholar]
- 19.Nemoto K., Baniya B., Minami M., Ujihara A. Grain amaranth research in Nepal. J. Fac. Agric. Shinshu Univ. 1998;34:49–58. [Google Scholar]
- 20.Chan K.-L., Ho C.-L., Namasivayam P., Napis S. A simple and rapid method for RNA isolation from plant tissues with high phenolic compounds and polysaccharides. Protoc. Exch. 2007 doi:10.1038/nprot.2007.184. [Google Scholar]
- 21.Li R., Zhu H., Ruan J., et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome. Res. 2010;20:265–72. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogg C.D., Patel B.K.C. Draft genome sequence of Caloramator australicus strain RC3T, a thermoanaerobe from the Great Artesian Basin of Australia. J. Bacteriol. 2011;193:2664–5. doi: 10.1128/JB.00193-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grabherr M.G., Haas B.J., Yassour M., et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011;29:644–52. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 25.Soderlund C., Nelson W., Shoemaker A., Paterson A. SyMAP: a system for discovering and viewing syntenic regions of FPC maps. Genome Res. 2006;16:1159–68. doi: 10.1101/gr.5396706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H., Handsaker B., Wysoker A., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lamesch P., Berardini T.Z., Li D., et al. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr1090. doi:10.1093/nar/gkr1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burge C., Karlin S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- 30.Stanke M., Steinkamp R., Waack S., Morgenstern B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 2004;32:W309–12. doi: 10.1093/nar/gkh379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Besemer J., Borodovsky M. GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005;33:W451–4. doi: 10.1093/nar/gki487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duvick J., Fu A., Muppirala U., et al. PlantGDB: a resource for comparative plant genomics. Nucleic Acids Res. 2008;36:D959–65. doi: 10.1093/nar/gkm1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jurka J., Kapitonov V.V., Pavlicek A., Klonowski P., Kohany O., Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements, Cytogenet. Genome Res. 2005;110:462–7. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 34.Price A.L., Jones N.C., Pevzner P.A. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21(Suppl 1):i351–8. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- 35.Smit A., Hubley R. RepeatModeler Open-1.0. 2008. http://www.repeatmasker.org. (9 October 2013, date last accessed)
- 36.Tempel S. Using and understanding RepeatMasker. Methods Mol. Biol. 2012;859:29–51. doi: 10.1007/978-1-61779-603-6_2. [DOI] [PubMed] [Google Scholar]
- 37.Smit A., Hubley R., Green P. RepeatMasker Open-3.0. 1996. http://www.repeatmasker.org . (9 October 2013, date last accessed)
- 38.Lee T.-H., Tang H., Wang X., Paterson A.H. PGDD: a database of gene and genome duplication in plants. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks1104. doi:10.1093/nar/gks1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.R Development Core Team. R Foundation for Statistical Computing; 2008,. R: A language and environment for statistical computing. http://www.R-project.org . (2 June 2014, date last accessed) [Google Scholar]
- 40.Edgar R.C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saitou N., Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4:406–25. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 42.Felsenstein J. Confidence limits on phylogenies: an approach using Bootstrap. Evolution. 1985;39:783–91. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 43.Zuckerkandl E., Pauling L. In: Evol. Genes Proteins. Bryson V., Vogel H., editors. Academic Press; 1965. pp. 97–166. [Google Scholar]
- 44.Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011;28:2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Letunic I., Bork P. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2007;23:127–8. doi: 10.1093/bioinformatics/btl529. [DOI] [PubMed] [Google Scholar]
- 46.Letunic I., Bork P. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 2011;39:W475–8. doi: 10.1093/nar/gkr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smarda P., Bureš P., Smerda J., Horová L. Measurements of genomic GC content in plant genomes with flow cytometry: a test for reliability. New Phytol. 2012;193:513–21. doi: 10.1111/j.1469-8137.2011.03942.x. [DOI] [PubMed] [Google Scholar]
- 48.Garg R., Patel R.K., Tyagi A.K., Jain M. De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 2011;18:53–63. doi: 10.1093/dnares/dsq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- 50.Jaillon O., Aury J.-M., Noel B., et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–7. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- 51.Consortium, T.T.G. The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;485:635–41. doi: 10.1038/nature11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chagné D., Crowhurst R.N., Troggio M., et al. Genome-wide SNP detection, validation, and development of an 8K SNP array for apple. PLoS ONE. 2012;7:e31745. doi: 10.1371/journal.pone.0031745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shen Y.-J., Jiang H., Jin J.-P., et al. Development of genome-wide DNA polymorphism database for map-based cloning of rice genes. Plant Physiol. 2004;135:1198–205. doi: 10.1104/pp.103.038463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ching A., Caldwell K.S., Jung M., et al. SNP frequency, haplotype structure and linkage disequilibrium in elite maize inbred lines. BMC Genet. 2002;3:1–14. doi: 10.1186/1471-2156-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Velasco R., Zharkikh A., Troggio M., et al. A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PloS ONE. 2007;2:e1326. doi: 10.1371/journal.pone.0001326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ge A.J., Han J., Li X.D., et al. Characterization of SNPs in strawberry cultivars in China. Genet. Mol. Res. 2013;12:639–45. doi: 10.4238/2013.March.7.2. [DOI] [PubMed] [Google Scholar]
- 57.Xu X., Pan S., Cheng S., et al. Genome sequence and analysis of the tuber crop potato. Nature. 2011;475:189–95. doi: 10.1038/nature10158. [DOI] [PubMed] [Google Scholar]
- 58.Macas J., Kejnovský E., Neumann P., Novák P., Koblížková A., Vyskot B. Next generation sequencing-based analysis of repetitive DNA in the model Dioceous plant Silene latifolia. PLoS ONE. 2011;6:e27335. doi: 10.1371/journal.pone.0027335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Axelsen J.B., Yan K.-K., Maslov S. Parameters of proteome evolution from histograms of amino-acid sequence identities of paralogous proteins. Biol. Direct. 2007;2:32. doi: 10.1186/1745-6150-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Van de Peer Y., Fawcett J.A., Proost S., Sterck L., Vandepoele K. The flowering world: a tale of duplications. Trends Plant Sci. 2009;14:680–8. doi: 10.1016/j.tplants.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 61.Fawcett J.A., Maere S., Van de Peer Y. Plants with double genomes might have had a better chance to survive the Cretaceous-Tertiary extinction event. Proc. Natl. Acad. Sci. USA. 2009;106:5737–42. doi: 10.1073/pnas.0900906106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Westhoff P., Gowik U. Evolution of C4 phosphoenolpyruvate carboxylase. Genes and proteins: a case study with the genus Flaveria. Ann. Bot. 2004;93:13–23. doi: 10.1093/aob/mch003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Christin P.-A., Salamin N., Kellogg E.A., Vicentini A., Besnard G. Integrating phylogeny into studies of C4 variation in the grasses. Plant Physiol. 2009;149:82–7. doi: 10.1104/pp.108.128553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grass Phylogeny Working Group II. New grass phylogeny resolves deep evolutionary relationships and discovers C4 origins. New Phytol. 2012;193:304–12. doi: 10.1111/j.1469-8137.2011.03972.x. [DOI] [PubMed] [Google Scholar]
- 65.Christin P.-A., Sage T.L., Edwards E.J., Ogburn R.M., Khoshravesh R., Sage R.F. Complex evolutionary transitions and the significance of C3-C4 intermediate forms of photosynthesis in Molluginaceae. Evol. Int. J. Org. Evol. 2011;65:643–60. doi: 10.1111/j.1558-5646.2010.01168.x. [DOI] [PubMed] [Google Scholar]
- 66.Galili G. The aspartate-family pathway of plants: linking production of essential amino acids with energy and stress regulation. Plant Signal. Behav. 2011;6:192–5. doi: 10.4161/psb.6.2.14425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kanehisa M., Goto S., Sato Y., Kawashima M., Furumichi M., Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kanehisa M., Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Curien G., Laurencin M., Robert-Genthon M., Dumas R. Allosteric monofunctional aspartate kinases from Arabidopsis. FEBS J. 2007;274:164–76. doi: 10.1111/j.1742-4658.2006.05573.x. [DOI] [PubMed] [Google Scholar]
- 70.Galili G. Regulation of lysine and threonine synthesis. Plant Cell. 1995;7:899–906. doi: 10.1105/tpc.7.7.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Madden T. USA: National Center for Biotechnology Information; 2002. NCBI Handbook [Internet]. Chapter 16The BLAST Sequence Analysis Tool http://www.ncbi.nlm.nih.gov/books/NBK21097/ (26 December 2013, date last accessed) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.