

Abstract
The pufferfish Takifugu flavidus is an important economic species due to its outstanding flavour and high market value. It has been regarded as an excellent model of genetic study for decades as well. In the present study, three mate-pair libraries of T. flavidus genome were sequenced by the SOLiD 4 next-generation sequencing platform, and the draft genome was constructed with the short reads using an assisted assembly strategy. The draft consists of 50,947 scaffolds with an N50 value of 305.7 kb, and the average GC content was 45.2%. The combined length of repetitive sequences was 26.5 Mb, which accounted for 6.87% of the genome, indicating that the compactness of T. flavidus genome was approximative with that of T. rubripes genome. A total of 1,253 non-coding RNA genes and 30,285 protein-encoding genes were assigned to the genome. There were 132,775 and 394 presumptive genes playing roles in the colour pattern variation, the relatively slow growth and the lipid metabolism, respectively. Among them, genes involved in the microtubule-dependent transport system, angiogenesis, decapentaplegic pathway and lipid mobilization were significantly expanded in the T. flavidus genome. This draft genome provides a valuable resource for understanding and improving both fundamental and applied research with pufferfish in the future.
Keywords: Takifugu flavidus, draft genome, NGS
1. Introduction
Pufferfish of the genus Takifugu are considered as one of the most delicious and expensive dishes in Japan, China and Korea, and they have been artificially cultivated since 1960s.1 Nowadays, a big industry of pufferfish aquaculture is already well established in the East Asia. Takifugu flavidus, also known as the tawny puffer, is one representative economic species that is mainly distributed in the shore waters of the East China Sea, the Yellow Sea and the Bohai Bay. The type locality of T. flavidus is in Qingdao, China. In comparison with another common economic pufferfish T. rubripes (torafugu, the tiger puffer), T. flavidus is incapable of long-distance migration, but only short-distance seasonal migration.2 In addition, T. flavidus is morphologically different from T. rubripes in the skin colour pattern, anal fin colour, body size and other characteristics. There is obvious variation in the skin colour pattern during the growth of T. flavidus. The juvenile T. flavidus is light yellow mixed with dark in the dorsal side with irregular white polka dots. As it grows, the dorsal side turns into dark yellow with elongated black spots with daisy-like edge.3 On the contrary, T. rubripes is black in the dorsal side and has particular black marks with white margins above the pectoral fins (colour pattern like tiger skin). The skin colour pattern basically remains unchanged during the growth of T. rubripes.4 Moreover, the body size of T. flavidus is significantly smaller than that of T. rubripes, as well as the growth rate is much slower. The maximum total lengths of T. flavidus and T. rubripes are 40 and 80 cm, respectively. To reach the weight of 300 g, it will take >30 months for T. flavidus but only 12 months for T. rubripes.5 Takifugu flavidus has the highest transaction price among all pufferfish in China, because the taste flavour is believed to be more superior.
Pufferfish have been regarded as excellent models for novel gene discovery and evolution study of vertebrates for decades because of their particularly small and compact genomes.6,7 Their genomes are the smallest of all known vertebrate genomes, while still retain a similar gene repertoire with vertebrates at the same time. There is no significant difference in the quantity and length of genes between pufferfish and other vertebrates, except the sizes of introns and repetitive regions are notably reduced.8 The draft genome of T. rubripes was sequenced and released as the second vertebrate genome in 2002,9 and the draft genome of Tetraodon nigroviridis (green spotted puffer) was released in 2004.10 However, the available genetic information of the T. flavidus is still very limited until now. The sequencing of T. flavidus genome is undoubtedly necessary to understand the genetic basis of the characteristic phenotypes of T. flavidus, and it is also helpful to assist the molecular breeding.
The second-generation sequencing (next-generation sequencing, NGS) technologies have successfully been applied for many research fields in recent years because of the extraordinary high-throughput sequencing capacity and the low cost per base, such as transcriptome analysis, genotyping and target resequencing.11 However, the short reads of NGS make it still a tough task to de novo assemble higher organism genomes.12 A common strategy for the genome construction of higher organism is hybrid assembling with longer reads, for instance, the reads of bacterial artificial chromosome sequencing or 454 sequencing,13 with the inevitable consequence of more workload and longer turnaround time.
In the present study, the draft genome of T. flavidus was sequenced only with the short reads of SOLiD 4 platform and assembled using an assisted assembly approach. This draft was the first genome assembly of T. flavidus, and the third genome of pufferfish. Information on the T. flavidus genome obtained in this study will enhance both fundamental and applied research with T. flavidus and related pufferfish.
2. Materials and methods
2.1. Biological material
The T. flavidus pufferfish used in the present study was from inbred lines produced through several generations of sister–brother mating at the Ocean and Fishery Sciences Research Institute of Hebei Province, China. One individual was randomly selected from the population of inbred for dissection and sampling.
2.2. Genome sequencing
Genomic DNA was extracted from the muscle sample of a 2-yr-old male T. flavidus using the DNeasy Blood & Tissue Kit (Qiagen), and fragmented using the HydroShear DNA Shearing system (Digilab). Mate-pair libraries with average insert sizes of 1, 3 and 7 kb were constructed following the SOLiD 4 Library Preparation Guide (Applied Biosystems), respectively. Templated bead preparation was performed using the SOLiD EZ Bead system (Applied Biosystems). P2-enriched beads were quantified using Nanodrop-2000 (Thermo) and sequenced on the SOLiD 4 Analyzer (Applied Biosystems). The raw sequencing reads were submitted to NCBI Short Read Archive under the accession number SRA059136.
2.3. Genome assembly
Raw reads were error-corrected by the SOLiD Accuracy Enhancer Tool and mapped to the genome of T. rubripes (release 4.66 from Ensembl) using the Bioscope software (Applied Biosystems). The intersection of all sequence variants [reported as single nucleotide polymorphisms (SNPs) and Indels] was extracted from the alignment results of three libraries. Home-made Perl scripts were used for revising the genome of T. rubripes to obtain an ‘intermediate reference’ (IR), which was used as the reference in the next run of alignment step from which new SNPs and Indels were extracted from. The procedure was iterated until no more SNPs and Indels were found. From the result of the latest alignment, consecutive regions that meet the threshold values (length >100 bp, mismatch bases of seed ≤2 bp, mismatch bases of alignment ≤4 bp and coverage >7×) were extracted for contig construction. HAPS (Hybrid Assembly Pipeline with SOLiD reads, http://abcommunity.lifetechnologies.com/docs/DOC-1316) was used for the following scaffolding and gap-filling steps. This whole-genome shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession number AOOT00000000. The assembly described in this paper is the first version, AOOT01000000.
2.4. Genome sequence synteny
A home-made Perl script was used to extract all scaffolds with length above 10 kb in the genomes of T. flavidus, T. rubripes and Tetraodon nigroviridis. The scaffold sequences were aligned by MUMmer (version 3.0).14 The alignment command was ‘nucmer --mum -c 400 -l 150’ for T. flavidus versus T. rubripes and ‘promer --mum -c 40 -g 50 -l 15’ for T. flavidus versus Tetraodon nigroviridis. Alignment was repeated for every combination of scaffolds, and scaffolds were allowed to be rearranged and inverted to achieve max matches. The resulting alignment data were filtered with the delta-filter program of MUMmer.
2.5. Repetitive sequence and non-coding RNA gene identification
All repetitive sequences were identified by the RepeatMasker (version 3.3.0) with the latest RepBase libraries.15 Tandem repeat sequences were also determined using the Tandem Repeat Finder,16 whose results were subsequently summarized by the Tandem Repeats Analysis Program (TRAP).17 Non-coding RNAs were annotated in both the ab initio prediction way and the homologous alignment way. Rfam18 and miRBase19 were searched using BLAST+20 for the ncRNA annotation, and tRNAscan-SE21 was applied for the transfer RNA (tRNA) annotation.
2.6. Potential protein-encoding gene prediction and functional analysis
Gene prediction was performed by automatic gene assignment programmes that combined ab initio gene finding and extrinsic evidence prediction. Augustus22 was trained by the genome annotation of T. rubripes and fed with the transcriptome sequencing data of T. flavidus as ‘hints’ for the protein-encoding gene prediction. The protein-encoding gene prediction was also performed by GENSCAN23 with the parameter file of HumanIso.smat. All gene predictions were evaluated and combined into consensus gene structures by the EVidenceModeler.24 The predicted peptide sequences were aligned to the ‘nr’ database of NCBI using Blastp with an E-value of 1e−5. Sequences with length <50 aa or poorly supported by the Blast hit were discarded. The predicted genes were searched against the InterPro database using InterProScan25 for gene families and functional domain assignment. The local database of Blast2GO26 was used for the GO annotation and the KEGG pathway retrieve.
2.7. Related genome reference information
The genome and all protein sequences of T. rubripes (v4.66) and Tetraodon nigroviridis (v8.66) were downloaded from Ensembl ftp and used for comparison (http://www.ensembl.org/info/data/ftp/index.html).
3. Results and discussion
3.1. Assembly and sequence analysis of the T. flavidus genome
Three mate-pair libraries with average insert sizes of 1, 3 and 7 kb were sequenced and generated 426.3, 154.9 and 177.9 million paired-end reads with read length of 2 × 50 bp, respectively. The genome size was estimated to be ∼390 Mb using K-mer counting. The total sequencing length was 75,920 Mb, covering ∼195-fold of the whole genome.
During the assisted assembling (Supplementary Data), 779,694 SNPs and 156,376 Indels with combined length of 1,779,694 and 1,075,154 bp, respectively, were used as a guide of the genome revision to obtain IR. A total of 511.6 million reads, corresponding to 131.2-fold of the genome, were successfully mapped to IR. Based on the alignment result, 241,314 contigs longer than 100 bp were constructed (Table 1). The longest contig was of 36,127 bp, and the total length of contigs was of 313,590,395 bp. The N50 size of contigs was 2,774 bp.
Table 1.
Assembly statistics
| Total number | N50 (bp) | N90 (bp) | Maximum length (bp) | Mean length (bp) | Total length (Mb) | |
|---|---|---|---|---|---|---|
| Contigs | 241,314 | 2,774 | 577 | 36,127 | 1,299.5 | 313.6 |
| Scaffolds | 50,947 | 305,741 | 10,816 | 2,799,124 | 7,546.1 | 384.5 |
Scaffolding and gap filling were performed using the mate-pair information to interlink the contigs and cover the gaps as many as possible. As a result, 31,086 gaps from low similarity regions were successfully filled among the total 208,550 gaps (Supplementary Table S1). There were 50,947 scaffolds with the combined length of 384,451,902 bp and N50 size of 305,741 bp, which covered >95% of the whole genome (Table 1 and Supplementary Table S2). Thirty-five scaffolds were with length longer than 1 Mb, among which the longest was 2,799,124 bp. The average GC content of T. flavidus genome was 45.2% (Fig. 1), which was very close to that of T. rubripes genome (45.5%) and slightly lower than that of Tetraodon nigroviridis genome (46.3%). In addition, the GC-rich and GC-poor regions in the genome of T. flavidus were slightly less than those of T. rubripes and Tetraodon nigroviridis (Fig. 1).
Figure 1.
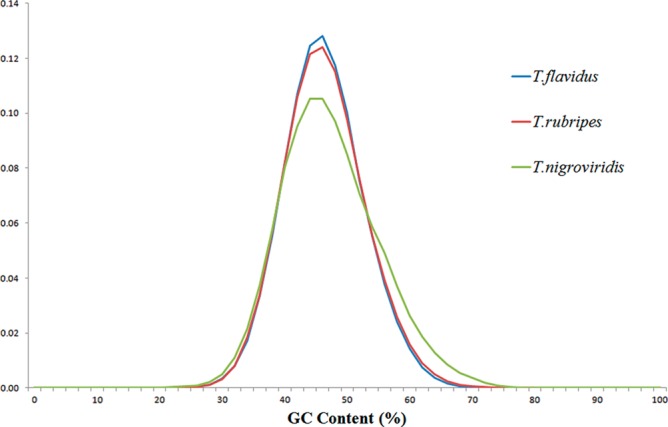
The GC contents of the T. flavidus genome. The size of sliding window was 500 bp. The horizontal axis was the percentage of GC in the windows, whereas the vertical axis showed the frequency of windows. The GC contents of genomes of T. rubripes and Tetraodon nigroviridis were shown for comparison. This figure appears in colour in the online version of DNA Research.
The sequence synteny was evaluated by comparing the T. flavidus assembly with the genome of T. rubripes and Tetraodon nigroviridis, respectively. There were 2,469 scaffolds of T. flavidus (total length was 348.0 Mb) aligned to the 1,996 scaffolds of T. rubripes (total length was 366.9 Mb) and 4,027 scaffolds of Tetraodon nigroviridis (total length was 284.9 Mb), respectively. For T. flavidus versus T. rubripes, there was a high level of colinearity (Fig. 2A). Most scaffolds of T. flavidus were aligned with scaffolds of T. rubripes, whereas only a small number of them had no homologous sequences. For T. flavidus versus Tetraodon nigroviridis, the colinearity was not obvious at DNA level, but clearly visible at protein level (Fig. 2B). The result demonstrated the conservation among the genomes of pufferfish, especially that between T. flavidus and T. rubripes from the same genus.
Figure 2.
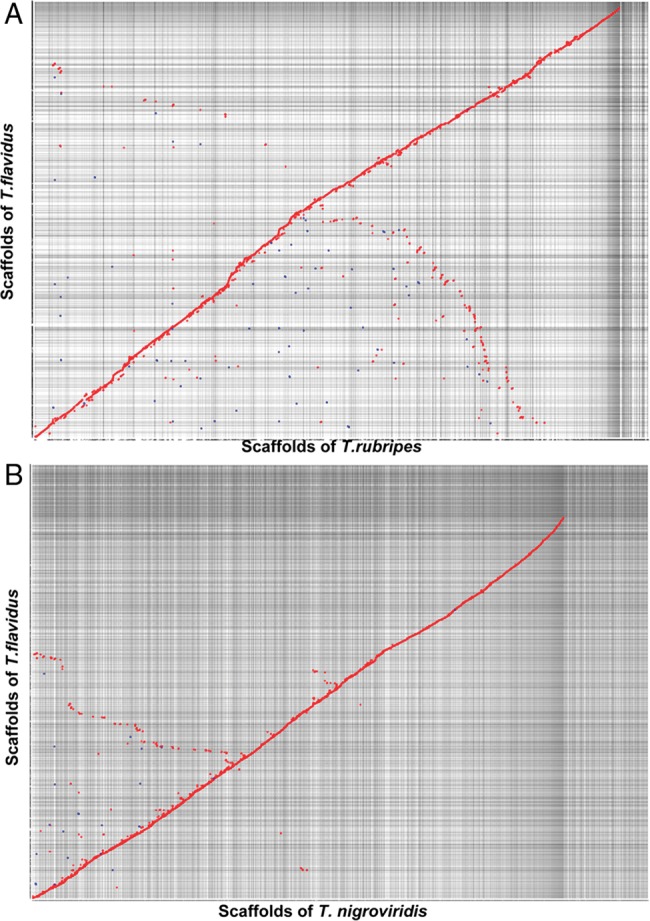
The scaffolds synteny between genomes. Scaffolds from T. flavidus were aligned with that from T. rubripes (A) and Tetraodon nigroviridis (B). The units on the X/Y axis represented the number of scaffolds. Forward matches were shown as red dots, whereas reverse matches were shown in blue dots. This figure appears in colour in the online version of DNA Research.
It is usually computationally demanding and even impossible to assemble the genomes of higher organisms with short reads due to the larger genome size and repetitive sequences.27 Hybrid assembly was a widely used strategy that extensively used longer sequencing reads with higher cost.28,29 In the present study, the genome of T. flavidus was assembled only with the SOLiD short reads following the assisted assembly strategy. The N50 size of contigs was 2.8 kb, N50 size of scaffolds was 305.7 kb and >95% of genome was covered by gap-including scaffolds, exhibiting that the assembly effect was comparable with that of the hybrid assembly strategy.
3.2. Repetitive sequences
A total of 26.5 Mb sequences were identified as repetitive sequences, which accounted for 6.87% of the total genome of T. flavidus. There were 46.9% repetitive sequences with length from 2 to 10 bp, and their combined length was 12.4 Mb. The di-, tri- and tetranucleotide simple sequence repeats (SSRs) took up 26.1, 7.9 and 11.0% of the identified SSRs, respectively. (CA)n, (GCA)n and (ATCC)n were the most common SSR patterns, each representing 83.0% of dinucleotide, 24.4% of trinucleotide and 26.6% of tetranucleotide SSRs, respectively (Supplementary Table S3). RepeatMasker analysis showed 4.81% of genome matched interspersed repeats (Table 2). The most abundant repeats classification was the retroelement [class I transposable elements (TE)], of which the major components were the L2/CR1/Rex type LINEs (1.64%) and the RTE/Bov-B type LINEs (0.50%).
Table 2.
Repetitive sequences in the genome sequences of T. flavidus
| Repeat classification | Element number | Length occupied (bp) | Percentage of sequence |
|---|---|---|---|
| Retroelements (Class I TE) | 35,453 | 13,617,164 | 3.53 |
| SINEs | 4,624 | 682,986 | 0.18 |
| Penelope | 2,472 | 584,662 | 0.15 |
| LINEs | 25,330 | 10,437,024 | 2.71 |
| L2/CR1/Rex | 16,149 | 6,329,913 | 1.64 |
| R2/R4/NeSL | 1,448 | 698,863 | 0.18 |
| RTE/Bov-B | 3,644 | 1,909,961 | 0.50 |
| L1/CIN4 | 851 | 392,390 | 0.10 |
| LTR elements | 5,499 | 2,497,154 | 0.65 |
| BEL/Pao | 84 | 54,053 | 0.01 |
| Ty1/Copia | 93 | 62,584 | 0.02 |
| Gypsy/DIRS1 | 3,811 | 1,780,914 | 0.46 |
| Retroviral | 1,152 | 485,775 | 0.13 |
| DNA transposons (Class II TE) | 12,414 | 4,024,945 | 1.04 |
| hobo-activator | 3,856 | 1,056,562 | 0.27 |
| Tc1-IS630-Pogo | 6,383 | 2,277,388 | 0.59 |
| MuDR-IS905 | 51 | 19,538 | 0.01 |
| PiggyBac | 209 | 47,311 | 0.01 |
| Tourist/Harbinger | 1,286 | 505,502 | 0.13 |
| Unclassified | 5,792 | 887,474 | 0.23 |
| Satellites | 132 | 45,255 | 0.01 |
| Simple repeats | 100,212 | 6,348,631 | 1.65 |
| Low complexity | 30,479 | 1,536,650 | 0.40 |
The proportion of repeats in the T. flavidus genome was greater than that in the Tetraodon nigroviridis genome, which was the most compact vertebrate genome (4.51%), and slightly less than that in the T. rubripes (7.40%). It indicated that the compactness of the T. flavidus genome was close to the genome of T. rubripes, but less than the genome of Tetraodon nigroviridis. The results also showed that class I TE was the major repetitive sequences in all the three pufferfish genomes, and the L2/CR1/Rex type LINEs constituted the major component of the class I, whereas the Tc1-IS630-Pogo type transposons constituted the major component of the class II TE. The high diversity of retrotransposons has been observed in the genomes of other fishes like zebrafish Danio rerio and regarded as a characteristic feature of fish genomes.30 The repetitive sequence repertoires of the three pufferfish species were conserved not only in the diversity but also in the copy number, which indicated that the divergence of the pufferfish lineage occurred not long ago and the evolutionary pressures might lead to the preservation of the repeats identified in the present study.
3.3. Non-coding RNA genes
There were 659 putative tRNA genes identified in the genomic sequences. Among them, 54 genes were probable pseudogenes due to incomplete open reading frames (ORFs), and the remaining 605 tRNA genes corresponding to 50 species of anticodons were sufficient for translation of all amino acids (Table 3 and Supplementary Table S4). In addition, there were 75 genes for 69 species of snoRNA, 86 for 12 species of snRNA (U-RNA), 115 for 5S RNA and 19 for SSU rRNA in the genome of T. flavidus.
Table 3.
Non-coding RNA genes in the genome sequences of T. flavidus
| Non-coding RNA genes | Number of species | Number of genes | Percentage of RNA genes |
|---|---|---|---|
| tRNAs | 50 | 659 | 52.6 |
| tRNAs for standard AA | 49 | 597 | 47.6 |
| tRNAs for Sel-Cys | 1 | 4 | 0.3 |
| tRNAs of unknown isotypes | 4 | 0.3 | |
| Pseudogenes for tRNAs | 54 | 4.3 | |
| 5S rRNAs | 1 | 115 | 9.2 |
| SSU rRNAs | 1 | 19 | 1.5 |
| snoRNAs | 69 | 75 | 6.0 |
| snRNAs | 12 | 86 | 6.9 |
| miRNAs | 143 | 183 | 14.6 |
| Others | 20 | 116 | 9.3 |
| All | 296 | 1,253 | 100 |
Additionally, 183 genes for 143 miRNA species were identified in the genome of T. flavidus (Supplementary Table S5). Among them, 93 miRNA species were also identified in the T. rubripes genome, whereas only 19 miRNA species were identified in the Tetraodon nigroviridis genome. In other words, >53% of the miRNA species of T. flavidus were uniquely conserved within the Takifugu genus, indicating that these genes might emerge after the appearance of Takifugu pufferfish. Meanwhile, the copy numbers of the 20 most abundant miRNA species in the T. flavidus genome were compared with that in the T. rubripes genomes. There were six miRNA species with the same copy number (mir-124-3, mir-135a-1, mir181b-1, mir190, mir190b and mir-375). However, the copy number variations were detected for the remaining 14 miRNA species, and all of them were more abundant in the T. flavidus genome. Since miRNAs played important regulatory roles by targeting mRNAs for cleavage or translational repression,31 the higher copy number of the conserved miRNA species in T. flavidus indicated that its gene expressions were under more rigorous monitoring. It could also be deduced that the recruitment of miRNA genes had been ongoing with speciation in the Takifugu lineage.
3.4. Protein-encoding genes
3.4.1. Prediction of protein-encoding genes
The current T. flavidus gene catalogue was composed of 30,285 protein-encoding genes. Among them, 29,192 genes were complete models with both the start codon and the stop codon, whereas the remaining 1,093 genes were with incomplete ORFs. The longest protein product was of 13,255 aa, and the shortest was of 50 aa. More than 33.7% of the protein products (10,206/30,285) had the sizes above the average length of 517.9 aa. The number of exons per gene was 7.2 on average. In addition, 75% of introns were below 543 bp in size (Q3 value), and the most common intron size was 76 bp (the modal value). By comparing the predicted gene repertoire with gene sets assembled from transcriptome sequencing data,32 only 16 predicted exons (∼0.0%) and 336 predicted introns (∼0.2%) were missed in the transcriptome. In addition, a total of 29,710 novel exons and 2,839 novel introns were present on the transcriptome. The comparison results confirmed the accuracy of gene prediction and also indicated that there were more transcript isoforms in T. flavidus than expected.
The set of protein-encoding genes in T. flavidus was more similar in the number and size distribution as that in T. rubripes (33,609 protein products with an average length of 501.0 aa) than that in Tetraodon nigroviridis (22,400 protein products with an average length of 545.8 aa).9,10 Although the modal values of intron sizes in T. flavidus and T. rubripes were quite similar (76 and 79 bp, respectively), there was an interesting diversity that the Q3 value of intron sizes in T. flavidus was greater than that in T. rubripes and Tetraodon nigroviridis by 68 and >300 bp, respectively, suggesting that T. flavidus possessed slightly longer introns than the other two pufferfish.
3.4.2. Protein-encoding gene components
A similarity search of protein sequences of the 30,285 presumptive protein-encoding genes was performed with the protein sequences of T. rubripes and Tetraodon nigroviridis, respectively. The alignment result indicated that 27,337 genes (90.3%) and 24,088 genes (79.5%) shared significant similarity with those in the T. rubripes and Tetraodon nigroviridis, respectively (E-value <1e−20). Moreover, a total of 270,803 InterPro annotations that were corresponding to 21,848 protein signatures (protein motifs, functional sites and domains) were retrieved for 26,344 protein-encoding genes (87.0%). There were 957 and 1,285 protein signatures of T. flavidus with >10 gene copy number variance comparing with T. rubripes and Tetraodon nigroviridis, respectively (Supplementary Table S6). Among them, 60.4% (578/957) were more abundant than that of T. rubripes, whereas only 28.1% (361/1,285) were more abundant than that of Tetraodon nigroviridis. There were 221 protein signatures in T. flavidus, which were more abundant than that in both T. rubripes and Tetraodon nigroviridis, such as the fibronectin type III superfamily (SSF49265) and SET domain superfamily (SSF82199), whereas 253 protein signatures were less abundant than that in both T. rubripes and Tetraodon nigroviridis such as the neurotransmitter-gated ion-channel transmembrane superfamily (SSF90112). The majority of protein signatures in T. flavidus have a similar copy number with that in T. rubripes and Tetraodon nigroviridis. Since the genome of the genus Takifugu is very compact, the functional genes are supposed to be under high evolutionary pressures, and the pseudogenes often with copy number variation only account for a very small proportion. As for the more or less abundant genes, the copy number variance might be related to unique characteristic such as the muscle composition of T. flavidus.
A total of 51,990 GO annotations were assigned, and 15,813 of all protein-encoding genes were classified into the biological process, molecular function and cellular component categories (Fig. 3). In the level 2 biological process category, cellular process (19.0%), metabolic process (17.2%) and biological regulation (14.0%) accounted for the major portion, whereas the molecular function category and cellular component category were mainly consisted of binding (51.7%) and cell (54.5%), respectively. There were 117 metabolic pathways affiliated by the presumptive protein-encoding genes of T. flavidus in the KEGG database (data not shown).
Figure 3.

GO category classification. The number of GO terms for the biological process, molecular function and cellular component categories was shown. (A) GO-level distribution. P, F and C were short form for biological process, molecular function and cellular component, respectively. (B) Level 2 GO terms in biological process category. (C) Level 2 GO terms in molecular function category. (D) Level 2 GO terms in cellular component category. This figure appears in colour in the online version of DNA Research.
3.5. Characteristic features of the genes in T. flavidus
3.5.1. Genes involved in colour pattern variation
In the present study, 16 genes were identified to be involved in the pigment biosynthesis, including 3 CSF-1 receptor orthologs, 3 tyrosinase orthologs, 8 tetrahydrobiopterin synthase orthologs and 4 GTP cyclohydrolase orthologs (two genes were also tetrahydrobiopterin synthase orthologs). They were important components of the two major pigment synthesis pathways in teleost fish: the melanin and the pteridine pathways, which played important roles in the generation of black and yellow pigments, respectively.33 In teleost fish, the pigment granules were distributed in either microtubule-dependent or microtubule-independent ways.34,35 In the T. flavidus genome, there were 116 genes involved in the microtubule-dependent transport system, including the plectin and dynein orthologous genes (Supplementary Table S7), and 108 genes were with complete ORFs. It was interesting that 12 dynein orthologous genes were aligned in the SCAFFOLD_0437, suggesting the tandem duplication of these genes. The corresponding sequences that shared sequence similarity with the dynein orthologous genes distributed in at least five scaffolds (scaffold_144, scaffold_214, scaffold_286, scaffold_371 and scaffold_440) of the T. rubripes genome, but none of them contains more than two copies of dynein orthologous genes. On the contrary, there were only 58 microtubule-related genes in the genome of T. rubripes. The identified genes confirmed the mechanisms of the pigment development and colour pattern formation in teleost fish, and the expansion of microtubule-related genes in T. flavidus suggested that the enhanced transport capacity provided by the microtubule-dependent pigment transport system might play an important role in the colour pattern variation during the growth of T. flavidus.
3.5.2. Growth- and development-related genes
There were a batch of genes involved in the growth and development through hormonally mediated pathways in the teleost fish, such as insulin-like growth factors (IGFs) and growth hormone (GH) axis.36 The growth rate and appetite were obviously enhanced in the GH-transgenic coho salmon, according to previous studies.37 In the genome of T. flavidus, there were 718 genes from 9 species of growth factors and corresponding receptors, including vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), fibroblast growth factor (FGF), transforming growth factor (TGF), platelet-derived growth factor (PDGF), nerve growth factor (NGF), insulin-like growth factor (IGF), granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), as well as 8 genes of GHs and 7 genes of the somatostatin (GH-inhibiting hormone) (Table 4). Comparing with the T. rubripes and Tetraodon nigroviridis genomes, the gene copy numbers of VEGF and VEGF receptors in T. flavidus were significantly greater, which participated in blood vessel formation (angiogenesis). It might hint that the angiogenesis function was more activated in T. flavidus, but conclusion can only be made upon further evidence. Surprisingly, there was no significant difference in most growth factors/hormone-related gene copies among the three pufferfish. EGF and PDGF even had more gene copies in Tetraodon nigroviridis, which had the smallest body size. It was reasonable to believe that the body size was determined and regulated by alternative genes. At the same time, there was an interesting finding that the genes potentially encoding proteins with the Brinker DNA-binding domain (BrkDBD) existed in both the T. flavidus and Tetraodon nigroviridis genomes, whereas none was found in T. rubripes. In T. flavidus, 40 of the 42 Brinker-encoding genes were with complete ORFs and distributed in 38 scaffolds of T. flavidus. Structure comparison between BrkDBD coding genes of T. flavidus (including exons and introns) and genome sequences of T. rubripes was also performed (Fig. 4). The data showed that even sharing a high level of sequence similarity in total, there is no complete homologous sequences of the BrkDBD coding gene distributed in a single scaffold of T. rubripes. Instead, the homologous sequences distribute into multiple scaffolds of T. rubripes. It is most likely due to the species difference, not assembly artefacts, since the BrkDBD coding regions are supported by transcriptome data as well. Among them, 30 BrkDBD-encoding regions were hit by the transcriptome sequencing data of T. flavidus,32 whereas none was observed in the transcriptome of T. rubripes. In Tetraodon nigroviridis, there were five BrkDBD-encoding genes located in the same chromosome. The Brinker is a transcription repressor of the activated targets of decapentaplegic (Dpp), whose gradient resulted in significantly increased tissue size and cell amounts.38 The Dpp/Brinker system was a well-known regulator of tissue growth and size.39,40 There was likelihood that the smaller body size and slow growth rate of T. flavidus and Tetraodon nigroviridis were regulated by the Brinker-encoding genes.
Table 4.
The growth- and development-related genes in T. flavidus
| Genes | T. flavidus | T. rubripes | Tetraodon nigroviridis |
|---|---|---|---|
| VEGF | 15 | 5 | 12 |
| EGF | 488 | 381 | 513 |
| FGF | 37 | 40 | 31 |
| TGF | 91 | 78 | 86 |
| PDGF | 24 | 17 | 30 |
| NGF | 7 | 6 | 7 |
| IGF | 23 | 28 | 26 |
| G/GM-CSF | 5 | 5 | 4 |
| Growth hormone | 8 | 8 | 7 |
| BrkDBD | 42 | 0 | 5 |
| Somatostatin | 7 | 8 | 8 |
The copy number of growth factors/hormones and their receptors was exhibited. T. rubripes and Tetraodon nigroviridis were also shown for comparison.
Figure 4.

Homologous sequences of BrkDBD coding genes in T. flavidus and T. rubripes. BrkDBD coding genes in T. flavidus were aligned to the T. rubripes genome and are shown in (B). The gene names were suffixed with responding scaffold ID. Detailed comparison of one BrkDBD gene between T. flavidus and analogous sequences of T. rubripes is shown as an example in (A). This figure appears in colour in the online version of DNA Research.
3.5.3. Genes involved in lipid metabolism
Lipids, along with proteins, are the major organic constituents of fish and play an indispensable role in maintaining the structure and function of cellular membranes. More importantly, they are the major source of metabolic energy for growth, reproduction, swimming and migration in teleost fish rather than carbohydrates which are mainly utilized in mammals.41 In the genome of T. flavidus, there were a total of 394 genes involved in the lipid metabolism, including 185 genes in the lipid digestion, 12 in the lipid absorption, 154 in the extracellular and intracellular transport, 38 in the biosynthesis and catabolism and 5 in the mobilization (Supplementary Table S8). No significant variation was observed for the copy numbers of the most lipid metabolism-related genes in T. flavidus (short-distance migration puffer) comparing with that in the T. rubripes (long-distance migration puffer). In the genome of T. flavidus, there were one adiponectin receptor gene and one noradrenaline transporter gene both involved in the mobilization of the lipids, whereas none was found in the genome of T. rubripes. The potential coding sequences found in T. flavidus were then aligned to the T. rubripes genome with blastn (E-value = 1e−20), revealing 5 and 7 scaffolds of T. rubripes sharing significant similarity, respectively (Fig. 5). Taken the adiponectin receptor gene for example, the homologous sequences for the first ∼6 kb are distributed in scaffold_202 of T. rubripes, and the majority of the left region (after ∼7 kb) is distributed in scaffold_8 of T. rubripes. The dispersed distribution led to the fact that no complete coding DNA sequence (CDS) of these two genes can be found in T. rubripes. However, the adiponectin receptor gene was identified in both transcriptomes when mapped onto the T. flavidus genome with a similar expression level in T. flavidus and T. rubripes. One reasonable explanation is that the T. rubripes assembly is still incomplete, which caused difficulty of CDS searching and protein prediction, so the adiponectin receptor gene should exist and be activated in both T. flavidus and T. rubripes. In addition, 20 of 24 exons of the potential noradrenaline transporter-encoding gene were found in the transcriptome of T. flavidus, but only 2 exons were hit by the transcriptome of T. rubripes. It was possibly only caused by mismatch, because only two exons were hit and expression levels were quite low. The transcriptome data confirmed the possibility that the noradrenaline transporter gene has a higher expression level in T. flavidus, even if there is complete CDS in T. rubripes. It was known that noradrenaline regulated the energy supply by stimulating the triacylglycerol hydrolysis and fatty acid oxidation.41 Therefore, it might be an implication of the particular lipid utilization mechanisms for the short-distance migration of T. flavidus, comparing with the long-distance migration pufferfish.
Figure 5.
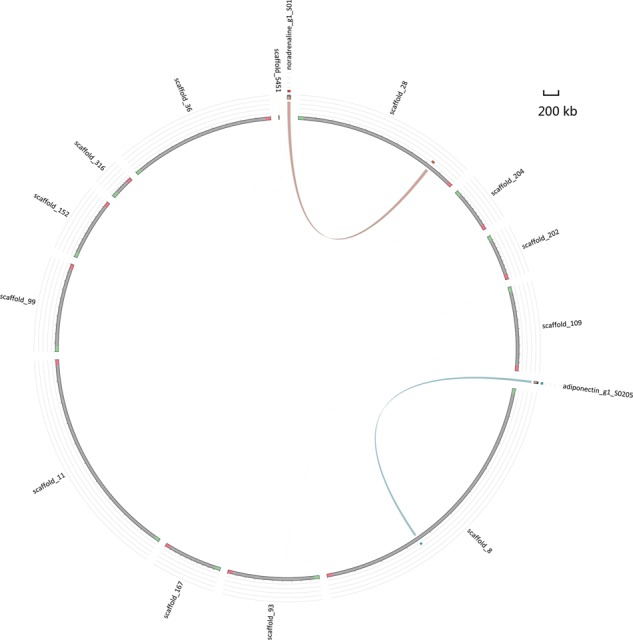
Homologous sequences of adiponectin receptor gene and noradrenaline transporter gene in T. flavidus and T. rubripes. The sequences of adiponectin receptor gene and noradrenaline transporter gene in T. flavidus were aligned to scaffolds of T. rubripes and shown respectively. This figure appears in colour in the online version of DNA Research.
3.6. Conclusions
The genome of T. flavidus pufferfish was sequenced by the SOLiD 4 platform, and the short reads were assembled using the assisted assembly strategy. The draft genome was of 384.5 Mb, comprising 50,947 scaffolds with an N50 value of 305.7 kb. The assisted assembly in the present study achieved a rival result comparing with the hybrid assembly in cooperation with longer reads, but at much lower cost. It can be applied to the genome assembling of species whichever had a closely related reference. The genome analysis revealed that T. flavidus has a lack of repetitive sequences, and the compactness was similar with the genome of T. rubripes. There were 30,285 presumptive protein-encoding genes in the genome. Through functional analysis of genes, the presumable genes involving in the characteristic features of T. flavidus including the colour pattern variation during growth, the relatively slow growth rate and the possible lipid metabolism mechanisms for short-distance migration were identified. The genome information reported in the present study provides a valuable platform for further studies of pufferfish and possesses significant importance in aquaculture industry and scientific research.
Supplementary Data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
This research was supported by High Technology Project (863 Program, No. 2014AA093501) from the Chinese Ministry of Science and Technology. Funding to pay the Open Access publication charges for this article was provided by the Ministry of Science and Technology of the People's Republic of China.
Supplementary Material
Acknowledgements
We thank Zhao Xu and Hongshan Jiang for the assistance of data analysis and assembly pipeline development and also thank Ping Zhang and Zengfang Zhao from High Performance Computing Center, Institute of Oceanology, Chinese Academy of Sciences and the Supercomputing Center of Chinese Academy of Sciences for the computing resources and technical support.
References
- 1.Watabe S., Ikeda D. Diversity of the pufferfish Takifugu rubripes fast skeletal myosin heavy chain genes. Comp. Biochem. Physiol. 2006;1:28–34. doi: 10.1016/j.cbd.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Zhang G., Shi Y., Zhu Y., Liu J., Zang W. Effects of salinity on embryos and larvae of tawny puffer Takifugu flavidus. Aquaculture. 2010;302:71–5. [Google Scholar]
- 3.Shi Y., Zhang G., Zhu Y., Liu J., Zang W. Embryonic development in cultured tawny puffer, Takifugu flavidus in an estuary. J. Dalian Ocean Univ. 2010;25:238–42. [Google Scholar]
- 4.Reza M.S., Furukawa S., Mochizuki T., Matsumura H., WataBe S. Genetic comparison between torafugu Takifugu rubripes and its closely related species karasu Takifugu chinensis. Fish. Sci. 2008;74:743–54. [Google Scholar]
- 5.Shi Y., Zhang G., Zhu Y., Yan Y., Liu J., Zhu J. Study on artificial breeding techniques of Takifugu flavidus in estuary area of Hangzhou Bay. Mod. Fish. Sust. Dev. 2009;1:165–72. [Google Scholar]
- 6.Brenner S., Elgar G., Sandford R., Macrae A., Venkatesh B., Aparicio S. Characterization of the pufferfish (Fugu) genome as a compact model vertebrate genome. Nature. 1993;366:265–8. doi: 10.1038/366265a0. [DOI] [PubMed] [Google Scholar]
- 7.Venkatesh B., Gilligan P.,, Brenner S. Fugu: a compact vertebrate reference genome. FEBS Lett. 2000;476:3–7. doi: 10.1016/s0014-5793(00)01659-8. [DOI] [PubMed] [Google Scholar]
- 8.Poulter R., Butler M. A retrotransposon family from the pufferfish (fugu) Fugu rubripes. Gene. 1998;215:241–9. doi: 10.1016/s0378-1119(98)00296-0. [DOI] [PubMed] [Google Scholar]
- 9.Aparicio S., Chapman J., Stupka E., et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science. 2002;297:1301–10. doi: 10.1126/science.1072104. [DOI] [PubMed] [Google Scholar]
- 10.Jaillon O., Aury J.-M., Brunet F., et al. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature. 2004;431:946–57. doi: 10.1038/nature03025. [DOI] [PubMed] [Google Scholar]
- 11.Metzker M.L. Sequencing technologies—the next generation. Nat. Rev. Genet. 2009;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 12.Flicek P., Birney E. Sense from sequence reads: methods for alignment and assembly. Nat. Methods. 2009;6:S6–12. doi: 10.1038/nmeth.1376. [DOI] [PubMed] [Google Scholar]
- 13.Schatz M.C., Delcher A.L., Salzberg S.L. Assembly of large genomes using second-generation sequencing. Genome Res. 2010;20:1165–73. doi: 10.1101/gr.101360.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurtz S., Phillippy A., Delcher A.L., et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jurka J., Kapitonov V., Pavlicek A., Klonowski P., Kohany O., Walichiewicz J. Repbase update: a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005;110:462–7. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 16.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–80. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sobreira T.J.P., Durham A.M., Gruber A. TRAP: automated classification, quantification and annotation of tandemly repeated sequences. Bioinformatics. 2006;22:361–2. doi: 10.1093/bioinformatics/bti809. [DOI] [PubMed] [Google Scholar]
- 18.Gardner P.P., Daub J., Tate J., et al. Rfam: wikipedia, clans and the ‘decimal’ release. Nucleic Acids Res. 2011;39:D141–5. doi: 10.1093/nar/gkq1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozomara A., Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–7. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Camacho C., Coulouris G., Avagyan V., et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lowe T.M., Eddy S.R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keller O., Kollmar M., Stanke M., Waack S. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics. 2011;27:757–63. doi: 10.1093/bioinformatics/btr010. [DOI] [PubMed] [Google Scholar]
- 23.Burge C., Karlin S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- 24.Haas B.J., Salzberg S.L., Zhu W., et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9:R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hunter S., Apweiler R., Attwood T.K., et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–5. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Conesa A., Götz S., García-Gómez J.M., Terol J., Talón M., Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–6. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 27.Ye L., Hillier L., Minx P., et al. A vertebrate case study of the quality of assemblies derived from next-generation sequences. Genome Biol. 2011;12:R31. doi: 10.1186/gb-2011-12-3-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takeuchi T., Kawashima T., Koyanagi R., et al. Draft genome of the Pearl Oyster Pinctada fucata: a platform for understanding bivalve biology. DNA Res. 2012;19:117–30. doi: 10.1093/dnares/dss005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato S., Hirakawa H., Isobe S., et al. Sequence analysis of the genome of an oil-bearing tree, Jatropha curcas L. DNA Res. 2010;18:65–76. doi: 10.1093/dnares/dsq030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Volff J.-N., Bouneau L., Ozouf-Costaz C., Fischer C. Diversity of retrotransposable elements in compact pufferfish genomes. Trends Genet. 2003;19:674–8. doi: 10.1016/j.tig.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 31.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 32.Yang G., Huan Z., Qiang G., et al. Transcriptome analysis of artificial hybrid pufferfish Jiyan-1 and its parental species: implications for pufferfish heterosis. PLoS ONE. 2013;8:e58453. doi: 10.1371/journal.pone.0058453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Braasch I., Schartl M., Volff J.N. Evolution of pigment synthesis pathways by gene and genome duplication in fish. BMC Evol. Biol. 2007;7:74. doi: 10.1186/1471-2148-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murphy D.B., Tilney L.G. The role of microtubules in the movement of pigment granules in teleost melanophores. J. Cell Biol. 1974;61:757–79. doi: 10.1083/jcb.61.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schliwa M., Euteneuer U. A microtubule-independent component may be involved in granule transport in pigment cells. Nature. 1978;273:556–8. doi: 10.1038/273556a0. [DOI] [PubMed] [Google Scholar]
- 36.Duan C. Nutritional and developmental regulation of insulin-like growth factors in fish. J. Nutr. 1998;128:306S–14S. doi: 10.1093/jn/128.2.306S. [DOI] [PubMed] [Google Scholar]
- 37.Devlin R., Johnsson J., Smailus D., Biagi C., Jönsson E., Björnsson B.T. Increased ability to compete for food by growth hormone-transgenic coho salmon Oncorhynchus kisutch (Walbaum) Aquaculture Res. 1999;30:479–82. [Google Scholar]
- 38.Rogulja D., Irvine K.D. Regulation of cell proliferation by a morphogen gradient. Cell. 2005;123:449–61. doi: 10.1016/j.cell.2005.08.030. [DOI] [PubMed] [Google Scholar]
- 39.Schwank G., Restrepo S., Basler K. Growth regulation by Dpp: an essential role for Brinker and a non-essential role for graded signaling levels. Development. 2008;135:4003–13. doi: 10.1242/dev.025635. [DOI] [PubMed] [Google Scholar]
- 40.Jaźwińska A., Kirov N., Wieschaus E., Roth S., Rushlow C. The Drosophila gene Brinker reveals a novel mechanism of Dpp target gene regulation. Cell. 1999;96:563. doi: 10.1016/s0092-8674(00)80660-1. [DOI] [PubMed] [Google Scholar]
- 41.Tocher D.R. Metabolism and functions of lipids and fatty acids in teleost fish. Rev. Fish. Sci. 2003;11:107–84. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.