

Abstract
Pericytes play a pivotal role in contraction, mediating inflammation and regulation of blood flow in the brain. In this study, changes of pericytes in the neurovascular unit (NVU) were examined in relation to the effects of exogenous tissue plasminogen activator (tPA) and a free radical scavenger, edaravone. Immunohistochemistry and Western blot analyses showed that the overlap between platelet-derived growth factor receptor β-positive pericytes and N-acetylglucosamine oligomers (NAGO)-positive endothelial cells increased significantly at 4 days after 90 min of transient middle cerebral artery occlusion (tMCAO). The number of pericytes and the overlap with NAGO decreased with tPA but recovered with edaravone 4 days after tMCAO with proliferation. Thus, tPA treatment damaged pericytes, resulting in the detachment from astrocytes and a decrease in glial cell line-derived neurotrophic factor secretion. However, treatment with edaravone greatly improved tPA-induced damage to pericytes. The present study demonstrates that exogenous tPA strongly damages pericytes and destroys the integrity of the NVU, but edaravone treatment can greatly ameliorate such damage after acute cerebral ischemia in rats. © 2014 The Authors. Journal of Neuroscience Research Published by Wiley Periodicals, Inc.
Keywords: pericyte, neurovascular unit, edaravone, tPA, cerebral ischemia
Ischemic brain damage can be effectively ameliorated if cerebral blood flow (CBF) is restored by thrombolytic agents, such as tissue plasminogen activator (tPA), within a short space of time (Group, 1997). Although the ECASS III trial showed that the therapeutic time frame of intravenous thrombolysis with tPA for acute ischemic stroke was extended from 3 hr to 4.5 hr after the onset of symptoms, the trial simultaneously showed that tPA was more frequently associated with symptomatic intracranial hemorrhage (Hacke et al., 2008). Endogenous tPA is a major parenchymal serine protease in the brain that regulates physiological tissue remodeling and plasticity (Tsirka et al., 1997; Gravanis and Tsirka, 2005). However, a high dose of exogenous tPA could also cause hemorrhagic transformation by disturbing the neurovascular unit (NVU; Yamashita et al., 2009) and by direct neurotoxicity (Lukic-Panin et al., 2010), presenting a threat to the safe use for thrombolytic therapy.
Recently, brain pericytes have received increasing attention in various neurological disorders, such as ischemic stroke, Alzheimer's disease, and diabetic retinopathy (Hamilton et al., 2010). Brain pericytes are present in isolated preparations in the ratio of one pericyte per three brain capillary endothelial cells (Pardridge, 1999). Brain pericytes have functional properties consisting of contraction, mediation of inflammation, and regulation of endothelial cell activity (Nag, 2003). Yemisci et al. (2009) showed that ischemia induces sustained contraction of pericytes on microvessels in the intact mouse brain and that suppression of oxidative–nitrative stress relieved pericyte contraction.
Our previous study showed a marked dissociation between the basement membrane and astrocyte foot processes in the peri-ischemic lesion with tPA, which was improved by the addition of the free radical scavenger edaravone (Yamashita et al., 2009). The present study focuses on the relationship between NVU pericytes in association with a neuroprotective effect and angiogenesis and the effects of tPA and edaravone after transient middle cerebral artery occlusion (tMCAO).
MATERIALS AND METHODS
Animals and Focal Cerebral Ischemia
All experimental protocols and procedures were approved by the Animal Committee of the Okayama University Graduate School of Medicine and Dentistry. Adult male Wistar rats (SLC, Shizuoka, Japan), 12 weeks old (body weight 250–280 g), were anesthetized with an intraperitoneal (i.p.) injection of pentobarbital (10 mg/250 g). A burr hole (2 mm in diameter) was carefully made in the skull to measure the regional cerebral blood flow (rCBF), with the dura mater preserved at this time. The location of the burr hole was 3 mm dorsal and 5 mm lateral to the right from bregma, which is located in the upper part of the middle cerebral artery (MCA) territory.
On the following day, an inhalation mask was used to anesthetize the animals with a mixture of nitrous oxide/oxygen/isoflurane (69%/30%/1%) during surgical preparation. Body temperature was monitored and maintained at 37°C ± 0.3°C by using a heating pad during the surgical procedure. The right MCA was occluded by insertion of a 4-0 surgical nylon thread with silicon coating through the common carotid artery, as described by Abe et al. (1992). After 90 min of tMCAO, the nylon thread was gently removed to restore blood flow in the MCA territory. Vehicle (physiologic saline [PS], i.v., 0.5 ml) or edaravone (Mitsubishi Tanabe Pharma, Osaka, Japan; i.v., 3 mg/kg in 0.5 ml PS) was injected twice at the beginning of transient cerebral ischemia and reperfusion, followed by vehicle (i.v., 0.5 ml PS) or tPA (Grtpa; Mitsubishi Tanabe Pharma; i.v., 10 mg/kg in 0.5 ml PS) at the time of reperfusion. This dose of tPA was selected based on the report of a tenfold difference in fibrin-specific activity between humans and rodents (Korninger and Collen, 1981). The incision was then closed. Sham control (SC) animals were incised simultaneously, but the incision was closed without inserting the thread. rCBF of the right frontoparietal cortex region was measured before, during, and after tMCAO through the burr hole by using a laser blood flowmeter (Flo-C1; Omegawave, Tokyo, Japan), as described previously (Kitagawa et al., 1999). After the incision was closed, the animals were allowed to recover at ambient temperature until sampling, with free access to water and food.
After reperfusion, rats were decapitated under deep anesthesia by i.p. injection of pentobarbital (20 mg/250 g) at each time point for the experimental groups. The brains were removed quickly, frozen immediately in powdered dry ice, and stored at −80°C until use. Coronal sections 12 μm thick at the caudate level were made by using a cryostat at −18°C and mounted onto silane-coated glass slides.
Experimental Groups and Drug Treatment
Rats were sacrificed at 1, 4, and 14 days after reperfusion (each group; n = 5), including the SC (n = 5), to investigate, by immunohistochemistry, the chronological changes to pericytes after 90 min of tMCAO. Next, another set of rats was divided into four groups to investigate the effect of tPA and edaravone on pericytes. The V + V control group (n = 5) received only the vehicle twice at the beginning and end of tMCAO, followed by the same vehicle at reperfusion. The V + tPA group (n = 5) received a similar vehicle treatment twice during tMCAO, followed by tPA treatment at reperfusion. The E + V group (n = 5) received edaravone at the beginning and end of tMCAO, followed by the vehicle at reperfusion. The E + tPA group (n = 5) received edaravone twice, followed by tPA treatment at reperfusion. Edaravone was injected twice because of its short half-life (T1/2 = 5.4 min; Watanabe et al., 1994) and because free radicals are generated during and after cerebral ischemia (Abe et al., 1988). These rats were simultaneously sacrificed at 4 days after reperfusion when the expression of the pericytic marker platelet-derived growth factor receptor β (PDGFRβ) had peaked.
Histology and Immunohistochemistry
To determine the area of ischemic lesions affected by tPA and edaravone at 4 days after tMCAO, brain sections were stained with hematoxylin–eosin staining and examined with a light microscope (SZX-12; Olympus Optical, Tokyo, Japan). Sections were made at 2, 0, −2, −4, and −6 mm from bregma. The infarct area was measured in these five sections by counting pixels in Photoshop CS5, and infarct volume was calculated by multiplying the infarct area by 2 mm thickness (Kawai et al., 2011).
Double immunohistochemical analysis was performed between PDGFRβ and N-acetylglucosamine oligomers (NAGO), Ki67, glial fibrillary acidic protein (GFAP), or glial cell line-derived neurotrophic factor (GDNF) to analyze the pericytes surrounding the endothelial cells. Lycopersicon esculentum lectin (LEL) is a glycoprotein with positive affinity for NAGO that is expressed in mature vascular endothelial cells (Deguchi et al., 2006). PDGFRβ, Ki67, and GFAP are markers of pericyte cells, proliferating cells, and differentiated astroglial cells, respectively. GDNF has been identified as a potent neurotrophic factor that enhances the survival of neurons (Abe et al., 1997).
Frozen brain sections were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer (PB; pH 7.4) for 10 min and rinsed with phosphate-buffered saline (PBS; pH 7.4). After blocking of the nonspecific reaction with 5% bovine serum albumin solution for 2 hr at room temperature, the brain sections were incubated with rabbit anti-PDGFRβ antibody (1:500; Cell Signaling Technology, Danvers, MA), biotinylated LEL (1:200; Vector Laboratories, Burlingame, CA), goat anti-Ki67 antibody (1:200; Santa Cruz Biotechnology, Santa Cruz, CA), mouse anti-GFAP (1:500; Millipore, Temecula CA), or goat anti-GDNF antibody (1:200; Santa Cruz Biotechnology) overnight at 4°C. Slides were washed with PBS, then incubated with Alexa Fluor 488-labeled donkey anti-rabbit IgG (1:200; Invitrogen, Eugene, OR), streptavidin DyLight 488 conjugate (1:200; Vector Laboratories), or Alexa Fluor 555-labeled donkey anti-mouse and goat IgG (1:200; Invitrogen) for 2 hr at room temperature. The slides were washed in PBS, then covered with Vectashield mounting medium with 4′,6′-diamidino-2-phenylindole (Vector Laboratories).
The treated sections were scanned with a confocal microscope equipped with an argon and HeNe laser (LSM-510; Carl Zeiss, Jena, Germany). Sets of fluorescent images were acquired sequentially for the red and green channels to prevent the crossover of signals from green to red or from red to green channels. A set of sections was stained in a similar way without primary antibodies to confirm the specificity of the primary antibody. Photoshop CS5 was used to measure the area of vascular endothelial cells from NAGO staining, whereas the area of pericytes was estimated from PDGFβ staining in the same region of double immunohistochemical staining. The coverage ratio of pericyte area to endothelial cell area was then calculated.
Western Blot Analysis
For Western blot analysis, 90 min of tMCAO was performed in a different set of animals, which were decapitated under deep anesthesia by i.p. administration of pentobarbital (40 mg/kg) at 1, 2, 4, 7, 14, and 28 days after reperfusion (each group, n = 5). The brains were removed, and the peri-ischemic region of the cerebral cortices (about 100 mg) was quickly frozen in liquid nitrogen. These brain tissue samples were sonicated in ice-cold lysis buffer containing 50 mM Tris-HCl, pH 7.2, 250 mM NaCl, 1% NP-40, and the Complete Mini Protease Inhibitor Cocktail (Roche, Basel, Switzerland). Protein concentrations were determined by the Lowry assay (Bio-Rad Laboratories, Hercules, CA).
Total protein extract (20 μg) was loaded onto an 8% polyacrylamide gel, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to a polyvinylidene fluoride membrane (Millipore). The membranes were washed with PBS, pH 7.4, containing 4% skim milk and 1% Tween 20, then incubated overnight at 4°C with rabbit anti-PDGFRβ antibody (1:1,000 dilution in skimmed milk). The membranes were washed with PBS, then probed with a horseradish peroxidase-conjugated secondary antibody, and immunodetection was performed with an enhanced chemiluminescent (ECL) substrate (Pierce, Rockford, IL). After the detection of ECL, membranes were incubated in stripping buffer (62.5 mM Tris-HCl, pH 6.7, 2% SDS, 0.7% β-mercaptoethanol) at 50°C for 10 min, then reprobed with a monoclonal anti-β-tubulin antibody (1:5,000; Sigma, Tokyo, Japan) as a loading control for protein quantification. The signals were quantified with a luminoimage analyzer (LAS 1000-Mini; Fuji Film, Tokyo, Japan), and quantitative densitometric analysis was performed in ImageJ.
Statistical Analysis
Statistical differences in pericyte coverage by endothelial cells, the number of Ki67-positive cells, and the results of Western blot analysis among the four groups were evaluated using one-way ANOVA with a post hoc test. Statistical significance was assumed at P < 0.05.
RESULTS
CBF
rCBF immediately dropped to less than 30% of the basal level after MCAO. After reperfusion, rCBF quickly recovered to about 90% of the basal level in all experimental groups, as previously described (Zhang et al., 2000). There were no significant differences in rCBF between all groups throughout the tMCAO experiments (data not shown).
Chronological Change of Pericyte
Color panels in Figure 1 show the peri-ischemic brain regions. PDGFRβ-positive pericyte (red) partially overlapped (merged, yellow) with NAGO-positive endothelial cells (green). The coverage of PDGFRβ-positive pericytes with NAGO-positive endothelial cells was significantly reduced at 1 day after tMCAO (54.0% ± 2.2%, P < 0.01 vs. SC) compared with that of SC (61.7% ± 2.7%) and then increased significantly at 4 days (69.9% ± 1.3%, P < 0.01 vs. SC; Fig. 1B).
Fig 1.
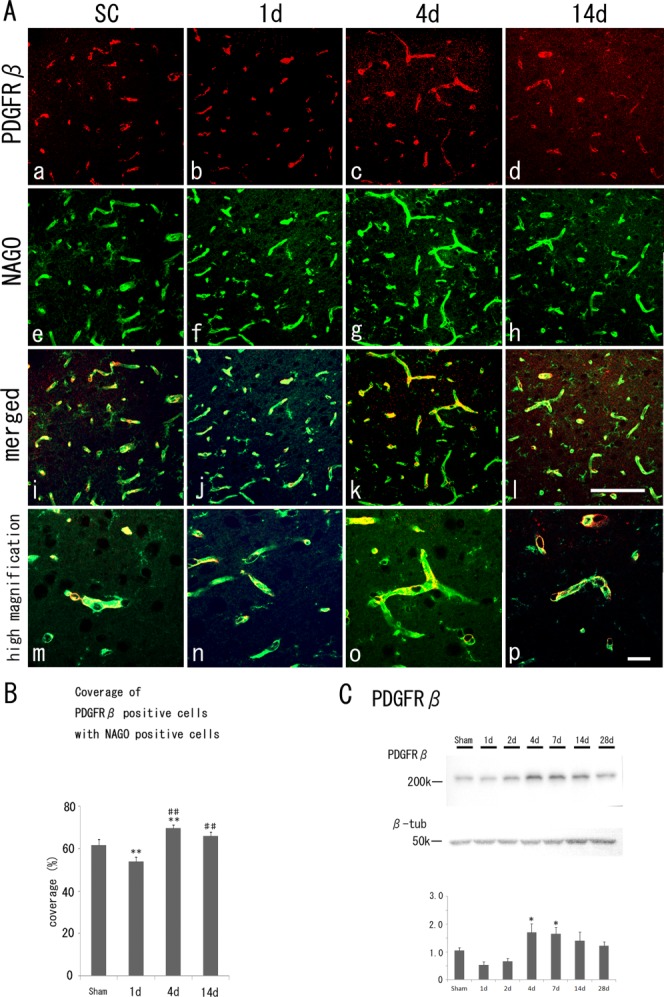
A: Fluorescent immunohistochemistry of PDGFRβ-positive pericytes (a–d, red), NAGO-positive endothelial cells (e–h, green), merged images (i–l) with their high magnification images (m–p) in the cerebral cortex of the sham control (SC) and the border zone of ischemic regions at 1, 4, and 14 days after tMCAO. B: Quantitative coverage ratio of pericytes to endothelial cells shows a transient reduction (1 day) and overshoot recovery (4 days) after tMCAO. C: Western blot analysis shows an increase of PDGFRβ (210 kDa) level at 4 days and 7 days. *P < 0.05 vs. SC, **P < 0.01 vs. SC. ##P < 0.01 vs. 1 day. Scale bars = 100 μm in l (applies to a–l); 20 μm in p (applies to m–p).
Western blot analysis (Fig. 1C) showed that PDGFRβ (210 kDa) levels of the peri-ischemic regions decreased slightly at 1 and 2 days after reperfusion; nevertheless, they increased significantly more than levels of the peri-ischemic regions in the SC group from 4 days, with a gradual decrease until 28 days (P < 0.05 vs. SC at 4 days and 7 days).
Proliferation of Pericyte After Cerebral Ischemia
The double-fluorescence study showed that PDGFRβ-positive cells (Fig. 2, red) were double positive with a small number of Ki67-positive cells (Fig. 2, green) in the SC group (7.5 ± 10.4/mm2) at 1 day (18.9 ± 13.4/mm2). The number of double-positive cells increased significantly, peaking at 4 days (Fig. 2, arrowheads; 132.3 ± 29.8/mm2, P < 0.01 vs. SC) and 14 days (Fig. 2, arrowheads; 71.8 ± 15.8/mm2, P < 0.01 vs. SC).
Fig 2.
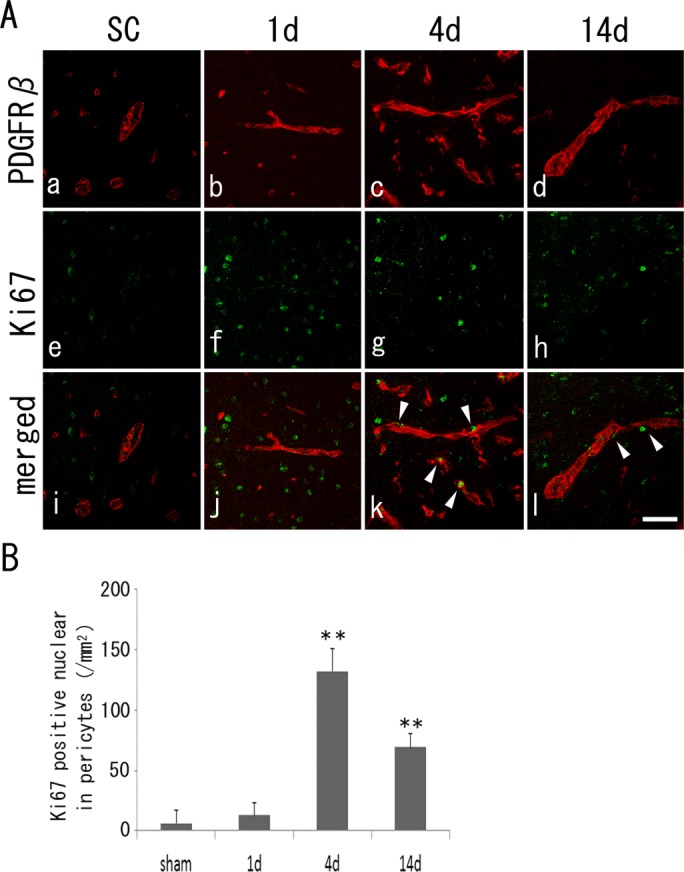
A: Double immunohistochemistry of PDGFRβ-positive pericytes (a–d, red), Ki67-positive cells (e–h, green), and merged images (i–l, arrowheads in double-positive cells) with a peak at 4 days (B). **P < 0.01 vs. SC. Scale bar = 100 μm.
Cerebral Infarct Volume With or Without Edaravone and tPA
The V + tPA group showed a small and nonsignificant increase in infarct size. In contrast, infarct volume decreased significantly in the E + V group compared with in the V + V and V + tPA groups. The E + tPA group also showed lower infarct volume compared with the V + tPA group but not the V + V group. These results were described in a previous article (Deguchi et al., 2012).
Change in Pericyte Coverage With Edaravone and tPA
As shown in Figure 3, the area of PDGFRβ-positive pericyte overlap, or coverage, was significantly reduced in the V + tPA group (62.9% ± 4.2%, P < 0.05) compared with in the V + V group (68.9 ± 2.6%). Compared with that in the V + V and the V + tPA groups, the coverage of the E + V group (79.3% ± 3.1%, P < 0.01 vs. V + V and V + tPA groups) increased significantly. In addition, coverage of the E + tPA group (73.5 ± 1.7%, P < 0.05 vs. V + tPA and P < 0.01 vs. E + V groups) increased significantly more than that of the V + tPA group but was lower than that of the E + V group (Fig. 3B).
Fig 3.
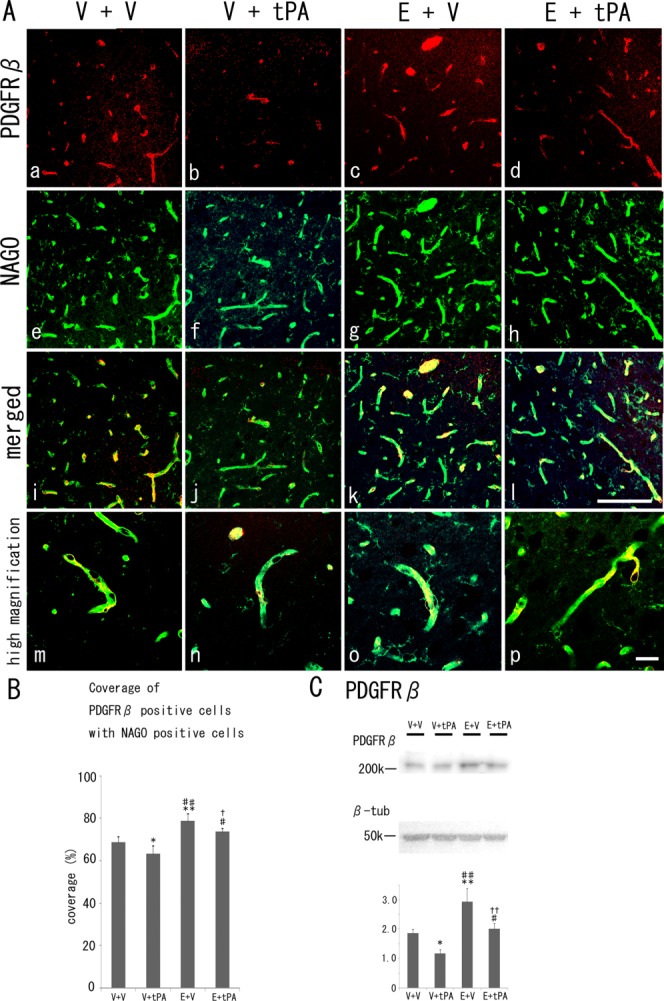
A: Fluorescent immunohistochemistry of PDGFRβ-positive pericytes (a–d, red), NAGO-positive endothelial cells (e–h, green), and merged images (i–l) with their high magnification images (m–p) in the border zone of ischemic regions at 4 days after tMCAO with or without edaravone and tPA. B: Quantitative coverage ratio of pericytes to endothelial cells shows a reduction in the V + tPA group and overshoot in the E + V and E + tPA groups 4 days after tMCAO. C: Western blot analysis shows a decrease in the level of PDGFRβin the V + tPA group and an increase in the E + V and E + tPA groups. V, vehicle; E, edaravone. *P < 0.05, **P < 0.01 vs. V + V group, #P < 0.05, ##P < 0.01 vs. V + tPA group, and †P < 0.05, ††P < 0.01 vs. E + V group. Scale bars = 100 μm in l (applies to a–l); 20 μm in p (applies to m–p).
Western blot analysis (Fig. 3C) showed that the level of PDGFRβ of the V + tPA group decreased significantly (P < 0.05) and that of the E + V group increased significantly (P < 0.01) compared with the V + V group. In addition, the level of PDGFRβ of the E + tPA group increased much more (P < 0.01) than the V + tPA group but decreased (P < 0.01) relative to the E + V group.
Proliferation of Pericytes With Edaravone and tPA
The double-fluorescence study showed that PDGFRβ-positive cells (Fig. 4, red) were double positive (arrowheads), with a smaller number of Ki67-positive cells (Fig. 4, green) in the V + tPA group (68.1 ± 10.4/mm2, P < 0.01) than in the V + V group (128.5 ± 15.8/mm2). On the other hand, the double-positive cells increased greatly in the E + V group (204.2 ± 8.5/mm2, P < 0.01 vs. V + V and V + tPA groups) compared with the V + V and the V + tPA groups. In addition, the double-positive cells in the E + tPA group (139.9 ± 10.4/mm2) increased significantly (P < 0.01) more than the V + tPA group and decreased (P < 0.01) relative to the E + V group (Fig. 4B).
Fig 4.
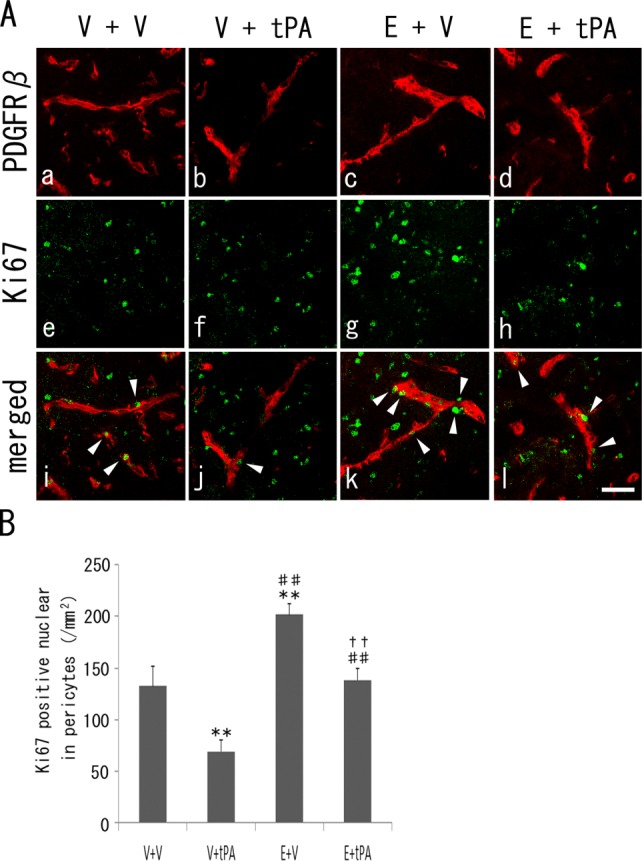
A: Double immunohistochemistry of PDGFRβ-positive pericytes (a–d, red), Ki67-positive cells (e–h, green), and merged images (i–l, arrowheads in double-positive cells), with a decrease in the V + tPA group and an increase in the E + V and E + tPA groups (B). **P < 0.01 vs. V + V group, ##P < 0.01 vs. V + tPA group, and ††P < 0.01 vs. E + V group. Scale bar = 100 μm.
Detachment of the Astrocyte From Pericytes
Figure 5 shows the detachment of the astrocyte endfeet surrounding the PDGFRβ-positive pericytes in a portion of vessels in the peri-ischemic brain regions of the V + V group (arrowhead). In addition, detachment was more widely observed in the V + tPA group (arrowheads), and astrocyte endfeet and pericytes were well merged in the vessels of the E + tPA group and especially in the E + V group (arrows).
Fig 5.
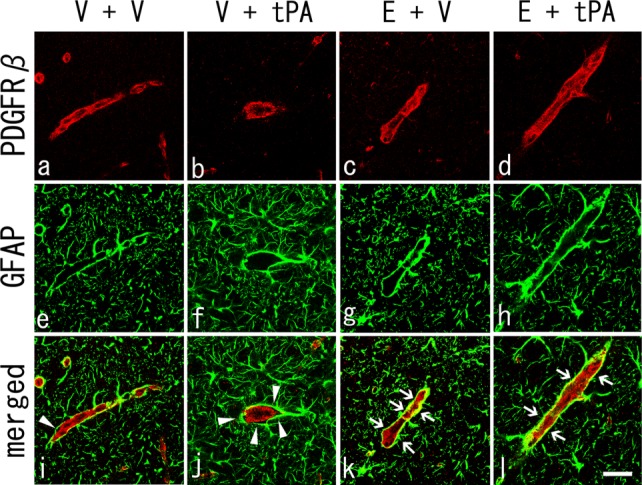
A: Double immunohistochemistry of PDGFRβ-positive pericytes (a–d, red), GFAP-positive astrocytes (e–h, green), and merged images (i–l) 4 days after tMCAO. Note the detachment of the astrocyte endfeet surrounding the pericytes in the V + V group (i, arrowhead) and a wide detachment observed in the V + tPA group (j, arrowheads). Also note the preserved astrocyte endfeet and overlapping pericytes in E + tPA and E + V groups (k,l, arrows). Scale bar = 100 μm.
Double Immunofluorescence Analyses of PDGFRβ and GDNF
Figure 6 shows that fluorescent signals for GDNF (green) were partially observed surrounding the PDGFRβ-positive pericyte (red) in the peri-ischemic region of the V + V group (merged). In addition, GDNF signals obviously decreased in the V + tPA group, whereas strong overlapping yellow signals were observed in the peri-ischemic region of the E + tPA group and especially the E + V group (arrowheads).
Fig 6.
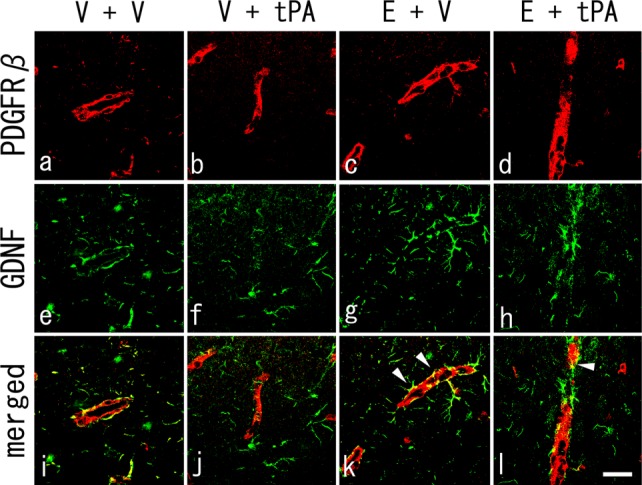
Double immunohistochemistry of PDGFRβ-positive pericytes (a–d, red), GDNF-positive cells (e–h, green), and merged images (i–l) at 4 days after tMCAO. Note that the fluorescent signals for GDNF were partially observed surrounding the PDGFRβ-positive pericytes in the V + V group (i), with weaker signals observed in the V + tPA group (j). Also note the preserved GDNF secretion and overlapping pericytes in E + tPA and E + V groups (k,l, arrowheads). Scale bar = 100 μm
DISCUSSION
The role of pericytes, which encircle capillary vessels, is remarkably important for the maturation and stabilization of capillary vessels during angiogenesis. Ischemia and reperfusion-induced injury may impair microcirculatory reflow, which negatively affects the survival of pericytes (Yemisci et al., 2009). The present study demonstrates that the coverage of endothelial cells by pericytes in the peri-ischemic brain regions is transiently reduced shortly after transient ischemia and then increased, peaking at 4–7 days with a gradual return until 28 days (Fig. 1). This study also shows that coverage of the pericytes is significantly reduced by intravenous administration of tPA but can be recovered with edaravone (Fig. 3). In addition, this study demonstrates that intravenous administration of tPA induces the detachment of astrocyte endfeet surrounding the pericytes in parts of vessels in peri-ischemic brain regions, although edaravone recovered this condition (Fig. 5).
Endothelial coverage by pericytes varies among organs and is correlated with the degree of tightness of the interendothelial junctions (Shepro and Morel, 1993). The endothelial coverage ratio by pericytes was 30–80% in brain regions and did not vary in the cerebral cortex, caudate, or hippocampus (Armulik et al., 2010; Bell et al., 2010; Daneman et al., 2010; Dalkara et al., 2011). The coverage of pericytes was 61.7% ± 2.7% before cerebral ischemia (Fig. 1), confirming previous reports. In addition, this study shows a decrease of endothelial coverage by pericytes at 1 day after tMCAO (Fig. 1), which then increased significantly as the number of Ki67 double-positive pericytes increased at 4 days after ischemia (Fig. 2). These results might correspond to the first step of angiogenesis in which pericytes degenerate and detach from the base membrane to form a new vessel (Hayashi et al., 2006; Duz et al., 2007; Yemisci et al., 2009). Arimura et al. (2012) revealed that the expression of PDGFRβ increased gradually over the 5 days following permanent MCAO in vascular walls in peri-infarct areas. Bone marrow-derived cells may also play a crucial role as a source of pericytes in vessel maturation after cerebral ischemia (Kokovay et al., 2006).
In the present study, administration of tPA decreased both pericyte coverage and PDGFRβ protein expression at 4 days after tMCAO (Fig. 3). A small amount of endogenous tPA is normally produced in neurons, astrocytes, microglia, and endothelial cells of the rodent brain (Tsirka et al., 1997; Schreiber et al., 1998; Kim et al., 2006; Xin et al., 2010). This tPA is secreted into the extracellular space (Samson and Medcalf, 2006) and is regulated by neuroserpin and plasminogen activator inhibitor-1 (PAI-1; Lawrence et al., 1990; Yepes and Lawrence, 2004). By means of this mechanism, tPA physiologically modulates learning (Seeds et al., 2003), synaptic plasticity (Krystosek and Seeds, 1981), cell death (Tsirka et al., 1997), and permeability of the NVU (Polavarapu et al., 2007; Park et al., 2008). In addition, pericytes regulate coagulation and fibrinolysis by releasing PAI-1 and antithrombotic serpin protease nexin-1 (Kim et al., 2006). However, a high dose of exogenous tPA administered for thrombolytic therapy in acute stroke damages brain tissue by activating NMDA receptor, disrupting the extracellular matrix, disturbing the NVU, and causing direct neurotoxicity (Kaur et al., 2004; Zhang et al., 2004; Mannaioni et al., 2008; Girouard et al., 2009; Yamashita et al., 2009). This study suggests a new aspect of the deleterious effect of tPA on pericytes by reducing the proliferation of, and suppressing, PDGFRβ protein even after 4 days (Figs. 3, 4).
On the other hand, the administration of edaravone restored both pericyte coverage and PDGFRβ protein expression at 4 days after tMCAO (Fig. 3). Yemisci et al. (2009) demonstrated that pericyte contraction induced by oxidative–nitrative stress impairs capillary reflow after MCAO and that suppression of oxidative–nitrative stress relieves pericyte contraction, reduces erythrocyte entrapment, and restores microvascular patency, hence improving tissue survival. The administration of the free radical scavenger edaravone might prevent pericyte contraction, induce the proliferation of pericytes, and reduce infarct volume. We previously demonstrated that neurotoxic effects of tPA were ameliorated by i.v. edavarone treatment (Zhang et al., 2004) and that edaravone decreased MMP-9 activation and oxidative stress induced by tPA (Lukic-Panin et al., 2010). This study confirms that pericytes were damaged and that proliferation of pericytes was prevented by tPA and that edaravone ameliorated these damaging effects, allowing pericytes to recover after tMCAO.
The present study shows a marked dissociation between astrocyte foot processes and pericytes in the peri-ischemic lesion with tPA, which was improved in the edaravone-treated group (Fig. 5). Previous studies showed that pericytes increased nerve growth factor (NGF) and neurotrophin-3 (NT-3) production after cerebral ischemia (Arimura et al., 2012; Ishitsuka et al., 2012). Pericytes also secreted GDNF in the NVU (Shimizu et al., 2012). In this study, the expression of GDNF decreased considerably in the tPA-treated group but was restored in the edaravone-treated group (Fig. 6). The dissociation between astrocyte foot processes and pericytes could induce a dysfunctional NVU and decrease the secretion of neuroprotective and angiogenesis-related proteins such as GDNF, NGF, and NT-3.
We previously reported that edaravone maintains the integrity of NVU and inhibits tPA-induced hemorrhagic transformation (Lukic-Panin et al., 2010; Yamashita et al., 2009). The present study re-emphasizes that exogenous tPA damaged proliferating pericytes (Figs. 3, 4), the attachment to astrocytes (Fig. 5), and the secretion of neuroprotective factor (Fig. 6). However, edaravone not only greatly ameliorated this damage to pericytes after tMCAO, it maintained the structure (Fig. 5) and function (Fig. 6) of the NVU. Additional studies are required to reveal a more detailed interaction between edaravone and pericytes in neuroprotection and angiogenesis after tMCAO.
REFERENCES
- Abe K, Yuki S, Kogure K. Strong attenuation of ischemic and postischemic brain edema in rats by a novel free radical scavenger. Stroke. 1988;19:480–485. doi: 10.1161/01.str.19.4.480. [DOI] [PubMed] [Google Scholar]
- Abe K, Hayashi T, Itoyama Y. Amelioration of brain edema by topical application of glial cell line-derived neurotrophic factor in reperfused rat brain. Neurosci Lett. 1997;231:37–40. doi: 10.1016/s0304-3940(97)00517-x. [DOI] [PubMed] [Google Scholar]
- Abe K, Kawagoe J, Araki T, Aoki M, Kogure K. Differential expression of heat shock protein 70 gene between the cortex and caudate after transient focal cerebral ischaemia in rats. Neurol Res. 1992;14:381–385. doi: 10.1080/01616412.1992.11740089. [DOI] [PubMed] [Google Scholar]
- Arimura K, Ago T, Kamouchi M, Nakamura K, Ishitsuka K, Kuroda J, Sugimori H, Ooboshi H, Sasaki T, Kitazono T. PDGF receptor beta signaling in pericytes following ischemic brain injury. Curr Neurovasc Res. 2012;9:1–9. doi: 10.2174/156720212799297100. [DOI] [PubMed] [Google Scholar]
- Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, Johansson BR, Betsholtz C. Pericytes regulate the blood–brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalkara T, Gursoy-Ozdemir Y, Yemisci M. Brain microvascular pericytes in health and disease. Acta Neuropathol. 2011;122:1–9. doi: 10.1007/s00401-011-0847-6. [DOI] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deguchi K, Tsuru K, Hayashi T, Takaishi M, Nagahara M, Nagotani S, Sehara Y, Jin G, Zhang H, Hayakawa S, Shoji M, Miyazaki M, Osaka A, Huh NH, Abe K. Implantation of a new porous gelatin–siloxane hybrid into a brain lesion as a potential scaffold for tissue regeneration. J Cereb Blood Flow Metab. 2006;26:1263–1273. doi: 10.1038/sj.jcbfm.9600275. [DOI] [PubMed] [Google Scholar]
- Deguchi K, Miyazaki K, Tian F, Liu N, Liu W, Kawai H, Omote Y, Kono S, Yunoki T, Deguchi S, Abe K. Modifying neurorepair and neuroregenerative factors with tPA and edaravone after transient middle cerebral artery occlusion in rat brain. Brain Res. 2012;1436:168–177. doi: 10.1016/j.brainres.2011.12.016. [DOI] [PubMed] [Google Scholar]
- Duz B, Oztas E, Erginay T, Erdogan E, Gonul E. The effect of moderate hypothermia in acute ischemic stroke on pericyte migration: an ultrastructural study. Cryobiology. 2007;55:279–284. doi: 10.1016/j.cryobiol.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Girouard H, Wang G, Gallo EF, Anrather J, Zhou P, Pickel VM, Iadecola C. NMDA receptor activation increases free radical production through nitric oxide and NOX2. J Neurosci. 2009;29:2545–2552. doi: 10.1523/JNEUROSCI.0133-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravanis I, Tsirka SE. Tissue plasminogen activator and glial function. Glia. 2005;49:177–183. doi: 10.1002/glia.20115. [DOI] [PubMed] [Google Scholar]
- Group TNt-PSS. Intracerebral hemorrhage after intravenous t-PA therapy for ischemic stroke. Stroke. 1997;28:2109–2118. doi: 10.1161/01.str.28.11.2109. [DOI] [PubMed] [Google Scholar]
- Hacke W, Kaste M, Bluhmki E, Brozman M, Davalos A, Guidetti D, Larrue V, Lees KR, Medeghri Z, Machnig T, Schneider D, von Kummer R, Wahlgren N, Toni D. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008;359:1317–1329. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- Hamilton NB, Attwell D, Hall CN. Pericyte-mediated regulation of capillary diameter: a component of neurovascular coupling in health and disease. Front Neuroenerget. 2010;2 doi: 10.3389/fnene.2010.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Deguchi K, Nagotani S, Zhang H, Sehara Y, Tsuchiya A, Abe K. Cerebral ischemia and angiogenesis. Curr Neurovasc Res. 2006;3:119–129. doi: 10.2174/156720206776875902. [DOI] [PubMed] [Google Scholar]
- Ishitsuka K, Ago T, Arimura K, Nakamura K, Tokami H, Makihara N, Kuroda J, Kamouchi M, Kitazono T. Neurotrophin production in brain pericytes during hypoxia: a role of pericytes for neuroprotection. Microvasc Res. 2012;83:352–359. doi: 10.1016/j.mvr.2012.02.009. [DOI] [PubMed] [Google Scholar]
- Kaur J, Zhao Z, Klein GM, Lo EH, Buchan AM. The neurotoxicity of tissue plasminogen activator? J Cereb Blood Flow Metab. 2004;24:945–963. doi: 10.1097/01.WCB.0000137868.50767.E8. [DOI] [PubMed] [Google Scholar]
- Kawai H, Deguchi S, Deguchi K, Yamashita T, Ohta Y, Shang J, Tian F, Zhang X, Liu N, Liu W, Ikeda Y, Matsuura T, Abe K. Synergistic benefit of combined amlodipine plus atorvastatin on neuronal damage after stroke in Zucker metabolic rat. Brain Res. 2011;1368:317–323. doi: 10.1016/j.brainres.2010.10.046. [DOI] [PubMed] [Google Scholar]
- Kim JA, Tran ND, Li Z, Yang F, Zhou W, Fisher MJ. Brain endothelial hemostasis regulation by pericytes. J Cereb Blood Flow Metab. 2006;26:209–217. doi: 10.1038/sj.jcbfm.9600181. [DOI] [PubMed] [Google Scholar]
- Kitagawa H, Sasaki C, Sakai K, Mori A, Mitsumoto Y, Mori T, Fukuchi Y, Setoguchi Y, Abe K. Adenovirus-mediated gene transfer of glial cell line-derived neurotrophic factor prevents ischemic brain injury after transient middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab. 1999;19:1336–1344. doi: 10.1097/00004647-199912000-00007. [DOI] [PubMed] [Google Scholar]
- Kokovay E, Li L, Cunningham LA. Angiogenic recruitment of pericytes from bone marrow after stroke. J Cereb Blood Flow Metab. 2006;26:545–555. doi: 10.1038/sj.jcbfm.9600214. [DOI] [PubMed] [Google Scholar]
- Korninger C, Collen D. Studies on the specific fibrinolytic effect of human extrinsic (tissue-type) plasminogen activator in human blood and in various animal species in vitro. Thromb Haemost. 1981;46:561–565. [PubMed] [Google Scholar]
- Krystosek A, Seeds NW. Plasminogen activator release at the neuronal growth cone. Science. 1981;213:1532–1534. doi: 10.1126/science.7197054. [DOI] [PubMed] [Google Scholar]
- Lawrence DA, Strandberg L, Ericson J, Ny T. Structure-function studies of the SERPIN plasminogen activator inhibitor type 1. Analysis of chimeric strained loop mutants. J Biol Chem. 1990;265:20293–20301. [PubMed] [Google Scholar]
- Lukic-Panin V, Deguchi K, Yamashita T, Shang J, Zhang X, Tian F, Liu N, Kawai H, Matsuura T, Abe K. Free radical scavenger edaravone administration protects against tissue plasminogen activator induced oxidative stress and blood brain barrier damage. Curr Neurovasc Res. 2010;7:319–329. doi: 10.2174/156720210793180747. [DOI] [PubMed] [Google Scholar]
- Mannaioni G, Orr AG, Hamill CE, Yuan H, Pedone KH, McCoy KL, Berlinguer Palmini R, Junge CE, Lee CJ, Yepes M, Hepler JR, Traynelis SF. Plasmin potentiates synaptic N-methyl-D-aspartate receptor function in hippocampal neurons through activation of protease-activated receptor-1. J Biol Chem. 2008;283:20600–20611. doi: 10.1074/jbc.M803015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag S. Morphology and molecular properties of cellular components of normal cerebral vessels. Methods Mol Med. 2003;89:3–36. doi: 10.1385/1-59259-419-0:3. [DOI] [PubMed] [Google Scholar]
- Pardridge WM. Blood–brain barrier biology and methodology. J Neurovirol. 1999;5:556–569. doi: 10.3109/13550289909021285. [DOI] [PubMed] [Google Scholar]
- Park L, Gallo EF, Anrather J, Wang G, Norris EH, Paul J, Strickland S, Iadecola C. Key role of tissue plasminogen activator in neurovascular coupling. Proc Natl Acad Sci U S A. 2008;105:1073–1078. doi: 10.1073/pnas.0708823105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polavarapu R, Gongora MC, Yi H, Ranganthan S, Lawrence DA, Strickland D, Yepes M. Tissue-type plasminogen activator-mediated shedding of astrocytic low-density lipoprotein receptor-related protein increases the permeability of the neurovascular unit. Blood. 2007;109:3270–3278. doi: 10.1182/blood-2006-08-043125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson AL, Medcalf RL. Tissue-type plasminogen activator: a multifaceted modulator of neurotransmission and synaptic plasticity. Neuron. 2006;50:673–678. doi: 10.1016/j.neuron.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Schreiber SS, Tan Z, Sun N, Wang L, Zlokovic BV. Immunohistochemical localization of tissue plasminogen activator in vascular endothelium of stroke-prone regions of the rat brain. Neurosurgery. 1998;43:909–913. doi: 10.1097/00006123-199810000-00107. [DOI] [PubMed] [Google Scholar]
- Seeds NW, Basham ME, Ferguson JE. Absence of tissue plasminogen activator gene or activity impairs mouse cerebellar motor learning. J Neurosci. 2003;23:7368–7375. doi: 10.1523/JNEUROSCI.23-19-07368.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepro D, Morel NM. Pericyte physiology. FASEB J. 1993;7:1031–1038. doi: 10.1096/fasebj.7.11.8370472. [DOI] [PubMed] [Google Scholar]
- Shimizu F, Sano Y, Saito K, Abe MA, Maeda T, Haruki H, Kanda T. Pericyte-derived glial cell line-derived neurotrophic factor increase the expression of claudin-5 in the blood–brain barrier and the blood–nerve barrier. Neurochem Res. 2012;37:401–409. doi: 10.1007/s11064-011-0626-8. [DOI] [PubMed] [Google Scholar]
- Tsirka SE, Rogove AD, Bugge TH, Degen JL, Strickland S. An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J Neurosci. 1997;17:543–552. doi: 10.1523/JNEUROSCI.17-02-00543.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Yuki S, Egawa M, Nishi H. Protective effects of MCI-186 on cerebral ischemia: possible involvement of free radical scavenging and antioxidant actions. J Pharmacol Exp Ther. 1994;268:1597–1604. [PubMed] [Google Scholar]
- Xin H, Li Y, Shen LH, Liu X, Wang X, Zhang J, Pourabdollah-Nejad DS, Zhang C, Zhang L, Jiang H, Zhang ZG, Chopp M. Increasing tPA activity in astrocytes induced by multipotent mesenchymal stromal cells facilitate neurite outgrowth after stroke in the mouse. PLoS One. 2010;5:e9027. doi: 10.1371/journal.pone.0009027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Kamiya T, Deguchi K, Inaba T, Zhang H, Shang J, Miyazaki K, Ohtsuka A, Katayama Y, Abe K. Dissociation and protection of the neurovascular unit after thrombolysis and reperfusion in ischemic rat brain. J Cereb Blood Flow Metab. 2009;29:715–725. doi: 10.1038/jcbfm.2008.164. [DOI] [PubMed] [Google Scholar]
- Yemisci M, Gursoy-Ozdemir Y, Vural A, Can A, Topalkara K, Dalkara T. Pericyte contraction induced by oxidative–nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med. 2009;15:1031–1037. doi: 10.1038/nm.2022. [DOI] [PubMed] [Google Scholar]
- Yepes M, Lawrence DA. Neuroserpin: a selective inhibitor of tissue-type plasminogen activator in the central nervous system. Thromb Haemost. 2004;91:457–464. doi: 10.1160/TH03-12-0766. [DOI] [PubMed] [Google Scholar]
- Zhang WR, Hayashi T, Kitagawa H, Sasaki C, Sakai K, Warita H, Wang JM, Shiro Y, Uchida M, Abe K. Protective effect of ginkgo extract on rat brain with transient middle cerebral artery occlusion. Neurol Res. 2000;22:517–521. doi: 10.1080/01616412.2000.11740713. [DOI] [PubMed] [Google Scholar]
- Zhang W, Sato K, Hayashi T, Omori N, Nagano I, Kato S, Horiuchi S, Abe K. Extension of ischemic therapeutic time window by a free radical scavenger, edaravone, reperfused with tPA in rat brain. Neurol Res. 2004;26:342–348. doi: 10.1179/016164104225014058. [DOI] [PubMed] [Google Scholar]