

Abstract
Eukaryotic microbial communities play key functional roles in soil biology and potentially represent a rich source of natural products including biocatalysts. Culture-independent molecular methods are powerful tools to isolate functional genes from uncultured microorganisms. However, none of the methods used in environmental genomics allow for a rapid isolation of numerous functional genes from eukaryotic microbial communities. We developed an original adaptation of the solution hybrid selection (SHS) for an efficient recovery of functional complementary DNAs (cDNAs) synthesized from soil-extracted polyadenylated mRNAs. This protocol was tested on the Glycoside Hydrolase 11 gene family encoding endo-xylanases for which we designed 35 explorative 31-mers capture probes. SHS was implemented on four soil eukaryotic cDNA pools. After two successive rounds of capture, >90% of the resulting cDNAs were GH11 sequences, of which 70% (38 among 53 sequenced genes) were full length. Between 1.5 and 25% of the cloned captured sequences were expressed in Saccharomyces cerevisiae. Sequencing of polymerase chain reaction-amplified GH11 gene fragments from the captured sequences highlighted hundreds of phylogenetically diverse sequences that were not yet described, in public databases. This protocol offers the possibility of performing exhaustive exploration of eukaryotic gene families within microbial communities thriving in any type of environment.
Keywords: metatranscriptomics, soil RNA, soil eukaryotes, sequence capture, glycoside hydrolase family GH11
1. Introduction
A common objective of many studies in the field of environmental microbiology is to evaluate the functional diversity of the complex microbial communities colonizing natural or man-made environments, fresh or marine waters, sediments, soils, digestive tracts or food products. This diversity can be apprehended through the systematic sequencing and functional annotation of DNA (metagenomics) or RNA (metatranscriptomics) molecules directly extracted from environmental samples.1,2 However, as a result of the extreme taxonomic richness of most microbial communities, high-throughput shotgun sequencing of environmental nucleic acids is far from covering their full gene repertoire.3 Alternatively, many studies focus on specific environmental processes which, for some of them, are controlled by a limited and defined set of genes encoding key enzymes. The diversity of the corresponding gene families and of the organisms that possess and express them is classically evaluated by the systematic sequencing and taxonomic annotation of polymerase chain reaction (PCR)-amplified gene fragments from environmental DNA or RNA (metabarcoding).4–7 This latter approach has itself well-documented limitations. One of the limitations is that the use of a single pair of degenerate primers, designed to hybridize to internal gene consensus sequences, usually fails to amplify all homologous sequences present in an environmental sample.8 Another, often underestimated limitation is that metabarcoding does not allow amplification of full-length functional genes. Besides limiting the number of phylogenetically informative nucleotide positions for precise phylogenetic assignment of environmental sequences, obtaining partial sequences also prevents their functional study by expression in a heterologous microbial host. Full-length functional genes are yet of importance (i) in ecology to establish potential relationships between enzyme catalytic properties (substrate range, sensitivity to physicochemical parameters) and prevailing environmental conditions and (ii) in environmental biotechnology to isolate novel biocatalysts for industrial purpose.
Recently, Denonfoux et al.9 developed an alternative strategy to explore microbial communities from complex environments. Based on solution hybrid selection (hereafter referred to as SHS), this method allows for the specific recovery of large DNA fragments harbouring biomarkers of interest even from rare or unknown microorganisms. Indeed, SHS is based on the design of several oligonucleotide probes which can cover the whole gene of interest as opposed to PCR strategies targeting internal regions. Moreover, explorative probe design strategies using appropriate software such as HiSpOD10 or KASpOD11 allow recovering not yet described homologous sequences.9 These probes are synthesized as biotinylated RNA oligonucleotides and hybridized, in solution, to the target gene sequences diluted among a majority of non-target DNA fragments. The hybrid molecules (biotinylated probes + target sequences) are then specifically captured by affinity binding on streptavidin-coated paramagnetic beads. SHS can be repeated several times successively to increase the enrichment in desired sequences by a factor of up to 1.7 × 105 times.9 In environmental microbiology, the captured DNA fragments can be subjected to high-throughput sequencing. In silico assembly of the reads not only leads to the reconstruction of the full-length sequences of the different members of the targeted gene family, but also of their genomic environment and could therefore facilitate operon reconstructions.9
In microbial ecology, SHS has thus far been successfully used to capture archaeal protein-coding genes from environmental DNA.9 As previously discussed,12 environmental DNA is however not the most appropriate matrix to recover full-length functional genes of eukaryotic origin, which could be easily expressed in a heterologous microbial host. Environmental polyadenylated messenger RNAs, devoid of introns, represent a better source of eukaryotic genes which, following their conversion into complementary DNAs (cDNAs), can be expressed in either bacteria or yeasts.12–16
Soil eukaryotes such as fungi are highly diverse,17,18 play essential roles in soil biology as, for example, the main agents in plant organic matter degradation19,20 and represent a rich source of enzymes and biomolecules used in industry.21 Despite these obvious interests, very few environmental genomics studies specifically focus on soil eukaryote functional diversity.22
To promote such studies, we developed and evaluated in the present report an original adaptation of the SHS for the efficient recovery of full-length functional fungal cDNAs synthesized from soil RNA. Successful development of this technique was favoured by the ever increasing number of available fungal genomes that provide a correspondingly large number of members of specific gene families for the design of hybridization probes.23 The fungal gene family targeted in this study is the Glycoside Hydrolase 11 (GH11) family which encode endo-β-1,4-xylanases (E.C. 3.2.1.8) (CAZY Carbohydrate Active Enzymes database, http://www.cazy.org).24 As xylan is the second most abundant polysaccharide in nature and one of the major structural polysaccharide in the plant cell wall, such enzymes have an obvious importance for soil ecology and for the degradation of plant hemicelluloses. A recent study also suggested that fungi contributed to most xylanase activity in soils.25 Furthermore, GH11 enzymes are also abundantly used in different industrial processes.26 GH11 genes are present in the genomes of numerous fungi, mainly Ascomycota and Basidiomycota, and at the start of this study, >300 sequences were publicly available. Furthermore, in a random shotgun sequencing of forest soil eukaryotic polyA-mRNAs, it was shown that GH11 transcripts occurred at a low frequency ranging from 0 to 1 per 104 sequences obtained.22,27
2. Materials and methods
2.1. Soil RNA extraction and cDNA synthesis
Four different forest soils from France and Italy were used in this study (see Supplementary Table S1 for sites and soils characteristics). At each site, between 30 (BEW) and 60 (BRH) sieved (2 mm) soil cores were mixed together to constitute composite samples which were stored at −75°C prior to RNA extraction. RNA was extracted from 4 to 48 g of soil using protocols adapted to each soil. RNA from the Puéchabon (PUE) sample was extracted according to Luis et al.28 RNA from the Breuil Spruce (BRE) and Breuil Beech (BRH) samples was extracted according to Damon et al.29 RNA from the Berchidda (BEW) sample was extracted using the PowerSoil® Total RNA Isolation Kit (Mo Bio Laboratories), according to the manufacturer's instructions. All RNA samples were treated with RNase-free DNase I to remove residual DNA contaminations and quantified by spectrophotometry (ND-1000 NanoDrop®, Thermo Scientific).
Eukaryotic cDNAs were synthesized from 2 µg of total soil RNA by using the Mint-2 cDNA synthesis and amplification kit according to the manufacturer's instructions (Evrogen). First-strand synthesis was initiated at the RNA 3′ poly-A end using a modified poly-dT primer (CDS-4M). The number of PCR cycles (between 22 and 30) necessary for optimal synthesis of the double-stranded cDNA (dscDNA) was evaluated for each cDNA sample. As a result of using the Mint-2 kit, all amplified cDNAs were bordered at their 5′ end by the M1 sequence (AAGCAGTGGTATCAACGCAGAGT) and the SfiIA restriction site (GGCCATTACGGCC) while, at their 3′ end, they were bordered by the SfiIB restriction site (GGCCGAGGCGGCC) and the M1 sequence. dscDNA was purified by phenol–chloroform extraction, precipitated by 2.5 volume of ethanol and 0.1 volume of sodium acetate, resuspended in ultrapure water and quantified.
2.2. Capture probe design and synthesis
As in July 2012, all publicly available GH11 DNA coding sequences of eukaryotic origin were identified by BLAST searches30 and collected from GenBank (http://www.ncbi.nlm.nih.gov/genbank/), the Joint Genome Institute database (http://jgi.doe.gov/), the Broad Institute genome database (http://www.broadinstitute.org/) and CAZy (http://www.cazy.org/). A set of thirty-five 31-mers, degenerate capture probes, targeting the catalytic domain of the encoded proteins (pfam no. PF00457, ∼540 nucleotide long; Supplementary Fig. S1), was designed from a collection of 342 coding DNA sequences using the KASpOD software.11 Individual probe coverage ranged from 7 to 54% of the 342 sequences, leading to a probe set coverage of 90% (four allowed mismatches).
The 35 oligonucleotide probes included the specific sequences (X)31 targeting cDNAs encoding GH11 and adaptor sequences at each extremities for PCR amplification: ATCGCACCAGCGTGT-(X)31-CACTGCGGCTCCTCA (Supplementary Table S2 and Fig. S1). Biotinylated RNA capture probes were prepared according to the two-step procedure of Gnirke et al.31 In the first step, each single-stranded DNA probe was amplified by PCR using primers complementary to the 5′ and 3′ adaptors to allow double-strand DNA formation. In the second step, agarose gel-purified double-stranded DNA probes were converted into biotinylated RNA probes by in vitro transcription using the MEGAScript®T7 kit (Ambion) and biotin-dUTP (TeBu Bio). RNA probes were then mixed together in equimolar amounts.
2.3. cDNA capture
cDNA capture was carried out as described by Denonfoux et al.9 and summarized in Supplementary Fig. S2. Briefly, 500 ng of heat denatured PCR-amplified cDNAs were hybridized to the equimolar mix of biotinylated RNA probes (500 ng) for 24 h at 65°C. Probe/cDNA hybrids were trapped by streptavidin-coated paramagnetic beads (Dynabeads® M-280 Streptavidin, Invitrogen). After different washing steps to remove unbound cDNAs, the captured cDNAs were eluted from the beads using 50 µl of 0.1 M NaOH at room temperature, neutralized with 70 µl of 1 M Tris–HCl, pH 7.5, and purified using the Qiaquick PCR purification kit (Qiagen).
Captured cDNAs were PCR amplified using primer M1 that binds at both 5′ and 3′ ends of the cDNAs. PCRs were set up using 5 µl of eluate, 200 µM of deoxynucleotides (dNTPs), 400 nM primer M1, 5 µl of reaction buffer 10× (Evrogen) and 1 µl of 50× Encyclo DNA polymerase (Evrogen) in a final volume of 50 µl. After an initial denaturation at 95°C for 1 min, cDNAs were amplified for 25 cycles comprising 15 s at 95°C, 20 s at 66°C and 3 min at 72°C. Ten independent amplifications were conducted for each sample. PCR products of the same sample were purified on QIAquick columns (Qiagen) and pooled. A second round of hybridization and PCR amplification was performed using each of the amplified cDNA samples obtained after the first hybridization capture. Purified products originating from the same cDNA sample were pooled together and quantified by spectrophotometry (NanoDrop™ 2000, Thermo Scientific). The DNA quality and size distribution of captured cDNA were assessed on an Agilent 2100 Bioanalyzer DNA 12000 chip (Agilent Technologies).
2.4. Semi-quantitative PCR
Enrichment in GH11 sequences at each step of the capture protocol was evaluated by semi-quantitative PCR using different quantities of cDNAs and GH11-fungal-specific degenerate primers GH11-F (GGVAAGGGITGGAAYCNNGG) and GH11-R (TGKCGRACIGACCARTAYTG) amplifying a ±281-bp fragment (Luis P. et al., unpublished). PCRs were performed using 10, 1, 0.1 or 0.01 ng cDNAs obtained before and after one or two cycles of hybridization capture. Twenty-five microlitres of PCR mixes contained 1 µl of template cDNA, 2.5 µl of 10× PCR buffer without Mg (Invitrogen), 1.5 mM MgCl2, 0.8 mM of each dNTP, 0.5 µM of each primer and 1 U of Taq DNA polymerase (Invitrogen). After an initial denaturation at 94°C for 3 min, GH11 gene fragments were amplified for 45 cycles comprising 45 s at 94°C, 45 s at 50°C and 2 min at 72°C. After a final elongation at 72°C for 10 min, 10 µl of PCR products were run in a 1.5% ethidium bromide-stained agarose gel.
2.5. High-throughput sequencing
Diversity of GH11 sequences at each step of the capture protocol was evaluated by high-throughput sequencing of GH11 PCR products obtained, as described above, using primers GH11-F and GH11-R. PCRs were performed using cDNAs obtained before and after one or two cycles of hybridization capture. Twenty-five microlitres of PCR mixes contained 10 ng of template cDNA, 2.5 µl of 10× PCR buffer without Mg (Invitrogen), 1.5 mM MgCl2, 0.8 mM of each dNTP, 0.5 µM of each primer and 1.25 U of DNA polymerase (a 24:1 mix of Invitrogen Taq DNA polymerase and Biorad iProof polymerase). PCR cycling conditions were as described above. Five different PCRs were prepared and run in parallel for each cDNA sample. PCR products were first checked on 1.5% agarose gel before pooling together the five replicates and purification using the QIAquick PCR purification kit (Qiagen). Paired-end sequencing (2 × 250 bp) was carried out on an Illumina MiSeq sequencer (Fasteris, Switzerland).
Paired-end reads were assembled using PandaSeq v.2.5,32 and all sequences containing unidentified nucleotide positions (‘N’) were filtered out. Primers and barcodes were removed using MOTHUR v.1.30.2.33 UCHIME34 was used for chimera detection, and sequence clusters were constructed at a 95% nucleotide sequence identity threshold. The most abundant representative sequence of each of the most abundant clusters, altogether encompassing >90% of the sequences, was translated into amino acid sequence using the ORF Finder tool of the Sequence Manipulation Suite35 (http://www.bioinformatics.org/sms2/). Shannon diversity indices (H′) were calculated after rarefying the different data sets from the same soil to the same sequencing depth (i.e. the lowest sequencing depth of the three samples of each soil, Table 2). Venn diagrams were drawn using the BioVenn tool (http://www.cmbi.ru.nl/cdd/biovenn/).
Table 2.
Summary statistics from Illumina MiSeq sequencing of GH11 PCR fragments amplified, for each four cDNA samples, before (H0) or after one (H1) or two (H2) hybridization capture
| Sample | Total no. of sequences | Total no. of clustersa (95%) | No. of clusters encompassing ≥90% of the sequences | Shannon diversity index (H′)b | No. of shared clusters between H0–H1–H2b |
|---|---|---|---|---|---|
| PUE_H0 | 12,960 | 298 | 52 (17%) | 3.819 | 70 (11%) |
| PUE_H1 | 24,565 | 227 | 51 (22%) | 4.015 | |
| PUE_H2 | 25,053 | 291 | 46 (16%) | 3.912 | |
| BRE_H0 | 13,538 | 87 | 9 (10%) | 2.254 | 11 (5%) |
| BRE_H1 | 42,000 | 140 | 5 (4%) | 1.651 | |
| BRE_H2 | 46,626 | 112 | 6 (5%) | 1.73 | |
| BRH_H0 | 2,765 | 26 | 3 (12%) | 1.061 | 5 (4%) |
| BRH_H1 | 28,366 | 51 | 3 (6%) | 1.234 | |
| BRH_H2 | 17,322 | 159 | 18 (11%) | 2.135 | |
| BEW_H0 | 41,799 | 214 | 15 (7%) | 2.761 | 38 (6%) |
| BEW_H1 | 42,308 | 249 | 10 (4%) | 2.496 | |
| BEW_H2 | 36,859 | 205 | 6 (3%) | 2.196 |
aIncluding singletons.
bShannon diversity indices and shared clusters were calculated after rarefying the different data sets from the same soil to the same sequencing depth of 12,960, 13,538, 2,765 and 36,859 sequences for PUE, BRE, BRH and BEW, respectively.
2.6. Full-length cDNA cloning and sequencing
Amplified cDNAs obtained after two rounds of hybridization capture were digested by SfiI (Fermentas), which recognizes two distinct SfiIA and SfiIB sites located at the 5′ and 3′ ends of the cDNAs, respectively. Digested cDNAs were then ligated to the SfiI-digested pDR196-SfiI-Kan yeast expression vector36 modified to contain two SfiIA and SfiIB sites, downstream of the Saccharomyces cerevisiae PMA1 promoter, thus allowing the directional cloning and potential constitutive expression of the cDNAs in yeast.
Several transformed, kanamycin-resistant Escherichia coli (One Shot® TOP10 strain, Invitrogen) colonies from each sample were first randomly selected and subjected to colony PCR using the GH11-F and GH11-R primers to detect the presence of a GH11 cDNA insert. cDNA inserts from PCR-positive bacterial colonies were entirely sequenced by BIOFIDAL (Villeurbanne, France) using a PMA1 primer (CTCTCTTTTATACACACATTC) and additional internal primers when necessary.
2.7. Plasmid library construction, yeast transformation and functional screening
For each cDNA sample, a minimum of 2,000 independent kanamycin-resistant transformed E. coli colonies were pooled together for plasmid extraction using the alkaline lysis method.37 Aliquot samples of each plasmid library were used to transform the S. cerevisiae strain DSY-5 (MATα leu2 trp1 ura3-52 his3::PGAL1-GAL4 pep4 prb1-1122; Dualsystems Biotech) using a standard lithium acetate protocol.38 Transformed yeasts were selected on a solid yeast nitrogen base (YNB) minimal medium supplemented with glucose (2%) and amino acids, but lacking uracil. YNB agar plates were overlaid by a thin layer of the same medium containing 4 mg l−1 of AZCL-xylan (Megazyme), a substrate specific for endo-xylanases. Plates were incubated at 30°C. Yeast colonies producing a secreted endo-xylanase were surrounded by a dark blue halo resulting from the hydrolysis of AZCL-xylan.
For each sample, several yeast colonies positive for endo-xylanase activity were picked, lysed at 95°C for 10 min in 3 µl of 20 mM NaOH and the pDR196 insert amplified by PCR using primers PMA1 and ADH (GCGAATTTCTTATGATTTATG). PCR products were sequenced by BIOFIDAL using the PMA1 primer.
2.8. Phylogenetic analyses
Sequences obtained from plasmid inserts were manually edited and corrected. Deduced amino acid sequences were aligned using MUSCLE39 to GH11 amino acid sequences obtained from public databases. Maximum likelihood phylogeny analyses were generated with the PhyML 3.0 program using the WAG substitution model as implemented in SeaView v. 4.40 Phylogenetic trees were drawn in MEGA v. 6.41
2.9. Sequence accessibility
Sequences from plasmid inserts are available in the EBI/DDJB/GenBank databases under accession Nos. LK932029-LK932091. Illumina MiSeq sequence reads have been deposited in the Sequence Read Archive of the EBI database under study no. PRJEB6672.
3. Results
3.1. GH11 cDNA capture
As in July 2012, we identified and collected 342 full-length eukaryotic GH11 DNA coding sequences from public databases, from 113 fungal species and from 2 non-fungal ones. Seventy-two percent of these sequences were from Ascomycotina (85 species), 20% from Basidiomycotina (26 species) and 7% from other taxonomic groups. Prevalence of sequences from Ascomycotina is likely to reflect a greater genome sequencing effort in this taxonomic group, rather than a higher occurrence of the GH11 family among Ascomycotina.23 Among the publicly available sequences, those putatively full-length sequences ranged in size from 639 to 2,099 bp. Occurrence of carbohydrate-binding motives or of C-terminal, non-catalytic extensions in the encoded polypeptides accounted for most of these size variations. The 35 degenerate capture probes were exclusively designed on the shared ca. 540-bp-long conserved catalytic domain and were susceptible to hybridize to 90% of the collected sequences.
SHS was performed on cDNAs synthesized from polyadenylated mRNAs extracted from four different forest soils. Electrophoregrams of all cDNAs recovered after two successive rounds of capture were characterized by a background smear of which emerged discrete bands ranging in size from 300 to 1,500 bp (Fig. 1).
Figure 1.

Electrophoretic separation of cDNAs obtained following two consecutive solution hybridization selection. Captured cDNAs from the four soil samples PUE, BRH, BRE and BEW were run on an Agilent DNA 12000 microfluidic chip. Each band could encompass one or several unique but abundant GH11 cDNAs.
Successful enrichment in GH11 sequences along the capture protocol was demonstrated by semi-quantitative PCR using GH11-specific PCR primers and different quantities of cDNA in the PCRs (from 10 to 0.01 ng). As illustrated in Fig. 2 for the Breuil beech forest (BRH sample) and for the other soil samples discussed in Supplementary Fig. S3, clear positive amplification of a GH11 fragment after two rounds of capture was always obtained using the lowest quantity of cDNA (0.01 ng), whereas no amplification could be observed for the same amount of cDNA prior to SHS.
Figure 2.
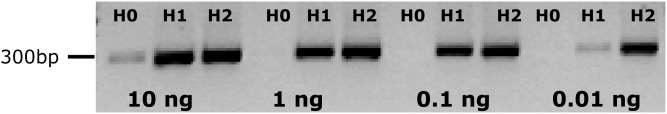
Semi-quantitative PCR amplification of a 281-bp GH11 fragment using different quantities (from 10 to 0.01 ng) of BRH cDNA obtained before (H0) and after one (H1) or two (H2) cycles of hybridization. Before capture, PCR products could only be obtained using 10 ng of input cDNA. Amplifications of the PUE, BRE and BEW samples are illustrated in Supplementary Fig. S3.
3.2. Cloning, sequencing and heterologous expression of captured cDNA
Captured cDNAs in the range of 700–1,500 bp were cloned into the pDR196 E. coli/S. cerevisiae shuttle expression vector to constitute four soil-specific GH11-enriched plasmid libraries (Table 1). Forty recombinant colonies per library were randomly screened by PCR using GH11-specific primers to evaluate the percentage of GH11-containing recombinant plasmids. Efficient enrichment occurred for all libraries with 80 to >90% of positive clones (Table 1). Among the 55 fully sequenced plasmid inserts from PCR- positive colonies, all but two indeed corresponded to GH11 sequences (Table 1). Seventy-two percent of the sequences encoded putatively full-length GH11 polypeptides based on alignment length to known GH11 polypeptides and the presence of in-frame putative start and stop codons. Out of them, 15% were characterized by the presence of a family 1 carbohydrate-binding domain (CBM1) in a C-terminal position.
Table 1.
Cloning and characterization of captured GH11 cDNAs
| Samples | PUE | BRH | BRE | BEW |
|---|---|---|---|---|
| No. of captured cDNAs cloned in Escherichia coli | 6,770 | 2,020 | 5,720 | 5,880 |
| No. of E. coli colonies screened by PCR | 40 | 40 | 40 | 40 |
| Positive amplification of a GH11 fragment (%) | 37 (92.5) | 33 (82.5) | 35 (87.5) | 36 (90) |
| No. of inserts sequenced | 12 | 13 | 16 | 14 |
| No. of GH11 inserts (%) | 11 (92) | 12 (92) | 16 (100) | 14 (100) |
| No. of putative full-length GH11 (%) | 9 (82) | 9 (75) | 11 (69) | 9 (64) |
| % of endo-xylanase-positive yeast colonies | 1.5 | 25 | 12 | 6 |
Functional screening using S. cerevisiae was conducted on the four GH11-enriched plasmid libraries by plating the recombinant yeasts onto a medium supplemented with an endo-xylanase-specific colour reagent (AZCL-xylan). Depending on the library, between 1.5 (sample PUE) and 25% (sample BRH) of the transformed yeast colonies developed a dark blue halo demonstrating secretion of a functional endo-xylanase (Supplementary Fig. S4). All 11 sequenced plasmid inserts from these xylanase-positive yeast colonies encoded GH11 proteins (ranged between 221 and 289 amino acids in length); 5 of them had already been identified among sequences obtained from bacterial colonies and 4 had a C-terminal CBM1 domain. The percent sequence identity between the catalytic domain of the selected functional proteins and the catalytic domain of their closest Blastp hits in GenBank ranged between 69% (81% similarity) and 87% (94% similarity).
3.3. Selectivity of the SHS GH11 capture
To evaluate the diversity of GH11 sequences at each step of the capture protocol, we performed a high-throughput Illumina MiSeq sequencing of GH11 amplicons obtained from all four cDNA samples, prior (H0) and after one (H1) or two (H2) cycles of SHS capture. Paired-end sequence reads were assembled to reconstitute the ca. 281-bp-long amplicons. Altogether, the total data set contained 334,161 full-length amplicon sequences that were clustered at a 95% nucleotide sequence identity threshold to produce a total number of 1,458 clusters, of which 1,001 (69%) were singletons (data summarized in Table 2 for each sample). Each of the 12 sequence data sets (4 cDNA samples × the 3 steps of the SHS) was characterized by few dominant clusters encompassing most of the sequences and a large number of clusters each containing a few, or even a single, sequences (illustrated in Fig. 3A for the PUE sample). None of the sequences obtained were identical to sequences deposited in databases. Only 17 of the sequence clusters, of which 14 exclusively from the BEW site, were >90% identical (maximum value of 97.5%) at the nucleotide level over their entire length to GH11 genes from either the Basidiomycota Tulasnella calospora or the Ascomycota Nectria haematococca and Pyrenophora teres.
Figure 3.

Selectivity of the SHS capture. (A) Rank-abundance distribution of the most abundant GH11 nucleotide sequence clusters identified before (H0), or after one (H1) or two (H2) cycles of hybridization on the PUE cDNAs. Only clusters encompassing 80% of the sequences in the H0, H1 or H2 samples are shown. ‘C’ or ‘Y’ letters above bars indicate sequences obtained by random sequencing of plasmid inserts or which could be functionally expressed in yeast, respectively. (B) Venn diagram showing the number of unique or shared GH11 sequence clusters, before (H0), or after one (H1) or two (H2) cycles of hybridization on the PUE cDNAs. As in (A), only the most abundant clusters, encompassing 90% of the sequences, were used for the calculation. GH11 PCR sequences were clustered using a nucleotide sequence identity threshold of 95%. Similar Venn diagrams for the BRH, BRE and BEW samples are illustrated in Supplementary Fig. S5.
Figure 3 also showed that the most abundant sequence clusters obtained after one (H1) and two (H2) cycles of capture did not, for a majority of them, correspond to the most abundant clusters present before capture (H0). Venn diagrams drawn using only these most prominent sequence clusters, encompassing altogether 90–93% of sample sequences, showed that there existed a larger overlap between the post-capture samples H1 and H2 than between the pre-capture samples H0 and H1 or H2 (Fig. 3B). This trend was observed, to some extent, for samples BEW, BRE and PUE, but not for the BRH one which differed from the others by the dominance of only three clusters in the H0 cDNA pool which encompassed 90% of the sequenced reads (Supplementary Fig. S5). Despite these apparent differences in sequence distribution between the pre-capture H0 and the post-capture H1 and H2 samples, sequence diversity indices, such as the Shannon index, did not differ between the pre- and post-capture sequence pools (Table 2, with the exception of the BRH sample). Between 2.7% (BRE and BEW) and 15% (PUE and BRH) of the sequence clusters were shared between two sites. Eight sequence clusters were identified in all four studied sites.
To address the phylogenetic diversity of the captured sequences, we first produced an amino acid sequence alignment of 62 known GH11 proteins representative of the phylogenetic diversity of this gene family. To this alignment, we added the GH11 sequences obtained by the random sequencing of plasmid inserts, the sequences producing a functional enzyme in yeast and the sequences representative of the most abundant Illumina sequence clusters before (H0) or after (H1 and H2) SHS capture. The GH11 family is a highly diversified and fast-evolving gene family and phylogenies based either on full-length protein sequence alignments or on partial alignments, as in the present case, clearly do not reflect the species phylogenies and comprise very few well-supported internal branches (Fig. 4). Phylogenetic trees obtained for sequences from the four studied soils (Fig. 4; Supplementary Fig. S6) all clearly showed that the captured sequences were distributed over the entire reference tree.
Figure 4.

Phylogenetic diversity of the GH11 partial amino acid sequences obtained from PUE cDNA samples. 0, 1 and 2 translated PCR sequences obtained before or after one or two cycles of hybridization. PUE sequences are scattered over the entire tree that includes representative reference sequences from Ascomycota and Basidiomycota. c, sequences obtained from Escherichia coli clones; y, sequences functionally expressed in yeast clones. PhyML tree calculation was based on an alignment of ca. 80-amino-acid-long GH11 partial sequences. Thicker internal black branches indicate bootstrap value ≥60% (1,000 replications). Full species names and accession numbers of the reference sequences are given in Supplementary Fig. S6A. Similar trees drawn using the sequences from sites BRE, BRH and BEW are illustrated in Supplementary Fig. S6 B, C and D, respectively. This figure appears in colour in the online version of DNA Research.
4. Discussion
The results obtained clearly demonstrate that SHS represents a powerful strategy to select full-length cDNAs, representative of a specific gene family, originally diluted in a highly complex metatranscriptomic sequence pool. This protocol was successfully implemented on four different forest soil RNA samples. Based on previous estimates of the frequency of GH11 sequences among eukaryotic cDNA for two of the soils used in this study (BRE and BRH),22 two successive cycles of SHS have the potential to enrich specific cDNA sequences by a factor of at least 104. As suggested by the results of the semi-quantitative PCR, in some cases (e.g. the PUE sample, Supplementary Fig. S3), one cycle of capture may be sufficient to get a maximum level of enrichment, while in other cases two cycles seem required (e.g. the BRH sample, Fig. 2).
Sequence analysis of PCR fragments amplified from pre- or post-capture cDNAs demonstrated that capture succeeded in selecting both a large number and phylogenetically diverse representatives of the selected gene family. Furthermore, none of the captured sequences appeared to be identical to already known ones which we originally used for probe design. Capture could however preferentially select sequences that were not necessarily among the most abundant in the original cDNA pool. This should be evaluated in the future by quantitative PCR. Despite explorative probe design strategy, publicly available homologous sequences at the start of the study greatly influence the capture selectivity. Probe sets utilized to capture a given biomarker should therefore be upgraded regularly, taking into account newly deposited sequences.
Thanks to the ever increasing number of published fungal genomes, representative of the phylogenetic diversity of this taxonomic group; explorative probe design strategies could be carried out to unravel the metabolic capacities of these microorganisms within different ecosystems. Besides GH11 sequences, SHS capture can be implemented for any other gene family of interest, allowing a comprehensive taxonomic or functional description of the studied microbial community. As mentioned in the introduction, sequence capture presents the advantage over PCR to give access to the full-length gene sequence, including facultative modules, not always associated to the studied catalytic domain. This was indeed the case for the GH11, for which we estimated that 72% of the captured sequences were full length and that 15% of them processed a C-terminal, fungal-specific, CBM1 module (see the CAZy database, http://www.cazy.org). A discrepancy however existed between the estimated fraction of full-length captured GH11 cDNA and the systematically lower fraction of cDNAs which produced a functional enzyme upon expression in S. cerevisiae. The absence of expression in yeast can be attributed to a number of independent factors such as bias in codon usage, non-recognition by S. cerevisiae of the protein signal peptide necessary for correct secretion, protein misfolding or hyperglycosylation. Some of these problems could be addressed by using expression plasmids including a yeast signal peptide downstream of the cloning site and/or by using a different yeast species for protein production.
Sequencing of PCR fragments amplified from captured cDNAs also indicate that altogether the four captured cDNA samples obtained in this single study encompass a greater number of novel and different GH11 sequences than have been deposited and are available in public databases over several decades. This observation should promote the use of cDNA sequence capture (i) as a complementary approach to PCR to explore and quantify the extent of eukaryotic functional diversity in complex environments, but also (ii) as a powerful tool in environmental biotechnology to efficiently screen for enzyme variants with novel biochemical properties.
Supplementary Data
Supplementary Data are available at www.dnaresearch.oxfordjournals.org.
Funding
C.B. was supported by the University of Turin and the région Rhône-Alpes (CMIRA program); C.R. received a graduate grant from the Ministère de l'Enseignement Supérieur et de la Recherche; N.P. was funded by the Direction Générale de l'Armement and E.P. by the Agence Nationale pour la Recherche. Work was financed by the CNRS-INSU ECCO Microbien program, the INRA métaprogramme M2E (project Metascreen), project ANR 09-GENM-033-001 (Eumetasol); EU-project ‘EcoFINDERS' No. 264465 and local funding by the University of Turin (ex-60%). Funding to pay the Open Access publication charges for this article was provided by the Centre national de la recherche scientifique (CNRS).
Supplementary Material
Acknowledgements
We would like to thank Richard Joffre, Jacques Ranger and Alberto Orgiazzi for soil sampling at the Puéchabon, Breuil and Berchidda sites, respectively. Audrey Dubost and Stefano Ghignone contributed to bioinformatic analyses and Jérémie Denonfoux to gene capture. We acknowledge the JGI of the US Department of Energy and the Broad Institute for making available genome data prior to their publication.
References
- 1.Tringe S.G., Rubin E.M. Metagenomics: DNA sequencing of environmental samples. Nat. Rev. Genet. 2005;6:805–14. doi: 10.1038/nrg1709. [DOI] [PubMed] [Google Scholar]
- 2.Simon C., Daniel R. Metagenomic analyses: past and future trends. Appl. Environ. Microbiol. 2011;77:1153–61. doi: 10.1128/AEM.02345-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Howe A.C., Jansson J.K., Malfatti S.A., Tringe S.G., Tiedje J.M., Brown C.T. Tackling soil diversity with the assembly of large, complex metagenomes. Proc. Natl. Acad. Sci. USA. 2014;111:4904–9. doi: 10.1073/pnas.1402564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Voříšková J., Baldrian P. Fungal community on decomposing leaf litter undergoes rapid successional changes. ISME J. 2013;7:477–86. doi: 10.1038/ismej.2012.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luis P., Kellner H., Zimdars B., Langer U., Martin F., Buscot F. Patchiness and spatial distribution of laccase genes of ectomycorrhizal, saprotrophic, and unknown basidiomycetes in the upper horizons of a mixed forest cambisol. Microb. Ecol. 2005;50:570–9. doi: 10.1007/s00248-005-5047-2. [DOI] [PubMed] [Google Scholar]
- 6.Kellner H., Luis P., Pecyna M.J., et al. Widespread occurrence of expressed fungal secretory peroxidases in forest soils. PLoS ONE. 2014;9:e95557. doi: 10.1371/journal.pone.0095557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kellner H., Zak D.R., Vandenbol M. Fungi unearthed: transcripts encoding lignocellulolytic and chitinolytic enzymes in forest soil. PLoS ONE. 2010;5:e10971. doi: 10.1371/journal.pone.0010971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hong S., Bunge J., Leslin C., Jeon S., Epstein S.S. Polymerase chain reaction primers miss half of rRNA microbial diversity. ISME J. 2009;3:1365–73. doi: 10.1038/ismej.2009.89. [DOI] [PubMed] [Google Scholar]
- 9.Denonfoux J., Parisot N., Dugat-Bony E., et al. Gene capture coupled to high-throughput sequencing as a strategy for targeted metagenome exploration DNA. DNA Res. 2013;20:185–96. doi: 10.1093/dnares/dst001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dugat-Bony E., Missaoui M., Peyretaillade E., et al. HiSpOD: probe design for functional DNA microarrays. Bioinformatics. 2011;27:641–8. doi: 10.1093/bioinformatics/btq712. [DOI] [PubMed] [Google Scholar]
- 11.Parisot N., Denonfoux J., Dugat-Bony E., Peyret P., Peyretaillade E. KASpOD-a web service for highly specific and explorative oligonucleotide design. Bioinformatics. 2012;28:3161–2. doi: 10.1093/bioinformatics/bts597. [DOI] [PubMed] [Google Scholar]
- 12.Bailly J., Fraissinet-Tachet L., Verner M.C., et al. Soil eukaryotic functional diversity, a metatranscriptomic approach. ISME J. 2007;1:632–42. doi: 10.1038/ismej.2007.68. [DOI] [PubMed] [Google Scholar]
- 13.Kellner H., Luis P., Portetelle D., Vandenbol M. Screening of a soil metatranscriptomic library by functional complementation of Saccharomyces cerevisiae mutants. Microbiol. Res. 2011;166:360–8. doi: 10.1016/j.micres.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 14.Damon C., Vallon L., Zimmermann S., et al. A novel fungal family of oligopeptide transporters identified by functional metatranscriptomics of soil eukaryotes. ISME J. 2011;5:1871–80. doi: 10.1038/ismej.2011.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lehembre F., Doillon D., David E., et al. Soil metatranscriptomics for mining eukaryotic heavy metal resistance genes. Environ. Microbiol. 2013;15:2829–40. doi: 10.1111/1462-2920.12143. [DOI] [PubMed] [Google Scholar]
- 16.Takasaki K., Miura T., Kanno M., et al. Discovery of glycoside hydrolase enzymes in an avicel-adapted forest soil fungal community by a metatranscriptomic approach. PLoS ONE. 2013;8:e55485. doi: 10.1371/journal.pone.0055485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bates S.T., Clemente J.C., Flores G.E., et al. Global biogeography of highly diverse protistan communities in soil. ISME J. 2013;7:652–9. doi: 10.1038/ismej.2012.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor D.L., Hollingsworth T.N., McFarland J.W., Lennon N.J., Nusbaum C., Ruess R.W. A first comprehensive census of fungi in soil reveals both hyperdiversity and fine-scale niche partitioning. Ecol. Monogr. 2014;84:3–20. [Google Scholar]
- 19.Schneider T., Keiblinger K.M., Schmid E., et al. Who is who in litter decomposition? Metaproteomics reveals major microbial players and their biogeochemical functions. ISME J. 2012;6:1749–62. doi: 10.1038/ismej.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stursová M., Zifčáková L., Leigh M.B., Burgess R., Baldrian P. Cellulose utilization in forest litter and soil: identification of bacterial and fungal decomposers. FEMS Microbiol. Ecol. 2012;80:735–46. doi: 10.1111/j.1574-6941.2012.01343.x. [DOI] [PubMed] [Google Scholar]
- 21.Demain A.L., Dana C.A. The business of biotechnology. Ind. Biotechnol. 2007;3:269–83. [Google Scholar]
- 22.Damon C., Lehembre F., Oger-Desfeux C., et al. Metatranscriptomics reveals the diversity of genes expressed by eukaryotes in forest soils. PloS ONE. 2012;7:e28967. doi: 10.1371/journal.pone.0028967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grigoriev I.V., Nikitin R., Haridas S., et al. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014;42:D699–704. doi: 10.1093/nar/gkt1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lombard V., Golaconda Ramulu H., Drula E., Coutinho P.M., Henrissat B. The Carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Margesin R., Minerbi S., Schinner F. Long term monitoring of soil microbiological activities in two forest sites in South tyrol in the italian alps. Microb. Environ. 2014;29:277–85. doi: 10.1264/jsme2.ME14050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paës G., Berrin J.G., Beaugrand J. GH11 xylanases: structure/function/properties relationships and applications. Biotechnol. Adv. 2012;30:564–92. doi: 10.1016/j.biotechadv.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 27.Kuramae E.E., Hillekens R.H., de Hollander M., et al. Structural and functional variation in soil fungal communities associated with litter bags containing maize leaf. FEMS Microbiol. Ecol. 2013;84:519–31. doi: 10.1111/1574-6941.12080. [DOI] [PubMed] [Google Scholar]
- 28.Luis P., Kellner H., Martin F., Buscot F. A molecular method to evaluate Basidiomycete laccase gene expression in forest soils. Geoderma. 2005;128:18–27. [Google Scholar]
- 29.Damon C., Barroso G., Férandon C., Ranger J., Fraissinet-Tachet L., Marmeisse R. Performance of the COX1 gene as a marker for the study of metabolically active Pezizomycotina and Agaricomycetes fungal communities from the analysis of soil RNA. FEMS Microbiol. Ecol. 2010;74:693–705. doi: 10.1111/j.1574-6941.2010.00983.x. [DOI] [PubMed] [Google Scholar]
- 30.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 31.Gnirke A., Melnikov A., Maguire J., et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 2009;27:182–9. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masella A.P., Bartram A.K., Truszkowski J.M., Brown D.G., Neufeld J.D. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics. 2012;13:31. doi: 10.1186/1471-2105-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schloss P.D., Westcott S.L., Ryabin T., et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009;75:7537–41. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edgar R.C., Haas B.J., Clemente J.C., Quince C., Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stothard P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques. 2000;28:1102–4. doi: 10.2144/00286ir01. [DOI] [PubMed] [Google Scholar]
- 36.Rentsch D., Laloi M., Rouhara I., Schmelzer E., Delrot S., Frommer W.B. NTR1 encodes a high affinity oligopeptide transporter in Arabidopsis. FEBS Lett. 1995;370:264–8. doi: 10.1016/0014-5793(95)00853-2. [DOI] [PubMed] [Google Scholar]
- 37.Sambrook J., Russell D.W. Molecular cloning: a laboratory manual. 3rd edition. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 38.Rose M., Winston F., Hieter P. Methods in yeast genetics: a laboratory course manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1990. [Google Scholar]
- 39.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gouy M., Guindon S., Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010;27:221–4. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- 41.Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013;30:2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.