

Abstract
A hallmark of penicillin-binding protein 2 (PBP2) from penicillin-resistant strains of Neisseria gonorrhoeae is insertion of an aspartate after position 345. The insertion resides on a loop near the active site and is immediately adjacent to an existing aspartate (Asp346) that forms a functionally important hydrogen bond with Ser363 of the SxN conserved motif. Insertion of other amino acids, including Glu and Asn, can also lower the rate of acylation by penicillin, but these insertions abolish transpeptidase function. Although the kinetic consequences of the Asp insertion are well-established, how it impacts the structure of PBP2 is unknown. Here, we report the 2.2 Å resolution crystal structure of a truncated construct of PBP2 containing all five mutations present in PBP2 from the penicillin-resistant strain 6140, including the Asp insertion. Commensurate with the strict specificity for the Asp insertion over similar amino acids, the insertion does not cause disordering of the structure, but rather induces localized flexibility in the β2c−β2d loop. The crystal structure resolves the ambiguity of whether the insertion is Asp345a or Asp346a (due to the adjacent Asp) because the hydrogen bond between Asp346 and Ser362 is preserved and the insertion is therefore Asp346a. The side chain of Asp346a projects directly toward the β-lactam-binding site near Asn364 of the SxN motif. The Asp insertion may lower the rate of acylation by sterically impeding binding of the antibiotic or by hindering breakage of the β-lactam ring during acylation because of the negative charge of its side chain.
Neisseria gonorrhoeae is the causative agent of the sexually transmitted infection gonorrhea. Penicillin was the primary treatment for gonorrhea for more than 40 years, but in 1987 was withdrawn by the Centers for Disease Control and Prevention (CDC) as a recommended treatment because of the increasing prevalence of N. gonorrhoeae strains exhibiting resistance. Extended-spectrum cephalosporins and fluoroquinolones then became the mainstay for treatment, but again, because of increasing resistance, fluoroquinolones were withdrawn in 2007; this was followed by cefixime in 2012.1 The current recommendation from the CDC for treatment of gonorrhea is dual therapy with ceftriaxone and either azithromycin or doxycycline. However, strains of N. gonorrhoeae have been identified with high-level resistance to azithromycin,2 and together with the recent isolation of strains with high-level resistance to ceftriaxone,3−5 this portends that strains exhibiting resistance to essentially all antibiotics will soon emerge.
The lethal targets for penicillin and other β-lactams are the penicillin-binding proteins (PBPs), which function as transpeptidases (TPases), carboxypeptidases, or endopeptidases during the latter stages of cell-wall synthesis.6−8 As structural analogues of the acyl-d-Ala-d-Ala peptide substrate for PBPs, β-lactams bind to the active site of PBPs and acylate a serine nucleophile, forming a long-lived covalent intermediate that renders the active site unavailable to bind peptide substrate. There are four PBPs in the N. gonorrhoeae genome. PBP1 and PBP2 are high-molecular mass (HMM) PBPs that are essential for growth; PBP1 is a bifunctional glycosyl transferase and TPase important for peptidoglycan biosynthesis during cell growth, whereas PBP2 is a monofunctional TPase involved in cell division.9 In contrast, PBP3 and PBP4 are nonessential low-molecular mass PBPs that catalyze carboxypeptidase and endopeptidase activity in vitro.10,11 While PBP4 does not appear to be expressed, when assessed by binding of a fluorescent β-lactam, a strain with a double deletion of PBP3 and PBP4 exhibits a much stronger morphological phenotype and grows slower than either of the strains with individual deletions.10
In the stepwise transformation of N. gonorrhoeae from a penicillin-susceptible strain to a strain exhibiting high-level resistance, acquisition of a mutated allele of PBP2 is the first and prerequisite step.12,13 These variants of PBP2 contain mutations that lower the second-order rate of acylation by penicillin without any apparent impairment of the essential TPase function of the PBP. Examination of the sequence of penA, the gene encoding PBP2, from a number of penicillin-resistant strains of N. gonorrhoeae reveals that there are generally five to eight amino acid changes in PBP2 compared to wild-type penA from the penicillin-susceptible strains, FA19 and LM306.14−17 These changes include insertion of an aspartate codon after position 345 (termed Asp345a) and a variable number of substitutions toward the C-terminal end of the protein. The Asp insertion is a consistent feature of penA sequences obtained from penicillin-resistant strains15 and is the only amino acid selected for in random insertional mutagenesis experiments at position 345a.16 The crystal structure of PBP2 is known,17 and the insertion is positioned on the β2a−β2d hairpin loop that is in the proximity of the active site. This loop is connected to the conserved SxN active-site motif via a hydrogen bond between Asp346 and Ser363 (the x of the SxN motif).17 The curiosity of this insertion is that because it is adjacent to an existing aspartate (Asp346), the actual location of the insertion is ambiguous; it could reside before or after Asp346, i.e., Asp345a or Asp346a.18 Interestingly, there is a strict requirement for an Asp-Asp pair to achieve penicillin resistance. Insertion of nearly all other amino acids at either position, including the similarly charged glutamate or similarly shaped asparagine, lowers the rate of acylation by penicillin but cannot transform N. gonorrhoeae to higher resistance, suggesting that these insertions disrupt the essential TPase function of PBP2.16,18 These data also indicate that the Asp insertion, whether at position 345a or 346a, contributes to penicillin resistance in a very specific way.
PBP2 derived from the penicillin-resistant gonococcal strain FA614019 contains the Asp insertion along with four C-terminal substitutions, which together lower the rate of acylation of PBP2 by 15-fold.17 In a previous study, we assessed the relative contributions of the four C-terminal substitutions versus the Asp insertion and found that they each lowered the rate of acylation of PBP2 with penicillin G by 6-fold. Surprisingly, a crystal structure of PBP2 containing the four C-terminal substitutions (PBP2-6140CT) showed very little difference when compared with that of wild-type PBP2 from FA19, suggesting a subtle mechanism of penicillin resistance.17 To date, however, full-length constructs of PBP2 containing the Asp insertion have failed to crystallize, thus restricting our understanding of how this insertion contributes to penicillin resistance.
Here we report the crystal structure of the TPase/β-lactam-binding domain of PBP2 harboring the Asp insertion and the four C-terminal mutations, determined at 2.2 Å resolution. The structure shows that functionally the Asp insertion comes after Asp346, as the first Asp of the pair retains its interaction with Ser363, while the second Asp points directly toward the β-lactam-binding site where it interferes with binding of the antibiotic or the chemistry of acylation.
Experimental Procedures
Cloning, Expression, and Purification of a Truncated PBP2 Construct
Truncated constructs comprising only the TPase/β-lactam-binding domain of PBP2 (residues 237–581) were generated by cloning nucleotides 709–1746 of the wild-type penA gene from FA19 or nucleotides 709–1749 of the penA gene from the N. gonorrhoeae penicillin-resistant strain 6140 into pMALC2KV, a derivative of pMAL-C2 (New England Biolabs, Beverly, MA). This construct fuses PBP2 to maltose-binding protein (MBP) containing a hexahistidine tag at its N-terminus and an intervening tobacco etch virus (TEV) protease site between the two proteins. A segment of the protein that protrudes from the TPase domain, comprising residues 283–297, was also removed, and fusion of Pro282 and Arg298 was made with an intervening glycine (with the goal of introducing a β-turn). The final purified constructs encoded proteins of 331 (termed PBP2-t3-wt) and 332 amino acids (termed PBP2-t3-6140).
The expression constructs were transformed into Escherichia coli GW6011 cells; 2 L of cell culture was grown at 37 °C, and protein expression was induced by addition of 0.3 mM isopropyl β-d-thiogalactoside, followed by overnight incubation at 20 °C. Cells were harvested by centrifugation, lysed in 20 mM Tris-HCl (pH 8.0), 500 mM NaCl, and 10% glycerol (TNG), and the MBP–PBP2 fusion protein was purified on a 5 mL HisTrap FF column (GE Healthcare, Piscataway, NJ). The fusion protein was pooled, concentrated to 2–3 mL by ultrafiltration, mixed with His6-tagged TEV protease at a molar ratio of 10:1, and dialyzed overnight at 4 °C against TNG to allow cleavage of the fusion protein and to remove imidazole. The resulting digest was then passed over a 5 mL HisTrap HP column equilibrated with TNG. Purified PBP2-t3-wt and PBP2-t3-6140 did not elute in the flow-through but instead were eluted by a TNG/15 to 250 mM imidazole linear gradient. Uncleaved fusion protein, His6-TEV, and His6-maltose-binding protein remained bound to the column and were eluted with a TNG/250 mM imidazole wash step. The purified proteins were pooled, dialyzed into 20 mM Tris (pH 8.0), 500 mM NaCl, and 10% glycerol, and concentrated by ultrafiltration.
Acylation Rates of PBP2-t3 Constructs
The reaction of β-lactam antibiotics with PBPs is described by eq 1:
| 1 |
where E·S is the noncovalent enzyme–antibiotic complex, E–S′ is the acyl–enzyme complex, and P is the hydrolyzed antibiotic.20 The second-order rate constant (k2/Ks) is a direct measure of the reactivity of an antibiotic for a PBP. k2/Ks constants for the reaction of PBP2 variants with the fluorescent penicillin Bocillin-FL (Life Technologies Inc., Grand Island, NY) were determined by measuring time-dependent decreases in the intrinsic fluorescence of PBP2. Under subsaturating conditions, when [S] < Km, the kinetics are described by eq 2:
| 2 |
where [S] is the β-lactam concentration, ka is the pseudo-first-order rate constant of formation of the E–S complex at a given [S] value, v is the initial rate of formation of the E–S complex, and Et is the total enzyme concentration. k2/Ks is the slope of a plot of ka versus [S].20 The deacylation rate (k3) is very slow compared to the acylation rate and can be ignored for these experiments.
Acylation of PBP2 by Bocillin-FL quenches the intrinsic fluorescence of the protein, thus providing a real-time measurement of formation of the acyl–enzyme complex. The fluorescence quenching curves of PBP2 variants were recorded at 25 °C on a Quanta Master fluorimeter (QM4, Photon Technology International, Inc., Birmingham, NJ) with fluorescence excitation at 295 nm and emission at 331 nm (slit widths of 2 and 4 nm, respectively). PBP2 was diluted into 2 mL of binding buffer [50 mM sodium phosphate (pH 8)] to a final concentration of 1 μM and then mixed with Bocillin-FL. The time-dependent decrease in intrinsic fluorescence was recorded immediately after the addition of Bocillin-FL in the subsaturating concentration range of 5–10 μM. At least two independent experiments were performed with a new protein sample each time. Data were corrected for the buffer and ligand background signals. The quench curves of intrinsic fluorescence were then used to calculate pseudo-first-order rate constants of acylation (ka) at each Bocillin-FL concentration. Using eq 2, k2/Ks values were derived from plots of ka versus [S], where the slope of the line defines the second-order acylation rate constant. The reported value for k2/Ks is the average of at least two such plots. All curve-fitting analyses and calculations were performed using GraphPad Prism version 4.00 for Windows (GraphPad Software, Inc., San Diego, CA).
Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis (SDS–PAGE)-Based Binding Assay
k2/Ks constants for the reaction of truncated constructs of wild-type PBP2 and PBP2-6140 with meropenem were determined by a SDS–PAGE-based competition assay using Bocillin-FL. Each PBP2 variant (1 μM) was incubated with 10 μM Bocillin-FL in the presence of increasing concentrations (0.05–100 μM) of meropenem to a final volume of 50 μL at 25 °C for 30 min. The reaction was stopped by adding 10 μL of 5× SDS–PAGE sample buffer, and the mixture was boiled at 98 °C for 2 min. Acylated PBP2 was then separated from free β-lactam using 10% Mini-Protean TGX SDS–PAGE gels (Bio-Rad Laboratories, Inc.). Gels were scanned using a Kodak EDAS 290 UV imaging system (Scientific Imaging Systems, Eastman Kodak, New Haven, CT), followed by staining with Coomassie R-250 to confirm equivalent loading. Bocillin-FL-bound PBP2 was quantified by densitometry using ImageJ version 1.48 (National Institutes of Health, Bethesda, MD). Data points were normalized to the maximum of the fluorescence intensity (defining the complete saturation of PBP2 by Bocillin-FL). Data from three independent experiments (each in duplicate) were used to determine IC50 values of meropenem for each PBP2 variant, from which second-order acylation rate constants were derived using eq 3:20
| 3 |
where (k2/Ks)U is the rate constant of an unlabeled compound, (k2/Ks)L is the rate constant of Bocillin-FL, and [SL] is the concentration of Bocillin-FL used in the competition experiment.
All curve-fitting analyses and calculations were performed using GraphPad Prism version 4.00 for Windows (GraphPad Software, Inc.).
Proteolytic Susceptibility of PBP2-6140 Mutants
The susceptibility of purified full-length (i.e., residues 44–581) PBP2 and PBP2-6140 mutants to proteolysis was determined by conducting a time course of trypsin digestion, separating the resulting fragments on SDS–10% polyacrylamide gels, and staining with Coomassie R-250, as described previously.18
Crystallization and Data Collection
Crystallization trials of PBP2-t3-6140 were performed using a Gryphon liquid-handling robot (Art-Robbins, Sunnyvale, CA) in 96-well sitting-drop plates (INTELLI-PLATE, Art-Robbins). The protein was screened at a concentration of 9.2 mg/mL using the vapor-diffusion method against a number of commercially available sparse matrix screens, including JCSG+ Suite and JCSG Cores I–IV Suite (Qiagen, Germantown, MD). For each well, 200 nL of protein was mixed with 200 nL of well solution. Conditions under which crystals were obtained were later optimized in 24-well Limbro plates (Hampton Research, Aliso Viejo, CA) using 2 μL hanging drop volumes.
Structure Determination
Crystals were cryoprotected in mother liquor, flash-frozen in liquid nitrogen, and diffraction data were collected at SER-CAT beamline ID22 at the Advanced Photon Source at the Argonne National Laboratory (Argonne, IL) in 1° oscillations with an exposure time of 3 s per frame and a crystal–detector distance of 200 mm. Data were processed with HKL2000.21 The structure was determined by molecular replacement using the CCP4 program PHASER22 with the TPase domain of wild-type PBP2 as the search model [molecule A of Protein Data Bank (PDB) entry 3EQU].17 The structure was determined with two molecules in the asymmetric unit and then refined by alternating rounds of REFMAC refinement23 and manual building using O.24 Residues 542–545 of molecule A and residues 504–511 and 543–545 of molecule B could be not be modeled because of weak or absent electron density. Alternative conformations were modeled for the side chain of Ser551 in molecule A. A non-native alanine at the N-terminus resulting from the construct was modeled as Ala236 in both molecules of the asymmetric unit.
The stereochemistry of the final model was assessed by PROCHECK.25 The φ and ψ angles for Leu447 in both molecules of the asymmetric unit occupy the disallowed region of the Ramachandran plot. Both of these residues, however, exhibit excellent electron density. These residues are present on the α9−α10 loop; its conformation is influenced by the absence of the N-terminal domain of PBP2 in the truncated construct (see Results and Discussion). Asp346a and Thr347 of molecule B both occupy the generously allowed region of the Ramachandran plot, but these findings can be explained by the weak electron density at the Asp insertion site.
Results and Discussion
A Truncated Construct of PBP2 Exhibits Wild-Type Rates of Acylation by β-Lactams
Despite extensive screening, full-length constructs of PBP2 containing the Asp345a insertion have failed to crystallize to date; therefore, we designed, expressed, and purified a truncated construct of PBP2 to provide a new vehicle for crystallization (PBP2-t3-wt). This construct contains only the TPase/β-lactam-binding domain of the enzyme (amino acids 237–581) minus a segment of 15 residues between Pro282 and Arg298. This segment was excluded because, in the absence of the N-terminal domain, these residues would form a distinct protrusion from the TPase domain that might impede crystallization (see below). A similar construct was also made using penA from the penicillin-resistant strain 6140 (PBP2-t3-6140).
Before we proceeded with crystallization trials, it was important to determine whether the absence of the N-terminal domain impacted the acylation rate of the truncated PBP2 constructs. The second-order rates of acylation (k2/Ks) of (full-length) wild-type PBP2 and PBP2-t3-wt for the fluorescent penicillin, Bocillin-FL, were nearly identical, and the corresponding k2/Ks acylation rates for PBP2-6140 (full-length PBP2 containing five mutations) and PBP2-t3-6140 are also very similar (Table 1 and Figure 1). For both full-length and truncated constructs, the fold difference between the wild type (14-fold) and 6140 (9-fold) was also similar. These data indicate that removal of the N-terminal domain does not significantly impact the β-lactam-binding activity of the TPase domain and that mutations associated with penicillin resistance have the same impact on the acylation kinetics.
Table 1. k2/Ks Constants for Full-Length and Truncated Constructs of Wild-Type PBP2 and PBP2-6140.
| PBP2-wt | PBP2-t3-wt | PBP2-6140 | PBP2-t3-6140 | |
|---|---|---|---|---|
| k2/Ks | 78380 ± 2820 (n = 3)a | 80010 ± 4515 (n = 2)a | 5596 ± 140 (n = 4)a | 8785 ± 184 (n = 3)a |
n is the number of measurements of k2/Ks.
Figure 1.

Second-order rates of acylation of Bocillin-FL against full-length and truncated constructs of wild-type PBP2 and PBP2 from the penicillin-resistant strain 6140. For each experiment, the pseudo-first-order rates of acylation (ka) were plotted vs the concentration of Bocillin-FL. Shown is the average plot of at least two or more independent experiments, where the slope of the line yields the second-order k2/Ks value.
Because the only acylated structure of PBP2 determined to date is in complex with meropenem (see below), we also measured k2/Ks acylation constants of PBP2-t3-wt and PBP2-t3-6140 for meropenem (see Experimental Procedures and Figure 1 of the Supporting Information). These were 313400 ± 63500 M–1 s–1 for PBP2-t3-wt and 68100 ± 14500 M–1 s–1 for PBP2-t3-6140, corresponding to a nearly 5-fold decrease in acylation rate. This shows that mutations contributing to penicillin resistance can also discriminate against a carbapenem.
Structure Determination
Crystals of PBP2-t3-6140 were obtained at 18 °C over wells containing 21% PEG 6000 buffered with 0.1 M HEPES (pH 7.8). The space group of the crystals is P21, and there are two molecules in the asymmetric unit. The crystal structure was determined by molecular replacement using the TPase domain of wild-type PBP2 as a search model17 and refined to 2.2 Å resolution with a crystallographic R factor of 21.3% with excellent stereochemistry (Table 2).
Table 2. X-ray Diffraction Data and Model Refinement Statistics for the Crystal Structure of PBP2-t3-6140a.
| Data Collection | |
| space group | P21 |
| cell dimensions [a, b, c (Å); β (deg)] | 44.5, 77.4, 88.0; 92.5 |
| resolution range (Å) | 50.0–2.20 (2.24–2.20) |
| Rmerge (%) | 7.7 (28.3) |
| completeness (%) | 96.9 (78.0) |
| redundancy | 5.3 (4.1) |
| ⟨I⟩/⟨σI⟩ | 31.5 (4.2) |
| no. of unique reflections | 29597 (1192) |
| Refinement | |
| resolution (Å) | 35.4–2.2 |
| no. of non-hydrogen protein atoms | 4825 |
| no. of water molecules | 92 |
| Rcryst/Rfree (%) | 0.195/0.243 |
| rmsd from ideal stereochemistry | |
| bond lengths (Å) | 0.010 |
| bond angles (deg) | 1.45 |
| B factor | |
| mean B factor (main chain) (Å2) | 40.8 |
| rmsd in main chain B factors (Å2) | 1.38 |
| mean B factor (side chains and waters) (Å2) | 45.3 |
| rmsd in side chain B factors (Å2) | 1.96 |
| Ramachandran plot (%) | |
| residues in most favored regions | 93.5 |
| residues in disallowed regions | 5.7 |
| residues in generously allowed regions | 0.4 |
| residues in disallowed regions | 0.4 |
| PDB entry | 4U3T |
Numbers in parentheses are for the outer resolution shells of data.
Comparison between Molecules A and B of the Asymmetric Unit
Overall, the two molecules in the asymmetric unit adopt similar structures and superimpose with a rmsd of 0.86 Å for all common main chain atoms (Figure 2). There are, however, some structural differences of note. In the crystal structures of both wild-type PBP2 and PBP2-6140CT (which contains the four C-terminal substitutions but lacks the Asp345a insertion), 10–11 residues of the β3−β4 loop could not be modeled because of flexibility.17 In molecule A of the crystal structure of PBP2-t3-6140, all residues of the loop are visible in the electron density, thus providing the first structural view of these residues (Figure 3). In contrast, residues 504–511 in molecule B exhibit weak or absent electron density and could not be modeled. This region is significant because two mutations associated with penicillin resistance (P504L and A510V) reside in this loop.17 The differences in this region between the two molecules of the asymmetric unit suggest the degree of flexibility in the β3−β4 loop is influenced directly by crystal packing interactions. Hence, the structural impact on this loop of mutations associated with antibiotic resistance must be assessed with caution. Other regions that differ in the two molecules of the asymmetric unit are the β2a−β2d hairpin, the loop between α9 and α10, the N-terminal end of α8, and the β5−α11 loop (Figure 2). As features on the surface of the molecule, all these differences could result from the different crystal packing environment around each molecule.
Figure 2.
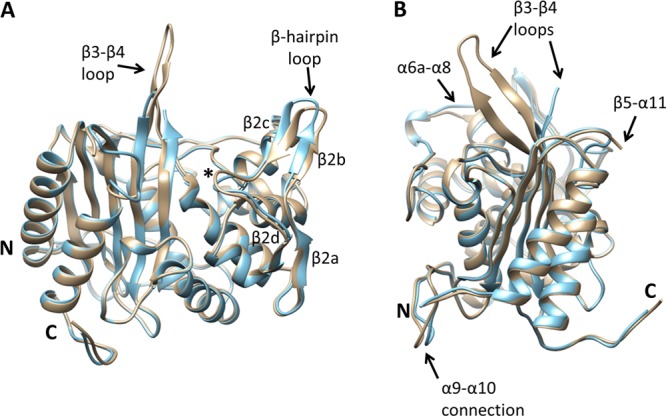
Superimposition of both molecules of the asymmetric unit of the crystal structure of PBP2-t3-6140. Each molecule is shown in cartoon representation in which molecule A is colored brown and molecule B blue. Regions that differ structurally between the two molecules are labeled. The position of the active-site nucleophile, Ser310, is indicated with an asterisk. The view in panel A is rotated approximately 90° with respect to panel B about the vertical axis.
Figure 3.
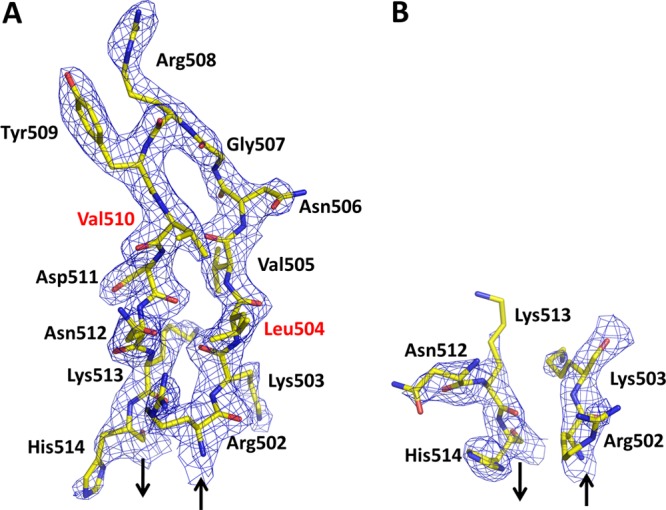
Electron density of the β3−β4 loop in each molecule of the asymmetric unit. The 2|Fo| – |Fc| electron density is contoured at 1σ. Sites of mutation compared with PBP2 from penicillin-susceptible strains of N. gonorrhoeae are labeled in red. Arrows indicate the preceding and following polypeptide chains: (A) molecule A and (B) molecule B.
Comparison with the Structure of Wild-Type PBP2
As shown in Figure 2 of the Supporting Information, the respective TPase domains of wild-type PBP2 (within the full-length structure) and PBP2-t3-6140 do not differ significantly in structure. The rmsds of 0.75 Å (molecule A to molecule A) and 0.77 Å (molecule B to molecule B) between all common main chain atoms in the TPase domains indicate a very close superimposition for both molecules of the asymmetric unit. Hence, the absence of the N-terminal domain in PBP2-t3-6140 does not have any significant impact on the structure of the TPase domain of PBP2 and is congruent with the nearly identical rates of acylation observed for the t3 constructs compared to the full-length proteins (see above). This result also reinforces the idea that the β domains of PBPs (whether N-terminal or C-terminal) serve mainly as pedestals either to project the catalytic domain toward its peptidoglycan substrates or to target the protein to appropriate sites of cell-wall synthesis, rather than having any direct effect on enzyme function.17,26
Aside from the insertion site and the aforementioned β3−β4 loop, the other differences are minimal (Figure 4). The shift in residues 388–403, which is a β-hairpin loop that connects α9 with α10, can be explained by the absence of the N-terminal domain because, in the structure of full-length PBP2, this loop packs directly against the N-terminal domain and those contacts are lost in PBP2-t3-6140. In fact, this is the only difference between the two structures that can be attributed directly to the absence of the N-terminal domain. As expected, there is also a difference at the Pro282-Arg298 “join” in PBP2-t3-6140. The removal of residues 283–297 (with addition of a Gly linker), however, was a very successful strategy because there is only a marginal difference in the structure immediately preceding and following the join. The join itself exhibits excellent density, even though the Gly linker is non-native (Figure 3 of the Supporting Information).
Figure 4.
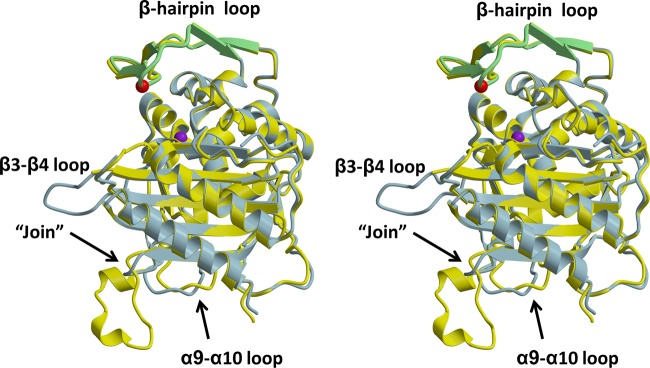
Superimposition of the structures of PBP2-t3-6140 and the TPase/β-lactam-binding domain from wild-type PBP2. In this stereoview, each molecule is displayed in cartoon form, with PBP2-t3-6140 colored blue and wild-type PBP2 colored yellow. Regions exhibiting structural differences are indicated. The β-hairpin region of PBP2-t3-6140 is colored green. The location of the Asp insertion is indicated by a red sphere, corresponding to the Cα position, and that of Ser310 by a purple sphere.
Structural Impact of the Asp Insertion
The primary goal of determining the structure of PBP2-t3-6140 was to determine the structural effects of the Asp insertion, which by itself causes a 6-fold decrease in the rate of acylation by penicillin G and, when combined with the four C-terminal substitutions, a 15-fold decrease.17 The first observation from the structure is that the electron density in the immediate vicinity of the insertion is relatively weak (Figure 5). This is more pronounced in molecule A than in molecule B and occurs C-terminal to the existing aspartate (Asp346). It leads to slight uncertainty in the position of several side chain residues, including the inserted Asp, Thr347, and His348. Nevertheless, it is clear that the insertion does not cause disordering of the β-hairpin loop region but rather introduces only localized flexibility at the insertion site.
Figure 5.
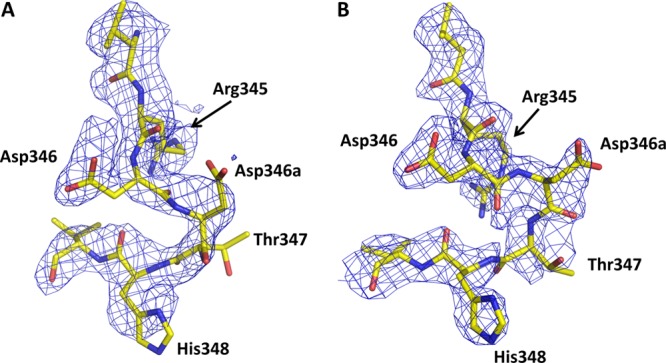
Electron density at the site of the Asp insertion in both molecules of the asymmetric unit of PBP-t3-6140. In both cases, unbiased |Fo| – |Fc| electron density is shown, contoured at 1σ: (A) molecule A and (B) molecule B.
A second observation is that the insertion is best described functionally as an Asp346a insertion. As shown by superimposition with the structure of wild-type PBP2 (Figure 6), the structural shift occurs after Asp346, which is essentially unchanged in position and makes the same hydrogen bonding interaction with Ser363 of the SxN motif that is present in wild-type PBP2. Finally, the β2a−β2d hairpin loop has shifted relatively little, with the biggest difference being a longer loop between β2b and β2c as a result of the additional residue. Overall, these structural observations agree closely with our previous mutagenesis data that show the effect of the Asp insertion to be highly specific.18
Figure 6.
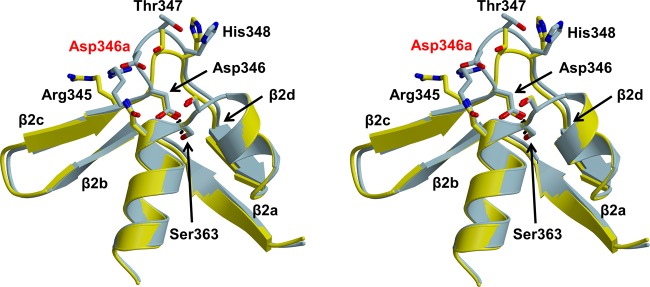
Stereoview showing the superimposition of wild-type PBP2 (yellow) and PBP2-t3-6140 (blue). Molecule A of each structure was superimposed. The hydrogen bond between Asp346 and Ser363 is shown as a dashed line, and the inserted Asp is labeled in red.
How Does Asp346a Lower the Rate of Acylation by Penicillin?
Having analyzed the structure, we must now consider how the insertion contributes to penicillin resistance, i.e., how Asp346a lowers the rate of acylation by penicillin. We envision two potential mechanisms that, on the basis of the crystal structure and biochemical data, could account for the effects of the Asp insertion.
By lengthening the β2c−β2d loop, the primary effect of the insertion is to project the new aspartate side chain directly toward the active site, potentially placing it within hydrogen bonding distance of Asn364. The latter is part of the SxN active-site motif that is found in all serine-based PBPs and β-lactamases. The mechanistic role of Asn364 in both transpeptidation and β-lactam binding is not entirely clear, but it is required for activity27 and participates in the rich hydrogen bonding environment within the active site. This network involves both Ser310, which is the nucleophile that forms the acyl–enzyme bond with peptide substrates and β-lactam antibiotics, and Lys313, which is believed to activate the serine nucleophile by functioning in PBPs as a general base.26,28−30 Although disruption of the hydrogen bonding network by the inserted Asp might be responsible for the decreased rates of acylation with β-lactams, the close overlap of active-site residues in both the wild-type and 6140 structures of PBP2 shows the hydrogen bonding is essentially the same in the presence of the insertion (Figure 7). The apparent flexibility around the Asp insertion site and with it the absence of a stable hydrogen bond with Asn364 also make this mechanism uncertain.
Figure 7.
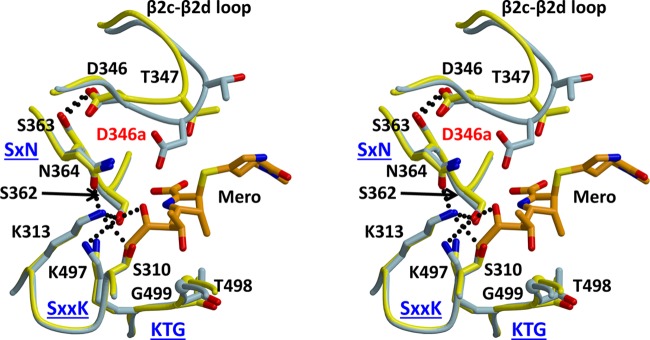
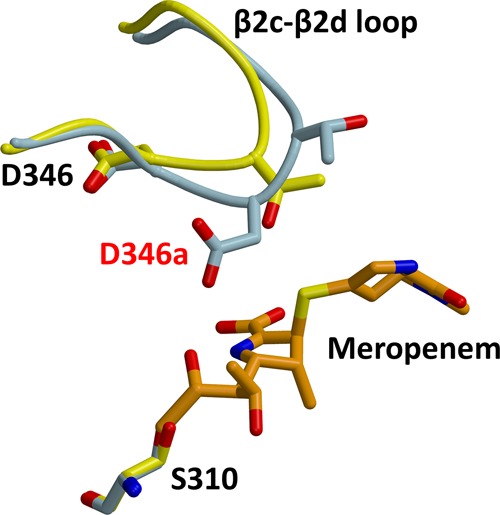
Stereoview of a superimposition of the structures of PBP2-t3-6140 and wild-type PBP2 in which a meropenem molecule was docked via superimposition with a crystal structure of PBP2 in complex with meropenem. Wild-type PBP2 is colored yellow, PBP2-t3-6140 blue, and meropenem orange. The hydrogen bond between Asp346 and Ser363 is shown as a dashed line, and the inserted Asp is labeled in red. The three conserved active-site motifs of PBPs are labeled in blue.
Another possibility is that the Asp insertion directly hinders the binding or acylation reaction with β-lactams. To examine this possibility, we docked meropenem into the structure of PBP2-t3-6140, using as a guide the crystal structure of the PBP2 acyl–enzyme complex with meropenem (A. Fedarovich, R. A. Nicholas, and C. Davies, unpublished observations) (Figure 7). To date, this is the only structure of an acylated complex of PBP2. The relevance of this model is supported by the 5-fold lowering of the acylation rate for meropenem against the 6140 variant of PBP2 compared to that of the wild type. It shows that although the inserted aspartate does not reach far enough to clash directly with meropenem, its side chain lies near the 6-hydroxyethyl group and the opened β-lactam ring. One possible consequence of the Asp346a mutation, therefore, is that its negative charge impedes binding of what is predominantly a hydrophobic region of the antibiotic, i.e., the thiazolidine ring and the ethyl of the hydroxyethyl group. The region equivalent to the hydroxyethyl in penicillin G is the benzyl R1 group, and thus, any unfavorable interaction between a negative charge and a hydrophobic region might be greater. In support of this, the difference in acylation rates between the wild type and 6140 variants of PBP2 is greater for penicillin (15-fold) than for meropenem (5-fold). Finally, it is equally possible that the proximity of a new negative charge in the active site interferes with the chemistry of acylation by altering the electrostatic environment of the active site.
Preservation of TPase Function
A key question to address is how TPase function is preserved in the presence of an insertion of Asp into the active site. Our previous studies have shown that there is a very fine line between gaining resistance via mutations that lower the rate of acylation by penicillin and maintaining essential TPase function in PBP2.18 It is clear that the integrity of the β2c−β2d loop and its interaction with the SxN motif via a hydrogen bond between Asp346 and Ser363 are vital for TPase function and that only subtle changes in this region can be tolerated. Our structure of PBP2-t3-6140 shows that the β-hairpin loop (β2a−β2d) is mostly unchanged in structure, with an only localized impact in the immediate vicinity of the Asp insertion, and that the hydrogen bond between Ser363 and Asp346 is preserved, even though the insertion is immediately after Asp346. The superimposition of wild-type PBP2 with PBP2-t3-6140 (Figure 7) shows that the amino acids comprising the three conserved motifs of the active site overlap remarkably closely. Thus, significant remodeling of the active site has not occurred as a result of the Asp insertion, and the key catalytic residues remain appropriately positioned for catalysis.
Mutations that contribute to resistance against β-lactams must not impair binding or acylation of the peptide substrate. There is a remarkable specificity for an aspartate insertion at position 346a because only this insertion can contribute to penicillin resistance without causing irreparable damage to TPase function.18 In a previous study, we showed that mutation to alanine of either residue mediating the Ser363–Asp346 hydrogen bond both ablated TPase activity and increased the proteolytic susceptibility of PBP2, revealing the importance of this connection for the integrity of the active site, whereas the proteolytic susceptibility of PBP2-6140 containing the Asp insertion was the same as that of wild-type PBP2. In a similar manner, we examined the proteolytic susceptibility of PBP2-6140 with insertions of Glu, Asn, and His at position 346a (Figure 4 of the Supporting Information). The proteolytic susceptibility of all three of these mutants is the same as that of wild-type PBP2, showing that, like Asp346a, these insertions do not decrease the stability of the active-site region. These data indicate that these mutations impair TPase function by a specific mechanism, with their side chains presumably interfering with either the binding or acylation of the peptide substrate. Elucidation of exactly how Asp346a preserves TPase activity when other insertions abrogate it, however, must await determination of the structure of PBP2 in complex with a cell-wall peptide or peptide mimetic.
Conclusion
We have determined the crystal structure of a truncated construct of N. gonorrhoeae PBP2 containing the Asp345a insertion implicated in penicillin resistance. The new aspartate is functionally an Asp346a insertion, as the position of Asp346 is essentially unchanged from that of the wild type. The inserted side chain points directly toward the active site, where its negative charge may impede binding of β-lactams via repulsion against hydrophobic regions of the antibiotic or more simply alters the acylation chemistry that results in opening of the β-lactam ring by perturbing the electrostatic balance around the serine nucleophile.
Acknowledgments
Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract W-31-109-ENG-38. Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html. The X-ray crystallography facility used for this work is supported by the Medical University of South Carolina’s Research Resource Facilities program. Figures were prepared using MOLSCRIPT31 and RASTER3D,32 Pymol (http://pymol.sourceforge.net), or Chimera.33
Glossary
Abbreviations
- PBP
penicillin-binding protein
- rmsd
root-mean-square deviation
- TEV
tobacco etch virus
- TPase
transpeptidase.
Supporting Information Available
Calculation of second-order rate constants for meropenem against truncated constructs of wild-type and 6140 PBP2 (Figure 1), superimposition of wild-type (full-length) PBP2 versus PBP2-t3-6140 (Figure 2), 2|Fo| – |Fc| electron density of the join between Pro282 and Arg298 with an intervening Gly linker (Figure 3), and proteolytic susceptibilities of PBP2 with different insertions at position 346a (Figure 4). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
A.F. and E.C. contributed equally to this work.
This work was supported by National Institutes of Health Grants GM66861 (to C.D.) and AI36901 (to R.A.N.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- (2012) Update to CDC’s Sexually Transmitted Diseases Treatment Guidelines, 2010: Oral Cephalosporins No Longer a Recommended Treatment for Gonococcal Infections. Morbidity and Mortality Weekly Report 61, 590–594. [PubMed] [Google Scholar]
- Allen V. G.; Seah C.; Martin I.; Melano R. G. (2014) Azithromycin resistance is co-evolving with reduced susceptibility to cephalosporins in Neisseria gonorrhoeae, Ontario, Canada. Antimicrob. Agents Chemother. 58, 2528–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi M.; Saika T.; Hoshina S.; Iwasaku K.; Nakayama S.; Watanabe H.; Kitawaki J. (2011) Ceftriaxone-resistant Neisseria gonorrhoeae, Japan. Emerging Infect. Dis. 17, 148–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi M.; Golparian D.; Shimuta K.; Saika T.; Hoshina S.; Iwasaku K.; Nakayama S.; Kitawaki J.; Unemo M. (2011) Is Neisseria gonorrhoeae initiating a future era of untreatable gonorrhea?: Detailed characterization of the first strain with high-level resistance to ceftriaxone. Antimicrob. Agents Chemother. 55, 3538–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unemo M.; Golparian D.; Nicholas R.; Ohnishi M.; Gallay A.; Sednaoui P. (2012) High-level cefixime- and ceftriaxone-resistant Neisseria gonorrhoeae in France: Novel penA mosaic allele in a successful international clone causes treatment failure. Antimicrob. Agents Chemother. 56, 1273–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheboeuf P.; Contreras-Martel C.; Job V.; Dideberg O.; Dessen A. (2006) Penicillin binding proteins: Key players in bacterial cell cycle and drug resistance processes. FEMS Microbiol. Rev. 30, 673–691. [DOI] [PubMed] [Google Scholar]
- Sauvage E.; Kerff F.; Terrak M.; Ayala J. A.; Charlier P. (2008) The penicillin-binding proteins: Structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32, 234–258. [DOI] [PubMed] [Google Scholar]
- Nicholas R. A., and Davies C. (2012) Structural Mechanisms of β-Lactam Antibiotic Resistance in Penicillin-Binding Proteins. In Antibiotic Discovery & Development (Dougherty T. J., and Pucci M. J., Eds.) pp 397–425, Springer-Verlag, Berlin. [Google Scholar]
- Barbour A. G. (1981) Properties of penicillin-binding proteins in Neisseria gonorrhoeae. Antimicrob. Agents Chemother. 19, 316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanova M. E.; Tomberg J.; Olesky M.; Holtje J. V.; Gutheil W. G.; Nicholas R. A. (2003) Neisseria gonorrhoeae penicillin-binding protein 3 exhibits exceptionally high carboxypeptidase and β-lactam binding activities. Biochemistry 42, 14614–14625. [DOI] [PubMed] [Google Scholar]
- Stefanova M. E.; Tomberg J.; Davies C.; Nicholas R. A.; Gutheil W. G. (2004) Overexpression and enzymatic characterization of Neisseria gonorrhoeae penicillin-binding protein 4. Eur. J. Biochem. 271, 23–32. [DOI] [PubMed] [Google Scholar]
- Cannon J. G.; Sparling P. F. (1984) The genetics of the gonococcus. Annu. Rev. Microbiol. 38, 111–133. [DOI] [PubMed] [Google Scholar]
- Shafer W. M., Folster J. P., and Nicholas R. A. (2010) Molecular mechanisms of antibiotic resistance expressed by the pathogenic Neisseriae. In Neisseria: Molecular Mechanisms of Pathogenesis (Genco C. A., and Wetzler L., Eds.) p 245, Caister Academic Press, Norfolk, U.K. [Google Scholar]
- Spratt B. G. (1988) Hybrid penicillin-binding proteins in penicillin-resistant strains of Neisseria gonorrhoeae. Nature 332, 173–176. [DOI] [PubMed] [Google Scholar]
- Dowson C. G.; Jephcott A. E.; Gough K. R.; Spratt B. G. (1989) Penicillin-binding protein 2 genes of non-β-lactamase-producing, penicillin-resistant strains of Neisseria gonorrhoeae. Mol. Microbiol. 3, 35–41. [DOI] [PubMed] [Google Scholar]
- Brannigan J. A.; Tirodimos I. A.; Zhang Q. Y.; Dowson C. G.; Spratt B. G. (1990) Insertion of an extra amino acid is the main cause of the low affinity of penicillin-binding protein 2 in penicillin-resistant strains of Neisseria gonorrhoeae. Mol. Microbiol. 4, 913–919. [DOI] [PubMed] [Google Scholar]
- Powell A. J.; Tomberg J.; Deacon A. M.; Nicholas R. A.; Davies C. (2009) Crystal structures of penicillin-binding protein 2 from penicillin-susceptible and -resistant strains of Neisseria gonorrhoeae reveal an unexpectedly subtle mechanism for antibiotic resistance. J. Biol. Chem. 284, 1202–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomberg J.; Temple B.; Fedarovich A.; Davies C.; Nicholas R. A. (2012) A highly conserved interaction involving the middle residue of the SXN active-site motif is crucial for function of class B penicillin-binding proteins: Mutational and computational analysis of PBP 2 from N. gonorrhoeae. Biochemistry 51, 2775–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faruki H.; Sparling P. F. (1986) Genetics of resistance in a non-β-lactamase-producing gonococcus with relatively high-level penicillin resistance. Antimicrob. Agents Chemother. 30, 856–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frere J. M., Nguyen-Disteche M., Coyette J., and Joris B. (1992) Mode of action: Interaction with the penicillin-binding proteins. In The Chemistry of β-Lactams (Page M. I., Ed.) pp 148–196, Chapman & Hall, Glasgow, U.K. [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Jones T. A.; Zou J.-Y.; Cowan S. W.; Kjeldgaard M. (1991) Improved methods for building protein structures in electron-density maps and the location of errors in these models. Acta Crystallogr. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Moss D. S.; Thornton J. M. (1993) PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291. [Google Scholar]
- Davies C.; White S. W.; Nicholas R. A. (2001) Crystal structure of a deacylation-defective mutant of penicillin-binding protein 5 at 2.3 Å resolution. J. Biol. Chem. 276, 616–623. [DOI] [PubMed] [Google Scholar]
- van der Linden M. P.; de Haan L.; Dideberg O.; Keck W. (1994) Site-directed mutagenesis of proposed active-site residues of penicillin-binding protein 5 from Escherichia coli. Biochem. J. 303, 357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanova M. E.; Davies C.; Nicholas R. A.; Gutheil W. G. (2002) pH, inhibitor, and substrate specificity studies on Escherichia coli penicillin-binding protein 5. Biochim. Biophys. Acta 1597, 292–300. [DOI] [PubMed] [Google Scholar]
- Nicholas R. A.; Krings S.; Tomberg J.; Nicola G.; Davies C. (2003) Crystal structure of wild-type penicillin-binding protein 5 from E. coli: Implications for deacylation of the acyl-enzyme complex. J. Biol. Chem. 278, 52826–52833. [DOI] [PubMed] [Google Scholar]
- Silvaggi N. R.; Anderson J. W.; Brinsmade S. R.; Pratt R. F.; Kelly J. A. (2003) The crystal structure of phosphonate-inhibited d-Ala-d-Ala peptidase reveals an analogue of a tetrahedral transition state. Biochemistry 42, 1199–1208. [DOI] [PubMed] [Google Scholar]
- Kraulis P. J. (1991) MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24, 946–950. [Google Scholar]
- Merritt E. A.; Murphy M. E. P. (1994) Raster3D version 2.0. A program for photorealistic molecular graphics. Acta Crystallogr. D50, 869–873. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. (2004) UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.