

Abstract
Some cytochrome P450 enzymes epoxidize unsaturated substrates, but this activity has not been described for the steroid hydroxylases. Physiologic steroid substrates, however, lack carbon–carbon double bonds in the parts of the pregnane molecules where steroidogenic hydroxylations occur. Limited data on the reactivity of steroidogenic P450s toward olefinic substrates exist, and the study of occult activities toward alternative substrates is a fundamental aspect of the growing field of combinatorial biosynthesis. We reasoned that human P450c17 (steroid 17-hydroxylase/17,20-lyase, CYP17A1), which 17- and 16α-hydroxylates progesterone, might catalyze the formation of the 16α,17-epoxide from 16,17-dehydroprogesterone (pregna-4,16-diene-3,20-dione). CYP17A1 catalyzed the novel 16α,17-epoxidation and the ordinarily minor 21-hydroxylation of 16,17-dehydroprogesterone in a 1:1 ratio. CYP17A1 mutation A105L, which has reduced progesterone 16α-hydroxylase activity, gave a 1:5 ratio of epoxide:21-hydroxylated products. In contrast, human P450c21 (steroid 21-hydroxylase, CYP21A2) converted 16,17-dehydroprogesterone to the 21-hydroxylated product and only a trace of epoxide. CYP21A2 mutation V359A, which has significant 16α-hydroxylase activity, likewise afforded the 21-hydroxylated product and slightly more epoxide. CYP17A1 wild-type and mutation A105L do not 21- or 16α-hydroxylate pregnenolone, but the enzymes 21-hydroxylated and 16α,17-epoxidized 16,17-dehydropregnenolone (pregna-5,16-diene-3β-ol-20-one) in 4:1 or 12:1 ratios, respectively. Catalase and superoxide dismutase did not prevent epoxide formation. The progesterone epoxide was not a time-dependent, irreversible CYP17A1 inhibitor. Our substrate modification studies have revealed occult epoxidase and 21-hydroxylase activities of CYP17A1, and the fraction of epoxide formed correlated with the 16α-hydroxylase activity of the enzymes.
Cytochromes P450c17 (steroid 17-hydroxylase/17,20-lyase, CYP17A1) and P450c21 (steroid 21-hydroxylase, CYP21A2) catalyze key reactions in adrenal and gonadal steroid biosynthesis.1 CYP17A1 catalyzes 17α-hydroxylation needed for cortisol synthesis and the 17,20-lyase reaction needed for androgen production. CYP21A2 specifically catalyzes the 21-hydroxylation of steroids required for the synthesis of mineralocorticoids and glucocorticoids, such as aldosterone and cortisol. Both P450s share common substrates, including progesterone and 17-hydroxyprogesterone, but each enzyme yields a different product profile. Human CYP17A1 catalyzes the 17- and 16α-hydroxylase reactions with progesterone as substrate in a 3:1 ratio, as well as a trace of 21-hydroxylase activity.2 The small side chain of A105 favors 16α-hydroxylation, and CYP17A1 mutation A105L reduces the portion of 16α-hydroxylation from 25% to 10%.2−4 Conversely, human CYP21A2 catalyzes progesterone 21-hydroxylation and a trace of 16α-hydroxylation, but for CYP21A2 mutations V359A and V359G, 16α-hydroxylation accounts for 40% and 90% of the products, respectively.5
Besides hydroxylation activities, cytochromes P450 catalyze a variety of other oxidative reactions including epoxidation, dehydrogenation, deamination, dehalogenation, and heteroatom oxygenation/dealkylation.6,7 Under appropriate conditions, P450s also catalyze atypical reactions such as cleavage of C–C bonds,8,9 Wagner–Meerwein type rearrangements,10 Paterno–Buchi cyclization,11 cyclopropanations,12 imidation of sulfides,13 and other chemistries. For example, CYP17A1 also catalyzes the oxidative cleavage of the C17–C20 carbon–carbon bond in 17-hydroxysteroids, yielding 19-carbon steroids, and cytochrome b5 selectively stimulates the rate of these 17,20-lyase reactions.14 In the presence of cytochrome b5, CYP17A1 also metabolizes about 10% of pregnenolone to androsta-5,16-diene-3β-ol,15 which acts as a pheromone in some species. We envisioned that these steroidogenic P450s with 17- and/or 16α-hydroxylase activity might also demonstrate epoxidation activity with steroid analogues containing an unsaturated carbon–carbon bond at C-16. Here, we have introduced an alkene in the position of the steroid substrate where these enzymes normally hydroxylate to probe for epoxidation chemistry with pregna-4,16-diene-3,20-dione (16,17-dehydroprogesterone, compound 1, Scheme 1) and pregna-5,16-diene-3β-ol-20-one (16,17-dehydropregnenolone, compound 5, Scheme 2). In addition, we employed mutations with different product distributions, CYP17A1-A105L and CYP21A2-V359A, to determine the influence of regiochemical preferences on epoxidase activity and product distributions.
Scheme 1. Epoxidation and Alternative Hydroxylation Chemistry Probed in This Study.

Scheme 2. Synthetic Pathway for 16,17-Dehydroprogesterone and Product Standards.

Compound 7 was synthesized as previously reported.16 DMP: Dess–Martin periodinane. NIS: N-iodosuccinimide.
Experimental Procedures
General Methods and Reagents
NMR spectra were obtained using Varian instruments at frequencies for 1H and 13C as specified in the experimental detail. Chemical shifts were referenced to the chloroform peak in the 1H NMR assigned at 7.26 ppm and in the 13C NMR assigned at 77.16 ppm. NMR spectra are provided in the Supporting Information. Reaction progress was monitored with TLC and detection under UV light or staining with a solution of ammonium molybdate tetrahydrate [CAS: 12054-85-2] (12 g), ammonium cerium sulfate dihydrate [CAS: 10378-47-9] (0.5 g), and concentrated H2SO4 (15 mL) in 235 mL of water. Alternatively, an aliquot of the reaction was taken and analyzed by NMR. Progesterone and all other reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO), Steraloids (Newport, RI), ThermoFisher Scientific (Pittsburgh, PA), or as specified. Protein determinations used the Coomassie Plus Reagent (Pierce, Rockford, IL). Silica gel (flash grade, 60 Å) was purchased from Dynamic Adsorbents Inc. (Norcross, GA).
Steroid Syntheses
Pregna-4,16-diene-3,20-dione (16,17-Dehydroprogesterone, Compound 1)
To a solution of pregna-5,16-dien-3β-acetoxy-20-one (16,17-dehydropregnenolone-3β-acetate (compound 4, 4.65 g, 13.0 mmol) [Waterstone Technology, Carmel, IN] in tetrahydrofuran (50 mL) was added water (10 mL) and HCl (12 N, 5 mL). After being stirred at room temperature for 7 days, the reaction was filtered through a cotton-plugged funnel, and the white solid was washed with hexanes. The solid precipitate was concentrated via reduced pressure to yield compound 5 (3.5 g, 11.1 mmol, 85% yield) as the product. 1H NMR (400 MHz, CDCl3): δ 6.71 (m, 1H), 5.35 (m, 1H), 3.56–3.48 (m, 1H), 2.42–2.38 (m, 1H), 2.34–2.25 (m, 2H), 2.26 (s, 3H), 2.08–2.00 (m, 2H), 1.86–1.82 (m, 2H), 1.74–1.28 (m, 9H), 1.11–1.04 (m, 1H), 1.04 (s, 3H), 0.92 (s, 3H).
Crude alcohol 5 (1.05 g, 3.34 mmol) was weighed in a 200 mL oven-dried round-bottom flask equipped with a stirrer. Dicholoromethane (50 mL) was added to dissolve the alcohol, and Dess–Martin periodinane (1.48 g, 3.49 mmol, 1.04 mol eq) was added to the reaction mixture, which was then stirred at room temperature for 1 h. The reaction mixture was diluted with ether and washed with NaHCO3 (saturated aq. solution, 50 mL). During purification via flash column chromatography (100% hexanes to 50% ethyl acetate in hexanes), some of the Δ5,6-double bond isomerized to the Δ4,5-position, which yielded a mixture of the less polar pregna-5,16-diene-3,20-dione (0.45 g, 1.44 mmol, 43% yield) and the more polar pregna-4,16-diene-3,20-dione (compound 1, 0.18 g, 0.57 mmol, 17% yield). 1H NMR for pregna-5,16-dien-3-one (500 MHz, CDCl3): δ 6.71 (m, 1H), 5.35 (m, 1H), 3.30 (d, J = 13.2 Hz, 1H), 2.83 (d, J = 12.8 Hz, 1H), 2.51–2.42 (m, 2H), 2.32–2.30 (m, 2H), 2.27 (s, 3H), 2.09–2.04 (m, 3H), 1.77–1.72 (m, 1H), 1.70–1.61 (m, 3H), 1.50–1.42 (m, 2H), 1.38–1.34 (m, 1H), 1.23 (s, 3H), 1.15–1.10 (m, 1H), 0.95 (s, 3H). 1H NMR for compound 1 (500 MHz, CDCl3): δ 6.70–6.69 (m, 1H), 5.72 (m, 1H), 2.46–2.37 (m, 3H), 2.35–2.27 (m, 3H), 2.26 (s, 3H), 2.09–2.05 (m, 1H), 2.04–1.98 (m, 1H), 1.89–1.84 (m, 1H), 1.78–1.65 (m, 2H), 1.62–1.48 (m, 2H), 1.44–1.38 (m, 1H), 1.35–1.29 (m, 1H), 1.20 (s, 3H), 1.17–1.07 (m, 1H), 1.04–0.95 (m, 1H), 0.92 (s, 3H); 13C NMR for compound 1 (125 MHz, CDCl3): δ 199.7, 196.9, 171.1, 155.2, 144.3, 124.1, 55.8, 54.2, 46.2, 38.8, 35.6, 34.5, 34.0, 33.9, 32.8, 32.2, 31.9, 27.2, 20.8, 17.3, 15.9.
16α,17-Epoxypregn-4-ene-3,20-dione (16α,17-Epoxyprogesterone, Compound 2)
Compound 4 (2.5 g, 7.0 mmol) was weighed in a 100 mL round-bottom flask equipped with stirrer and treated with NaOH (1.0 g, 25 mmol, 3.6 mol eq), followed by hydrogen peroxide (30% aq., 20 mL), then methanol (5 mL). The reaction flask was equipped with rubber septum and a balloon, and it was stirred for 36 h. The resulting mixture was diluted with ethyl acetate (100 mL) and washed with brine. The combined organic extracts were dried with MgSO4 and concentrated via reduced pressure. The crude material was used for the next step (compound 6, 2.1 g, 6.4 mmol, 92% yield). 1H NMR (400 MHz, CDCl3): δ 5.34–5.32 (m, 1H), 3.68 (s, 1H), 3.52–3.45 (m, 1H), 2.32–2.17 (m, 2H), 2.06–2.01 (m, 1H), 2.03 (s, 3H), 2.0–1.9 (m, 2H), 1.9–1.8 (m, 2H), 1.70–1.40 (m, 6H), 1.32 (apparent t, J = 12.0 Hz, 1H), 1.2–0.9 (m, 2H), 1.04 (s, 3H), 1.02 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 205.1, 141.3, 121.2, 71.8, 71.2, 60.7, 50.4, 45.7, 42.4, 41.7, 37.3, 36.8, 31.7, 31.6, 31.5, 29.9, 27.7, 26.2, 20.6, 19.5, 15.4.
The crude epoxide (compound 6, 2.0 g, 6.05 mmol) was weighed in an oven-dried 100 mL round-bottom flask equipped with stirrer. Dichloromethane (50 mL) was added to dissolve the starting material, and Dess–Martin periodinane (2.8 g, 6.6 mmol, 1.1 mol eq) was added to the reaction mixture, which was then stirred at room temperature and monitored by TLC until completion. Upon completion, the mixture was diluted with ether (100 mL) and washed with water (50 mL) and NaHCO3 (saturated aqueous solution, 3 × 50 mL). The organic layer was concentrated, and the crude material was purified via flash column chromatography (100% hexanes to 50% hexanes/ethyl acetate) to afford the less polar 3-keto-Δ5,6-product (0.4 g, 1.2 mmol, 20%) and the isomerized (more polar) 3-keto-Δ4,5-product (compound 2, 0.6 g, 1.8 mmol, 30%). 1H NMR for 3-keto-Δ5,6-product (500 MHz, CDCl3): δ 5.32–5.31 (m, 1H), 3.69 (s, 1H), 3.30–3.24 (m, 1H), 2.85–2.78 (m, 1H), 2.46 (ddd, J1 = 12.0 Hz, J2 = 12.0 Hz, J3 = 4.0 Hz, 1H), 2.33–2.25 (m, 1H), 2.13–2.00 (m, 2H), 2.02 (s, 3H), 1.99–1.94 (m, 2H), 1.70–1.55 (m, 3H), 1.52–1.40 (m, 2H), 1.38–1.30 (m, 1H), 1.23–1.15 (m, 1H), 1.19 (s, 3H), 1.07 (s, 3H) 1.11–1.02 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 210.1, 205.0, 139.0, 122.3, 71.1, 60.6, 49.5, 48.5, 45.6, 41.7, 37.7, 37.2, 37.0, 31.5, 31.4, 29.9, 27.6, 26.1, 20.8, 19.2, 15.4. 1H NMR for compound 2 (500 MHz, CDCl3): δ 5.68–5.67 (m, 1H), 3.67 (s, 1H), 2.43–2.21 (m, 4H), 2.1–1.9 (m, 5H), 1.98 (s, 3H), 1.79–1.70 (m, 1H), 1.70–1.55 (m, 3H), 1.50–1.28 (m, 3H), 1.16 (s, 3H), 1.04 (s, 3H), 0.95–0.86 (m, 1H); 13C NMR (125 MHz, CDCl3): δ 204.8, 199.5, 170.7, 124.1, 70.7, 60.5, 53.9, 44.9, 41.6, 38.7, 35.6, 34.0, 33.3, 32.7, 31.6, 31.2, 27.4, 26.0, 20.4, 17.3, 15.3.
Pregna-5,16-diene-3β-formyloxy-21-acetoxy-20-one (Compound 8).16
Formic acid (50 mL) was added to compound 5 (5 g, 19.2 mmol) in a 100 mL round-bottom flask equipped with stirrer, and the reaction was stirred at reflux for 1 h and monitored by TLC (1:1 v/v ethyl acetate:hexanes). The reaction mixture was cooled to room temperature, diluted with water, and extracted with ether (2 × 100 mL). The formylated intermediate (1.22 g, 3.56 mmol) and p-toluenesulfonic acid monohydrate (0.15 g, 0.79 mmol, 0.2 mol eq) were weighed in a 100 mL round-bottom flask equipped with a stirrer. Isopropenyl acetate (50 mL) was added, and the flask was fitted with a Dean–Stark apparatus and reflux condenser. The reaction was refluxed, and the solution was never evaporated completely. After 20 h, the reaction was cooled to room temperature and diluted with diethyl ether (100 mL). The solution was washed with NaHCO3 (saturated aq. solution, 2 × 50 mL), and the organic layer was washed with brine (2 × 25 mL). The combined organic extracts were dried with MgSO4 and concentrated via reduced pressure. A solid formed under a vacuum, and the solid was washed with ice-cold methanol to yield the enol acetate intermediate (0.9 g, 2.34 mmol, 66% yield). 1H NMR (400 MHz, CDCl3): δ 8.04 (s, 1H), 5.82 (m, 1H), 5.40 (m, 1H), 5.06 (s, 1H), 4.78 (2, 1H), 4.65–4.78 (m, 1H), 2.36–2.39 (m, 2H), 2.18 (s, 3H), 2.10–2.15 (m, 1H), 1.99–2.04 (m, 1H), 1.86–1.95 (m, 3H), 1.47–1.70 (m, 8H), 1.13–1.19 (m, 1H), 1.07 (s, 3H), 0.98 (s, 3H).
To the crude enol acetate (0.33 g, 0.86 mmol) in dichloromethane (50 mL) was added N-iodosuccinimide (0.29 g, 1.29 mmol, 1.5 mmol). The reaction was stirred at room temperature for 1 h, and the reaction mixture was directly purified via flash column chromatography to afford iodide 7 (0.30 g, 0.64 mmol, 74% yield).16 Compound 7 (92 mg, 0.20 mmol) in a 100 mL round-bottom flask equipped with stirrer was dissolved in 20 mL of acetonitrile and treated with silver acetate (60 mg, 0.36 mmol, 1.8 mol eq). The reaction was stirred for 24 h at room temperature, and the reaction mixture was directly loaded on a silica gel column for purification (100% hexanes to 50% ethyl acetate/hexanes) to yield compound 8 (70 mg, 0.18 mmol, 89% yield). 1H NMR (400 MHz, CDCl3): δ 8.04 (s, 1H), 6.75 (s, 1H), 5.35 (m, 1H), 4.99 (d, J = 16.0 Hz, 1H), 4.84 (d, J = 16.0 Hz, 1H), 4.65–4.78 (m, 1H), 2.31–2.38 (m, 4H), 2.18 (s, 3H), 2.00–2.13 (m, 2H), 1.87–1.90 (m, 2H), 1.59–1.71 (m, 4H), 1.34–1.47 (m, 2H), 1.11–1.18 (m, 1H), 1.06 (s, 3H), 0.92 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 190.5, 170.4, 160.6, 151.9, 144.1, 139.9, 122.2, 104.8, 73.7, 65.6, 55.9, 50.3, 46.7, 38.0, 36.8, 36.7, 34.3, 32.7, 31.5, 30.0, 27.7, 20.6, 19.2, 15.8.
Pregna-4,16-diene-21-acetoxy-3,20-dione (Compound 10)
To a stirring solution of compound 8 (16 mg, 0.04 mmol) in 10 mL of each dichloromethane and methanol was added HCl (12 N, 0.2 mL). The reaction was stirred for 1 h at room temperature, and the resulting reaction mixture was purified via flash column chromatography (100% hexanes to 50% ethyl acetate/hexanes) to yield compound 9 (14 mg, 0.038 mmol, 94% yield). 1H NMR (400 MHz, CDCl3): δ 6.75 (s, 1H), 5.35 (broad s, 1H), 5.03, 4.88 (ABq, 2H, JAB = 16.0 Hz), 3.60–3.49 (m, 1H), 2.42–2.22 (m, 2H), 2.18 (s, 3H), 2.20–1.85 (m, 2H), 0.90–0.80 (m, 2H), 1.04 (s, 3H), 0.94 (s, 3H).
Dess–Martin periodinane (0.43 g, 1.0 mmol) was added to a solution of alcohol 9 (0.27 g, 0.72 mmol) in dichloromethane (20 mL), and the reaction was stirred for 1 h followed by filtration through a short pad of silica. During purification via flash column chromatography (100% hexanes to 50% ethyl acetate in hexanes), isomerization of the 3-keto-Δ5,6-product to the 3-keto-Δ4,5-product occurred on the column, yielding compound 10 (0.13 g, 0.35 mmol, 49%) as a white solid. 1H NMR (400 MHz, CDCl3): δ 6.74 (s, 1H), 5.74 (s, 1H), 5.03, 4.87 (ABq, 2H, JAB = 16.0 Hz), 2.48–2.25 (m, 6H), 2.18 (s, 3H), 2.12 (dd, J1 = 17.0 Hz, J2 = 13.4 Hz, 1H), 2.03–2.00 (m, 1H), 1.88–1.85 (m, 1H), 1.80–1.60 (m, 3H), 1.53 (ddd, J1 = 17.0 Hz, J2 = 13.5 Hz, J3 = 4.0 Hz, 1H), 1.46–1.32 (m, 2H), 1.21 (s, 3H), 1.13 (ddd, J1 = 16.5 Hz, J2 = 13.0 Hz, J3 = 4.0 Hz, 1H), 1.01 (ddd, J1 = 15.5 Hz, J2 = 12.0 Hz, J3 = 5.0 Hz, 1H), 0.96 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 199.6, 190.6, 170.8, 170.6, 151.9, 143.9, 124.2, 65.7, 55.4, 54.2, 46.8, 38.8, 35.7, 34.3, 34.1, 33.9, 32.8, 32.7, 31.9, 20.8, 20.7, 17.3, 16.1.
Pregna-4,16-diene-21-ol-3,20-dione (Compound 3)
To a flask containing compound 10 (25 mg, 0.068 mmol) was added methanol (5 mL) and HCl (12 N, 0.1 mL). The reaction mixture was stirred for 10 h at room temperature, concentrated via reduced pressure, and purified via flash column chromatography (gradient from 100% hexanes to 50% ethyl acetate/hexanes). Compound 3 (10 mg, 0.030 mmol, 45% yield) was isolated as a white solid. 1H NMR (500 MHz, CDCl3): δ 6.74 (dd, J1 = 3.6 Hz, J2 = 1.6 Hz, 1H), 5.74 (m, 1H), 4.54 (dd, J1 = 18.0, J2 = 4.8 Hz, 1H), 4.31 (dd, J1 = 18.0, J2 = 4.8 Hz, 1H), 3.28 (apparent t, J = 4.8 Hz, 1H), 2.29–2.49 (m, 5H), 2.13 (ddd, J1 = 16.0 Hz, J2 = 8.0 Hz, J3 = 1.6 Hz, 1H), 2.00–2.06 (m, 1H), 1.85–1.92 (m, 1H), 1.61–1.80 (m, 3H), 1.41–1.55 (m, 1H), 1.35–1.39 (m, 1H), 1.24–1.28 (m, 1H), 1.23 (s, 3H), 1.12–1.20 (m, 1H), 1.00–1.09 (m, 1H), 0.98 (s, 3H); 13C NMR (125 MHz, CDCl3): 199.61, 199.58, 170.8, 151.4, 144.6, 124.2, 65.2, 54.2, 46.7, 38.8, 35.7, 34.4, 34.1, 34.0, 33.9, 32.8, 32.7, 31.9, 20.8, 17.3, 16.2.
General Procedure for Converting 3-Keto-Δ5,6-steroids to 3-Keto-Δ4,5-steroids
To increase the yield of the desired 3-keto-Δ4,5-steroids after oxidation of 3β-hydroxy-Δ5,6-steroids with Dess–Martin periodinane, the 3-keto-Δ5,6 intermediates (less polar) are dissolved to 0.1 M in a 1:1 (v/v) mixture of methanol:dichloromethane and treated with 0.001% (v/v) of concentrated HCl. The reaction is stirred at room temperature for up to 1 h and purified by flash column chromatography to yield the more polar 3-keto-Δ4,5-steroid products (compounds 1, 2, and 3).
Enzymology Studies
Plasmids
The expression plasmids were generous gifts obtained from the following investigators: human CYP17A1 in pCW17 and N-27-human P450-oxidoreductase (POR) in pET2218 from Professor Walter L. Miller (University of California, San Francisco, CA); human CYP21A2 in pET-17b from Professor Michael R. Waterman (Vanderbilt University, Nashville, TN). The construct pCW-CYP17A1-A105L was previously described.2 For the CYP21A2-V359A construct, a PCR product was obtained using hC21V359A_S: 5′-CCA GGA GTT CTG TGA GCG CAT GA-3′, hC21V359A_AS: 5′-CCC CCA TCC CCC GGG GCT GCA GCC G-3′ and V60-C21V359A5 as a template. This amplicon was digested with EcoN1and Stu1, and the resulting 700-base pair fragment containing the desired mutation (valine GTG codon changed to alanine GCG codon) was ligated into pET17b-CYP21A2, which was digested with the same enzymes. All constructs were sequenced to ensure accurate mutagenesis.
Protein Expression in Escherichia coli and Purification
Modified human CYP17A1, WT and A105L, were expressed in E. coli JM109 cells and purified to homogeneity as described.2 For human CYP21A2, the membrane anchor and basic region were replaced with MAKKTSSKGK from CYP2C3, and both WT and mutation V359A were expressed in E. coli BL21(DE3) cells and purified to homogeneity using Ni-NTA affinity resin (Qiagen, Valencia, CA) after solubilization with 1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate as described for bovine CYP21A2.19 GroEL/ES chaperones (pGro7 plasmid) were coexpressed with the P450s to increase expression of active enzyme. Modified human POR was expressed in E. coli C41(DE3) cells and purified according to the previously published procedure.2
Activity Assays with Yeast Microsomal Enzymes
Microsomes were prepared from a yeast strain that expresses human CYP17A1 (or CYP21A2 or variants) and human POR, according to methods previously described.20 Yeast microsomes (3–10 μL, 10–30 pmol) containing CYP17A1 or CYP21A2 or mutations were incubated in 0.25 mL of 50 mM potassium phosphate (pH 7.4) with 20 μM steroid and 1 mM NADPH at 37 °C for 40 min. When required, superoxide dismutase (SOD; 100 U, Sigma) and catalase (1000 U, Sigma) were added to the incubations prior to the addition of the NADPH. The reaction was stopped by addition of 1 mL of dichloromethane, and the organic phase was collected and dried under a nitrogen flow. Steroids were analyzed using the Agilent 1260 Infinity HPLC system with UV detector. Steroids were dissolved in 20 μL of methanol, and 5 μL injections were resolved with a 50 mm × 2.1 mm, 2.6 μm particle size C8 Kinetex column (Phenomenex, Torrence, CA) equipped with a guard column at a flow of 0.4 mL/min. Aqueous methanol linear gradients were employed (27% methanol from 0 to 0.5 min, jump to 39% methanol, and gradient from 39 to 75% methanol over 30 min). Products were identified by retention times of external standards chromatographed at the beginning of the experiment.
Reconstituted Activity Assays Using Purified Proteins
For CYP17A1 or CYP21A2 reconstitution with POR, yeast microsomal lipids were used as the source of phospholipid. Control yeast microsomes (CYMS) were prepared from strain YiV(B) transformed with empty V60 vector as described.20 CYP17A1 (20 pmol) or CYP21A2 (30 pmol), POR (80–120 pmol), and CYMS (20 μg protein) were added to a 2 mL polypropylene tube in 0.2 mL total reaction volume. The contents were gently swirled and set at room temperature for 5 min. The mixture was dissolved in 50 mM potassium phosphate buffer (pH 7.4) with 20 μM substrate. The resulting mixture was mixed gently and set at 37 °C for 3 min. NADPH (1 mM) was added, and the incubation was continued at 37 °C for 30 min. The products were extracted and chromatographed as for assays with yeast microsomes. For incubations with the 3β-hydroxy-Δ5,6-steroid compound 5, the products were converted to their UV-active, 3-keto-Δ4,5-homologues using cholesterol oxidase as described.2
Mass Spectrometry Analyses
The metabolites were analyzed with an ion trap mass spectrometer (LCQ DecaXP, Thermo Finnigan) coupled with a Shimadzu HPLC system. The products 21-hydroxy-16,17-dehydroprogesterone (compound 3), 16,17-epoxyprogesterone (compound 2), and substrate 16,17-dehydroprogesterone (compound 1) were separated on a Kinetex XB C18 column (2.1 × 50 mm, Phenomenex) with a binary mobile phase of 10% methanol (Solvent A) and 50% methanol:49.9% acetonitrile:0.1% formic acid (Solvent B). The flow rate was 0.3 mL/min. The MS was operated at a positive electrospray mode with the following settings: capillary temperature, 325 °C; spray voltage, 5 kV; sheath gas flow, 60 (arbitrary units); auxiliary gas, 20 (arbitrary units).
Results
Chemical Synthesis of 16,17-Dehydroprogesterone Substrate (Compound 1) and Product Standards
The synthetic approaches for substrate pregna-4,16-diene-3,20-dione (16,17-dehydroprogesterone, compound 1) and for products 16α,17-epoxypregn-4-ene-3,20-dione (16α,17-epoxyprogesterone, compound 2) and pregna-4,16-diene-21-ol-3,20-dione (21-hydroxy-16,17-dehydroprogesterone, compound 3) are outlined in Scheme 2. The synthesis of compound 1 started with commercially available pregna-5,16-dien-3β-acetoxy-20-one (compound 4). The 16-position was found to be electrophilic in acidic (and basic) conditions with a Lewis-base solvent such as methanol. Therefore, the 3β-acetate was cleanly deprotected by stirring in a 1:1 mixture of tetrahydrofuran:water in the presence of concentrated HCl for 7 days to yield compound 5 (16,17-dehydropregnenolone), which was also tested as a substrate. The subsequent oxidation of the 3β-alcohol with Dess–Martin periodinane, followed by incomplete isomerization during purification on silica gel, yielded compound 1. Isomerization with HCl in methanol/dicholoromethane increases yields of 3-keto-Δ4,5-steroids (see Experimental Procedures), but yields were not optimized. This compound would be tested as a substrate for CYP17A1 and CYP21A2, which might epoxidize the 16,17-olefin.
The synthesis of the epoxide product, compound 2, was achieved by epoxidizing compound 4 with basic hydrogen peroxide, which simultaneously epoxidized the 16,17-olefin and cleaved the 3β-acetate to afford compound 6.21 The resulting 3-hydroxy group was oxidized with Dess–Martin periodinane, and the 5,6-alkene was isomerized during the purification process to yield compound 2.
The synthesis of pregna-4,16-diene-21-ol-3,20-dione (21-hydroxy-16,17-dehydroprogesterone, compound 3) was more challenging, because the last step of these syntheses usually involves the oxidation of the 3β-alcohol derived from the pregnenolone backbone. The presence of a 21-hydroxyl group creates the problem of selectively deprotecting and oxidizing the 3β-hydroxyl group in the presence of a 21-hydroxy group. Thus, the synthesis of compound 3 was accomplished by employing an orthogonal protecting group strategy. The 3β-hydroxyl was protected as the formate, iodinating the enol acetate, and acetolysis of the iodide with silver acetate in acetonitrile to afford acetate 8. The 3β-formate, which is more reactive than the 21-acetate, was selectively methanolyzed to alcohol 9 with careful monitoring by TLC. The 3β-hydroxy moiety was oxidized with Dess–Martin periodinane to afford compound 10, and under acidic conditions in methanol, the 21-acetate was cleaved to yield compound 3.
Metabolism of 16,17-Dehydroprogesterone (1) by Steroid Hydroxylases
CYP17A1-Catalyzed Activities with Compound 1
The incubation of compound 1 with yeast microsomes containing wild-type CYP17A1 and POR led to the formation of two products in a nearly 1:1 ratio (Figure 1). We predicted one of the products to be 16α,17-epoxyprogesterone (compound 2), and its identification was confirmed by coelution with authentic standard at 21 min and identical MS/MS fragmentation patterns (Figure 2 and Figures S1–S3 of the Supporting Information). The mass spectrum of epoxide 2 (Figure S1) contains a molecular ion [M + H]+ at m/z = 329.2, with a single dominant fragment ion at m/z = 269.1, derived from loss of CH3COOH. Furthermore, the second, earlier-eluting product at 15.1 min coeluted with the 21-hydroxylated product (compound 3), a type of metabolic switching introduced by blocking hydroxylation chemistry at C16 and C17. The molecular ion [M + H]+ of compound 3 also gave m/z = 329.2, but the major fragments of m/z = 311.1 and 293.1 readily distinguished this product from compound 2 (Figure S1). Although human CYP17A1 21-hydroxylates progesterone, this 21-hydroxylase activity accounts for <5% of products,2 far less than the large fraction of 21-hydroxylated product observed using compound 1 as substrate.
Figure 1.

HPLC chromatograms of products derived from incubations of compound 1 (16,17-dehydroprogesterone) with wild-type CYP17A1, CYP17A1 mutation A105L, wild-type CYP21A2, or CYP21A2 mutation V359A and POR in yeast microsomes. Products were identified by retention times of standards chromatographed before and after samples: 1, substrate 16,17-dehydroprogesterone; 2, epoxide product 16α,17-epoxyprogesterone; 3, 21-hydroxy product 21-hydroxy-16,17-dehydroprogesterone. Ordinate scales are 0.05–0.10 AU full scale. The integrated UV-detector responses with equal amounts of compounds 2 and 3 were within 30% (not shown); therefore, peak integrals were not corrected to determine product ratios.
Figure 2.

Molecular ion [M + H]+ (A) and collision-induced dissociation (CID) fragmentation pattern (B) of enzymatically generated epoxide (compound 2), with mass per unit charge (m/z) shown as a function of ion intensity.
To probe the regiochemical determinants of 16α,17-epoxidase activity, we used CYP17A1 mutation A105L, which has reduced 16α-hydroxylase activity4 but enhanced 21-hydroxylase activity with progesterone substrate.2 Incubation of compound 1 with yeast microsomes containing CYP17A1-A105L and POR afforded the same products as wild-type CYP17A1, but the ratio of products corresponding to compounds 2 and 3 had changed from 1:1 to 1:5 (Figure 1). Because mutation A105L has poor progesterone 16α-hydroxylase activity, this result suggests that epoxidation activity at the C16–C17 olefin derives primarily from the capacity of the enzyme to perform 16α-hydroxylation. In addition, this result confirmed that CYP17A1 has intrinsic 21-hydroxylase activity, which is normally suppressed by the presence of CYP17A1 residue A105 and the more reactive substrate C–H bonds at C16 and C17.
CYP21A2-Catalyzed Activities with Compound 1
We have demonstrated that CYP21A2 is a progesterone 16α-hydroxylase, which is markedly increased with mutations that reduce the bulk of V359.5 We therefore explored whether CYP21A2 and mutation V359A also catalyze the conversion of a Δ16-steroid olefin to its epoxide metabolite. Incubation of compound 1 with yeast microsomes containing CYP21A2 and POR yielded primarily the 21-hydroxylated product 3, as well as a trace of epoxide 2 for both wild-type enzyme and mutation V359A (Figure 1 and Figures S4–S5 of the Supporting Information). The total yield of products for CYP21A2 was lower than for CYP17A1, and no other metabolites were formed. Thus, all four enzymes in this study 21-hydroxylated compound 1, whereas the fraction of epoxide product correlated with the progesterone 16α-hydroxylase activity and was best for enzymes with progesterone 17-hydroxylase activity as well.
Exclusion of Nonenzymatic Reactions
Uncoupling of NADPH oxidation from product formation during cytochrome P450 catalysis can release hydrogen peroxide, superoxide, and other reactive oxygen species into solution, which might then react with substrates to form products via nonenzymatic reactions. To confirm that 2 and 3 are enzyme-catalyzed products and not artifacts from reactions with released reactive oxygen species, catalase and superoxide dismutase were added to the incubations. The inclusion of catalase and superoxide dismutase had no effect on product formation (Table 1). In addition, the inhibitors ketoconazole and abiraterone blocked product formation via CYP17A1 (Figure S6 of the Supporting Information). These data confirm that product formation was enzyme-catalyzed.
Table 1. Activity of Recombinant Yeast Microsomal P450s in Catalyzing Epoxide Formation from Compound 1a.
| activity, % product conversion |
|||
|---|---|---|---|
| system | SOD/catalase | epoxide (2) | 21-hydroxy (3) |
| CYP17A1-WT | – | 29.6 ± 1.8 | 29.6 ± 0.8 |
| + | 29.5 ± 1.5 | 29.5 ± 1.5 | |
| CYP17A1-A105L | – | 14.0 ± 0.5 | 70.4 ± 1.3 |
| + | 14.3 ± 0.6 | 73.7 ± 1.3 | |
| CYP21A2-WT | – | 0.1 ± 0.0 | 3.6 ± 0.2 |
| + | 0.1 ± 0.0 | 4.3 ± 0.1 | |
| CYP21A2-V359A | – | 0.2 ± 0.0 | 3.4 ± 0.3 |
| + | 0.3 ± 0.1 | 2.9 ± 0.2 | |
All data are expressed as percent conversion of compound 1 to epoxide 2 or 21-hydroxy product 3 by recombinant yeast microsomes containing specified P450 and POR (60–200 μg of protein, 10–30 pmol of P450) with or without SOD and catalase after 40 min. Data represent the mean ± standard deviation for three experiments.
Reconstituted Assays with Purified Proteins
To confirm that these products were not artifacts of the yeast microsome system, particularly for CYP21A2 with low activity, incubations were repeated with purified proteins in reconstituted assay systems. Descriptions of the expression and purification of human CYP21A2 using E. coli have been limited, yet the X-ray crystal structure of bovine CYP21A2 has been reported.22 The distribution of products from compound 1 with CYP17A1, CYP17A1-A105L, CYP21A2, and CYP21A2-V359A were not significantly different in reconstituted assays (Table 2) than using yeast microsomes. The higher activity afforded with the reconstituted assay system confirmed the small but reproducible 16α,17-epoxidase activity of CYP21A2 with compound 1, the higher epoxidase activity with mutation V359A, and the correlation of progesterone 16α-hydroxylase activity with epoxidation chemistry for compound 1 (Table 2). Preincubation with the 3-keto-Δ4,5-analogue of abiraterone, which binds with high affinity to and inhibits both CYP17A123 and CYP21A2 (Figure S7A of the Supporting Information), also abrogated CYP21A2-catalyzed metabolism of compound 1 (Figure S7B).
Table 2. Epoxidation and Hydroxylation of Steroids by Purified Human P450s and PORa.
| compound 1 |
progesterone |
||||
|---|---|---|---|---|---|
| P450 | epoxide (2) | 21-hydroxy (3) | 16α-hydroxy | 17-hydroxy | 21-hydroxy |
| CYP17A1-WT | 41.0 ± 1.0 | 41.5 ± 1.4 | 21.7 ± 0.1 | 66.9 ± 1.4 | 3.0 ± 0.2 |
| CYP17A1-A105L | 16.2 ± 0.7 | 81.6 ± 1.2 | 6.6 ± 0.1 | 85.6 ± 0.5 | 6.5 ± 0.1 |
| CYP21A2-WT | 0.07 ± 0.02 | 41.5 ± 2.5 | 0.27 ± 0.04 | ND | 54.9 ± 1.9 |
| CYP21A2-V359A | 0.25 ± 0.01 | 43.5 ± 0.7 | 8.46 ± 0.64 | ND | 11.8 ± 0.6 |
All data are expressed as percent conversion of substrate (20 μM) to products by reconstituted system containing purified P450 (20–30 pmol), POR (80–120 pmol), and CYMS lipid (20 μg of protein) after 30 min. Data represent the mean ± standard deviation for three experiments; ND, not detectable.
Metabolism of 16,17-Dehydropregnenolone (5) by CYP17A1
While CYP17A1 hydroxylates the Δ4,5-steroid progesterone at three sites, the enzyme converts the Δ5,6-steroid pregnenolone exclusively to the 17-hydroxylated product, and CYP21A2 does not use pregnenolone as a substrate. To investigate whether CYP17A1 retains activity with a Δ5,6-pregnenolone analogue when 17-hydroxylation is prevented, compound 5 was incubated with purified CYP17A1 and POR reconstituted with CYMS, followed by enzymatic conversion to their 3-keto-Δ4-homologues with cholesterol oxidase. These experiments yielded both the 16α,17-epoxide and the 21-hydroxylated product in a 1:4 ratio (Table 3). CYP17A1 mutation A105L also metabolized compound 5 to the same two products, and the ratio decreased to 1:12. The product distribution reflects results with compound 1, for which CYP17A1 mutation A105L also gave a greater proportion of 21-hydroxylated product. These results also demonstrate that CYP17A1 is capable of epoxidizing and 21-hydroxylating substrates with a double bond at the C16–C17 position, even if the enzyme lacks 16α- and 21-hydroxylase activities toward the homologous, endogenous substrate lacking the extra double bond.
Table 3. Epoxidation and Hydroxylation of Compound 5 by CYP17A1-WT and A105La.
| compound 5 |
pregnenolone |
|||
|---|---|---|---|---|
| P450 | 16,17-epoxy | 21-hydroxy | androstenedione | 17-hydroxy |
| CYP17A1-WT | 11.0 ± 0.8 | 37.5 ± 2.7 | 2.3 ± 0.2 | 97.4 ± 0.3 |
| CYP17A1-A105L | 4.4 ± 0.3 | 50.8 ± 1.0 | 4.7 ± 0.4 | 94.9 ± 0.3 |
All data are expressed as percent conversion of substrate (20 μM) to epoxidation or hydroxylation product by reconstituted system containing purified P450 (20 pmol), POR (80 pmol), and CYMS lipid (20 μg of protein) after 30 min. Data represent the mean ± standard deviation for three experiments. Androstenedione is derived from dehydroepiandrosterone, the subsequent 17,20-lyase metabolite of 17-hydroxypregnenolone, following cholesterol oxidase treatment.
Substrate Binding and Product Inhibition Properties
Substrate-Binding Spectroscopy
Titration of CYP17A1 with compound 1 elicited a typical type I substrate-binding spectrum similar to progesterone (Figure 3A). The presence of 16,17-double bond yielded a lower spectral change than for progesterone, with ΔAmax about half that of progesterone (ε = 46 ± 2 mM–1 cm–1 for compound 1; ε = 94 ± 3 mM–1 cm–1 for progesterone), although with a higher affinity (Ks = 540 nM for compound 1; Ks = 740 nM for progesterone). With CYP21A2, spectral titrations gave the opposite pattern. Progesterone bound with high affinity (Ks = 60 nM) but gave a weak spectral change (ε = 38 ± 1 mM–1 cm–1), as was previously reported.24 With compound 1, the magnitude of the spectral change was about twice as great (ε = 77 ± 3 mM–1 cm–1), yet the affinity was poorer (Ks = 100 nM, Figure 3B). Compound 1 is thus similar to progesterone in its affinity for CYP17A1 and CYP21A2 but somewhat different in its capacity to displace the axial water molecule from the resting enzymes and to convert the heme irons to the high-spin state.
Figure 3.
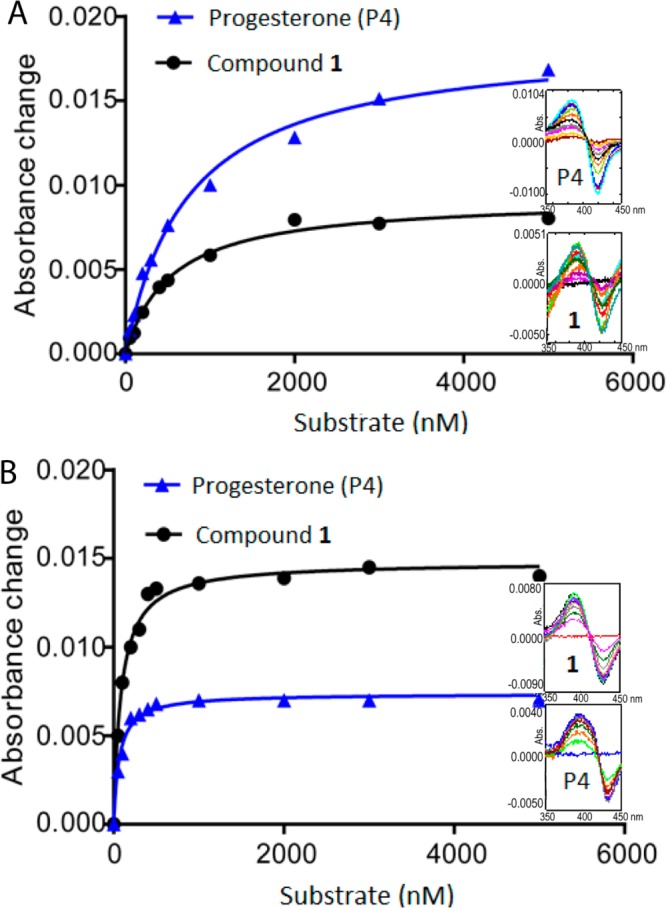
Spectrophotometric equilibrium binding titrations of (A) CYP17A1 or (B) CYP21A2 with progesterone and compound 1. Difference spectra were obtained upon titration of 200 nM purified P450 in 0.1 M potassium phosphate buffer, pH 7.4, containing 20% glycerol (v/v) with substrates at 50–5000 nM. Plots of Δ(A392 – A420) for compound 1 and Δ(A386 – A420) for progesterone versus concentration of substrate were fit to Michaelis–Menten equation using GraphPad Prism (see Experimental Procedures). Insets show individual spectra for progesterone (P4) and compound 1 at concentrations below plateau phase. The resulting Ks values were 540 nM and 60 nM for compound 1 and 740 nM and 100 nM for progesterone with CYP17A1 and CYP21A2, respectively.
Product Inhibition with Compound 2
Given the electrophilic properties of epoxides, an enzyme-generated epoxide in the active site might rapidly react with an enzymic nucleophile and afford time-dependent enzyme inactivation during turnover. To test for covalent binding and loss of enzyme activity, substrate 1 and epoxide 2 or progesterone control were preincubated with CYP17A1 before dilution and activity assay with pregnenolone substrate. After up to 40 min of preincubation, similar to the conditions of experiments using compound 1 as substrate, only 7–8% activity loss was observed for preincubations with all three steroids (Figure S8 of the Supporting Information). These results show negligible inactivation of CYP17A1 by epoxide 2 under the experimental conditions.
Discussion
The introduction of a double bond in our substrate analogue at the normal site of steroid hydroxylations revealed a novel 16α,17-epoxidase activity for CYP17A1 and CYP21A2 (Figure 1). Control experiments in the presence of catalase and superoxide dismutase to scavenge reactive oxygen species or with abiraterone, ketoconazole, and the Δ4,5-analogue of abiraterone to inhibit enzyme activity confirm that these enzymes catalyze the epoxidation reactions.
By employing CYP17A1 mutations A105L and CYP21A2 mutation V359A, we gleaned how regiochemical patterns based on progesterone substrate correlate with product distribution. The 16α,17-epoxidase activity toward compound 1 correlated with progesterone 16α-hydroxylase activity and was greatly enhanced when the enzyme also catalyzed progesterone 17-hydroxylation. Consequently, the 16α,17-epoxidase activity is greatest for wild-type CYP17A1 and lowest for wild-type CYP21A2 (Tables 1 and 2). Nevertheless, 16α-hydroxylase activity toward the C16–C17 saturated substrate was not an essential requirement for epoxidase activity, as CYP17A1 also epoxidized the Δ16,17-homologue of pregnenolone (16,17-dehydropregnenolone, compound 5, Table 3).
For other P450-catalyzed epoxidation reactions, the nature of the intermediate(s) in the catalytic cycle responsible for epoxidation has been debated. Both the iron-hydroperoxide and the iron-oxene have been proposed as the epoxidizing species. Vaz and Coon studied the epoxidation activity of CYP2E1 and mutation T303A, where the conserved proton-delivery threonine has been replaced by an alanine residue.25 On the basis of the preserved epoxidation activity of mutation T303A, they proposed that the hydroperoxy complex of the P450 mediates the epoxidation of unactivated olefinic bonds. Dawson and Sligar have also demonstrated that the T252A variant of P450cam, a mutant unable to hydroxylate camphor, was able to epoxidize olefins when treated with a second oxidant.26 These data have been interpreted as evidence of the Fe3+-OOH intermediate, which is proposed to be active in both electrophilic and nucleophilic reactions; nevertheless, alternate explanations for these observations are plausible.27,28 In fact, density functional theory (DFT) calculations by Shaik and co-workers favored the electrophilic iron-oxene species (“compound I”) in the epoxidation mechanism of styrene.29 Because the substrate used in this study involves an electrophilic carbon at the β-position of the enone involving C-16 to C-20 of compound 1, our data are consistent with the Fe3+-OOH as the intermediate,25,30 which might start the reaction with a nucleophilic attack at C-16. We do not have any data, however, to exclude concerted or multistep epoxidation via the iron-oxene species favored as the canonical hydroxylating species.
The alternative site of reactivity for CYP17A1 in the vicinity of the double bond between C-16 and C-17 in compound 1 is C-21. All enzymes studied readily 21-hydroxylated compound 1, even those with low progesterone 21-hydroxylase activity (Tables 1 and 2). It is likely that the reduced 16α,17-epoxidase activity of CYP17A1 mutation A105L also reflects the enhanced progesterone 21-hydroxylase activity of this enzyme, leading to greater metabolic switching to C-21. These data are consistent with a model in which substrate-binding trajectories for CYP17A1 render the hydrogen atoms at C-16, C-17, and C-21 all accessible for abstraction, but the regiochemical activities toward any given substrate reflect the reactivity of these positions, as well as the residence times within a critical distance from the heme oxygen. We previously suggested that the regiochemistry of CYP17A1 primarily reflects the stability of the carbon-based radicals generated after hydrogen atom abstraction in this part of the molecule: C-17 > C-16 > C-21, as carbon radical stability follows 3° > 2° > 1°.5 With this model, reactivity at C-21 is not sterically precluded but rather suppressed by the greater reactivity at C-16 and C-17. Metabolic switching to C-21 emerged as the primary reaction pathway when the substrate was engineered to prevent 16α- and 17-hydroxylation, and all enzymes also epoxidized the double bond at C-16. Our data do not allow us to determine the relative contributions of changes in substrate trajectories and of differences in the activation barrier for the epoxidation pathway versus the predominant 21-hydroxylation pathway. The similar but more disparate product distributions with compound 5 favor the conclusion that substrate modifications are primarily uncovering occult reactivities rather than markedly altering substrate-binding trajectories. Energy minimization with Chem3D Software demonstrates that the hydrogen atoms of C-21 in compound 1 are moved slightly (0.16 Å) closer to C-17 compared to progesterone (Figure 4), but this change is small compared to the marked increase in 21-hydroxylation.
Figure 4.
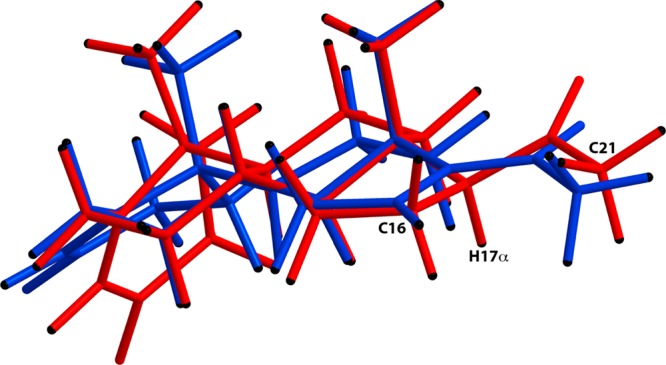
Chem3D Software was used to minimize the energy of the 16-dehydroprogesterone (compound 1) and progesterone (MM2) structures. Progesterone is shown in red, and compound 1 is shown in blue. The “Fast Overlay” method was used to overlay the two structures. After the “Fast Overlay” technique, the optimal distance measurement for the C13, C14, and C18 carbon atoms on both progesterone and 16-dehydroprogesterone structures was set to 0 Å, and the structures were manually minimized under the overlay option. This manual overlay based on atoms C13, C14, and C18 indicates that the introduction of the double bond between C16 and C17 positions the C21-carbon atom slightly closer to the original 17-position of progesterone. The distances between the progesterone 17-hydrogen atom and the C21-carbon atom of progesterone and of compound 1 are 3.032 and 2.872 Å, respectively. The distances between the progesterone 17-hydrogen atom and the C16-carbon atom of progesterone and of compound 1 are 2.185 and 2.327 Å, respectively.
Although compound 1 is not a typical steroid in conventional pathways, rat intestinal anaerobic bacteria possess 16-dehydratase activity, which catalyzes the formation of compound 1 from the CYP17A1 product 16α-hydroxyprogesterone.31 Compound 1 has been found in human feces as well.32 The traces of epoxides formed from enteral steroid metabolites are unlikely to result in physiologic consequences; however many xenobiotic compounds and steroidal drugs contain carbon–carbon double or triple bonds and might be substrates for CYP17A1 or CYP21A2. Mifepristone (RU486), for example, contains a 17α-propynyl group. In addition to being a progesterone and cortisol antagonist, mifepristone has been shown to inhibit monkey33 and rat34 17-hydroxylase activities. The CYP17A1 and CYP21A2 activities described herein might generate reactive epoxides and/or oxirenes from compounds such as mifepristone, which could then alkylate the enzymes and cause activity loss or react with other intracellular molecules, leading to adrenal toxicities.
Many steroidogenic enzymes of the mammalian adrenal cortex have two or more major activities, which are required to yield specific biologically active hormones. The intrinsic catalytic plasticity and liberal substrate specificity of these enzymes are not only essential properties for their physiologic functions but also vulnerabilities, enabling molecular diseases to arise from even conservative mutations in these enzymes. In the current study, we employed synthetic organic chemistry, site-directed mutagenesis, and enzymology studies to reveal the occult epoxidase chemistry of these steroid hydroxylases. Moreover, in a different context, CYP17A1, which normally 17-hydroxylates pregnenolone and both 17- and 16α-position of progesterone, was shown to have significant 21-hydroxylase activity presented with olefinated substrate analogues, in which the normal sites of reactivity are blocked. The immediate information gained from this report is that the steroidogenic enzymes can oxidize analogues of the natural substrates in a different fashion from traditionally known activities. Furthermore, using techniques such as linear free energy relationship (LFER) with broader substrate sets, we can further enhance our understanding of the active sites of these important enzymes.
Acknowledgments
We gratefully acknowledge Dr. Jiayan Liu for assistance with yeast microsome preparations and Dr. Dario Mizrachi for the site-directed mutagenesis of CYP17A1. F.K.Y. acknowledges the financial support of an Interface of Chemistry and Biology Fellowship from the University of Texas Southwestern Medical Center.
Glossary
Abbreviations:
- CID
collision-induced dissociation
- CYMS
control yeast microsomes
- CYP17A1
cytochrome P450c17
- CYP21A2
cytochrome P450c21
- DFT
density functional theory
- DMP
Dess–Martin periodinane
- HPLC
high-performance liquid chromatography
- MS
mass spectrometry
- NADPH
nicotinamide adenine dinucleotide phosphate (reduced form)
- Ni-NTA
nickel-nitrilotriacetic acid
- NIS
N-iodosuccinimide
- NMR
nuclear magnetic resonance spectroscopy
- POR
cytochrome P450 oxidoreductase
- SOD
superoxide dismutase
- TLC
thin-layer chromatography
- UV
ultraviolet light
Supporting Information Available
Tandem mass spectrometry spectra, negative control experiments, inhibition experiments, and NMR spectra of new compounds are provided in Figures S1–S10. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This work was supported by Grant R01-GM086596 from the National Institutes of Health and Grant I-1493 from the Robert A. Welch Foundation.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Miller W. L.; Auchus R. J. (2011) The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 32, 81–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto F. K.; Zhou Y.; Peng H. M.; Stidd D.; Yoshimoto J. A.; Sharma K. K.; Matthew S.; Auchus R. J. (2012) Minor activities and transition state properties of the human steroid hydroxylases cytochromes P450c17 and P450c21, from reactions observed with deuterium-labeled substrates. Biochemistry 51, 7064–7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swart P.; Swart A. C.; Waterman M. R.; Estabrook R. W.; Mason J. I. (1993) Progesterone 16α-hydroxylase activity is catalyzed by human cytochrome P450 17α-hydroxylase. J. Clin. Endocrinol. Metab. 77, 98–102. [DOI] [PubMed] [Google Scholar]
- Swart A. C.; Storbeck K. H.; Swart P. (2010) A single amino acid residue, Ala 105, confers 16α-hydroxylase activity to human cytochrome P450 17α-hydroxylase/17,20 lyase. J. Steroid Biochem. Mol. Biol. 119, 112–120. [DOI] [PubMed] [Google Scholar]
- Mizrachi D.; Wang Z.; Sharma K. K.; Gupta M. K.; Xu K.; Dwyer C. R.; Auchus R. J. (2011) Why human cytochrome P450c21 is a progesterone 21-hydroxylase. Biochemistry 50, 3968–3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coon M. J.; Ding X. X.; Pernecky S. J.; Vaz A. D. (1992) Cytochrome P450: progress and predictions. FASEB J. 6, 669–673. [DOI] [PubMed] [Google Scholar]
- Guengerich F. P. (2001) Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 14, 611–650. [DOI] [PubMed] [Google Scholar]
- Nakajin S.; Shinoda M.; Haniu M.; Shively J. E.; Hall P. F. (1984) C21 steroid side-chain cleavage enzyme from porcine adrenal microsomes. Purification and characterization of the 17α-hydroxylase/C17,20 lyase cytochrome P450. J. Biol. Chem. 259, 3971–3976. [PubMed] [Google Scholar]
- Shyadehi A. Z.; Lamb D. C.; Kelly S. L.; Kelly D. E.; Schunck W. H.; Wright J. N.; Corina D.; Akhtar M. (1996) The mechanism of the acyl-carbon bond cleavage reaction catalyzed by recombinant sterol 14α-demethylase of Candida albicans (other names are: lanosterol 14α-demethylase, P-45014DM, and CYP51). J. Biol. Chem. 271, 12445–12450. [DOI] [PubMed] [Google Scholar]
- Zhu D.; Seo M. J.; Ikeda H.; Cane D. E. (2011) Genome mining in Streptomyces. Discovery of an unprecedented P450-catalyzed oxidative rearrangement that is the final step in the biosynthesis of pentalenolactone. J. Am. Chem. Soc. 133, 2128–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q.; Lamb D. C.; Kelly S. L.; Lei L.; Guengerich F. P. (2010) Cyclization of a cellular dipentaenone by Streptomyces coelicolor cytochrome P450 154A1 without oxidation/reduction. J. Am. Chem. Soc. 132, 15173–15175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho P. S.; Brustad E. M.; Kannan A.; Arnold F. H. (2013) Olefin cyclopropanation via carbene transfer catalyzed by engineered cytochrome P450 enzymes. Science 339, 307–310. [DOI] [PubMed] [Google Scholar]
- Farwell C. C.; McIntosh J. A.; Hyster T. K.; Wang Z. J.; Arnold F. H. (2014) Enantioselective imidation of sulfides via enzyme-catalyzed intermolecular nitrogen-atom transfer. J. Am. Chem. Soc. 136, 8766–8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auchus R. J.; Lee T. C.; Miller W. L. (1998) Cytochrome b5 augments the 17,20 lyase activity of human P450c17 without direct electron transfer. J. Biol. Chem. 273, 3158–3165. [DOI] [PubMed] [Google Scholar]
- Meadus W. J.; Mason J. I.; Squires E. J. (1993) Cytochrome P450c17 from porcine and bovine adrenal catalyses the formation of 5,16-androstadien-3β-ol from pregnenolone in the presence of cytochrome b5. J. Steroid Biochem. Mol. Biol. 46, 565–572. [DOI] [PubMed] [Google Scholar]
- Yoshimoto F. K.; Desilets M. C.; Auchus R. J. (2012) Synthesis of halogenated pregnanes, mechanistic probes of steroid hydroxylases CYP17A1 and CYP21A2. J. Steroid Biochem. Mol. Biol. 128, 38–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. H.; Tee M. K.; Miller W. L. (2010) Human cytochrome P450c17: single step purification and phosphorylation of serine 258 by protein kinase A. Endocrinology 151, 1677–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandee D.; Miller W. L. (2011) High-yield expression of a catalytically active membrane-bound protein: human P450 oxidoreductase. Endocrinology 152, 2904–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arase M.; Waterman M. R.; Kagawa N. (2006) Purification and characterization of bovine steroid 21-hydroxylase (P450c21) efficiently expressed in Escherichia coli. Biochem. Biophys. Res. Commun. 344, 400–405. [DOI] [PubMed] [Google Scholar]
- Sherbet D. P.; Tiosano D.; Kwist K. M.; Hochberg Z.; Auchus R. J. (2003) CYP17 mutation E305G causes isolated 17,20-lyase deficiency by selectively altering substrate binding. J. Biol. Chem. 278, 48563–48569. [DOI] [PubMed] [Google Scholar]
- Pandey A. K.; Tiwari V.; Srivastava S.; Sethi A. (2006) Synthesis of some novel pregnane derivatives and its glycoside as possible anticancer agents. Ind. J. Heterocycl. Chem. 15, 353–358. [Google Scholar]
- Zhao B.; Lei L.; Kagawa N.; Sundaramoorthy M.; Banerjee S.; Nagy L. D.; Guengerich F. P.; Waterman M. R. (2012) A three-dimensional structure of steroid 21-hydroxylase (Cytochrome P450 21A2) with two substrates reveals locations of disease-associated variants. J. Biol. Chem. 287, 10613–10622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido M.; Peng H. M.; Yoshimoto F. K.; Upadhyay S. K.; Bratoeff E.; Auchus R. J. (2014) A-Ring modified steroidal azoles retain similar potent and slowly reversible CYP17A1 inhibition as abiraterone. J. Steroid Biochem. Mol. Biol. 143, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auchus R. J.; Kumar A. S.; Boswell C. A.; Gupta M. K.; Bruce K.; Rath N. P.; Covey D. F. (2003) The enantiomer of progesterone (ent-progesterone) is a competitive inhibitor of human cytochromes P450c17 and P450c21. Arch. Biochem. Biophys. 409, 134–144. [DOI] [PubMed] [Google Scholar]
- Vaz A. D.; McGinnity D. F.; Coon M. J. (1998) Epoxidation of olefins by cytochrome P450: evidence from site-specific mutagenesis for hydroperoxo-iron as an electrophilic oxidant. Proc. Natl. Acad. Sci. U. S. A. 95, 3555–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S.; Makris T. M.; Bryson T. A.; Sligar S. G.; Dawson J. H. (2003) Epoxidation of olefins by hydroperoxo-ferric cytochrome P450. J. Am. Chem. Soc. 125, 3406–3407. [DOI] [PubMed] [Google Scholar]
- de Visser S. P.; Ogliaro F.; Sharma P. K.; Shaik S. (2002) What factors affect the regioselectivity of oxidation by cytochrome P450? A DFT study of allylic hydroxylation and double bond epoxidation in a model reaction. J. Am. Chem. Soc. 124, 11809–11826. [DOI] [PubMed] [Google Scholar]
- Ogliaro F.; de Visser S. P.; Cohen S.; Sharma P. K.; Shaik S. (2002) Searching for the second oxidant in the catalytic cycle of cytochrome P450: a theoretical investigation of the iron(III)-hydroperoxo species and its epoxidation pathways. J. Am. Chem. Soc. 124, 2806–2817. [DOI] [PubMed] [Google Scholar]
- Kumar D.; de Visser S. P.; Shaik S. (2005) Multistate reactivity in styrene epoxidation by compound I of cytochrome P450: mechanisms of products and side products formation. Chemistry 11, 2825–2835. [DOI] [PubMed] [Google Scholar]
- Coon M. J.; Vaz A. D.; McGinnity D. F.; Peng H. M. (1998) Multiple activated oxygen species in P450 catalysis: Contributions to specificity in drug metabolism. Drug Metab. Dispos. 26, 1190–1193. [PubMed] [Google Scholar]
- Winter J.; O’Rourke S.; Bokkenheuser V. D.; Hylemon P. B.; Glass T. L. (1982) 16α-Dehydration of corticoids by bacteria isolated from rat fecal flora. J. Steroid Biochem. 16, 231–237. [DOI] [PubMed] [Google Scholar]
- Zheng X.; Xie G.; Zhao A.; Zhao L.; Yao C.; Chiu N. H. L.; Zhou Z.; Bao Y.; Jia W.; Nicholson J. K.; Jia W. (2011) The footprints of gut microbial-mammalian co-metabolism. J. Proteome Res. 10, 5512–5522. [DOI] [PubMed] [Google Scholar]
- Albertson B. D.; Hill R. B.; Sprague K. A.; Wood K. E.; Nieman L. K.; Loriaux D. L. (1994) Effect of the antiglucocorticoid RU486 on adrenal steroidogenic enzyme activity and steroidogenesis. Eur. J. Endocrinol. 130, 195–200. [DOI] [PubMed] [Google Scholar]
- Sanchez P. E.; Ryan M. A.; Kridelka F.; Gielen I.; Ren S. G.; Albertson B.; Malozowski S.; Nieman L.; Cassorla F. (1989) RU-486 inhibits rat gonadal steroidogenesis. Horm. Metab. Res. 21, 369–371. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.