

Abstract
High-density lipoprotein (HDL) retards atherosclerosis by accepting cholesterol from the artery wall. However, the structure of the proposed acceptor, monomeric apolipoprotein A-I (apoA-I), the major protein of HDL, is poorly understood. Two published models for monomeric apoA-I used cross-linking distance constraints to derive best fit conformations. This approach has limitations. (i) Cross-linked peptides provide no information about secondary structure. (ii) A protein chain can be folded in multiple ways to create a best fit. (iii) Ad hoc folding of a secondary structure is unlikely to produce a stable orientation of hydrophobic and hydrophilic residues. To address these limitations, we used a different approach. We first noted that the dimeric apoA-I crystal structure, (Δ185–243)apoA-I, is topologically identical to a monomer in which helix 5 forms a helical hairpin, a monomer with a hydrophobic cleft running the length of the molecule. We then realized that a second crystal structure, (Δ1–43)apoA-I, contains a C-terminal structure that fits snuggly via aromatic and hydrophobic interactions into the hydrophobic cleft. Consequently, we combined these crystal structures into an initial model that was subjected to molecular dynamics simulations. We tested the initial and simulated models and the two previously published models in three ways: against two published data sets (domains predicted to be helical by H/D exchange and six spin-coupled residues) and against our own experimentally determined cross-linking distance constraints. We note that the best fit simulation model, superior by all tests to previously published models, has dynamic features of a molten globule with interesting implications for the functions of apoA-I.
Clinical and epidemiological studies show a robust, inverse association of high-density lipoprotein cholesterol (HDL-C) levels and the risk for coronary vascular disease (CVD).1,2 Moreover, hypercholesterolemic mice with genetically engineered deficiencies in proteins implicated in HDL metabolism have strongly atherosclerotic phenotypes,3,4 providing compelling evidence that HDL is a key modulator of atherosclerosis in animal models. These observations have triggered intense interest in targeting HDL for therapeutic intervention.
The relationship of HDL-C to CVD is complex, however, because lower HDL-C levels are not uniformly associated with an increased cardiovascular risk.5−7 Moreover, several recent clinical studies have failed to show any benefit from the pharmacological elevation of HDL-C levels in statin-treated humans with established CVD.8,9 These observations suggest that HDL-C levels provide limited information about HDL’s proposed cardioprotective effects. Therefore, the rational design of therapies and assessments of their benefits demand detailed knowledge of the assembly and molecular structure of the cardioprotective form of HDL.
The anti-atherogenic nature of HDL has been most firmly related to reverse cholesterol transport, in which apoA-I, HDL’s major protein, accepts cholesterol from artery wall cells, counteracting lipid loading.8 Thus, the level of atherosclerosis is markedly increased in hypercholesterolemic mice that lack apoA-I,11,12 while transgenic expression of apoA-I inhibits atherosclerosis in a wide variety of hypercholesterolemic animal models.13−16 ApoA-I is secreted into the bloodstream as a lipid-free or lipid-poor monomer (preβ HDL), which is widely believed to be the most common acceptor of sterol transported out of cells.17 The circulating form of mature human apoA-I contains 243 amino acids encoded by exon 3 (residues 1–43) and exon 4 (residues 44–243). The region encoded by exon 4 contains 10 tandem 11/22 residue repeats, termed helical repeats 1–10 (H1–H10, respectively), which form a series of lipid-binding class A amphipathic helices.
ApoA-I accepts sterol from ABCA1, a transmembrane ATPase, to become discoidal HDL. In this configuration, two antiparallel molecules of apoA-I form a belt around the edge of a disk of phospholipid and cholesterol.18−23 This dimerization of apoA-I is driven in part by the formation of salt bridges among the 10 helical repeats.20 The flexible nature of apoA-I plays a key role in the lipidation and assembly of HDL by ABCA1,24 but this property hampers high-resolution structural studies of the full-length protein under physiologically relevant conditions. Fortunately, molecular modeling provides an alternative approach to understanding HDL’s structure.
Two all-atom models for lipid-free full-length human apoA-I have been proposed by Silva et al.25 and Pollard et al.26 Both models are helix bundle structures constructed as best fit explanations of the cross-linked peptides that are identified when apoA-I is exposed to chemical cross-linkers and then analyzed by high-precision mass spectrometry. In both models, H5, the central helical repeat of apoA-I, forms a helical hairpin. The Silva model is essentially an antiparallel six-helix bundle structure, while the Pollard model is a more loosely packed helix bundle structure containing multiple short helical segments.
Two crystal structures have also been determined for lipid-free human apoA-I: the N-terminally truncated structure (Δ1–43) of Borhani et al.18 and the high-resolution C-terminally truncated (Δ185–243) crystal structure of Mei and Atkinson.27 The Borhani structure is 93% helical and contains an antiparallel four-helix bundle with an elliptical ring shape, a model more consistent with the structure of apoA-I bound to discoidal HDL particles.18 The structure of Mei and Atkinson is less helical and forms a more compact helix bundle, which is more consistent with the biophysical and biochemical properties of lipid-free apoA-I.27 There are, however, substantial similarities between the two structures, e.g., the pattern of salt bridges between helical repeats 4 and 6 and pairwise helical repeats 5.28
In their two models for monomeric apoA-I, Silva et al.25 and Pollard et al.26 determined distance constraints by measuring maximal potential distances between the α-carbons of cross-linked amino acids in two different peptides. While driven by data, this approach has several limitations. (i) Cross-linked peptides provide no information about secondary structure. (ii) A secondary structure can be folded in many ways to create a best fit. (iii) Ad hoc folding of a secondary structure may not produce a stable conformation in which the hydrophobic and hydrophilic residues are oriented correctly. In the studies presented here, we used a different approach. Starting from first principles based on known protein structures, we generated an initial model, which we then subjected to molecular dynamics (MD) simulations. We ranked the resulting models by how well they conformed to two published data sets: domains predicted to be helical by H/D exchange and six spin-coupled residues. We tested the best fit model by chemically cross-linking peptides. The proposed structure, a molten globule, has interesting implications for the function of apoA-I.
Materials and Methods
Initial Models
We created an initial crystal model by taking parts of the recent crystal structure for dimeric C-terminally truncated apoA-I by Mei and Atkinson27 [Protein Data Bank (PDB) entry 3R2P] and parts of the crystal structure for the tetrameric N-terminally truncated apoA-I of Borhani et al.18 (PDB entry 1AV1). We began by building a monomer that was as complete as possible from each crystal structure by selecting residues from different chains so that there would be a continuous backbone connection and the monomer was as compact as possible. The parts from dimeric C-terminally truncated apoA-I were residues 3–130 of chain A and residues 131–182 of chain B. This formed a C-terminally truncated monomer of residues 3–182. The parts from tetrameric N-terminally truncated apoA-I were residues 43–67 of chain A, residues 68–130 of chain D, residues 131–192 of chain C, and residues 193–243 of chain B. This formed an N-terminally truncated monomer of residues 43–243. The two terminally truncated monomers are shown in Figure S1 of the Supporting Information. An immediate observation is that the structures are quite similar, especially with respect to the main double helix consisting of residues 68–182.
To build the initial crystal model, we formed a complete monomer by joining residues 3–182 of our C-terminally truncated monomer to residues 183–243 from our N-terminally truncated monomer. To insert residues 193–243 into the hydrophobic cleft seen in residues 3–182 (Figure 1d), we repositioned residues 193–243 by bending them slightly and stretching them at several points along the backbone. This aligned them with the aromatic residues of the cleft, as explained in more detail in Results. The hairpin between residues 192 and 193 in the N-terminally truncated monomer was instrumental in joining the long helix of residues 131–182 of the C-terminally truncated monomer around the bottom of the hybrid crystal model to residues 193–243 of the N-terminally truncated monomer via the short helix of residues 183–192. Additionally, residues 1 and 2 were taken from a previous simulation of ours of monomeric apoA-I at 500 K. The two residues were in a random coil structure and were joined to residue 3.
Figure 1.

Creation of a lipid-free apoA-I monomer model from a combination of the C-terminally truncated (Δ185–243)apoA-I27 and N-terminally truncated (Δ1–43)apoA-I18 crystal structures. (a and b) Depth-cued ribbon images showing the creation of a monomer from the crystal structure of the (Δ185–243)apoA-I dimer of Mei and Atkinson:27 (a) crystal dimer27 and (b) monomeric (Δ185–243)apoA-I created by forming a hairpin from H5. Because residues 1 and 2 were missing from the crystal structure, they were added from a previous MD simulation. (c–i) Development of the initial crystal model. Color codes for ribbon models: H5, green; other helical domains, red; nonhelical domains, green; prolines in panels a–c, e–g, and i, space-filling yellow. Color codes for space-filling models: aromatic residues, magenta; nonaromatic hydrophobic residues, gold; prolines, yellow. (c) Reorientation of the ribbon model of the (Δ185–243)apoA-I monomer in panel b. (d) Space-filling model of the structure in panel c showing its deep hydrophobic cleft. (e) Ribbon model of residues 183–243 from the N-terminally truncated crystal structure.18 (f) Space-filling model of the structure in panel e. (g) Alignment of aromatic and hydrophobic residues between the (Δ185–243)apoA-I monomer model (1–182) and the N-terminally truncated fragment. (h) Initial crystal model (space-filling) for the full-length monomer. (i) Ribbon model of the structure in panel h. Residues 1–182 are colored cyan and residues 183–243 red.
Molecular Dynamics Simulations
All-atom MD was performed using NAMD29 after solvating and ionizing the initial structure as described previously.30,31 After minimization and heating to 500 K for 50 ps, the MD simulation consisted of a 30 ns equilibration at 500 K. Postprocessing of trajectories was analyzed by writing VMD-Tcl scripts.
ApoA-I Model Figures
All molecular model figures were created with the PyMOL Molecular Graphics System (version 1.3, Schrödinger).
Root-Mean-Square Deviation and Root-Mean-Square Fluctuation
The root-mean-square deviation (rmsd) of protein α-carbon atoms was calculated for the apoA-I monomer over the entire 30 ns duration of the simulation by using the RMSD Trajectory Tool extension in VMD. The root-mean-square fluctuation (rmsf) of protein α-carbon atoms was calculated for the apoA-I monomer over the entire 30 ns duration of the simulation by using the “measure rmsf” command in VMD.
Measurement of α Helicity
The fraction of all protein residues that were α-helical was measured every time increment over the course of the 30 ns trajectory, using VMD’s implementation of the protein secondary structure determination program, STRIDE.32
Average 10–20 and 15 ns Models
This average structure was created by averaging the protein atom coordinates over all frames of the simulation from 10 to 20 ns by using the “measure avpos” command in VMD after aligning over all atoms with respect to the 10 ns frame. Because averaging creates possible close contacts between atoms and deforms bond lengths, torsions, and dihedrals, the structure was then subjected to 10000 steps of steepest-descent energy minimization. The result is called the 10–20 ns model.
Because the protein structure at 15 ns is kinetically expanded by the 500 K temperature of the simulation, we performed a 0.1 ns linear MD simulation cooling of the 15 ns structure to 310 K. The result is called the 15 ns model.
Sum of Spin-Coupled Residue Distances of >20 Å and Number of Spin-Coupled Distances of >20 Å
To determine the sum of the spin-coupled residue distances of >20 Å and the number of spin-coupled distances of >20 Å, we measured distances between the protein α-carbons for the spin-coupled residues by writing VMD-Tcl scripts for the individual models as well as over the 30 ns trajectory. See Results for more details.
Relative Residue to Residue Proximities Shown as Protein Contact Maps
To obtain protein contact maps for the 15 ns model and over the 30 ns trajectory, we counted the number of times that α-carbon atoms were within 23 Å of each other for every time increment of that simulation. The results were then combined and are shown as a contour plot of one color (for the one frame) or four colors (for the trajectory) indicating the quartile of counts (except for a count of 0, which is white) relative to the maximal number over all pairs of residues, respectively, using the graphics software GNUPLOT (www.gnuplot.info).
Sum of the Violation Distance Algorithm
The violation distance algorithm attempts to measure, given a set of distance constraints such as experimental cross-links, the amount that a given structure violates those constraints. For each experimentally determined cross-link, xi,yi, where xi and yi are the residue numbers of the lysine pairs (and the N-terminus) of the ith cross-link, a violation will occur if the minimal distance between the Cα atoms of xi and yi, di(xi,yi), is greater than the 23 Å length of the cross-linking reagent MDA33 (a distance of <28 Å for the reagent used by Silva et al.25 and Pollard et al.26). The sum of violations, ∑[di(xi,yi) – r], where r is the length of the cross-linker, plotted against the number of individual violations, is the extent to which any given model is consistent with the cross-linking data. The best model will be closest to the x,y origin. Because we can also examine MD trajectories instead of given structures, any single Cα–Cα distance ≤ r for a given xi,yi in any MD trajectory frame indicates no violation distance. Finally, for each cross-link xi,yi pair, we count only the minimal violation distance to calculate the sum of violation distances.
Isolating HDL and ApoA-I
Plasma was prepared from EDTA-anticoagulated blood collected from healthy adult subjects who had fasted overnight. HDL (density of 1.125–1.210 g/mL) was isolated from plasma by sequential ultracentrifugation and depleted of apolipoproteins E and B100 by heparin-agarose chromatography.34 ApoA-I was purified from HDL by ion exchange chromatography.34 The protein concentration was determined with the Lowry assay (Bio-Rad, Hercules, CA), with albumin as the standard. The Human Studies Committees at the University of Washington School of Medicine approved all protocols involving human material.
Cross-Linking Reactions and Liquid Chromatography–Electrospray Ionization Mass Spectrometry (LC–ESI-MS)
Previous studies demonstrate that malondialdehyde (MDA), a major product of lipid peroxidation that is also a reactive carbonyl peptide cross-linker, markedly impairs the ability of lipid-free apoA-I to promote the efflux of cholesterol from cells by the ABCA1 pathway.35 To further test the fit of the models to experimental data, we used MS to detect apoA-I peptides that had been cross-linked with MDA, a major product of lipid peroxidation.35,2 Reactions of freshly prepared MDA with lipid-free apoA-I (5 μM, 0.14 mg/mL) were conducted at 37 °C for 24 h in 50 mM sodium phosphate buffer (pH 7.4) containing 100 μM diethyleneaminepentaacetic acid to chelate redox-active metal ions.36 Reactions were initiated by adding MDA and terminated by adding a 20-fold molar excess (relative to MDA) of aminoguanidine.35,37 The apoA-I monomer, dimer, and multimer were separated by high-resolution size exclusion chromatography using fast protein liquid chromatography (two Superdex 200 10/300 GL columns, GE Healthcare). The monomer fractions were collected, combined, and concentrated using Amicon Ultra-4 filters (Millipore) before being digested.
Concentrated fractions were digested with sequencing grade modified trypsin (Promega) or endoproteinase Glu-C (from Staphylococcus aureus V8, Roche Applied Science) in 50 mM NH4HCO3 (pH 7.8).35,38 Proteolytic digests were desalted by solid phase extraction (OASIS HLB cartridge, Waters Corp., Milford, MA) and then analyzed by LC–ESI-MS/MS in positive ion mode on a hybrid linear ion trap-Orbitrap instrument (Thermo Fisher, San Jose, CA) equipped with a nanoflow UPLC system (NanoAcquity, Waters Corp.). MS survey scans were performed from m/z 200 to 2000 at a resolution of 60000 and ion populations of 5 × 105. Data-dependent analyses were performed using MS survey scans followed by data-dependent selection of the five most abundant precursor ions for MS/MS. Every sample was analyzed twice. Singly charged precursors were rejected from the data-dependent scan in the first run, and singly, doubly, and triply charged precursors were rejected from data-dependent selection in the second run to obtain optimal MS/MS scans for cross-linked peptides that usually present as ≥4+ ions.
Potentially cross-linked peptides were identified using a spreadsheet containing the masses of each possible peptide pair (or a single peptide for intrapeptide cross-links) plus the mass of the MDA cross-linker (K+36+K).35,2 All peptide identifications were confirmed by inspection of the MS/MS spectra, using the following criteria: (i) mass being within 10 ppm of the calculated mass, (ii) corresponding MS/MS spectrum exhibiting at least 50% of the b and y series of theoretical fragment ions expected, and (iii) all major fragmentation ions being consistent with the peptide identification.
Results
The Crystal Structures of C-Terminally Truncated ApoA-I and N-Terminally Truncated ApoA-I Potentially Interact via a Hydrophobic Cleft
The similarity of the two models created by the selection of individual fragments of different chains from the structures of Borhani et al.18 and Mei and Atkinson27 shown in Figure S1a,b of the Supporting Information led us to consider the possibility that a combination of these two general conformations might provide a model for the structure of lipid-free apoA-I in solution. Figure 1a shows a ribbon model of the (Δ185–243)apoA-I crystal structure determined by Mei and Atkinson.27 The H5 region of the structure has been proposed to function as a hinge that allows the extended form of apoA-I to fold back on itself to interconvert the protein between a monomer and dimer in solution.27 This structure is topologically identical to a monomer in which helix 5 forms a helical hairpin, converting the intermolecular H4/H6 pairings of the dimer into monomeric intramolecular H4/H6 pairings (Figure 1b). We observed that the space-filling model of the monomer created this way has a remarkable hydrophobic cleft running the length of the molecule. The cleft contains only aliphatic (gold) and aromatic (magenta) residues (Figure 1c,d).
Inspection of the C-terminal portion of the (Δ1–43)apoA-I crystal structure determined by Borhani et al.18 suggested that residues 183–243 of the C-terminus (Figure 1e,f) [residues 183 and 184 are not resolved in the (Δ185–243)apoA-I crystal structure] would fit snuggly into the hydrophobic cleft (Figure 1d). Only small bending and stretching of residues 183–243 aligned four of the cleft’s five aromatic residues (Figure 1d) with the four aromatic residues in the fragment of Borhani et al. (Figure 1g). We therefore docked this peptide (Figure 1e,f) with the monomeric (Δ185–243)apoA-I structure in Figure 1b to create an all-atom model for full-length monomeric lipid-free apoA-I (Figure 1h,i) that we term the initial crystal model. Importantly, we generated this model of full-length apoA-I by combining the two crystal structures of opposite ends of the protein into a hybrid structure.
MD Simulations with a Temperature Jump Decrease the Helicity of the Initial Crystal Model to Physiologically Plausible Levels
The experimentally determined helicity for monomeric lipid-free human apoA-I is 55 ± 5%.25,39,40 Our hybrid structure had significantly more helicity (74%). We therefore used MD simulations of the initial crystal model to explore additional conformational space. As MD simulations at 310 K produced only minor structural changes, we subjected the initial crystal model to MD simulations at 500 K for 30 ns. The rmsd of this simulation is shown in Figure S2 of the Supporting Information; the rmsd of the full model flattens out between 15 and 28 ns but increases between 28 and 30 ns (Figure S2a of the Supporting Information), suggesting at least a portion of the model is not equilibrating. Restriction of the rmsd to the model’s core helical domains 1, 4 and 5 (see Figure 6a for the definition) shows that the core is equilibrated from 15 ns to the 30 ns end (Figure S2b of the Supporting Information). The rmsd of the noncore domains, however, increased between 28 and 30 ns (Figure S2c of the Supporting Information), and thus, these domains account for the corresponding increase in the rmsd of the full model.
Figure 6.

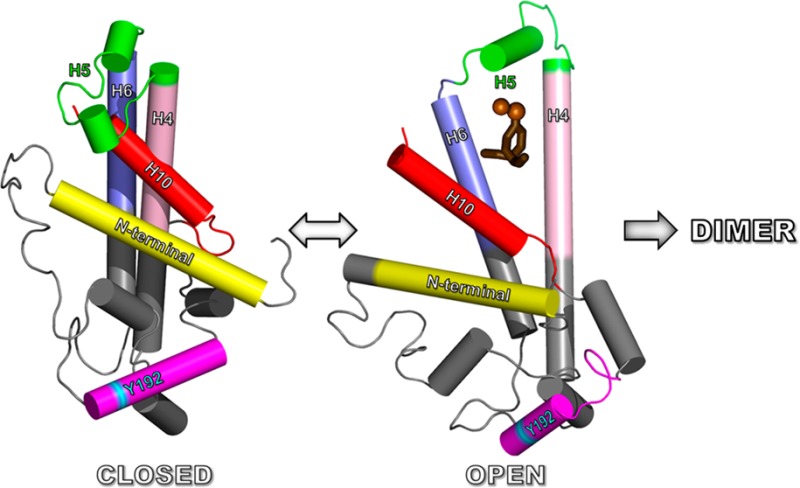
Ribbon comparison of the four alternatively open and closed structures: 15 ns (Closed1) → 20 ns (Open1) → 25 ns (Closed2) → 28 ns (Open2). Color code: H10, red; H5, green; H4, cyan; H6, magenta; remainder of structure, gray; N-termini, yellow arrowheads; C-termini, red arrowheads. In both transitions from open to closed structures, H5 and H10 move away from each other and from the main body of the protein exposing the H4–H5–H6 hairpin so that a second apoA-I rotated along its long axis by 180° can dimerize with the first through intermolecular interactions of the H4–H5–H6 region.
Changes in the helicity of the initial crystal model with simulation time decreased for the first 10 ns and then reached a relatively steady state between 10 and 20 ns (white dotted line in Figure 2a), indicating that the ensemble of structures reached some sort of conformational stability. The simulation between 10 and 20 ns exhibited ∼58% helicity, in excellent agreement with the value of 55 ± 5% reported in the literature for lipid-free monomeric apoA-I.25,39,40
Figure 2.

Plots of changes in properties of the initial crystal model during a 30 ns MD simulation at 500 K. The double-headed arrows mark the 10–20 ns intervals. The filled arrowheads mark the simulations at 15 ns. (a) Change in fraction helicity with time. The white dotted line is a linear trendline for changes in helicity between 10 and 20 ns. (b) Sum of the distances between Cα atoms of the 15-residue pair combinations from an average center for the six spin-coupled residues predicted by Lagerstedt et al.50 to define a central plane. (c). Total number of distances of >20 Å from the 15 combinations of distances between the six spin-coupled residues. In all panels, the empty diamonds represent the initial crystal model.
Testing Candidate MD Simulation Models against Published Experimental Data
Using site-directed spin-label electron paramagnetic resonance spectroscopy (EPR), Lagerstedt et al.41 identified six spin-coupled residues (G26, L44, L64, S167, G217, and K226) in lipid-free human apoA-I. In the initial crystal model, all but G26 are exposed to solvent and four (L44, L64, G217, and K226) are in loops on the surface of our 15 ns model (Figure 3c); the two that are not, G26 and S167, are small and in physical contact with each other.
Figure 3.

Goodness of fit of models to the helical regions predicted by Chetty et al.40 and the spin-coupled residues predicted by Lagerstedt et al.50 proposed to define a central plane. (a) Bar graph showing the goodness of fit of helicity of five models (initial crystal, 10–20 ns, 15 ns, Silva, and Pollard) to the position of the five helical regions (designated 1–5) predicted by Chetty et al.40 using H/D exchange. (b and c) Relaxed-eyed stereo ribbon images illustrating distances and positions of the six spin-coupled residues (Cα, magenta spheres) predicted by Lagerstedt et al.50 to define a central plane: (b) initial crystal model and (c) 15 ns model. Distances between residues are denoted by dotted lines. Color code: H5 (residues 121–143), green; prolines, yellow spheres; H10 (residues 221–243), red; remainder of the helical residues, cyan; remainder of random coil residues, orange.
To be spin-coupled, residue pairs must be within ∼20 Å of each other. The fact that six residues distributed along the full length of the apoA-I sequence, mostly on the surface, are spin-coupled suggests that the six form a spatially compact cluster in the solution structure of monomeric apoA-I.41 We therefore analyzed distances between the six spin-coupled residues over the 30 ns of MD simulation. As seen in Figure 2b, the sum of distances for the 15 combinations among the six residues (a measure of their spatial compactness) reaches its lowest value in the region of seeming helical equilibration (10–20 ns, double-headed arrow). In Figure 2c, the number of distances of ≥20 Å reaches a minimum of 2–3 near the 15 ns structure in the middle of the region of helical equilibration defined in Figure 2a.
On the basis of the best fit of our MD simulation of the initial crystal model with the spin-labeled data, we selected three models for additional analysis: (i) the initial crystal model, (ii) the average atom coordinates of the MD structural ensemble between 10 and 20 ns, termed the 10–20 ns model, and (iii) the 15 ns structure in the middle of the 10–20 ns region, termed the 15 ns model, based on the 15 ns frame of the full 30 ns ensemble. We selected the latter model because it was in the middle of both the zone of helix stability and the zone of spin-coupled spatial compactness. The rmsd between the 15 ns and 10–20 ns models is 5.4 Å, suggesting that the structures in the 10–20 ns range are not greatly different.
In the 15 ns model, cooled from 500 to 310 K, the radius of gyration of the apoA-I monomer was smaller than in the original structure, decreasing from 20.7 to 20.2 Å. This change reduced the monomer’s diameter of gyration by 1.08 Å. We term the 310 K cooled-down structure the 15 ns model. The average 10–20 ns structure, because there is no solvent present, could not be subjected to a similar cool-down procedure.
Chetty et al.40 used hydrogen/deuterium (H/D) exchange coupled with proteolysis and mass spectrometry to define the positions of helical domains in human full-length monomeric lipid-free apoA-I. The method quantifies the H/D exchange rate of measurable amide hydrogens at known positions throughout the native structure with the exchange rate expected for an unstructured chain. The authors reported three extended and two short α-helices (residues 7–44, 54–65, 70–78, 81–115, and 147–178, designated 1–5, respectively, in Figure 3a). The overall helical content of 52% predicted by the H/D exchange study agrees well with the values reported in the literature.25,39,40
We graphically compared the helical regions from the H/D exchange results of Chetty et al.40 with those predicted by our models (the initial crystal model, the 10–20 ns model, and the 15 ns model). We also compared the two all-atom models for human monomeric apoA-I reported by Silva et al.25 and Pollard et al.26 Inspection of Figure 3a suggests that our three models fit the five domains predicted to be helical by H/D exchange significantly better than the Silva or Pollard model. Of our models, the 15 ns model appeared to provide the best overall fit.
We used the secondary structure determination program STRIDE to quantify the goodness of fit between the experimental H/D exchange data40 and the different models. We determined the secondary structure “match” for each residue: only residues in the model that match residues in the H/D data (i.e., helix to helix or nonhelix to nonhelix) are counted. The results are reported as (i) the total number of matching residues and (ii) the fraction of matched residues. The 15 ns model matched the H/D data better than the initial crystal model or the 10–20 ns model (Table 1). Further, each of our models matched the H/D exchange data significantly better than either the Silva model or the Pollard model (Table 1). The total helicities of the 10–20 ns model and the 15 ns model were 56 and 59%, respectively, in good agreement with the value of 55 ± 5% reported in the literature.25,39,40
Table 1. Comparison of Helical Properties of Models to H/D Data.
| model | helical residues | fraction helicitya | H/D match | fraction match |
|---|---|---|---|---|
| H/D (Chetty et al.40) | 126 | 0.52 | 243 | 1.00 |
| initial crystal model | 180 | 0.74 | 171 | 0.70 |
| 10–20 ns model | 137 | 0.56 | 168 | 0.69 |
| 15 ns model | 144 | 0.59 | 175 | 0.72 |
| Silva model25 | 127 | 0.52 | 146 | 0.60 |
| Pollard model26 | 131 | 0.54 | 136 | 0.56 |
Monomeric apoA-I is 55 ± 5% helical.
We next compared the fit of our models and the Silva25 and Pollard26 models with the EPR spin-coupled data,41 using the following metrics: (i) the sum of distances (the total pairwise distances among Cα atoms of all six residues) and (ii) the number of distances between residues of >20 Å (the distance monitored by spin coupling). Each of our models fits the spin-coupled data better than either the Silva model or the Pollard model (Table 2); again, the 15 ns model provides the best fit. Panels b and c of Figure 3 compare the locations of the six spin-coupled residues in stereo images of two of our models, the initial crystal model and the 15 ns model. Each pair distance is denoted by a dotted line and distance in angstroms. In the initial crystal model, the six residues are scattered over the length of the monomer (Figure 3b). In the 15 ns model, the six residues form a spatially compact cluster. It is noteworthy that five of the six residues form a plane that is approximately equidistant from the long ends of the model (Figure 3c).41
Table 2. Fits of Models to Spin-Coupled Data.
To further test the fit of the models to experimental data, we used mass spectrometry to detect apoA-I peptides that we had cross-linked with MDA. We exposed full-length lipid-free human apoA-I (0.14 mg/mL, predominantly monomeric) to MDA (100 μM) in 50 mM sodium phosphate buffer (pH 7.4) containing 100 μM DTPA. Reactions were initiated by adding MDA and terminated by adding a 20-fold molar excess (relative to MDA) of aminoguanidine. Following high-resolution size exclusion chromatography, monomeric apoA-I was digested with trypsin. Then the peptide digest was analyzed by MS/MS on a high-mass resolution Orbitrap instrument. This approach identified 41 cross-linked pairs of peptides with a high degree of confidence (based on the mass accuracy of the peptide–peptide adduct and the tandem mass spectrum of the peptide). The cross-linked peptides we detected and their tandem mass spectra are summarized in Tables 3 and 4 and Figure S3 of the Supporting Information.
Table 3. Intrapeptide (adjacent) Cross-Links (K+36+K) Identified in the FPLC Monomer Fraction of ApoA-I Exposed to MDAa.
| X-linked Lys residues | peptide involved | theoretical mass (Da) | experimental mass (Da) | error (ppm) | protease | product yield (%) | relative intensity |
|---|---|---|---|---|---|---|---|
| NTb–K12 | 1–13 | 1604.73 | 1604.74 | 4.67 | Glu-C | 49.68 | 196.78 |
| K12–K23 | 11–27 | 1914.03 | 1914.04 | 4.51 | trypsin | 49.92 | 197.74 |
| K40–K45c | 28–59 | 3626.80 | 3626.82 | 4.76 | trypsin | 27.52 | 108.99 |
| K94–K96c | 93–111 | 2415.23 | 2415.24 | 4.26 | Glu-C | 7.42 | 29.39 |
| K106–K107c,d | 97–116 | 2681.27 | 2681.28 | 4.31 | trypsin | 44.42 | 175.93 |
| K133–K140d | 132–149 | 2201.14 | 2201.15 | 4.71 | trypsin | 47.53 | 188.23 |
| K195–K206d | 189–215 | 3057.60 | 3057.62 | 5.30 | trypsin | 25.10 | 99.43 |
| K206–K208c,e | 206–212 | 792.46 | 792.46 | 0.14 | Glu-C | 56.77 | 224.86 |
| K238–K239c,e | 236–243 | 1030.55 | 1031.55 | 0.10 | Glu-C | 25.93 | 102.68 |
ApoA-I (5 μM) was incubated with 100 μM MDA at 37 °C in phosphate buffer (pH 7.4) for 24 h. Reactions were initiated by adding MDA and terminated by adding a 20-fold molar excess (relative to MDA) of aminoguanidine. The reaction mixture was then concentrated, and the monomer was isolated by FPLC. The combined monomer fractions were further concentrated and digested with trypsin or Glu-C. Proteolytic digests were desalted by solid phase extraction prior to MS analysis and analyzed on a hybrid linear ion trap Orbitrap instrument.
N-Terminal amino group.
Cross-linked peptides can be considered short-range with respect to the protein sequences.
Also identified by Glu-C digestion.
Also identified by trypsin digestion.
Table 4. Interpeptide (distant) Cross-Links (K+36+K) Identified in the FPLC Monomer Fraction of ApoA-I Exposed to MDAa.
| X-linked Lys residues | peptide involved | theoretical mass (Da) | experimental mass (Da) | error (ppm) | protease | product yield (%) | relative intensity |
|---|---|---|---|---|---|---|---|
| Nt×K195 | 1–10 | 3289.58 | 3289.59 | 3.71 | trypsin | 3.85 | 15.25 |
| 189–206 | |||||||
| K12–K226 | 1–13 | 2960.44 | 2960.45 | 3.86 | Glu-C | 1.29 | 5.12 |
| 224–235 | |||||||
| K23–K59 | 13–27 | 3567.83 | 3567.85 | 5.13 | trypsin | 26.07 | 103.25 |
| 46–61 | |||||||
| K23×K239 | 13–27 | 2289.22 | 2289.22 | 0.60 | trypsin | 2.54 | 10.07 |
| 239–243 | |||||||
| K40–K94 | 28–45 | 2963.54 | 2963.55 | 3.86 | trypsin | 1.51 | 5.98 |
| 89–96 | |||||||
| K40×K133 | 28–45 | 3184.67 | 3184.68 | 4.90 | trypsin | 4.37 | 17.29 |
| 132–140 | |||||||
| K40×K239 | 28–45 | 2635.38 | 2635.38 | 0.85 | trypsin | 5.54 | 21.94 |
| 239–243 | |||||||
| K59–K195 | 46–61 | 3945.01 | 3945.02 | 3.43 | trypsin | 1.99 | 7.88 |
| 192–198 | |||||||
| K59–K239 | 46–61 | 2520.32 | 2520.32 | 2.26 | trypsin | 3.19 | 12.63 |
| 239–243 | |||||||
| K77–K195c | 71–78 | 1934.90 | 1934.90 | 1.12 | Glu-C | 3.98 | 15.76 |
| 192–198 | |||||||
| K77×K208 | 71–78 | 3665.85 | 3665.87 | 3.36 | trypsin | 0.69 | 2.71 |
| 207–215 | |||||||
| K88×K195 | 84–94 | 3398.69 | 3398.70 | 4.94 | trypsin | 0.72 | 2.85 |
| 192–198 | |||||||
| K94×K226 | 89–96 | 3564.92 | 3564.93 | 4.69 | trypsin | 1.27 | 5.03 |
| 216–238 | |||||||
| K94×K239 | 89–96 | 1569.86 | 1569.86 | 3.50 | trypsin | 5.87 | 23.25 |
| 239–243 | |||||||
| K96–K195 | 95–106 | 3514.79 | 3514.80 | 4.38 | trypsin | 5.88 | 23.29 |
| 189–206 | |||||||
| K96×K208 | 95–106 | 2499.33 | 2499.34 | 3.65 | trypsin | 3.52 | 13.96 |
| 207–215 | |||||||
| K96×K239 | 95–106 | 2090.10 | 2090.10 | 1.01 | trypsin | 5.64 | 22.33 |
| 239–243 | |||||||
| K107×K239 | 107–116 | 2050.01 | 2050.02 | 0.91 | trypsin | 1.88 | 7.45 |
| 239–243 | |||||||
| K118–K133c | 114–125 | 2475.36 | 2475.36 | 1.80 | Glu-C | 12.63 | 50.01 |
| 129–136 | |||||||
| K118–K140b | 117–123 | 2813.50 | 2813.50 | 0.60 | trypsin | 44.59 | 176.59 |
| 134–149 | |||||||
| K118×K195c | 114–125 | 2356.25 | 2356.25 | 2.37 | Glu-C | 2.03 | 8.03 |
| 192–198 | |||||||
| K118×K208 | 117–123 | 1917.10 | 1917.10 | 1.65 | trypsin | 0.92 | 3.64 |
| 207–215 | |||||||
| K118–K226 | 114–125 | 2893.55 | 2893.56 | 1.06 | Glu-C | 8.94 | 35.42 |
| 224–235 | |||||||
| K118×K239 | 114–125 | 1507.87 | 1507.87 | 0.05 | trypsin | 4.56 | 18.06 |
| 239–243 | |||||||
| K133×K239 | 132–140 | 1790.98 | 1790.98 | 0.50 | trypsin | 2.60 | 10.31 |
| 239–243 | |||||||
| K140–K239 | 134–149 | 2547.33 | 2547.34 | 1.68 | trypsin | 3.51 | 13.92 |
| 239–243 | |||||||
| K182–K195 | 180–183 | 1314.68 | 1314.68 | 0.15 | Glu-C | 9.48 | 37.55 |
| 192–198 | |||||||
| K182×K226 | 180–183 | 1851.97 | 1851.98 | 1.69 | Glu-C | 13.61 | 53.91 |
| 224–235 | |||||||
| K195–K226 | 192–198 | 2211.10 | 2211.11 | 1.36 | Glu-C | 6.03 | 23.89 |
| 224–235 | |||||||
| K195–K239b | 189–206 | 2666.39 | 2666.39 | 1.99 | trypsin | 4.45 | 17.63 |
| 239–243 | |||||||
| K206–K226 | 206–212 | 2148.17 | 2148.17 | 2.40 | Glu-C | 5.97 | 23.64 |
| 224–235 | |||||||
| K226–K239 | 224–235 | 2387.26 | 2387.26 | 1.76 | Glu-C | 3.31 | 13.11 |
| 236–243 |
ApoA-I (5 μM) was incubated with 100 μM MDA, and the cross-linked peptides were identified as described in footnote a of Table 3.
Also identified by Glu-C digestion.
Also identified by trypsin digestion.
We applied two methods to establish the goodness of fit of the models with the cross-linking data. First, we used the sum of violation distances algorithm recently described by Kalisman et al.42 to analyze the goodness of fit of different models to cross-link violation distances. We assumed a maximal Cα distance of linkable lysine residues of 23 Å, defined as 20 Å + 3.0 Å, the estimated coordinate error for two mobile surface residues.33 For each of the six models, the number of violations and the sum of minimal violations were plotted on the x- and y-axes, respectively (Figure 4a). In this plot, the initial crystal model is farthest from the origin, while the Pollard, Silva, and 10–20 ns models, in rank order, fall progressively closer to the origin. The best fit for any model (i.e., the one falling closest to the origin) was that of the 15 ns model. For comparison, a full trajectory analysis of the 0–30 ns MD simulation at 500 K, termed the full trajectory model, is also shown in Figure 4a. It should be noted that, in the latter analysis, every structure in the simulation was considered and the minimum of each individual violation distance was found among possibly different structures.
Figure 4.
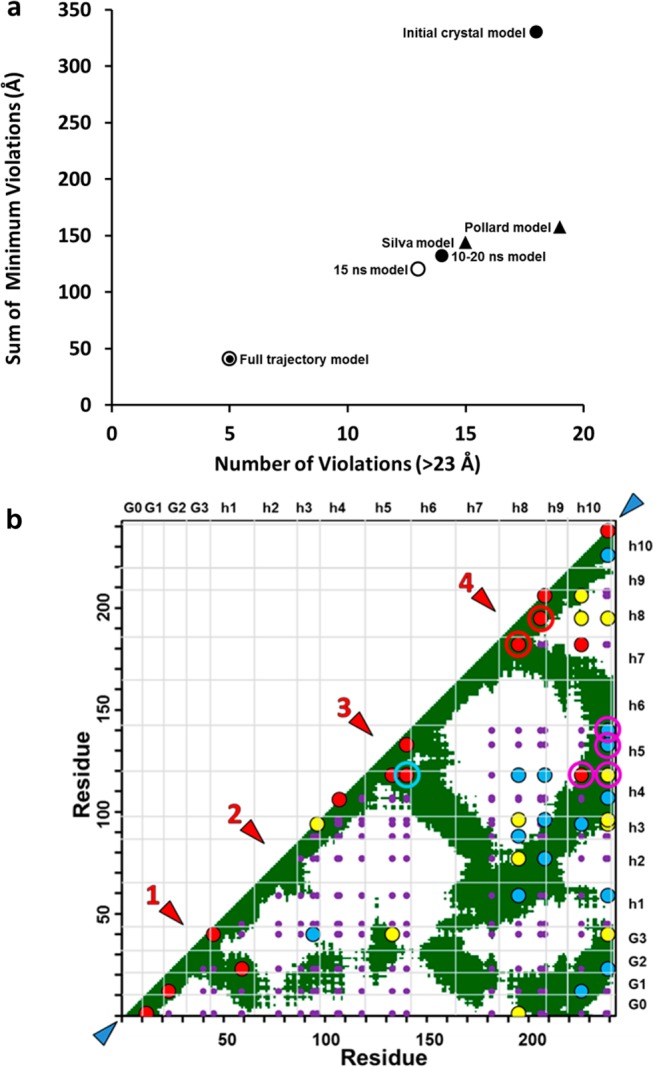
Graphic analyses of chemical cross-link data for different models. (a) Graphic analysis of the goodness of fit of the five models plus the entire 30 ns trajectory to cross-link violation distances, number vs sum, assuming a cross-linking distance cutoff of 23 Å. (b) Overlay of 41 chemical cross-linking data points onto a static contact map plot of Cα distances of ≤23 Å for the 15 ns model. Small purple circles represent the positions of all potential cross-links, red circles the most probable one-third of cross-links, yellow circles the median probable one-third of cross-links, and blue circles the least probable one-third of cross-links. Red arrowheads denote four loops and turns numbered from the N-terminus and blue arrowheads the N- and C-termini. Two cross-links (182–195 and 195–206) at turn/loop 4 are denoted with red bull’s-eyes and four cross-links of H5 to H10 (118–226, 118–239, 133–239, and 140–239) with magenta bull’s-eyes, and the cross-link supporting an H5 hairpin (118–140) is denoted with a blue bull’s-eye.
For the second method for analyzing the goodness of fit, we plotted each chemical cross-link as a colored dot onto the background of a two-dimensional contact map of all 23 Å residue-to-residue contacts, focusing on the static 15 ns model (Figure 4b). The apparent abundance of each cross-linked peptide (monitored as ion current) is indicated by the color of the corresponding dot. For comparison, we also plotted the cross-linking data on the contact map calculated over the full trajectory (Figure S4 of the Supporting Information).
Overall, the abundant peptides exhibited an excellent fit to the 15 ns model (red dots in Figure 4b). The majority of those cross-linked peptides were in the most mobile regions of the models: at the N- and C-termini (blue arrowheads) and near hairpins and loops represented by the junction of contact zones with (and at right angles to) the diagonal line of equality (red arrowheads).
Figure 5a is a stereo ribbon image of the 15 ns model. The five domains predicted to be helical by H/D exchange and the four domains that are helical in the 15 ns model but not helical as predicted by H/D exchange40 are colored red and green, respectively. Figure 5b illustrates the general conformation of the 15 ns model in the form of a stereo cartoon in which the 10 helical segments are shown as cylinders. The following are key features of the model. (i) The C-terminal H10 (red) is nestled under the folded H5 helical hairpin (green) covering the antiparallel H4–H6 array (residues 100–120 and 144–164). (ii) The C-terminal and N-terminal domains, residues 226–239 (red) and 7–31 (yellow), respectively, are in contact, forming an antiparallel helical array. (iii) H5 and the N- and C-terminal segments are in the proximity of each other on the side of the model facing the viewer, a feature also found in the Silva25 model.
Figure 5.

Relaxed-eyed stereo views of the 15 ns model. (a) Mapping of the location of the five helical domains predicted by Chetty et al. using H/D exchange40 onto a relaxed-eyed stereo ribbon representation of the 15 ns model. Helical domains [marked as 1–5 (see Figure 3a)] of the 15 ns model predicted to be helical are colored red and helical domains of the 15 ns model predicted not to be helical are green. The remainder of the nonhelical structure is colored gray. Prolines are shown as yellow spheres. The N-terminus is marked with a yellow N and the C-terminus with a red C. (b) Cartoon representation of the 15 ns model in same orientation as panel a. Helical domains are represented as cylinders and nonhelical domains as random coils. Color code: H5, green; H10, red; N-terminal helical domain, yellow; other helical domains, magenta; K40 and Y192, cyan and magenta spheres, respectively. The N- and C-termini are indicated as in panel a.
Discussion
By combining published crystal structures of the C-terminal and N-terminal portions of apoA-I, we derived an initial all-atom crystal model for the full-length monomeric protein. Using MD simulations, we modified that initial crystal model into a 10–20 ns model and a 15 ns model.
The 10–20 ns region was used to construct the 10–20 ns model because it represented a zone of helix stability and spin-coupled spatial compactness. The 15 ns frame was used to create the 15 ns model because it represented the middle of the zone of helix stability and maximal spin-coupled spatial compactness. This choice is supported by the cross-linking data (Figure 4a) and the observation that the core helical domains of the model, CH1, -4, and -5 [except for the spikes associated with the closed–open transitions (Figure 6)], are equilibrated at 15 ns (red circle in Figure S2b of the Supporting Information). The nonhelical core of the simulated structure reaches initial equilibration at 10 ns (cyan line in Figure S2c of the Supporting Information) but begins a steep increase in rmsd after 27 ns, in part because of significant changes in the position of H10 in the model (see below).
Examination of the simulation frames between 27 and 30 ns shows that (i) H/D helical domain 2 has become nonhelical (a particular problem because this domain was shown by Chetty et al.40 to be helical); (ii) H10 is trapped between CH1 and a portion of H6 and is beginning to separate from its previous interaction with H4 and H5 (Figure S5a of the Supporting Information); and (iii) for the first time, a gap has appeared between the H4 and H6 domains (Figure S5b of the Supporting Information). The gap and the loss of H/D helical domain 2 would suggest that the simulation is becoming unstable, although it is possible that the 30 ns structure represents a new transitional conformation for monomeric apoA-I. We will pursue the latter possibility at a later date.
We believe that the 15 ns model (Figure 5) represents the best available static structure for human lipid-free monomeric apoA-I; it fits all three experimentally determined parameters (helical domains, spin-coupled residues, and chemical cross-linking results) better than either of the two previously published all-atom models.25,26 Because the helical structure of monomeric human apoA-I is known to be very flexible and to unfold and refold in a dynamic manner,40 the 15 ns model, although static, illustrates in a clear fashion the most important conformational features of the ensemble of structures in thermal equilibrium (Figures 4a and 5).
The Chetty40 H/D exchange data suggest that residues 121–143 (H5) and 180–243 (H8–H10) are not helical. However, EPR studies led Oda et al.43 to conclude that H10 in monomeric apoA-I is at least partially helical. We propose that H5 and H8–H10 (green helical domains in Figure 5a) are in motion, forming and breaking helices sufficiently rapidly that H/D exchange cannot detect them reliably [perhaps similar to what is happening at 30 ns (Figure S5 of the Supporting Information)]. In support of this proposal, a comparison of the 15 ns model with the 20 ns structure (Figure 6) demonstrates that H5 and H10 have moved away from each other and from the main body of the protein, exposing tandem helical domains H4 and H6. This conformational transition happens twice during the 30 ns MD simulation. The two open structures can be recognized as two upward blips in the rmsd plot (empty arrowheads in Figure S2a of the Supporting Information). The closed structures appear at 15 and 23 ns and the open structures at 20 and 28 ns (Figure 6).
Although we performed the simulation at 500 K, we found little change in overall helicity between 10 and 30 ns (58 to 55%). We surmised from the H/D data40 that the structure’s core conformation remains relatively intact [red helical domains 1 (residues 7–31), 4 (residues 84–120), and 5 (residues 146–174) in Figure 5a]. Evidence of core stability is provided by the rmsd data (Figure S2 of the Supporting Information) and by the more extensive stability analyses shown in Figure 7. An rmsf plot of the fluctuation of each residue during the full simulation (Figure 7a) shows that core helix (CH) domains 1, 4, and 5 (along with smaller H/D helical domains 2 and 3) are more stable than most of the remainder of the model.
Figure 7.

Stability analysis of the apoA-I monomer core CH1, CH4, and CH5 domains. (a) Root-mean-square fluctuation (rmsf) of all residues of the initial crystal model of apoA-I over the entire course of the 30 ns MD simulation at 500 K. Red boxes show H/D helical regions 1, 4, and 5 (residues 7–44, 81–115, and 147–178, respectively) that represent the core helical domains of the monomer model. Colored circles (L22 and L163, yellow; G26 and S167, gray; E92, red; K96, blue; Y100, Y104, Y108, and Y166, magenta; L159, green) denote key residues in panel b that stabilize the crossover contacts among the three core helical domains. (b) Relaxed-eye stereo view of the CH1, CH4, and CH5 domains (transparent red ribbons identified by pink numerals) of the 15 ns model. Hydrophobic residues of the core are shown as green sticks. G26 and S167 (gray space-filling model) form a hydrogen-bonded pair at the crossover between CH1 and CH5. E92 (red space-filling model), K96 (blue space-filling model), and Y166 (magenta space-filling model) form a cation−π complex between CH4 and CH5. Y100, F104, and W108 (magenta, dotted space-filling model–stick combination) form an aromatic cup around L159 (green sticks) between CH4 and CH5. L22 on CH1 (yellow space-filling model) packs tightly against the G26–S167 hydrogen bond. L163 on CH5 (yellow space-filling model) is tightly packed into the angle between S167 and Y166. Y115 on CH4 associates with three surrounding hydrophobic residues to help hold the upper ends of CH4 and CH5 together. Similar hydrophobic interactions occur between CH4 and CH5 at the bottom.
We examined the molecular packing of the three core helical domains to identify stabilizing structural elements. The three helical domains are in contact at crossover zones of CH1 with CH5 and CH5 with CH4; CH1 makes no contact with CH4, leaving a cleft for interaction with H10. Hydrophobic residue contact is pronounced in the crossover zones of CH1 with CH5 and CH5 with CH4 (Figure 7b). Importantly, detailed examination of these two crossover zones shows several specific forms of molecular interactions (Figure 7b). (i) S167 in H5 forms a hydrogen bond with the backbone carbonyl of G26 in CH1, a common motif in helix–helix interaction in nonpolar environments.44 This hydrogen bond is not present in the initial crystal model. (ii) The formation of a salt bridge from E92 to K96 on CH4 creates a cation−π bond between K96 and Y166 of CH5.44 (iii) Three aromatic residues on CH4 (Y100, F104, and W108) form what we call a π cup around L159 on CH5. (iv) L22 on CH1 packs tightly against the G26–S167 hydrogen bond. (v) L163 is tightly packed into the angle between S167 and Y166 on CH5. These 11 residues involved in molecular interactions at the crossover zones of CH1 with CH5 and CH5 with CH4 when plotted onto the rmsf plot of Figure 7a define the least mobile regions of CH1, CH4, and CH5. Other interactions, e.g., between Y115 and three surrounding hydrophobic residues and several just below E92 (Figure 7b), hold the ends of CH4 and CH5 together.
Thus, in the MD simulation, the core remains relatively stable during three transitions involving H5 and H10: closed → open → closed → open (Figure 6). These observations are consistent with a wide range of biophysical studies24 demonstrating that lipid-free apoA-I exhibits a two-domain structure in solution: a more rigid N-terminal domain (residues 1–189, red helical domains in Figure 5a) and a less organized flexible C-terminal domain (residues 190–243, green helical domains in Figure 5a).
The stability of the initial crystal model’s secondary structure to MD simulations at 500 K is unusual because our 500 K MD simulations of the models of Silva25 and Pollard26 resulted in much greater changes in total helicity (Figure S6 of the Supporting Information): the Silva model went from 52% helicity at 0 ns to 28% at 30 ns (Figure S6a of the Supporting Information), while the Pollard model went from 54% helicity at 0 ns to 28% at 10 ns (Figure S6b of the Supporting Information). Our model for monomeric apoA-I is stable even when compared with one of the apolipophorin III proteins found in many insects.45 An MD simulation of the crystal structure of that four-helix bundle apolipoprotein showed no plateau in helix stability, which decreased linearly from 80% at 0 ns to 50% at 20 ns (Figure S6c of the Supporting Information).
A protein that conserves a nativelike secondary structure but has no tightly packed interior is defined as a molten globule, which supposedly is an intermediate thermodynamic state that clearly differs from either a tightly folded, highly stable structure or the denatured state.46 Our MD simulations, which suggest that lipid-free apoA-I maintains a core complex of intact folded helices with dynamic closing and opening of H5 and H10, are consistent with the definition of a molten globule. We suggest that apoA-I is highly dynamic, with a nativelike secondary structure that binds its targets tightly and specifically in the absence of side chain close packing.46 Consistent with our findings, biophysical studies suggest that apoA-I is a molten globule,39 a state suggested to mediate the incorporation of lipid into plasma.39
To sample the lipid affinity of our lipid-free apoA-I monomer model (Figure 5), we coarse grained the 15 ns model and performed a 20 μs coarse grained MD simulation of the model in a periodic solvent box containing 10 randomly distributed monomeric POPC; the results are shown in Figure S7 of the Supporting Information. After simulation, seven POPC bind, forming a small monolayer in the monomeric apoA-I “lipid cup”. This model shares similarities with a model for the preβ-apoA-I monomer recently proposed by Jayaraman et al.47 that is based upon lipid-poor monomeric apoA-I released by heating HDL. (i) Their particle contains 8 or 9 PCs and 1 CE. (ii) In their model, the C-terminal H10 folds back against a H4–H5–H6 “bucket” (see the cup formed by H4–H5–H6 and H10 in Figure S7d of the Supporting Information) derived from the crystal structure of (Δ185–243)apoA-I.27
The open conformation of apoA-I at 20 and 28 ns suggests a mechanism for dimerization and accumulation of lipid into the discoidal HDL form. The 20 and 28 ns structures expose the H4–H6 hairpin domains (Figure 6), so that two similarly structured apoA-I monomers rotated relative to one another along their long axes by 180° are able to dimerize. This association would be driven by the formation of intermolecular salt bridges among the H4, H5, and H6 domains of the two monomers and result in close planar proximity of the six spin-coupled residues in the dimer, as proposed by Lagerstedt et al.41 The addition of lipid (Figure S7 of the Supporting Information) and the unfolding of the helical remainder of the two apoA-I would provide the correct topology and lower the activation energy for dimerization. The dimerized lipid-poor apoA-I would then be poised to grow into a fully lipidated LL5/5 double-belt discoidal particle.20
As noted by Mei and Atkinson,27 the paired antiparallel H5 repeats between opposing chains of the apoA-I dimer form leucine zipper motifs. It seems likely that these are the principal motifs for aligning the pairwise antiparallel H5 repeats of the apoA-I dimer in the lipid-free state. For example, in the open 20 ns state (Figure 6), the leucine zippers might “zip-lock” the double belt into the H4–H6 antiparallel pair and ultimately the full LL5/520 rotamer registration. As noted elsewhere,28 however, this leucine zipper motif may not be important for maintaining the apoA-I dimer after apoA-I has acquired lipid, reflecting the marked increase in helicity and the formation of salt bridges between the proteins.20
A key element of our model is the loop/turn in the vicinity of Y192 that allows H10 to contact H5, a conformational motif whose formation and breaking we propose modulates protein dimerization and lipid binding. Our cross-linking data support all elements of this model. The existence of the loop/turn [loop 4 (Figure 4b)] is supported by cross-links 182–195 and 195–206. Contact of the H5 hairpin with H10 is supported by cross-links 118–226, 118–239, 133–239, and 140–239 (Figure 4b). The H5 hairpin itself is supported by cross-link 118–140 (Figure 4b). Table S1 of the Supporting Information compares cross-linked peptides identified in this study with those identified by Silva et al.25 and Pollard et al.26 Eight cross-links are common to all three studies (Figure S8a of the Supporting Information). Of the lysine cross-links identified in our study that support the H10–H5 contact model (Figure S8b of the Supporting Information), only one, cross-link 118–140 indicating a H5 hairpin, is common to all three studies. None of the other six lysine cross-links found in our study that support the H10–H5 contact model were reported by Silva et al.25 or Pollard et al.26 Although it does not directly support the Y192 loop, the ability of Pollard et al.26 to cross-link L200C and L233C mutations supports our contention that helix 10 is extremely mobile.
We tested the models experimentally by reacting monomeric apoA-I with MDA, which cross-links proteins and also impairs the ABCA1 activity of lipid-free apoA-I.35 Because MDA creates cross-links in apoA-I between H10 (C-terminus) and H5,35 cross-linking should hinder apoA-I’s ability to form discoidal HDL, a key step in reverse cholesterol transport, by locking H10 to H5, preventing the transition from the closed to open form (Figure 6). It is significant that HDL isolated from human atherosclerotic lesions is modified by MDA and that elevated levels of MDA-modified HDL have been detected in blood of subjects with established coronary artery disease. Thus, activation of the MDA pathway might impair sterol efflux from artery wall cells in vivo by locking apoA-I into an unfavorable conformation.35
Other studies suggest that Tyr192 of lipid-free apoA-I is also a key regulator of sterol efflux by the ABCA1 pathway.48 In our models, Y192 is the first aromatic residue in residues 183–243 of the C-terminal fragment of the Borhani crystal structure that aligns with the aromatic residues of the hydrophobic cleft of the crystal structure of Mei and Atkinson (Figure 1g). Thus, the π–π interactions between Y192 and W72 (Figure 1g) may be important in wrapping the C-terminal fragment around one end of the helix bundle structure (Figure 7b) to create the H10–H5 juxtaposition that we propose regulates interactions of apoA-I with lipids. Consistent with this possibility, we have shown that Y192 and the adjacent E191 are the only completely conserved adjacent pair of residues in apoA-I of the eight completely conserved residues found in 31 orthologs of apoA-I from mammals to fish.49
Finally, Pollard et al.26 have emphasized the degree of cross-linking connectivity of K40 to six other residues, K59, K118, K133, K140, K182, and K239. As shown in Figure 5b, K40 is within a long loop that extends from residue 32 to 55 and is expected to be highly mobile. In fact, three of the six connected residues, including K239 at the opposite end of the primary sequence from K40, are <23 Å from K40 in our 15 ns model, and all six are <17 Å from K40 at some point in the full trajectory model (Table 5).
Table 5. Minimal Distances (angstroms) from K40.
| residue | 15 ns model | full trajectory model |
|---|---|---|
| K59 | 22.3 | 11.6 |
| K118 | 32.4 | 16.1 |
| K133 | 18.4 | 16.0 |
| K140 | 28.4 | 13.9 |
| K182 | 24.7 | 15.2 |
| K239 | 17.4 | 6.7 |
In conclusion, we combined portions of the crystal structures of human apoA-I of Borhani et al.18 and Mei and Atkinson27 and used MD simulations to develop a refined, dynamic structure of lipid-free monomeric human apoA-I that is consistent with the molten globule state. This model robustly fits three experimentally determined parameters: (i) domains predicted to be helical by H/D exchange,40 (ii) spin-coupled residues defining a spatially compact cluster in the solution structure of monomeric apoA-I,50 and (iii) high-precision mass spectrometry/peptide analysis with cross-linking chemistry. Because our model, schematically illustrated in Figure S9 of the Supporting Information, cycles between closed and open states, it suggests a mechanism whereby monomeric apoA-I might dimerize and interact with lipid during the process of reverse cholesterol transport. Although our proposed model is consistent with the existing low-resolution data, it awaits ultimate experimental verification by higher-resolution methods.
Acknowledgments
Mass spectrometry experiments were performed by the Quantitative and Functional Proteomics Core of the Diabetes Research Center of the University of Washington. We thank Information Technology and the Department of Mechanical Engineering of the University of Alabama at Birmingham for use of the commodity cluster Cheaha that they jointly maintain. None of the sponsors supporting the work had any role in the trial design, data analysis, or reporting of the results.
Glossary
Abbreviations
- CH
core helix domain
- CVD
coronary vascular disease
- HDL-C
high-density lipoprotein cholesterol
- H/D
hydrogen/deuterium
- MDA
malondialdehyde
- MD
molecular dynamics
- rmsd
root-mean-square deviation
- rmsf
root-mean-square fluctuation
- VMD
Visual Molecular Dynamics.
Supporting Information Available
Figures S1–S9 and Table S1. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by National Institutes of Health (NIH) Grants P01HL34343 and R01HL102515 to J.P.S. and NIH Grants HL108897, HL112625, and P30DK017047 to J.W.H. B.S. is supported by National Heart, Lung and Blood Institute Award K99/R00 (R00HL091055) and a Beginning Grant-in-Aid (13BGIA17290026) from the American Heart Association (P30DK017047).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Wilson P. W.; Abbott R. D.; Castelli W. P. (1988) High density lipoprotein cholesterol and mortality. The Framingham Heart Study. Arteriosclerosis 8, 737–741. [DOI] [PubMed] [Google Scholar]
- Gordon D. J.; Rifkind B. M. (1989) High-density lipoprotein: The clinical implications of recent studies. N. Engl. J. Med. 321, 1311–1316. [DOI] [PubMed] [Google Scholar]
- Lewis G. F.; Rader D. J. (2005) New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ. Res. 96, 1221–1232. [DOI] [PubMed] [Google Scholar]
- Tall A. R.; Costet P.; Wang N. (2002) Regulation and mechanisms of macrophage cholesterol efflux. J. Clin. Invest. 110, 899–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulchenko Y. S.; Ripatti S.; Lindqvist I.; Boomsma D.; Heid I. M.; Pramstaller P. P.; Penninx B. W.; Janssens A. C.; Wilson J. F.; Spector T.; Martin N. G.; Pedersen N. L.; Kyvik K. O.; Kaprio J.; Hofman A.; Freimer N. B.; Jarvelin M. R.; Gyllensten U.; Campbell H.; Rudan I.; Johansson A.; Marroni F.; Hayward C.; Vitart V.; Jonasson I.; Pattaro C.; Wright A.; Hastie N.; Pichler I.; Hicks A. A.; Falchi M.; Willemsen G.; Hottenga J. J.; de Geus E. J.; Montgomery G. W.; Whitfield J.; Magnusson P.; Saharinen J.; Perola M.; Silander K.; Isaacs A.; Sijbrands E. J.; Uitterlinden A. G.; Witteman J. C.; Oostra B. A.; Elliott P.; Ruokonen A.; Sabatti C.; Gieger C.; Meitinger T.; Kronenberg F.; Doring A.; Wichmann H. E.; Smit J. H.; McCarthy M. I.; van Duijn C. M.; Peltonen L. (2009) Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat. Genet. 41, 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do R.; Willer C. J.; Schmidt E. M.; Sengupta S.; Gao C.; Peloso G. M.; Gustafsson S.; Kanoni S.; Ganna A.; Chen J.; Buchkovich M. L.; Mora S.; Beckmann J. S.; Bragg-Gresham J. L.; Chang H. Y.; Demirkan A.; Den Hertog H. M.; Donnelly L. A.; Ehret G. B.; Esko T.; Feitosa M. F.; Ferreira T.; Fischer K.; Fontanillas P.; Fraser R. M.; Freitag D. F.; Gurdasani D.; Heikkila K.; Hypponen E.; Isaacs A.; Jackson A. U.; Johansson A.; Johnson T.; Kaakinen M.; Kettunen J.; Kleber M. E.; Li X.; Luan J.; Lyytikainen L. P.; Magnusson P. K.; Mangino M.; Mihailov E.; Montasser M. E.; Muller-Nurasyid M.; Nolte I. M.; O’Connell J. R.; Palmer C. D.; Perola M.; Petersen A. K.; Sanna S.; Saxena R.; Service S. K.; Shah S.; Shungin D.; Sidore C.; Song C.; Strawbridge R. J.; Surakka I.; Tanaka T.; Teslovich T. M.; Thorleifsson G.; Van den Herik E. G.; Voight B. F.; Volcik K. A.; Waite L. L.; Wong A.; Wu Y.; Zhang W.; Absher D.; Asiki G.; Barroso I.; Been L. F.; Bolton J. L.; Bonnycastle L. L.; Brambilla P.; Burnett M. S.; Cesana G.; Dimitriou M.; Doney A. S.; Doring A.; Elliott P.; Epstein S. E.; Eyjolfsson G. I.; Gigante B.; Goodarzi M. O.; Grallert H.; Gravito M. L.; Groves C. J.; Hallmans G.; Hartikainen A. L.; Hayward C.; Hernandez D.; Hicks A. A.; Holm H.; Hung Y. J.; Illig T.; Jones M. R.; Kaleebu P.; Kastelein J. J.; Khaw K. T.; Kim E.; Klopp N.; Komulainen P.; Kumari M.; Langenberg C.; Lehtimaki T.; Lin S. Y.; Lindstrom J.; Loos R. J.; Mach F.; McArdle W. L.; Meisinger C.; Mitchell B. D.; Muller G.; Nagaraja R.; Narisu N.; Nieminen T. V.; Nsubuga R. N.; Olafsson I.; Ong K. K.; Palotie A.; Papamarkou T.; Pomilla C.; Pouta A.; Rader D. J.; Reilly M. P.; Ridker P. M.; Rivadeneira F.; Rudan I.; Ruokonen A.; Samani N.; Scharnagl H.; Seeley J.; Silander K.; Stancakova A.; Stirrups K.; Swift A. J.; Tiret L.; Uitterlinden A. G.; van Pelt L. J.; Vedantam S.; Wainwright N.; Wijmenga C.; Wild S. H.; Willemsen G.; Wilsgaard T.; Wilson J. F.; Young E. H.; Zhao J. H.; Adair L. S.; Arveiler D.; Assimes T. L.; Bandinelli S.; Bennett F.; Bochud M.; Boehm B. O.; Boomsma D. I.; Borecki I. B.; Bornstein S. R.; Bovet P.; Burnier M.; Campbell H.; Chakravarti A.; Chambers J. C.; Chen Y. D.; Collins F. S.; Cooper R. S.; Danesh J.; Dedoussis G.; de Faire U.; Feranil A. B.; Ferrieres J.; Ferrucci L.; Freimer N. B.; Gieger C.; Groop L. C.; Gudnason V.; Gyllensten U.; Hamsten A.; Harris T. B.; Hingorani A.; Hirschhorn J. N.; Hofman A.; Hovingh G. K.; Hsiung C. A.; Humphries S. E.; Hunt S. C.; Hveem K.; Iribarren C.; Jarvelin M. R.; Jula A.; Kahonen M.; Kaprio J.; Kesaniemi A.; Kivimaki M.; Kooner J. S.; Koudstaal P. J.; Krauss R. M.; Kuh D.; Kuusisto J.; Kyvik K. O.; Laakso M.; Lakka T. A.; Lind L.; Lindgren C. M.; Martin N. G.; Marz W.; McCarthy M. I.; McKenzie C. A.; Meneton P.; Metspalu A.; Moilanen L.; Morris A. D.; Munroe P. B.; Njolstad I.; Pedersen N. L.; Power C.; Pramstaller P. P.; Price J. F.; Psaty B. M.; Quertermous T.; Rauramaa R.; Saleheen D.; Salomaa V.; Sanghera D. K.; Saramies J.; Schwarz P. E.; Sheu W. H.; Shuldiner A. R.; Siegbahn A.; Spector T. D.; Stefansson K.; Strachan D. P.; Tayo B. O.; Tremoli E.; Tuomilehto J.; Uusitupa M.; van Duijn C. M.; Vollenweider P.; Wallentin L.; Wareham N. J.; Whitfield J. B.; Wolffenbuttel B. H.; Altshuler D.; Ordovas J. M.; Boerwinkle E.; Palmer C. N.; Thorsteinsdottir U.; Chasman D. I.; Rotter J. I.; Franks P. W.; Ripatti S.; Cupples L. A.; Sandhu M. S.; Rich S. S.; Boehnke M.; Deloukas P.; Mohlke K. L.; Ingelsson E.; Abecasis G. R.; Daly M. J.; Neale B. M.; Kathiresan S. (2013) Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat. Genet. 45, 1345–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight B. F.; Peloso G. M.; Orho-Melander M.; Frikke-Schmidt R.; Barbalic M.; Jensen M. K.; Hindy G.; Holm H.; Ding E. L.; Johnson T.; Schunkert H.; Samani N. J.; Clarke R.; Hopewell J. C.; Thompson J. F.; Li M.; Thorleifsson G.; Newton-Cheh C.; Musunuru K.; Pirruccello J. P.; Saleheen D.; Chen L.; Stewart A.; Schillert A.; Thorsteinsdottir U.; Thorgeirsson G.; Anand S.; Engert J. C.; Morgan T.; Spertus J.; Stoll M.; Berger K.; Martinelli N.; Girelli D.; McKeown P. P.; Patterson C. C.; Epstein S. E.; Devaney J.; Burnett M. S.; Mooser V.; Ripatti S.; Surakka I.; Nieminen M. S.; Sinisalo J.; Lokki M. L.; Perola M.; Havulinna A.; de Faire U.; Gigante B.; Ingelsson E.; Zeller T.; Wild P.; de Bakker P. I.; Klungel O. H.; Maitland-van der Zee A. H.; Peters B. J.; de Boer A.; Grobbee D. E.; Kamphuisen P. W.; Deneer V. H.; Elbers C. C.; Onland-Moret N. C.; Hofker M. H.; Wijmenga C.; Verschuren W. M.; Boer J. M.; van der Schouw Y. T.; Rasheed A.; Frossard P.; Demissie S.; Willer C.; Do R.; Ordovas J. M.; Abecasis G. R.; Boehnke M.; Mohlke K. L.; Daly M. J.; Guiducci C.; Burtt N. P.; Surti A.; Gonzalez E.; Purcell S.; Gabriel S.; Marrugat J.; Peden J.; Erdmann J.; Diemert P.; Willenborg C.; Konig I. R.; Fischer M.; Hengstenberg C.; Ziegler A.; Buysschaert I.; Lambrechts D.; Van de Werf F.; Fox K. A.; El Mokhtari N. E.; Rubin D.; Schrezenmeir J.; Schreiber S.; Schafer A.; Danesh J.; Blankenberg S.; Roberts R.; McPherson R.; Watkins H.; Hall A. S.; Overvad K.; Rimm E.; Boerwinkle E.; Tybjaerg-Hansen A.; Cupples L. A.; Reilly M. P.; Melander O.; Mannucci P. M.; Ardissino D.; Siscovick D.; Elosua R.; Stefansson K.; O’Donnell C. J.; Salomaa V.; Rader D. J.; Peltonen L.; Schwartz S. M.; Altshuler D.; Kathiresan S. (2012) Plasma HDL cholesterol and risk of myocardial infarction: A mendelian randomisation study. Lancet 380, 572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rader D. J.; Tall A. R. (2012) The not-so-simple HDL story: Is it time to revise the HDL cholesterol hypothesis?. Nat. Med. 18, 1344–1346. [DOI] [PubMed] [Google Scholar]
- Toth P. P.; Barter P. J.; Rosenson R. S.; Boden W. E.; Chapman M. J.; Cuchel M.; D’Agostino R. B. Sr.; Davidson M. H.; Davidson W. S.; Heinecke J. W.; Karas R. H.; Kontush A.; Krauss R. M.; Miller M.; Rader D. J. (2013) High-density lipoproteins: A consensus statement from the National Lipid Association. J. Clin. Lipidol. 7, 484–525. [DOI] [PubMed] [Google Scholar]
- Rubin E. M.; Krauss R. M.; Spangler E. A.; Verstuyft J. G.; Clift S. M. (1991) Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature 353, 265–267. [DOI] [PubMed] [Google Scholar]
- Voyiaziakis E.; Goldberg I. J.; Plump A. S.; Rubin E. M.; Breslow J. L.; Huang L. S. (1998) ApoA-I deficiency causes both hypertriglyceridemia and increased atherosclerosis in human apoB transgenic mice. J. Lipid Res. 39, 313–321. [PubMed] [Google Scholar]
- Benoit P.; Emmanuel F.; Caillaud J. M.; Bassinet L.; Castro G.; Gallix P.; Fruchart J. C.; Branellec D.; Denefle P.; Duverger N. (1999) Somatic gene transfer of human ApoA-I inhibits atherosclerosis progression in mouse models. Circulation 99, 105–110. [DOI] [PubMed] [Google Scholar]
- Liu A. C.; Lawn R. M.; Verstuyft J. G.; Rubin E. M. (1994) Human apolipoprotein A-I prevents atherosclerosis associated with apolipoprotein[a] in transgenic mice. J. Lipid Res. 35, 2263–2267. [PubMed] [Google Scholar]
- Plump A. S.; Scott C. J.; Breslow J. L. (1994) Human apolipoprotein A-I gene expression increases high density lipoprotein and suppresses atherosclerosis in the apolipoprotein E-deficient mouse. Proc. Natl. Acad. Sci. U.S.A. 91, 9607–9611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvan-Charvet L.; Pagler T.; Gautier E. L.; Avagyan S.; Siry R. L.; Han S.; Welch C. L.; Wang N.; Randolph G. J.; Snoeck H. W.; Tall A. R. (2010) ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 328, 1689–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane J. P.; Malloy M. J. (2012) Preβ-1 HDL and coronary heart disease. Curr. Opin. Lipidol. 23, 367–371. [DOI] [PubMed] [Google Scholar]
- Borhani D. W.; Rogers D. P.; Engler J. A.; Brouillette C. G. (1997) Crystal structure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc. Natl. Acad. Sci. U.S.A. 94, 12291–12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppaka V.; Silvestro L.; Engler J. A.; Brouillette C. G.; Axelsen P. H. (1999) The structure of human lipoprotein A-I. Evidence for the “belt” model. J. Biol. Chem. 274, 14541–14544. [DOI] [PubMed] [Google Scholar]
- Segrest J. P.; Jones M. K.; Klon A. E.; Sheldahl C. J.; Hellinger M.; De Loof H.; Harvey S. C. (1999) A detailed molecular belt model for apolipoprotein A-I in discoidal high density lipoprotein. J. Biol. Chem. 274, 31755–31758. [DOI] [PubMed] [Google Scholar]
- Panagotopulos S. E.; Horace E. M.; Maiorano J. N.; Davidson W. S. (2001) Apolipoprotein A-I Adopts a Belt-like Orientation in Reconstituted High Density Lipoproteins. J. Biol. Chem. 276, 42965–42970. [DOI] [PubMed] [Google Scholar]
- Tricerri M. A.; Behling Agree A. K.; Sanchez S. A.; Bronski J.; Jonas A. (2001) Arrangement of apolipoprotein A-I in reconstituted high-density lipoprotein disks: An alternative model based on fluorescence resonance energy transfer experiments. Biochemistry 40, 5065–5074. [DOI] [PubMed] [Google Scholar]
- Li H. H.; Lyles D. S.; Pan W.; Alexander E.; Thomas M. J.; Sorci-Thomas M. G. (2002) ApoA-I structure on discs and spheres. Variable helix registry and conformational states. J. Biol. Chem. 277, 39093–39101. [DOI] [PubMed] [Google Scholar]
- Rothblat G. H.; Phillips M. C. (2010) High-density lipoprotein heterogeneity and function in reverse cholesterol transport. Curr. Opin. Lipidol. 21, 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva R. A.; Hilliard G. M.; Fang J.; Macha S.; Davidson W. S. (2005) A three-dimensional molecular model of lipid-free apolipoprotein A-I determined by cross-linking/mass spectrometry and sequence threading. Biochemistry 44, 2759–2769. [DOI] [PubMed] [Google Scholar]
- Pollard R. D.; Fulp B.; Samuel M. P.; Sorci-Thomas M. G.; Thomas M. J. (2013) The conformation of lipid-free human apolipoprotein A-I in solution. Biochemistry 52, 9470–9481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei X.; Atkinson D. (2011) Crystal structure of C-terminal truncated apolipoprotein A-I reveals the assembly of high density lipoprotein (HDL) by dimerization. J. Biol. Chem. 286, 38570–38582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segrest J. P.; Jones M. K.; Catte A.; Thirumuruganandham S. P. (2012) Validation of previous computer models and MD simulations of discoidal HDL by a recent crystal structure of apoA-I. J. Lipid Res. 53, 1851–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalé L.; Skeel R.; Bhandarkar M.; Brunner R.; Gursoy A.; Krawetz N.; Phillips J.; Shinozaki A.; Varadarajan K.; Schulten K. (1999) NAMD2: Greater scalability for parallel molecular dynamics. J. Comput. Phys. 151, 283–312. [Google Scholar]
- Catte A.; Patterson J. C.; Jones M. K.; Jerome W. G.; Bashtovyy D.; Su Z.; Gu F.; Chen J.; Aliste M. P.; Harvey S. C.; Li L.; Weinstein G.; Segrest J. P. (2006) Novel changes in discoidal high density lipoprotein morphology: A molecular dynamics study. Biophys. J. 90, 4345–4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M. K.; Catte A.; Patterson J. C.; Gu F.; Chen J.; Li L.; Segrest J. P. (2009) Thermal stability of apolipoprotein A-I in high-density lipoproteins by molecular dynamics. Biophys. J. 96, 354–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frishman D.; Argos P. (1995) Knowledge-based protein secondary structure assignment. Proteins 23, 566–579. [DOI] [PubMed] [Google Scholar]
- Chen Z. A.; Jawhari A.; Fischer L.; Buchen C.; Tahir S.; Kamenski T.; Rasmussen M.; Lariviere L.; Bukowski-Wills J. C.; Nilges M.; Cramer P.; Rappsilber J. (2010) Architecture of the RNA polymerase II-TFIIF complex revealed by cross-linking and mass spectrometry. EMBO J. 29, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez A. J.; Oram J. F.; Bierman E. L. (1991) Protein kinase C as a mediator of high density lipoprotein receptor-dependent efflux of intracellular cholesterol. J. Biol. Chem. 266, 10104–10111. [PubMed] [Google Scholar]
- Shao B.; Pennathur S.; Pagani I.; Oda M. N.; Witztum J. L.; Oram J. F.; Heinecke J. W. (2010) Modifying apolipoprotein A-I by malondialdehyde, but not by an array of other reactive carbonyls, blocks cholesterol efflux by the ABCA1 pathway. J. Biol. Chem. 285, 18473–18484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinecke J. W.; Baker L.; Rosen H.; Chait A. (1986) Superoxide-mediated modification of low density lipoprotein by arterial smooth muscle cells. J. Clin. Invest. 77, 757–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Requena J. R.; Fu M. X.; Ahmed M. U.; Jenkins A. J.; Lyons T. J.; Baynes J. W.; Thorpe S. R. (1997) Quantification of malondialdehyde and 4-hydroxynonenal adducts to lysine residues in native and oxidized human low-density lipoprotein. Biochem. J. 322(Part 1), 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao B.; Heinecke J. W. (2008) Using tandem mass spectrometry to quantify site-specific chlorination and nitration of proteins: Model system studies with high-density lipoprotein oxidized by myeloperoxidase. Methods Enzymol. 440, 33–63. [DOI] [PubMed] [Google Scholar]
- Gursky O.; Atkinson D. (1996) Thermal unfolding of human high-density apolipoprotein A-1: Implications for a lipid-free molten globular state. Proc. Natl. Acad. Sci. U.S.A. 93, 2991–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetty P. S.; Mayne L.; Lund-Katz S.; Stranz D.; Englander S. W.; Phillips M. C. (2009) Helical structure and stability in human apolipoprotein A-I by hydrogen exchange and mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 106, 19005–19010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerstedt J. O.; Budamagunta M. S.; Liu G. S.; DeValle N. C.; Voss J. C.; Oda M. N. (2012) The “β-clasp” model of apolipoprotein A-I: A lipid-free solution structure determined by electron paramagnetic resonance spectroscopy. Biochim. Biophys. Acta 1821, 448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalisman N.; Adams C. M.; Levitt M. (2012) Subunit order of eukaryotic TRiC/CCT chaperonin by cross-linking, mass spectrometry, and combinatorial homology modeling. Proc. Natl. Acad. Sci. U.S.A. 109, 2884–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda M. N.; Forte T. M.; Ryan R. O.; Voss J. C. (2003) The C-terminal domain of apolipoprotein A-I contains a lipid-sensitive conformational trigger. Nat. Struct. Biol. 10, 455–460. [DOI] [PubMed] [Google Scholar]
- Lemmon M. A.; Flanagan J. M.; Hunt J. F.; Adair B. D.; Bormann B. J.; Dempsey C. E.; Engelman D. M. (1992) Glycophorin A dimerization is driven by specific interactions between transmembrane alpha-helices. J. Biol. Chem. 267, 7683–7689. [PubMed] [Google Scholar]
- Breiter D. R.; Kanost M. R.; Benning M. M.; Wesenberg G.; Law J. H.; Wells M. A.; Rayment I.; Holden H. M. (1991) Molecular structure of an apolipoprotein determined at 2.5-Å resolution. Biochemistry 30, 603–608. [DOI] [PubMed] [Google Scholar]
- Baldwin R. L.; Rose G. D. (2013) Molten globules, entropy-driven conformational change and protein folding. Curr. Opin. Struct. Biol. 23, 4–10. [DOI] [PubMed] [Google Scholar]
- Jayaraman S.; Cavigiolio G.; Gursky O. (2012) Folded functional lipid-poor apolipoprotein A-I obtained by heating of high-density lipoproteins: Relevance to high-density lipoprotein biogenesis. Biochem. J. 442, 703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao B.; Oda M. N.; Vaisar T.; Oram J. F.; Heinecke J. W. (2006) Pathways for oxidation of high-density lipoprotein in human cardiovascular disease. Curr. Opin. Mol. Ther. 8, 198–205. [PubMed] [Google Scholar]
- Bashtovyy D.; Jones M. K.; Anantharamaiah G. M.; Segrest J. P. (2011) Sequence conservation of apolipoprotein A-I affords novel insights into HDL structure-function. J. Lipid Res. 52, 435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerstedt J. O.; Budamagunta M. S.; Liu G. S.; Devalle N. C.; Voss J. C.; Oda M. N. (2012) The “β-clasp” model of apolipoprotein A-I: A lipid-free solution structure determined by electron paramagnetic resonance spectroscopy. Biochim. Biophys. Acta 1821, 448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.