

The ability to use CT and MR imaging with extracellular agents to identify and differentiate cirrhotic nodules, low-grade dysplastic nodules, high-grade dysplastic nodules, and early hepatocellular carcinomas (HCCs) is limited; hepatobiliary phase MR imaging shows promise for characterization of precursor lesions and for identifying high-grade dysplastic nodules and early HCCs prior to neoarterialization and progression to overt HCC.
Abstract
Computed tomography (CT) and magnetic resonance (MR) imaging play critical roles in the diagnosis and staging of hepatocellular carcinoma (HCC). The first article of this two-part review discusses key concepts of HCC development, growth, and spread, emphasizing those features with imaging correlates and hence most relevant to radiologists; state-of-the-art CT and MR imaging technique with extracellular and hepatobiliary contrast agents; and the imaging appearance of precursor nodules that eventually may transform into overt HCC.
© RSNA, 2014
Learning Objectives:
After reading the article and taking the test, the reader will be able to:
■ Discuss key concepts of hepatocellular carcinoma (HCC) development, growth, and spread
■ Discuss CT and MR imaging technique for diagnosis and staging of HCC
■ Describe characteristic CT and MR imaging features of precursor nodules
■ Discuss the limitations of CT and MR imaging for differentiation of precursor nodules
Accreditation and Designation Statement
The RSNA is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to provide continuing medical education for physicians. The RSNA designates this journal-based activity for a maximum of 1.0 AMA PRA Category 1 Credit TM. Physicans should claim only the credit commensurate with the extent of their participation in the activity.
Disclosure Statement
The ACCME requires that the RSNA, as an accredited provider of CME, obtain signed disclosure statements from the authors, editors, and reviewers for this activity. For this journal-based CME activity, author disclosures are listed at the end of this article.
Introduction
Hepatocellular carcinoma (HCC) is an epithelial tumor originating in the liver and composed of cells with characteristics similar to those of normal hepatocytes (1). It is the fifth most common tumor in the world, and its incidence is increasing, especially in Western nations (2). Cirrhosis is the most important clinical risk factor for HCC, with approximately 80% of cases of HCC developing in patients with a cirrhotic liver (3). In such patients, the annual incidence of HCC ranges from 2% to 8% (4,5). The exact incidence depends on the cause of cirrhosis (highest incidence in those infected with hepatitis C virus or hepatitis B virus), severity of cirrhosis (highest incidence in those with decompensated cirrhosis), geographic region (higher in Japan than in Europe or United States), and sex (higher in men than women). The risk is greater in individuals with multiple risk factors as well as in those coinfected with human immunodeficiency virus (6). Patients without cirrhosis also may develop HCC, especially those with long-standing chronic liver inflammation due to hepatitis B virus or hepatitis C virus infection (7) or nonalcoholic steatohepatitis (8), but at a much lower rate than those with cirrhosis. Other risk factors for HCC include heavy alcohol consumption, tobacco smoking (9), obesity, diabetes, hereditary hemochromatosis, high dietary consumption of aflatoxins, and family history of HCC (6). Importantly, cirrhosis and chronic hepatitis now are recognized as risk factors for intrahepatic cholangiocarcinoma (ICC) as well as HCC (10); thus, many patients at risk for HCC may develop ICC instead.
The prognosis of HCC depends largely on the stage at which the tumor is detected. Patients who present with symptoms generally have a dismal prognosis, as HCC usually does not produce symptoms beyond those of the underlying liver disease until it has become incurable; in such patients, median survival is less than 1 year and the 5-year survival is less than 10% (11). By comparison, patients in whom HCC is detected at an early stage may benefit from life-prolonging, potentially curative treatments.
The detection of HCC early in its development, therefore, is critical to improve the survival of affected patients. To this end, scientific societies have released clinical management guidelines that advocate surveillance of patients at risk due to cirrhosis or chronic viral hepatitis (4,12–14). While the surveillance strategies incorporated by the various guidelines differ, all current guidelines recommend ultrasonography (US) as the primary imaging test for surveillance, and two guidelines advocate the ancillary use of serum biomarkers (4,14). In general, neither computed tomography (CT) nor magnetic resonance (MR) imaging are advocated for surveillance, although three guidelines permit these modalities for surveillance of patients in whom US is limited by obesity or other factors (4,13,14) and for those at very high risk for HCC development (4). Once a surveillance test is positive (ie, an abnormality is detected that may represent HCC), a more definitive imaging examination is recommended for noninvasive diagnosis and staging of HCC. Currently, all guidelines endorse multiphasic CT and MR imaging with extracellular agents as first-line modalities for this purpose (4,12–14), although guidelines in Japan also advocate MR imaging with gadoxetate disodium (a hepatobiliary agent) as a second-line modality (4).
In this review, we discuss the current state of the art for the imaging-based diagnosis and staging of HCC, focusing on CT and MR imaging, as these are the most commonly used modalities for these purposes. The review is divided into two articles. This first article discusses key concepts of HCC development, growth, and spread, emphasizing those features with imaging correlates and hence most relevant to radiologists; CT and MR imaging technique; and the imaging appearance of precursor nodules that eventually may transform into overt HCC. The second article, to be presented in an upcoming issue, will build on these concepts and review in detail the diagnosis and staging of HCC with CT and MR imaging. Other modalities (eg, US, contrast-enhanced US, CT hepatic angiography, and CT during arterial portography) and other roles of imaging (eg, screening and surveillance of at risk-patients, guiding biopsy and local therapy, treatment and surgical planning, and assessing therapeutic response) are beyond the scope of this review and are discussed elsewhere (15,16).
HCC Development, Growth, and Spread
Hepatocarcinogenesis, the gradual transformation of nonmalignant liver cells into HCC, is a complex, multistep process characterized at the molecular and cellular level by the progressive accumulation of epigenetic and genetic alterations (17) and at the histologic level by the emergence and progression of successively more advanced precancerous, early cancerous, and overtly malignantly lesions (18,19).
Hepatocarcinogenesis: Molecular and Cellular Mechanisms
The molecular and cellular mechanisms underlying the transformation of initially nonmalignant cells into HCC in chronic liver disease are not yet fully elucidated (20). Growing evidence suggests that chronic inflammation plays a pivotal role by causing repeated cycles of cell injury, death, and regeneration–an environment that promotes aberrant cell signaling, epigenetic changes, mutational events, and accumulation of genetic damage (11,20–22). These molecular alterations begin during a prolonged preneoplastic phase, years or decades before cirrhosis is established, and progress in parallel with the evolution of fibrosis and cirrhosis (23,24). During this phase, the alterations are mainly due to epigenetic mechanisms (eg, changes in gene expression) with few or no structural changes in the genes or chromosomes (11). The earliest molecular changes of hepatocarcinogenesis are morphologically silent (24–26). Thus, the chronically diseased liver may contain scattered clonal populations of molecularly aberrant but phenotypically normal cells (24) that ultimately may progress to HCC. Subsequently, a neoplastic phase ensues in which structural alterations (eg, point mutations, allelic deletions, chromosomal gains and transpositions) in these aberrant cells escalate (11). During this phase, affected cells acquire progressively atypical phenotypic features and evolve through cellular intermediates to frank malignancy (22). The genomic changes underlying hepatocarcinogenesis are heterogeneous, and diverse combinations of aberrant genes and regulatory pathways may be involved (1,8,11,21,27). Several molecular variants of HCC may be produced–potentially with different growth properties and clinical course–between patients, between different tumors in the same liver, and within different regions in the same tumor (22,27). Although most HCCs develop in cirrhotic livers, cirrhosis is probably not a premalignant condition per se but rather a parallel process that develops over time in response to the same insults that promote hepatocarcinogenesis (6,24). The development of HCC usually is slower than that of cirrhosis; hence, most HCCs emerge after cirrhosis has been established.
The cell of origin of HCC is controversial. Historically, it was assumed that most HCCs arose from dedifferentiation of mature hepatocytes. Emerging data suggest that HCCs also may develop from intrahepatic stem cells (1,11). These stem cells reside in niches within the canals of Hering and, according to new models of liver carcinogenesis, can be activated to differentiate into hepatocytes or cholangiocytes while undergoing oncogenic stimulation in the context of chronic liver injury (28). The resulting tumors usually have predominantly hepatocellular (ie, HCC) or cholangiocellular (ie, ICC) phenotypic features, respectively, although some tumors may arise with combined or mixed features. If correct, these new models help to explain why patients with cirrhosis are at risk for developing ICCs and combined tumors in addition to HCCs.
Hepatocarcinogenesis: Pathologic Changes
Pathologically, multistep hepatocarcinogenesis is characterized by progressive dedifferentiation of phenotypically abnormal nodular lesions (18,19) (Fig 1). The evolution is driven by the repeated development and expansion of successively less differentiated clonal populations, often manifesting as subnodules within parent nodules (29). Over time, the less differentiated populations grow and completely replace the more differentiated surrounding tissues. Repeated cycles of clonal development and expansion eventually produce lesions with malignant phenotype. The process represents a biologic continuum but is arbitrarily divided into discrete steps for simplicity, clinical utility, and investigation (30). Importantly, the process may occur simultaneously at different rates in different parts of the liver (multicentric hepatocarcinogenesis). It should be emphasized that although most HCCs probably evolve from histologically abnormal precursor lesions, it is possible that many HCCs, especially those arising in noncirrhotic livers, may develop from transformed malignant cells without transitioning through histologically definable intermediate steps. The development of HCC without identifiable histologic precursors is termed “de novo hepatocarcinogenesis” (31).
Figure 1:

Hemodynamic and OATP expression changes during multistep hepatocarcinogenesis. Schematic drawing illustrates typical changes in intranodular hemodynamics and OATP expression during multistep hepatocarcinogenesis. As shown, multistep hepatocarcinogenesis is characterized by successive selection and expansion of less-differentiated subnodules within more well differentiated parent nodules. The subnodules grow and eventually replace (blue arrows) the parent nodules. Progressed HCCs show expansile growth (red arrows) and characteristically are encapsulated with fibrous septa. Earlier nodules lack these structures and show replacing growth. During hepatocarcinogenesis, the density of portal triads diminishes while the density of unpaired arteries increases. The net effect is that intranodular arterial supply diminishes initially and then increases (bottom graph); progressed HCCs typically show arterial hypervascularity compared with background liver, while earlier nodules typically do not. OATP expression usually diminishes progressively (top graph); progressed HCCs, early HCCs, many high-grade dysplastic nodules, and some low-grade dysplastic nodules show OATP underexpression compared with background liver. The shaded area in each graph represents the window of opportunity to detect nodules at different stages of tumor development based on net arterial flow or OATP expression; window of opportunity is larger and begins at earlier stages for OATP expression. Note that illustrations and graphs reflect typical changes in hemodynamics and OATP expression. Not all nodules exhibit the illustrated characteristics. Also note that during tumor development some stages may be skipped and not all HCCs arise from histologically definable precursor lesions. (Illustration by Matt Skalski, MD; copyright 2014, RSNA.)
Cirrhotic nodules.—Cirrhotic nodules, also known as cirrhosis-associated regenerative nodules, are innumerable well-defined rounded regions of the cirrhotic parenchyma surrounded by scar tissue and typically measuring 1–15 mm in diameter (30). Cirrhotic nodules larger than 1 cm are called “large cirrhotic nodules” or “large regenerative nodules.” Grossly and microscopically, cirrhotic nodules are indistinguishable from other cirrhotic nodules–in other words, all cirrhotic nodules in a given liver resemble each other and no cirrhotic nodule stands out as being distinctive from the others. Cirrhotic nodules lack clonal features histologically, and the cells are phenotypically normal (32). For these reasons, cirrhotic nodules usually are considered “benign.” The “benignity” of cirrhotic nodules is not unqualified, however. Based on molecular analyses, many cirrhotic nodules are clonal expansions of genomically aberrant cells (26), and hepatocytes within cirrhotic nodules may develop dysplastic features and thus give rise to dysplastic foci and nodules. As discussed below, most cirrhotic nodules are not discernible as individual lesions at in vivo imaging.
Dysplastic foci.—Dysplastic foci are microscopic lesions, arbitrarily less than 1 mm in diameter, composed of hepatocytes with precancerous features such as small cell change (6) arising within cirrhotic nodules or, if the liver is noncirrhotic, within single lobules (27). These lesions are identified incidentally at histologic evaluation and not detectable by means of in vivo imaging. As it is not possible to detect or follow them in vivo, their natural history is poorly understood. It is presumed that dysplastic foci may expand to become dysplastic nodules.
Dysplastic nodules.—Dysplastic nodules are nodular lesions, usually 1–1.5 cm in diameter, that differ in both macroscopic (size, color, or consistency) and microscopic appearance from background parenchyma (19). They are observed in up to 25% of cirrhotic livers but occasionally are detected in noncirrhotic livers and often are multiple (23). Dysplastic nodules are classified as low grade or high grade, depending on the presence of cytologic and architectural atypia (33).
Histologically, low-grade dysplastic nodules resemble cirrhotic nodules. The hepatocytes show no cytologic atypia (33), and neither expansile subnodules nor architectural alterations beyond those of cirrhotic nodules are observed (33,34). Findings that if present distinguish low-grade dysplastic nodules from cirrhotic nodules include unpaired arteries and clonelike populations (aggregates of cells with greater copper, iron, or fat accumulation than background liver) (6,19,33). High-grade dysplastic nodules resemble well-differentiated HCCs. The cells show cellular atypia, although the atypia is insufficient to establish a diagnosis of HCC, and may exhibit clonelike features. Architectural alterations, including arrangement of hepatocytes in thin trabeculae and pseudoglands, may be present (33,34). Expansile subnodules with varying degrees of atypia may be observed (6). Clinically, low-grade dysplastic nodules are considered preneoplastic lesions with slightly elevated risk of malignant transformation, while high-grade dysplastic nodules are considered advanced precursors of HCC with high risk of transformation (19,35). Some high-grade dysplastic nodules contain one or more subnodules of well-differentiated or moderately differentiated HCC (“nodule-in-nodule” configuration) (36); these are appropriately categorized as “HCC arising in high-grade dysplastic nodule” (6). As discussed below, CT and MR imaging have limited ability to identify and characterize dysplastic nodules.
Early HCCs
Early HCCs are an incipient stage of HCC development, analogous to “carcinoma in situ” or “microinvasive carcinoma” of other organs (37). Unlike overt progressed HCC, which displaces or destroys the liver parenchyma (see below), early HCCs grow by gradually replacing the parenchyma; as the cells spread, they surround neighboring portal tracts and central veins but do not displace or completely destroy these structures. Early HCCs typically measure 1–1.5 cm in diameter and rarely exceed 2 cm. Macroscopically, most early HCCs are vaguely nodular with indistinct margins and without a tumor capsule. Due to their small size and macroscopic appearance, these lesions frequently are termed “vaguely nodular small HCCs” or “small HCCs with indistinct margins.” (32). The lesions are indistinguishable from high-grade dysplastic nodule at gross pathologic examination (36). Histologically, early HCCs consist of small, well-differentiated neoplastic cells (36) arranged in irregular but thin trabeculae or pseudoglands (19). Thus the microscopic appearance closely resembles that of high-grade dysplastic nodules. The key distinguishing feature, present in early HCCs but not in high-grade dysplastic nodule, is stromal invasion, defined as infiltration of tumor cells into the fibrous tissue surrounding portal tracts retained within the nodule or into the stromal fibrous tissue surrounding the nodule (33). While stromal invasion is characteristic, vascular invasion is not observed (38), and intrahepatic metastasis is exceedingly rare. Early HCCs are considered precursors of progressed HCCs (19), although the rate at which they transform to progressed HCC has not been defined (39). Moreover, some progressed HCCs probably do not develop from early HCCs but rather arise as expansile subnodules within high-grade dysplastic nodules without transitioning through a vaguely nodular morphology. As will be discussed further in part 2 of this review, conventional CT and MR imaging have limited sensitivity for the detection of early HCCs, but hepatobiliary phase MR imaging shows promise for this purpose.
Progressed HCCs
Progressed HCCs are overtly malignant lesions with the ability to invade vessels and metastasize. Macroscopic and histologic features are variable, depending in part on lesion size.
Progressed HCCs smaller than 2 cm are distinctly nodular with well-defined margins; synonymous terms include “small and progressed HCCs” and “small distinctly nodular HCCs.” Unlike early HCCs, small and progressed HCCs grow by expanding into and compressing the adjacent parenchyma. Characteristically, they are surrounded by a tumor capsule and contain internal fibrous septa. Histologically, about 80% of small and progressed HCCs are moderately differentiated; the remaining 20% consist of both well-differentiated and moderately differentiated components (36). Architectural abnormalities include thickened plates more than three cells wide (30) and arrangement of hepatocytes in trabecular/platelike, pseudoglandular/acinar, or solid/compact patterns. A significant proportion of small and progressed HCCs are associated with vascular invasion and intrahepatic metastasis (19).
HCCs exceeding 2 cm in diameter are known as “large HCCs.” Compared with small and progressed HCCs, large HCCs tend to have higher histologic grade, more aggressive biologic behavior, and higher frequency of vascular invasion and metastasis. For these reasons, it is clinically important to develop imaging techniques that can be used to accurately diagnose small HCCs prior to their growth beyond 2 cm. Macroscopically, most large HCCs are expansile tumors with nodular morphology and surrounded by tumor capsules. Mosaic architecture is characteristic, defined by the presence of multiple internal tumor nodules separated by fibrous septations and areas of hemorrhage, necrosis, and occasionally fatty metamorphosis (40). Histologically, the internal tumor nodules may differ in grade, microscopic architectural pattern, and cytologic type (6). Molecularly, they may differ in epigenetic and genetic abnormalities (41). At least some of these nodules appear to arise through clonal divergence from common precursor clonal populations (42,43). About 5% of large HCCs have an infiltrative rather than an expansile growth pattern (11); these cancers usually are composed of poorly differentiated or undifferentiated cancer cells that spread into the surrounding sinusoids and cell plates, causing the tumor boundary to be ill defined (4,11).
Multifocal HCC
In more than one-third of patients, HCC is multifocal (44), defined by the presence of tumor nodules unmistakably separated by intervening nonneoplastic parenchyma (6). Multifocality may be due to synchronous development of multiple, independent liver tumors (multicentric hepatocarcinogenesis) or intrahepatic metastases from a primary tumor (11). In the former case, the tumors may vary in histologic grade and other features; in the latter case, all the tumors are progressed lesions with advanced tumor grade. The prognosis of patients with multifocal HCC due to intrahepatic metastasis tends to be worse than in those with multicentric development of independent tumors (45). Patients with HCC also are at high risk for future development of new tumors. Due to the high frequency with which multiple tumors may develop, patients with HCC sometimes cannot be cured by means of surgical resection or ablation; as long as the cancer has not spread outside the liver, hepatic transplantation may provide more prolonged survival.
Key Alterations during Hepatocarcinogenesis
Numerous pathophysiologic alterations accompany hepatocarcinogenesis, as summarized in Table 1 and discussed below.
Table 1.
Key Alterations during Hepatocarcinogenesis and Their Imaging Implications
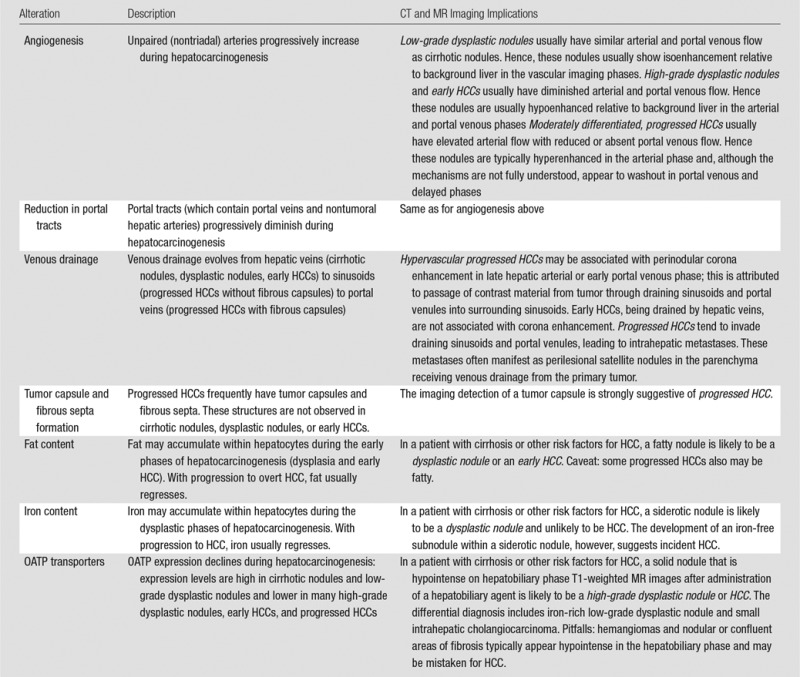
Angiogenesis
Angiogenesis, the formation and development of blood vessels, progresses during hepatocarcinogenesis. Histologically, angiogenesis is characterized by the presence of unpaired (or nontriadal) arteries and sinusoidal capillarization (28,29). Unpaired arteries are isolated arteries unaccompanied by bile ducts or portal veins. These abnormal arteries are absent in cirrhotic nodules, are sometimes present in small numbers in low-grade dysplastic nodules, and are present with increasing size and number in high-grade dysplastic nodules, early HCCs, and progressed HCCs (67) (Fig 1). Sinusoidal capillarization refers to alterations in the sinusoidal endothelium, including loss of fenestrae and deposition of a basement membrane, that make the sinusoids resemble systemic capillaries (46). Sinusoidal capillarization is minimal in cirrhotic nodules and low-grade dysplastic nodules and is successively more prominent in high-grade dysplastic nodules, early HCCs, and progressed HCCs. In parallel with these changes, the portal tracts (which contain portal veins and nontumoral hepatic arteries) progressively diminish: Portal tracts are normal in number in cirrhotic nodules and low-grade dysplastic nodules, reduced in number in high-grade dysplastic nodules and early HCCs, and virtually absent in progressed HCCs (47).
Physiologically, the diminution in portal tracts causes a gradual reduction in arterial and portal venous flow to the nodule, while the formation of unpaired neo-arteries increases arterial flow (28,29). The balance is such that in the early phases of hepatocarcinogenesis, there is a net decrease in intranodular arterial flow and preservation of portal venous flow, while in the later phases, portal blood flow declines and eventually becomes absent while net arterial flow increases (Fig 1). Thus, low-grade dysplastic nodules usually have a preserved vascular profile similar to that of background cirrhotic nodules; high-grade dysplastic nodules and early HCCs tend to have diminished arterial and portal venous flow; and moderately differentiated, progressed HCCs usually have elevated arterial flow with reduced or absent portal venous flow. With tumor growth beyond 5 cm or with further dedifferentiation and progression to poorly differentiated HCC, however, arterial flow may diminish (48). One explanation is that in very advanced HCCs, rapid cell proliferation in the tumor center elevates interstitial pressure, causing compression closure of tumor capillaries, and regression of neo-arteries (49).
Venous Drainage
Venous drainage evolves during hepatocarcinogenesis from hepatic veins (cirrhotic nodules, dysplastic nodules, early HCCs) to sinusoids (progressed HCCs without fibrous capsules) to portal veins (progressed HCCs with fibrous capsules) (50,51). The transition to portal venous drainage may explain in part the predilection of HCC to invade into and disseminate via portal compared with hepatic veins (50); as discussed further below, the metastases resulting from vascular invasion often manifest as satellite nodules within the venous drainage area of the primary tumor. The evolution in venous drainage also may explain the phenomenon of corona enhancement. As discussed in part II, corona enhancement is an imaging feature of hypervascular, progressed HCC. It refers to enhancement of the peritumoral parenchyma that begins a few seconds after enhancement of the tumor itself. It is attributed to passage of contrast material from the tumor through the draining sinusoids and portal venules into the surrounding parenchymal sinusoids, with which the drainage vessels communicate. Early HCCs drain via hepatic veins, not sinusoids or portal venules, and hence do not manifest corona enhancement (52).
Tumor Capsule and Fibrous Septa
Tumor capsules and fibrous septa are other features that develop during hepatocarcinogenesis. These structures are not observed in cirrhotic nodules, dysplastic nodules, or early HCCs but are characteristic of nodular progressed HCC, being observed in about 70% of these lesions (42). The capsule surrounds the tumor and consists of two layers. The inner layer is composed of tight, relatively pure fibrous tissue containing thin, slit-like vascular channels. The outer layer is composed of looser fibrovascular tissue containing portal venules, newly formed bile ducts, and prominent sinusoids (53,54). Blood drains from the tumor to the adjacent parenchyma through the capsule via complex communications between tumor sinusoids, intracapsular vessels and vascular channels, and perinodular portal venules and sinusoids. Fibrous septa are intratumoral fibrous bands at the interface between HCC subnodules or between areas of necrosis and HCC tissue (55). The mechanism of tumor capsule and fibrous septa formation is not fully understood. Studies suggest that host mesenchymal cells, not HCC cells, elaborate the extracellular matrix components of these structures (55), possibly in response to compression of liver parenchyma by expansile tumor (11,56) as well as host-tumor interactions (63).
Clinical investigations have shown that, after adjusting for tumor size and grade, HCCs with intact tumor capsules are associated with lower recurrence rates after resection or ablative therapy than HCCs without intact capsules (57), suggesting that the tumor capsule impedes HCC dissemination. The tight inner layer is thought to acts as a physical barrier that confines cells within the tumor margin, while the narrow transcapsular vascular channels may block tumor cells that have accessed the vascular lumen from embolizing downstream (58). It should be emphasized, however, that capsule formation is a feature of advanced HCC; thus while HCCs with intact capsules have a more favorable prognosis than HCCs of similar size and grade without capsules or with disrupted capsules, they have a worse prognosis than early HCCs, which are unencapsulated (59).
Fat Content
During the early phases of hepatocarcinogenesis, hepatocytes may accumulate fat, and low-grade dysplastic nodules, high-grade dysplastic nodules, and early HCCs may become more steatotic (60)—either focally within clonal-like subnodules or diffusely (19)—than background liver. The frequency of diffuse intranodular steatosis increases from low-grade dysplastic nodule to high-grade dysplastic nodule and then to early HCC, with 40% of early HCCs being diffusely steatotic (61). The frequency of diffuse steatosis peaks in early HCCs about 1.5 cm in diameter (61) and declines with increasing tumor size and grade (47,61). Thus, diffuse fatty change is uncommon in HCCs larger than 3 cm and in progressed HCCs (33,61); it usually is not observed in poorly differentiated HCC.
The mechanism underlying fat accumulation in early hepatocarcinogenesis is not fully understood. The prevailing hypothesis is that during the transition phase from portal to arterial supply, there is a period in which the development of unpaired arteries is not yet sufficient to compensate for the reduced portal venous and nontumoral arterial flow (61). The resulting ischemic/hypoxic environment is thought to induce hepatocellular fat accumulation. With tumor progression, unpaired arteries become more fully developed, the ischemic/hypoxic conditions resolve, and the steatosis regresses (42).
Recently, a steatohepatitic variant of HCC has been described. This variant has histologic features resembling those of steatohepatitis in nonneoplastic liver (eg, steatosis, inflammatory infiltrates, hepatocellular ballooning, Mallory-Denk bodies, and pericellular fibrosis) and appears to occur most commonly in patients with underlying steatohepatitis (62). In one study, all 16 steatohepatic HCCs showed moderate to severe steatosis, and 14 of 16 (87%) were poorly differentiated (62). Thus unlike conventional HCCs, in which steatosis regresses with tumor progression, steatosis may be a prominent feature in steatohepatitic HCC with advanced tumor grade.
Iron Content
In cirrhotic livers without diffuse iron deposition, iron may accumulate preferentially in low-grade dysplastic nodules and some high-grade dysplastic nodules (33). These iron-rich nodules commonly are described as “siderotic nodules.” The iron accumulation by these nodules is thought to reflect clonal expansion of hepatocytes with iron avidity (63). With further dedifferentiation, hepatocytes become “resistant” to iron accumulation, and most high-grade dysplastic nodules, early HCCs, and progressed HCCs are iron free, including high-grade dysplastic foci and subnodules of HCC developing within otherwise siderotic precursor nodules (64). This iron resistance has been attributed to greater iron utilization by neoplastic cells (65) or to higher cellular proliferation with consequent dilution of iron within progeny cells. Iron resistance also is observed in dysplastic foci, dysplastic nodules, and HCCs in livers with diffuse hepatic iron overload due to any cause (23). Thus in diffusely iron-overloaded livers, a solid nodule free of iron is likely to be dysplastic or malignant.
OATP Transporters
Organic anionic transporting polypeptides (OATP) are a family of proteins expressed in hepatocytes along the basolateral (sinusoidal) membrane and involved in transport of bile salts (66). One of these transporters, OATP 8 (also known as OATP1B1/3) (67), is thought to be responsible for uptake by human liver cells of two gadolinium-based contrast agents, gadoxetate disodium and gadobenate dimeglumine. Emerging evidence suggests that the expression of these transporters diminishes during hepatocarcinogenesis: Expression levels are high in cirrhotic nodules and low-grade dysplastic nodules and lower in many high-grade dysplastic nodules, early HCCs, and progressed HCCs (67). Importantly, this suggests that OATP8 expression level decreases during hepatocarcinogenesis prior to reduction in portal venous flow (68) and prior to complete neo-arterialization and to elevation of arterial flow (67) (Fig 1), with important implications for imaging-based detection of HCC using hepatobiliary agents as discussed later. Moreover, the degree of OATP8 expression correlates inversely with HCC tumor grade, such that expression levels tend to be lower in higher grade than in lower grade HCCs (67,69). The reduction in OATP8 expression during hepatocarcinogenesis may in part be attributable to expression of hepatocytes nuclear factor 3β, a liver-enriched transcription factor that is overexpressed in 70% of HCCs and that represses the transcription of OATP8 (100). Additionally, the OATP8 expression level in HCCs has been shown to inversely correlate with the presence of biliary phenotypic markers such biliary-type keratin 7 (K7) and keratin 19 (K19) (70); HCCs with these markers may be more aggressive and have worse prognosis (71). Paradoxically, about 5%–12% of moderately differentiated HCCs and rarely some well-differentiated HCCs overexpress OATP8; it has been speculated that the overexpression of OATP8 in these tumors may reflect a different cell of origin or may be due to genomic alterations during hepatocarcinogenesis (67).
MRP Transporters
Multidrug resistance–associated proteins (MRPs) are a family of transporters expressed in hepatocytes under physiologic conditions that play important roles in bile formation and excretion of toxic substances. The presence of cirrhosis upregulates the expression of these transporters to facilitate the elimination of various exogenous substances (72). Gadoxetate disodium and gadobenate dimeglumine are excreted by hepatocytes into the bile by these transporters, predominantly by MRP2 (73), and excreted back into the sinusoids, predominantly by MRP3 (73). Changes in expression levels of excretion transporters during hepatocarcinogenesis have not yet been systematically studied (67).
Spread of HCC
Intrahepatic Metastasis
Although progressed HCCs may spread contiguously into the surrounding liver by expansile or infiltrative growth, the most important mechanism of spread is intrahepatic metastasis. These metastases develop in progressed HCC (but rarely if ever in early HCC) when malignant cells enter portal venules draining the primary tumor and spread into the surrounding parenchyma. (As mentioned earlier, the venous drainage evolves during hepatocarcinogenesis from hepatic venules to portal venules; the portal venules are thought to be the conduits by which progressed HCC metastasizes). The resulting metastases usually manifest as small “satellites” within the venous drainage area around the primary tumor (74). Intrahepatic metastases also may form outside the drainage area, including in other segments or in the contralateral lobe. Some of these remote intrahepatic metastases may arise from tumor cells that entered the systemic circulation and then returned to the liver (75). Rarely, widespread intrahepatic metastases cause the liver to be diffusely studded with minute, uniformly sized, poorly differentiated tumor nodules that at gross pathologic examination may mimic the appearance of cirrhotic nodules (“diffuse” or “cirrhotomimetic” HCC) (6,76). Despite the tendency of HCCs to invade vessels and metastasize within the liver, extrahepatic metastases (lungs, lymph nodes, bones, adrenal glands) (6) are late manifestations (11).
Vascular Invasion
Vascular invasion, the entrance of tumor cells into the lumen of vessels, is a characteristic feature of progressed HCC (77). Such invasion distinguishes HCC from secondary liver cancers, which rarely invade intrahepatic vessels (76). Portal veins are invaded more commonly than hepatic veins; hepatic arteries are not invaded (77). Vascular invasion is classified as microvascular (visible only at microscopy) or macroscopic (visible at gross pathologic examination) (78). Both types of vascular invasion portent a poor prognosis, as they provide the route by which HCC cells access the circulation to metastasize through the liver or systemically. Thus, HCCs with vascular invasion frequently are multifocal and have higher recurrence rates after resection, ablation, and transplantation (79,80). Factors associated with vascular invasion include large tumor size and advanced histologic grade (74). The risk of recurrence is particularly high for patients with macrovascular invasion; for this reason, macrovascular invasion is regarded as a contraindication for surgical resection or liver transplantation (81).
Biliary Invasion
Bile duct invasion is uncommon clinically but reported in 5%–10% of autopsy series (6,42,82). Most cases are associated with infiltrative HCCs or HCCs with macrovascular invasion (42).
Tumor Capsule Invasion
HCC cells that elaborate metalloproteinases may infiltrate into and through the tumor capsule into the surrounding parenchyma (83). Such infiltration increases the risk of vascular invasion and intrahepatic metastasis, and so is associated with poorer prognosis (58,59,83,84).
CT and MR Imaging Technique
CT and MR Imaging with Extracellular Contrast Agents
As discussed further in part II, extracellular contrast agents permit diagnosis of HCC based mainly on the physiologic changes in intranodular blood flow that accompany hepatocarcinogenesis. To comprehensively evaluate these changes, multiphasic examinations are performed with acquisition of images before (precontrast) and dynamically after contrast agent administration (Figs 2, 3). For MR imaging, three-dimensional T1-weighted sequences usually are utilized for dynamic imaging. Typically, contrast agents are administered at rates of 4–6 mL/sec for CT (85) and 2 mL/sec for MR imaging (86) followed by saline infusion to clear residual contrast material from the intravenous tubing and injected vein. The contrast agent dose is usually based on body weight. For CT, 1.5–2 mL per kilogram of body weight (assuming a concentration of 350 mg iodine per milliliter) usually is appropriate to achieve a total iodine dose of 525 mg iodine per kilogram or more. For MR imaging, the dose varies from 0.025 to 0.1 mmol gadolinium per kilogram, depending on the agent and other factors, as discussed later.
Figure 2a:
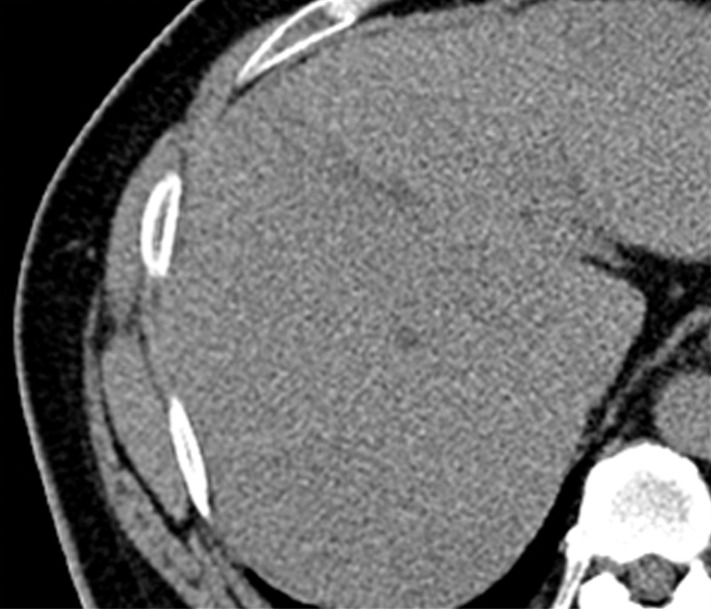
Images in a 51-year-old man with HCC and hepatitis B–related cirrhosis: multiphasic CT technique. (a) There is no discernible lesion on precontrast CT image. (b) Late hepatic arterial phase image shows heterogeneously hyperenhancing mass with mosaic architecture in segment VIII. Notice enhancement of hepatic artery and portal vein branches in late hepatic arterial phase. Hepatic veins are not enhanced. (c, d) Relative to liver, mass de-enhances on (c) portal venous and (d) 3-minute delayed phase images to become isoattenuating with background parenchyma. Mass has capsule appearance in venous phases, shown to best advantage in delayed phase. Notice that hepatic veins are enhanced in portal venous and delayed phases. (e) Gross pathology photograph of resected specimen confirms progressed, encapsulated HCC with expansile growth pattern. Histologic examination showed moderately differentiated tumor (Edmondson grade II). As illustrated in this case, delayed phase may show capsule appearance more clearly than portal venous phase.
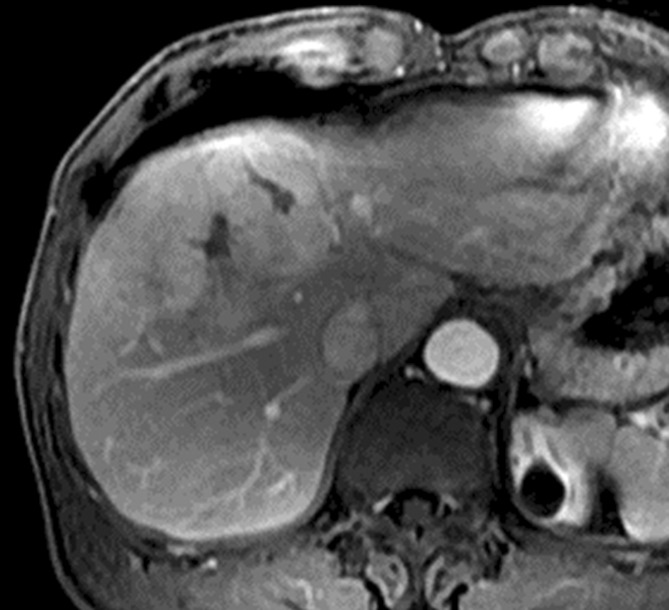
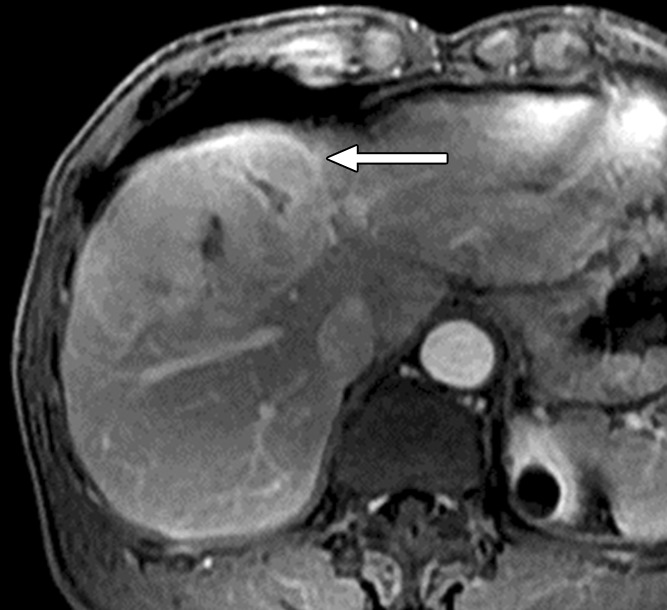
Figure 3a:
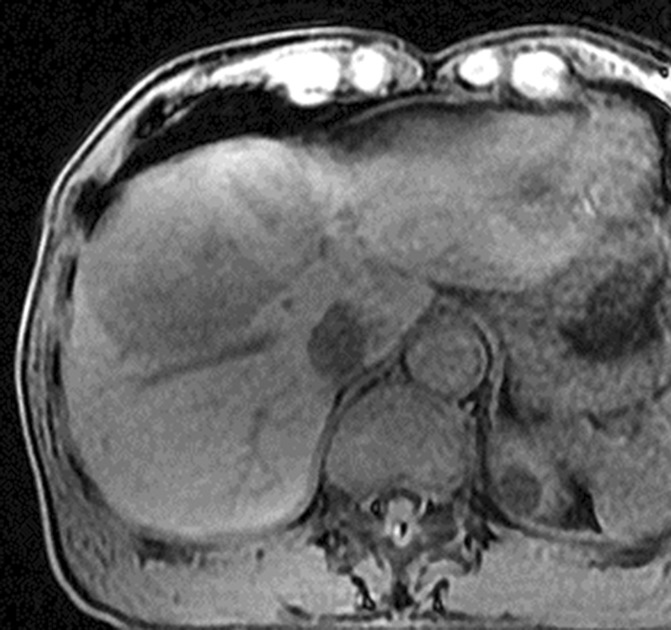
Images in 64-year-old man with HCC and hepatitis B–related cirrhosis: multiphasic MR technique with extracellular contrast agent. (a–d) Gadolinium-enhanced T1-weighted three-dimensional (3D) gradient-echo (GRE) images (repetition time msec/echo time msec, 6.3/1.9; flip angle, 15°) show large hypointense mass on (a) precontrast image with (b) hyperenhancement in late hepatic arterial phase. Notice enhancement of portal vein branches but not of hepatic vein branches in late hepatic arterial phase. (c) Portal venous and (d) 3-minute delayed phase images show persistent enhancement of tumor relative to liver. Persistent enhancement is atypical of large progressed HCCs, which characteristically appear to washout on venous phase images. Notice capsule appearance (arrow) on delayed phase image. Capsule appearance is seen to better advantage on delayed compared with portal venous phase image and permits confident diagnosis of HCC despite lack of washout appearance.
Figure 2b:
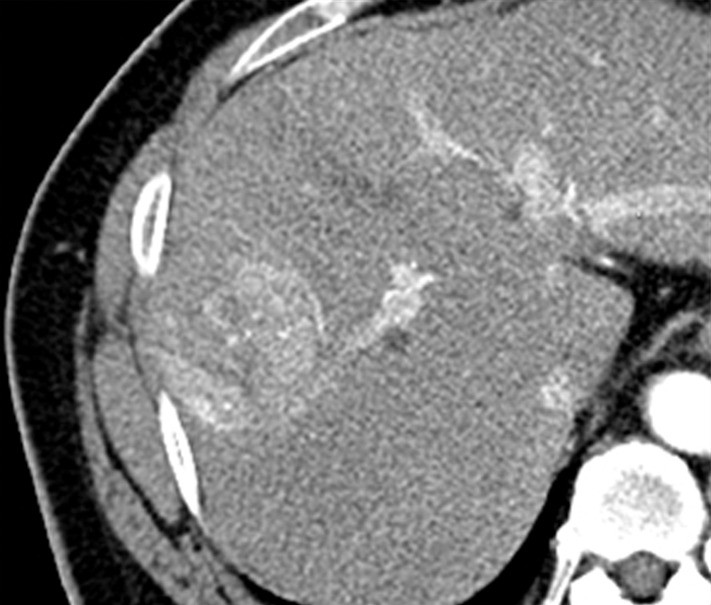
Images in a 51-year-old man with HCC and hepatitis B–related cirrhosis: multiphasic CT technique. (a) There is no discernible lesion on precontrast CT image. (b) Late hepatic arterial phase image shows heterogeneously hyperenhancing mass with mosaic architecture in segment VIII. Notice enhancement of hepatic artery and portal vein branches in late hepatic arterial phase. Hepatic veins are not enhanced. (c, d) Relative to liver, mass de-enhances on (c) portal venous and (d) 3-minute delayed phase images to become isoattenuating with background parenchyma. Mass has capsule appearance in venous phases, shown to best advantage in delayed phase. Notice that hepatic veins are enhanced in portal venous and delayed phases. (e) Gross pathology photograph of resected specimen confirms progressed, encapsulated HCC with expansile growth pattern. Histologic examination showed moderately differentiated tumor (Edmondson grade II). As illustrated in this case, delayed phase may show capsule appearance more clearly than portal venous phase.
Figure 2c:
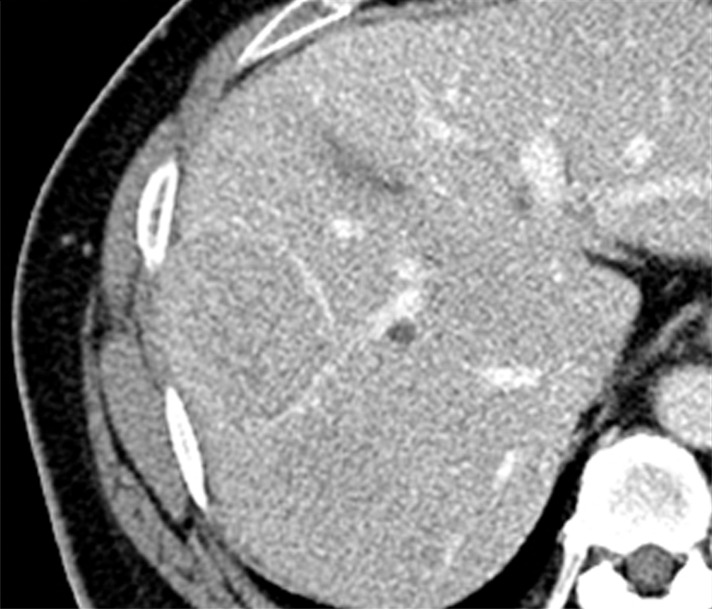
Images in a 51-year-old man with HCC and hepatitis B–related cirrhosis: multiphasic CT technique. (a) There is no discernible lesion on precontrast CT image. (b) Late hepatic arterial phase image shows heterogeneously hyperenhancing mass with mosaic architecture in segment VIII. Notice enhancement of hepatic artery and portal vein branches in late hepatic arterial phase. Hepatic veins are not enhanced. (c, d) Relative to liver, mass de-enhances on (c) portal venous and (d) 3-minute delayed phase images to become isoattenuating with background parenchyma. Mass has capsule appearance in venous phases, shown to best advantage in delayed phase. Notice that hepatic veins are enhanced in portal venous and delayed phases. (e) Gross pathology photograph of resected specimen confirms progressed, encapsulated HCC with expansile growth pattern. Histologic examination showed moderately differentiated tumor (Edmondson grade II). As illustrated in this case, delayed phase may show capsule appearance more clearly than portal venous phase.
Figure 2d:
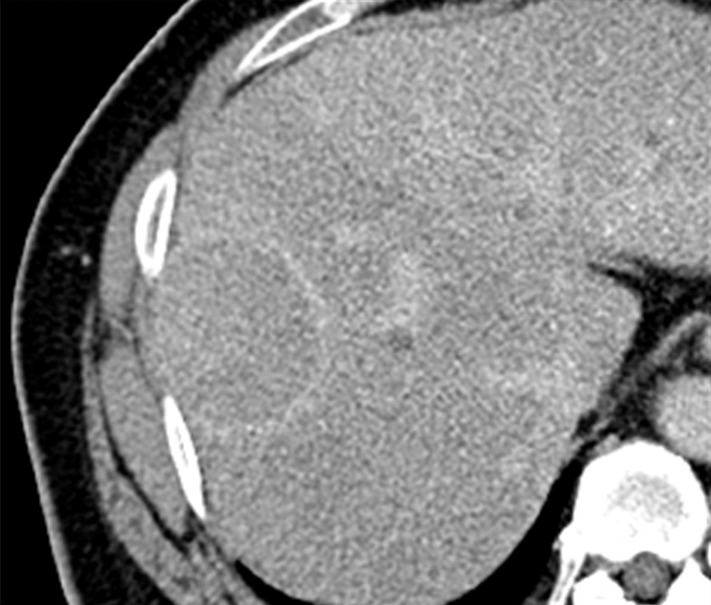
Images in a 51-year-old man with HCC and hepatitis B–related cirrhosis: multiphasic CT technique. (a) There is no discernible lesion on precontrast CT image. (b) Late hepatic arterial phase image shows heterogeneously hyperenhancing mass with mosaic architecture in segment VIII. Notice enhancement of hepatic artery and portal vein branches in late hepatic arterial phase. Hepatic veins are not enhanced. (c, d) Relative to liver, mass de-enhances on (c) portal venous and (d) 3-minute delayed phase images to become isoattenuating with background parenchyma. Mass has capsule appearance in venous phases, shown to best advantage in delayed phase. Notice that hepatic veins are enhanced in portal venous and delayed phases. (e) Gross pathology photograph of resected specimen confirms progressed, encapsulated HCC with expansile growth pattern. Histologic examination showed moderately differentiated tumor (Edmondson grade II). As illustrated in this case, delayed phase may show capsule appearance more clearly than portal venous phase.
Figure 2e:
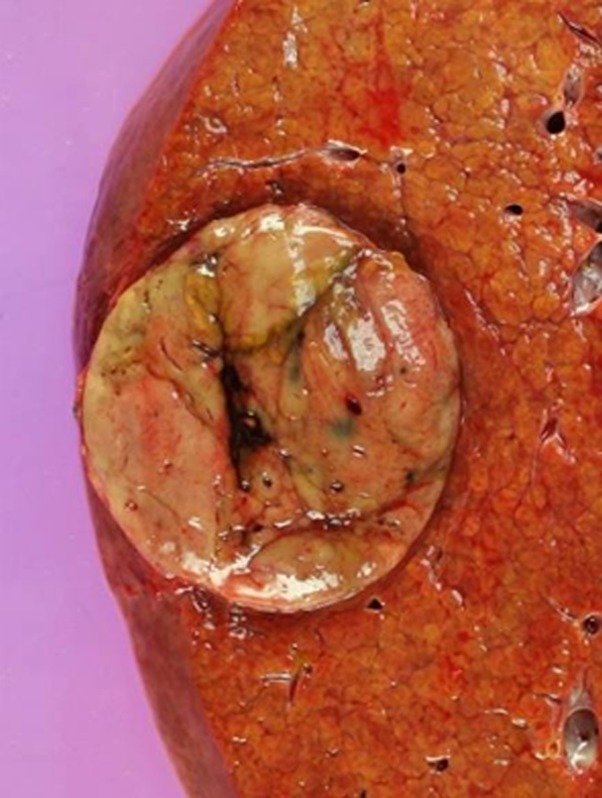
Images in a 51-year-old man with HCC and hepatitis B–related cirrhosis: multiphasic CT technique. (a) There is no discernible lesion on precontrast CT image. (b) Late hepatic arterial phase image shows heterogeneously hyperenhancing mass with mosaic architecture in segment VIII. Notice enhancement of hepatic artery and portal vein branches in late hepatic arterial phase. Hepatic veins are not enhanced. (c, d) Relative to liver, mass de-enhances on (c) portal venous and (d) 3-minute delayed phase images to become isoattenuating with background parenchyma. Mass has capsule appearance in venous phases, shown to best advantage in delayed phase. Notice that hepatic veins are enhanced in portal venous and delayed phases. (e) Gross pathology photograph of resected specimen confirms progressed, encapsulated HCC with expansile growth pattern. Histologic examination showed moderately differentiated tumor (Edmondson grade II). As illustrated in this case, delayed phase may show capsule appearance more clearly than portal venous phase.
Figure 3b:
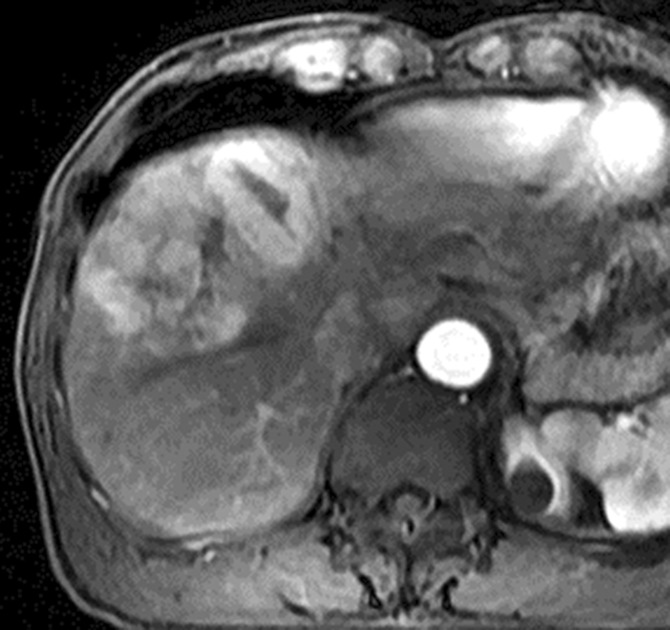
Images in 64-year-old man with HCC and hepatitis B–related cirrhosis: multiphasic MR technique with extracellular contrast agent. (a–d) Gadolinium-enhanced T1-weighted three-dimensional (3D) gradient-echo (GRE) images (repetition time msec/echo time msec, 6.3/1.9; flip angle, 15°) show large hypointense mass on (a) precontrast image with (b) hyperenhancement in late hepatic arterial phase. Notice enhancement of portal vein branches but not of hepatic vein branches in late hepatic arterial phase. (c) Portal venous and (d) 3-minute delayed phase images show persistent enhancement of tumor relative to liver. Persistent enhancement is atypical of large progressed HCCs, which characteristically appear to washout on venous phase images. Notice capsule appearance (arrow) on delayed phase image. Capsule appearance is seen to better advantage on delayed compared with portal venous phase image and permits confident diagnosis of HCC despite lack of washout appearance.
Figure 3c:
Images in 64-year-old man with HCC and hepatitis B–related cirrhosis: multiphasic MR technique with extracellular contrast agent. (a–d) Gadolinium-enhanced T1-weighted three-dimensional (3D) gradient-echo (GRE) images (repetition time msec/echo time msec, 6.3/1.9; flip angle, 15°) show large hypointense mass on (a) precontrast image with (b) hyperenhancement in late hepatic arterial phase. Notice enhancement of portal vein branches but not of hepatic vein branches in late hepatic arterial phase. (c) Portal venous and (d) 3-minute delayed phase images show persistent enhancement of tumor relative to liver. Persistent enhancement is atypical of large progressed HCCs, which characteristically appear to washout on venous phase images. Notice capsule appearance (arrow) on delayed phase image. Capsule appearance is seen to better advantage on delayed compared with portal venous phase image and permits confident diagnosis of HCC despite lack of washout appearance.
Figure 3d:
Images in 64-year-old man with HCC and hepatitis B–related cirrhosis: multiphasic MR technique with extracellular contrast agent. (a–d) Gadolinium-enhanced T1-weighted three-dimensional (3D) gradient-echo (GRE) images (repetition time msec/echo time msec, 6.3/1.9; flip angle, 15°) show large hypointense mass on (a) precontrast image with (b) hyperenhancement in late hepatic arterial phase. Notice enhancement of portal vein branches but not of hepatic vein branches in late hepatic arterial phase. (c) Portal venous and (d) 3-minute delayed phase images show persistent enhancement of tumor relative to liver. Persistent enhancement is atypical of large progressed HCCs, which characteristically appear to washout on venous phase images. Notice capsule appearance (arrow) on delayed phase image. Capsule appearance is seen to better advantage on delayed compared with portal venous phase image and permits confident diagnosis of HCC despite lack of washout appearance.
Three enhanced phases typically are acquired: late hepatic arterial, portal venous, and delayed phase. The late hepatic arterial phase is characterized by full enhancement of the hepatic artery and its branches as well as enhancement of the portal vein; the hepatic veins are not yet enhanced by antegrade flow (87). This phase coincides with peak arterial perfusion and enhancement of liver tumors, and it is critical for detection and characterization of hypervascular HCC (88) (Figs 2, 3). The early hepatic arterial phase, in which the hepatic artery is enhanced but the portal vein is not, is less effective, as tumor hypervascularity may be subtle or missed altogether (89). Most centers therefore omit the early hepatic arterial phase. Contrast agent bolus tracking or use of a test bolus is recommended for late hepatic arterial timing (90), since fixed delay is not reliable for this purpose. Another approach for achieving optimal timing is to obtain multiple high-temporal-resolution arterial-phase acquisitions, thereby ensuring that at least some images are captured during peak arterial perfusion and enhancement of the tumor (91). The portal venous phase coincides with peak parenchymal enhancement, is characterized by enhancement of hepatic veins as well as portal veins, and is acquired at around 60–80 seconds after the start of contrast agent injection (Figs 2, 3). The delayed phase is acquired at 3–5 minutes (92) (Figs 2, 3). As discussed further in part II, these latter phases are critical for characterizing key imaging features of HCC such as washout appearance and capsule appearance (93) (Figs 2, 3), and they help to differentiate small HCCs from small ICCs, which typically show prolonged central enhancement (94). For the above purposes, the delayed phase may be superior to the portal venous phase (95) (Fig 2). The portal venous and delayed phases also may be useful for measuring nodule diameter, depicting hypovascular nodules including early HCCs (96), and identifying vascular thrombosis. To reduce radiation dose, some centers skip the delayed phase at CT, but this practice is difficult to recommend because important diagnostic information may be lost. The precontrast image serves as a baseline to gauge subsequent enhancement. For observations that are hyperintense on precontrast MR images, subtraction images (postcontrast minus precontrast) may be helpful for detection of enhancement and evaluation of its degree (87). In theory, precontrast CT also might be helpful in patients with iron-rich nodules to detect hyperattenuation before contrast agent administration, thus avoiding misinterpretation of arterial-phase hyperenhancement, but there is little published evidence to our knowledge to support this benefit. Thus, except in patients previously treated with locoregional embolic or ablative therapies, precontrast CT adds little diagnostic value (93,97) and, to reduce radiation dose, usually may be omitted from routine multiphasic examinations without loss of significant diagnostic information. In the future, dual-energy CT may be of value by permitting the generation of virtual unenhanced images and/or iodine maps that depict the iodine concentration distribution in tumor and background liver.
In addition to the vascular phases, MR imaging examinations usually include T1-weighted in-phase and opposed-phase gradient-echo, T2-weighted fast-spin-echo or single-shot fast-spin-echo, and diffusion-weighted sequences. These sequences help to assess ancillary imaging features of HCC, as discussed in part II. For diffusion-weighted imaging, two or more b values typically are acquired, at least one of which is in the low (0–50 sec/mm2) and one in the intermediate to high (400–800 sec/mm2) range. T2-weighted and diffusion-weighted images can be acquired before or after contrast agent administration.
MR Imaging with Hepatobiliary Agents
A discussed further in part II, hepatobiliary agents permit diagnosis of HCC based not only on vascularity but also on hepatocellular function. Currently available hepatobiliary contrast agents include gadoxetate disodium and gadobenate dimeglumine, both of which are gadolinium-based agents. The approved dose of gadobenate dimeglumine is 0.1 mmol/kg, the same as for pure extracellular agents, and it is usually injected at 2 mL/sec (98). Some investigators use half dose (0.05 mmol/kg) in patients with renal impairment (99). The approved dose of gadoxetate disodium is 0.025 mmol/kg, a quarter that of extracellular agents, which usually corresponds to a volume of 5–10 mL, depending on patient weight (100). Some investigators use a higher-than-approved dose (up to 0.05 mmol/kg) to improve the quality of the vascular phases (101). On intravenous administration, these agents rapidly distribute in the vascular-interstitial compartment, enhance the extracellular space, and permit acquisition of dynamic images that allow for HCC diagnosis based on perfusion characteristics as discussed further in part II (102). After distribution in the extracellular space, these agents enter hepatocytes via OATP8 receptors, which recognize the agents’ ethoxybenzyl (gadoxetate disodium) (103) and benzyloxymethyl (gadobenate dimeglumine) (104) groups. The agents subsequently are excreted into the biliary canaliculi by MRP2 as well as back into the sinusoidal space by MRP3 (67). These transporter molecules are expressed only in “functioning” hepatocytes, including hepatic parenchyma and, as discussed later, some cirrhosis-associated hepatocellular nodules. They are not expressed in cells of nonhepatocyte origin such as vascular endothelium, cholangiocytes, fibrous tissue, or liver metastases from extrahepatic origins (105). Selective uptake of these agents by functioning hepatocytes and subsequent excretion into the biliary system permits acquisition of hepatobiliary phase T1-weighted images, typically at 1–3 hours for gadobenate dimeglumine (104) and at about 20 minutes for gadoxetate disodium (100). On such images, the hepatic parenchyma is strongly enhanced due to OATP-mediated intrahepatocellular uptake, the large bile ducts are enhanced due to biliary excretion, and blood vessels are hypoenhanced due to clearance of contrast material from the blood pool (Fig 4).
Figure 4a:
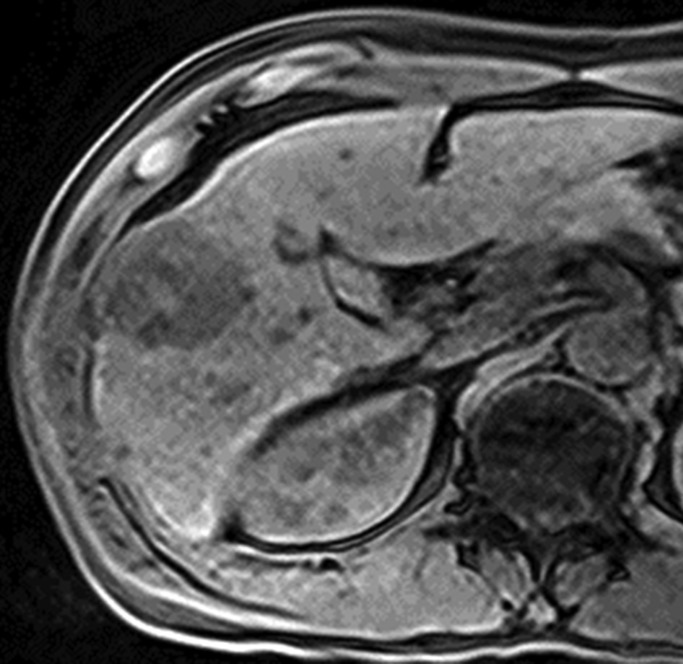
Images in 42-year-old man with HCC and hepatitis B–related cirrhosis: multiphasic MR technique with gadoxetate disodium. (a, b) Gadoxetate disodium–enhanced T1-weighted 3D GRE images (2.5/0.9; flip angle, 11°) show large hypointense mass on (a) precontrast image with (b) hyperenhancement in late hepatic arterial phase. (c) Portal venous and (d) transitional phase images show apparent washout of contrast material from tumor. (e) Mass is hypointense relative to strongly enhanced liver parenchyma on hepatobiliary phase image obtained at 20 minutes after injection. Surgical specimen confirmed progressed HCC with perilesional satellite metastases. Metastases were visible on other MR sections (not shown). Histologically, tumor was poorly differentiated (Edmondson grade III).
Figure 4b:
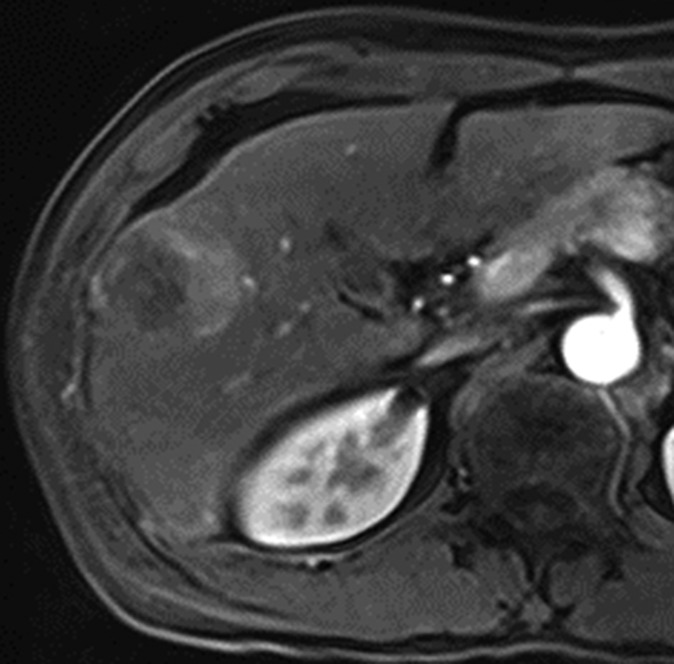
Images in 42-year-old man with HCC and hepatitis B–related cirrhosis: multiphasic MR technique with gadoxetate disodium. (a, b) Gadoxetate disodium–enhanced T1-weighted 3D GRE images (2.5/0.9; flip angle, 11°) show large hypointense mass on (a) precontrast image with (b) hyperenhancement in late hepatic arterial phase. (c) Portal venous and (d) transitional phase images show apparent washout of contrast material from tumor. (e) Mass is hypointense relative to strongly enhanced liver parenchyma on hepatobiliary phase image obtained at 20 minutes after injection. Surgical specimen confirmed progressed HCC with perilesional satellite metastases. Metastases were visible on other MR sections (not shown). Histologically, tumor was poorly differentiated (Edmondson grade III).
Figure 4c:
Images in 42-year-old man with HCC and hepatitis B–related cirrhosis: multiphasic MR technique with gadoxetate disodium. (a, b) Gadoxetate disodium–enhanced T1-weighted 3D GRE images (2.5/0.9; flip angle, 11°) show large hypointense mass on (a) precontrast image with (b) hyperenhancement in late hepatic arterial phase. (c) Portal venous and (d) transitional phase images show apparent washout of contrast material from tumor. (e) Mass is hypointense relative to strongly enhanced liver parenchyma on hepatobiliary phase image obtained at 20 minutes after injection. Surgical specimen confirmed progressed HCC with perilesional satellite metastases. Metastases were visible on other MR sections (not shown). Histologically, tumor was poorly differentiated (Edmondson grade III).
Figure 4d:
Images in 42-year-old man with HCC and hepatitis B–related cirrhosis: multiphasic MR technique with gadoxetate disodium. (a, b) Gadoxetate disodium–enhanced T1-weighted 3D GRE images (2.5/0.9; flip angle, 11°) show large hypointense mass on (a) precontrast image with (b) hyperenhancement in late hepatic arterial phase. (c) Portal venous and (d) transitional phase images show apparent washout of contrast material from tumor. (e) Mass is hypointense relative to strongly enhanced liver parenchyma on hepatobiliary phase image obtained at 20 minutes after injection. Surgical specimen confirmed progressed HCC with perilesional satellite metastases. Metastases were visible on other MR sections (not shown). Histologically, tumor was poorly differentiated (Edmondson grade III).
Figure 4e:
Images in 42-year-old man with HCC and hepatitis B–related cirrhosis: multiphasic MR technique with gadoxetate disodium. (a, b) Gadoxetate disodium–enhanced T1-weighted 3D GRE images (2.5/0.9; flip angle, 11°) show large hypointense mass on (a) precontrast image with (b) hyperenhancement in late hepatic arterial phase. (c) Portal venous and (d) transitional phase images show apparent washout of contrast material from tumor. (e) Mass is hypointense relative to strongly enhanced liver parenchyma on hepatobiliary phase image obtained at 20 minutes after injection. Surgical specimen confirmed progressed HCC with perilesional satellite metastases. Metastases were visible on other MR sections (not shown). Histologically, tumor was poorly differentiated (Edmondson grade III).
Key differences between the two agents are that gadoxetate disodium has greater hepatocellular uptake and biliary excretion (50% versus 5%) (106) than gadobenate dimeglumine as well as higher plasma relaxivity (r1 = 6.9/6.2 at 1.5 T/3 T versus 6.3/5.5 l/mmol/sec) (107). Consequently, hepatobiliary phase parenchymal enhancement peaks earlier and more strongly after injection of gadoxetate disodium, despite its smaller dose, than after injection of gadobenate dimeglumine (108). Due to the relatively long delay required for hepatobiliary phase imaging with gadobenate dimeglumine, many centers acquire hepatobiliary phase images using this agent only in select cases.
Although vascular-phase images acquired with gadobenate dimeglumine are qualitatively similar in appearance to those acquired with extracellular agents, images acquired with gadoxetate disodium differ. Due to the low volume of the injected dose, gadoxetate disodium provides a narrow imaging window for late hepatic arterial phase acquisition (109), which may complicate optimal timing. Additionally, rapid changes in arterial signal intensity during central k-space acquisition may introduce truncation artifacts (110). To compensate for these problems, many investigators administer the agent at 1 rather than mL/sec (111) and/or dilute the contrast material with saline to achieve a standard injection volume of 10 mL (86). These steps prolong the imaging window for the late hepatic arterial phase and may reduce truncation artifacts (112). Some patients also have transient breath-holding difficulty after gadoxetate disodium injection, predisposing to arterial-phase respiratory motion artifacts (113). A potential solution is to perform multiple acquisitions during the arterial phase with high temporal resolution; this not only helps achieve proper timing of the arterial phase as discussed earlier but–especially if a view-sharing technique is not used–also may help ensure that at least one arterial acquisition is free of motion artifact. The portal venous phase usually is acquired at about 60 seconds and is similar qualitatively to that acquired with extracellular agents (114). Unlike other agents, however, gadoxetate disodium does not provide a conventional delayed phase because hepatocellular uptake of the agent begins during its first pass through the hepatic sinusoids (115). Thus by the end of the portal venous phase, considerable hepatocellular uptake has occurred, and from about 2–5 minutes after injection, both the intracellular and extracellular pools of gadoxetate disodium contribute substantially to parenchymal enhancement (100,115). This phase, in which both the intracellular and extracellular pools are contributory, is sometimes termed the late dynamic phase (115) or, since it represents a transition from extracellular-dominant to intracellular-dominant enhancement, the transitional phase (116) (Fig 4).
CT and MR Imaging-based Characterization of Precursor Nodules
Table 2 summarizes the CT and MR imaging appearance of precursor nodules (cirrhotic nodules, low-grade dysplastic nodules, and high-grade dysplastic nodules), early HCCs, and progressed HCCs. Precursor nodules are discussed in depth below. Early and progressed HCC are discussed briefly here and in greater depth in the second article of this two-part review.
Table 2.
CT and MR Imaging Attenuation and Signal Intensity of Precursor Nodules, Early HCC, and Progressed HCC
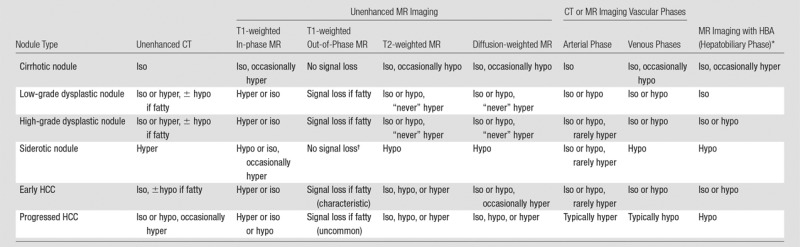
Note.—Table shows attenuation or signal intensity of each nodule type as depicted at various imaging sequences or phases. Hyper = hyperattenuating or hyperintense, hypo = hypoattenuating or hypointense, and iso = isoattenuating or isointense at CT or MR imaging, respectively.
HBA = hepatobiliary agent.
Some MR imagers acquire the out-of-phase T1-weighted image with a longer echo time than the in-phase image; in such cases, siderotic nodules show signal loss on the out-of-phase image, but the signal loss is due to T2* shortening, not presence of fat.
Cirrhotic Nodules
Although cirrhotic nodules are innumerable in cirrhosis, most cirrhotic nodules are imperceptible or only barely perceptible at CT and MR imaging. Relative to background parenchyma, these nodules usually are isoattenuating at unenhanced CT and isointense on unenhanced T1-, T2-, and diffusion-weighted MR imaging (117,118). Occasionally they may be hyperintense on T1-weighted in-phase images, without signal loss on out-of-phase images, and hypointense on T2-weighted images, similar to dysplastic nodules (119) (Fig 5). The exact cause for the T1 hyperintensity in cirrhotic nodules is unknown (119). Histologically, cirrhotic nodules do not contain paramagnetic materials such as copper or iron in amounts disproportionately greater than that of background liver (33,120), so it is difficult to attribute this imaging feature to the presence of such materials. Similarly, the cause of T2 hypointensity has not been delineated. If fibrosis is particularly prominent, cirrhotic nodules may be visible on unenhanced images as rounded lesions surrounded by cirrhotic scars, which owing to their high water content may be hypoattenuating at CT, hypointense at T1-weighted MR imaging, and hyperintense at T2-weighted MR imaging (121).
Figure 5a:
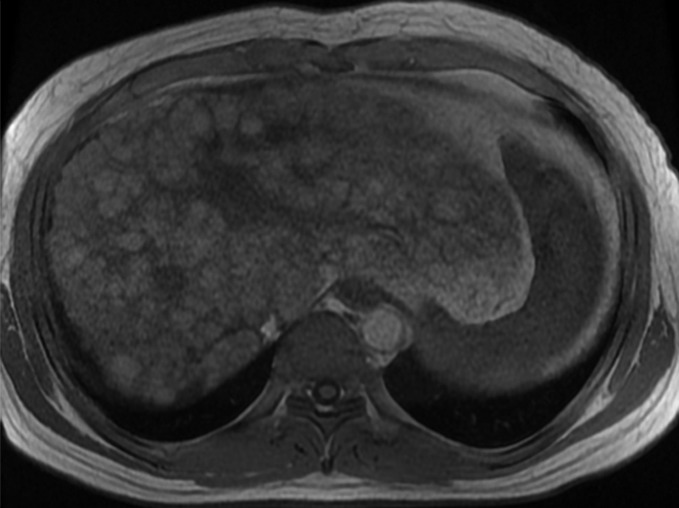
MR Images in 39-year-old man with liver cirrhosis and multiple cirrhotic nodules, some of which resemble dysplastic nodules at imaging. (a) Transverse T1-weighted 3D GRE in-phase image (6.6/4.4; flip angle, 12°) and (b) opposed-phase image (6.6/2.1; flip angle, 12°) show multiple iso- or hyperintense nodules in both lobes of liver. (c) Nodules are iso- or hypointense on fat-suppressed T2-weighted fast spin-echo image (3500/101). Some of the T1-hyperintense/T2-hypointense nodules are distinctive compared with background liver at MR imaging, suggesting the possibility of dysplastic nodules, but no dysplastic nodules were identified in the pathologic specimen (not shown). This case illustrates the difficulty in differentiating cirrhotic nodules and dysplastic nodules at imaging.
Figure 5b:
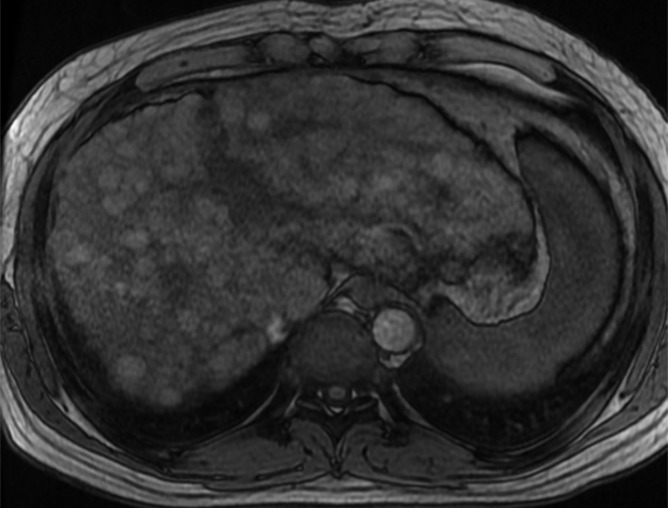
MR Images in 39-year-old man with liver cirrhosis and multiple cirrhotic nodules, some of which resemble dysplastic nodules at imaging. (a) Transverse T1-weighted 3D GRE in-phase image (6.6/4.4; flip angle, 12°) and (b) opposed-phase image (6.6/2.1; flip angle, 12°) show multiple iso- or hyperintense nodules in both lobes of liver. (c) Nodules are iso- or hypointense on fat-suppressed T2-weighted fast spin-echo image (3500/101). Some of the T1-hyperintense/T2-hypointense nodules are distinctive compared with background liver at MR imaging, suggesting the possibility of dysplastic nodules, but no dysplastic nodules were identified in the pathologic specimen (not shown). This case illustrates the difficulty in differentiating cirrhotic nodules and dysplastic nodules at imaging.
Figure 5c:
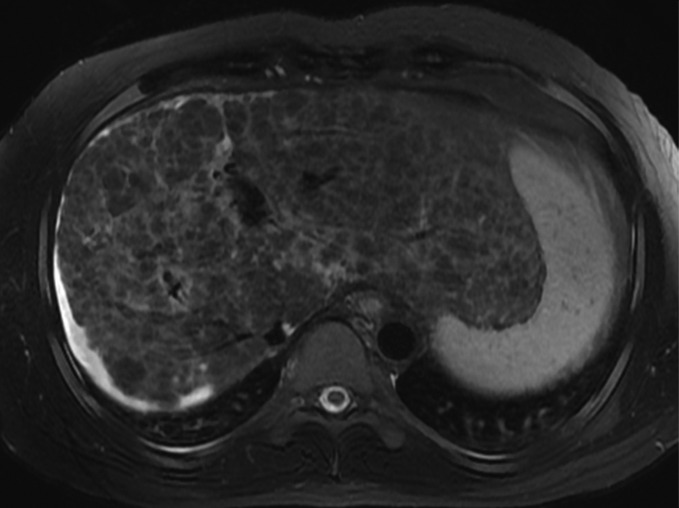
MR Images in 39-year-old man with liver cirrhosis and multiple cirrhotic nodules, some of which resemble dysplastic nodules at imaging. (a) Transverse T1-weighted 3D GRE in-phase image (6.6/4.4; flip angle, 12°) and (b) opposed-phase image (6.6/2.1; flip angle, 12°) show multiple iso- or hyperintense nodules in both lobes of liver. (c) Nodules are iso- or hypointense on fat-suppressed T2-weighted fast spin-echo image (3500/101). Some of the T1-hyperintense/T2-hypointense nodules are distinctive compared with background liver at MR imaging, suggesting the possibility of dysplastic nodules, but no dysplastic nodules were identified in the pathologic specimen (not shown). This case illustrates the difficulty in differentiating cirrhotic nodules and dysplastic nodules at imaging.
After extracellular contrast agent injection, most cirrhotic nodules enhance to the same degree as the adjacent liver or show slightly less enhancement, in which case they may be visible in the portal or delayed phases at CT or MR imaging as mildly hypoenhanced nodules relative to enhancing fibrosis (122). Cirrhotic nodules that are hyperenhanced in the arterial phase have been reported, although the mechanism is unclear because cirrhotic nodules do not show histologic evidence of neo-arterialization (123). Since OATP expression is preserved, cirrhotic nodules typically have similar signal intensity to surrounding liver parenchyma in the hepatobiliary phase after hepatobiliary contrast agent administration (124) or they may appear subtly hyperintense relative to cirrhotic scars, which do not contain hepatocytes and hence do not uptake the agents. Some cirrhotic nodules may be markedly more hyperintense in the hepatobiliary phase than background nodules, presumably because they have sufficient hepatocellular function to take up the agent but not to excrete it (125).
In summary, while most cirrhotic nodules are imperceptible or only barely perceptible at CT and MR imaging, some cirrhotic nodules may differ in appearance from background nodules due to unusual imaging features such T1 hyperintensity, T2 hypointensity, mild portal venous or delayed phase hypoenhancement, or hepatobiliary phase hyperintensity. Such cirrhotic nodules cannot be distinguished from low- or high-grade dysplastic nodules at imaging.
Dysplastic Nodules
Most dysplastic nodules are isoattenuating or hypoattenuating in the arterial, portal, and delayed phases at CT (126). Large dysplastic nodules may appear hyperattenuating relative to liver at unenhanced CT and become isoattenuating after contrast material administration (127).
Dysplastic nodules characteristically are hyperintense on T1-weighted images and isointense or hypointense on T2-weighted images (122,128) (Fig 6). As with cirrhotic nodules, the cause of these signal intensity characteristics is not well understood. Histologically, dysplastic nodules may contain more copper and/or iron than background liver, and the presence of such paramagnetic materials may contribute to T1 hyperintensity depending on the concentration of the materials and the image weighting (119,129).
Figure 6a:
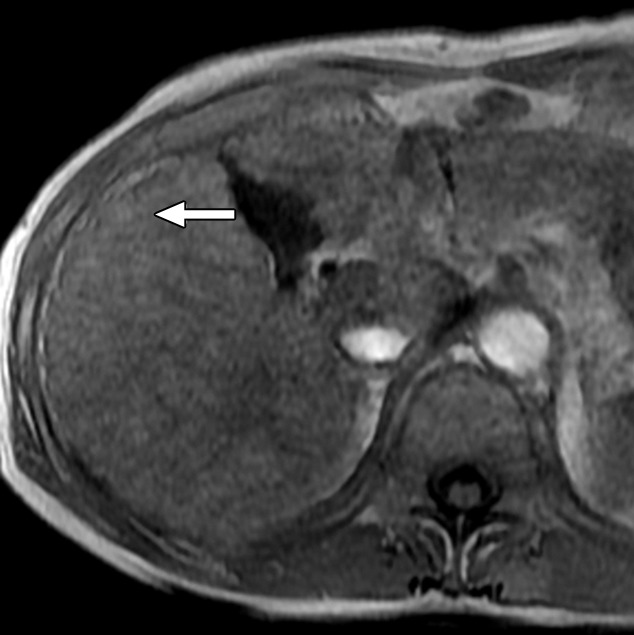
MR Images in 55-year-old man with low-grade dysplastic nodule and hepatitis B–related cirrhosis. A 1-cm nodule (arrow) near the hepatic capsule in segment V is minimally hyperintense relative to adjacent parenchyma on T1-weighted (a) in-phase image (7.8/5.2) and (b) opposed-phase image (7.8/2.3). (c) Nodule (arrow) is hypointense on T2-weighted fast-recovery fast spin-echo image (3500/101) and (d) isointense (arrow) on precontrast T1-weighted 3D GRE (4.5/2.2; 12° flip angle) image. (e) At gross pathologic examination, nodule (arrow) differs in size and color from background nodules, the gross pathologic definition of a dysplastic nodule. Histologically, cellular atypia was absent, and a diagnosis of low-grade dysplastic nodule was made.
Figure 6b:
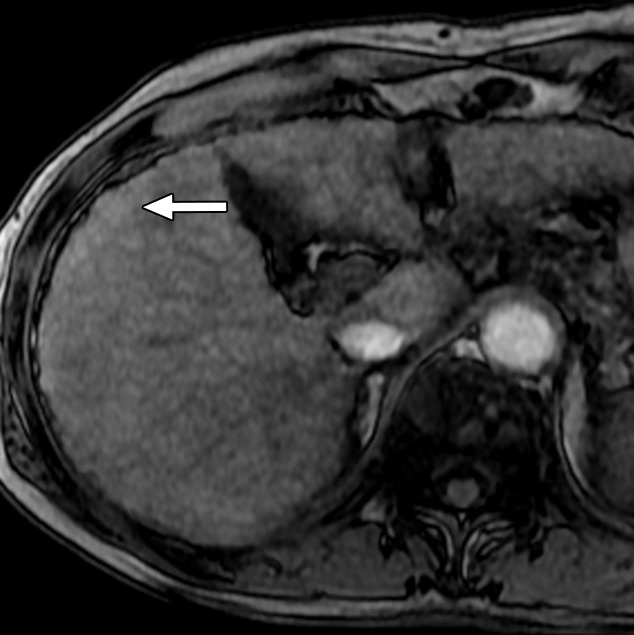
MR Images in 55-year-old man with low-grade dysplastic nodule and hepatitis B–related cirrhosis. A 1-cm nodule (arrow) near the hepatic capsule in segment V is minimally hyperintense relative to adjacent parenchyma on T1-weighted (a) in-phase image (7.8/5.2) and (b) opposed-phase image (7.8/2.3). (c) Nodule (arrow) is hypointense on T2-weighted fast-recovery fast spin-echo image (3500/101) and (d) isointense (arrow) on precontrast T1-weighted 3D GRE (4.5/2.2; 12° flip angle) image. (e) At gross pathologic examination, nodule (arrow) differs in size and color from background nodules, the gross pathologic definition of a dysplastic nodule. Histologically, cellular atypia was absent, and a diagnosis of low-grade dysplastic nodule was made.
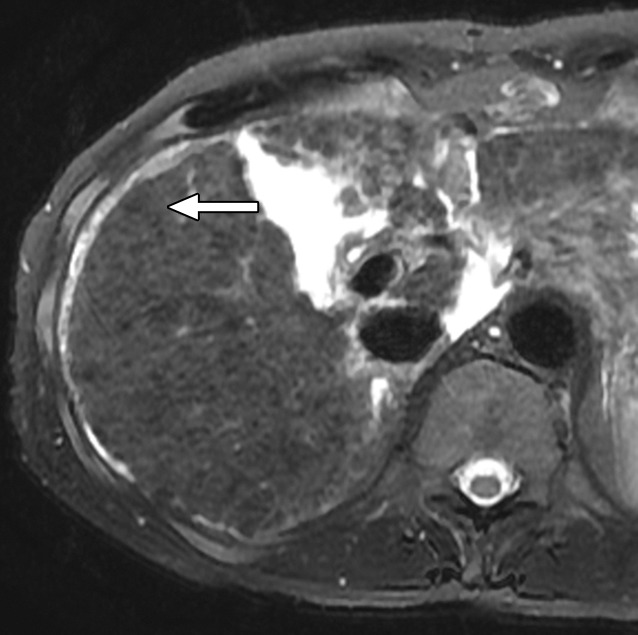
Figure 6c:
MR Images in 55-year-old man with low-grade dysplastic nodule and hepatitis B–related cirrhosis. A 1-cm nodule (arrow) near the hepatic capsule in segment V is minimally hyperintense relative to adjacent parenchyma on T1-weighted (a) in-phase image (7.8/5.2) and (b) opposed-phase image (7.8/2.3). (c) Nodule (arrow) is hypointense on T2-weighted fast-recovery fast spin-echo image (3500/101) and (d) isointense (arrow) on precontrast T1-weighted 3D GRE (4.5/2.2; 12° flip angle) image. (e) At gross pathologic examination, nodule (arrow) differs in size and color from background nodules, the gross pathologic definition of a dysplastic nodule. Histologically, cellular atypia was absent, and a diagnosis of low-grade dysplastic nodule was made.
Figure 6d:
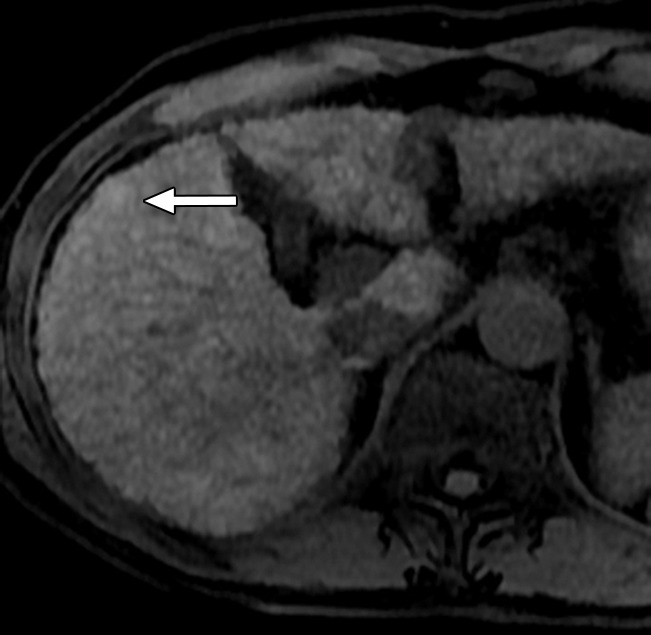
MR Images in 55-year-old man with low-grade dysplastic nodule and hepatitis B–related cirrhosis. A 1-cm nodule (arrow) near the hepatic capsule in segment V is minimally hyperintense relative to adjacent parenchyma on T1-weighted (a) in-phase image (7.8/5.2) and (b) opposed-phase image (7.8/2.3). (c) Nodule (arrow) is hypointense on T2-weighted fast-recovery fast spin-echo image (3500/101) and (d) isointense (arrow) on precontrast T1-weighted 3D GRE (4.5/2.2; 12° flip angle) image. (e) At gross pathologic examination, nodule (arrow) differs in size and color from background nodules, the gross pathologic definition of a dysplastic nodule. Histologically, cellular atypia was absent, and a diagnosis of low-grade dysplastic nodule was made.
Figure 6e:
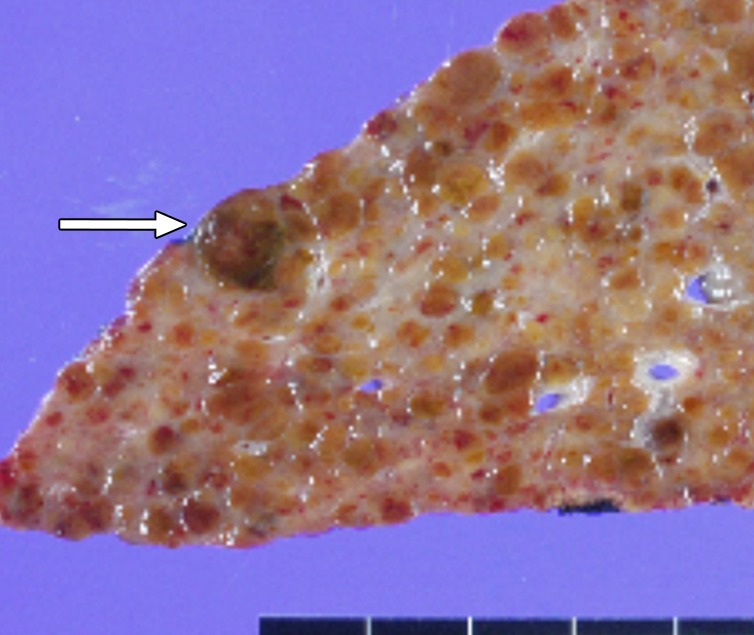
MR Images in 55-year-old man with low-grade dysplastic nodule and hepatitis B–related cirrhosis. A 1-cm nodule (arrow) near the hepatic capsule in segment V is minimally hyperintense relative to adjacent parenchyma on T1-weighted (a) in-phase image (7.8/5.2) and (b) opposed-phase image (7.8/2.3). (c) Nodule (arrow) is hypointense on T2-weighted fast-recovery fast spin-echo image (3500/101) and (d) isointense (arrow) on precontrast T1-weighted 3D GRE (4.5/2.2; 12° flip angle) image. (e) At gross pathologic examination, nodule (arrow) differs in size and color from background nodules, the gross pathologic definition of a dysplastic nodule. Histologically, cellular atypia was absent, and a diagnosis of low-grade dysplastic nodule was made.
Some dysplastic nodules may contain intracellular iron in high concentrations. Such iron-rich or siderotic nodules appear slightly hyperattenuating at unenhanced CT and, due to the T2- and T2*-shortening effects of iron, appear moderately to markedly hypointense on T2-weighted and T2*-weighted MR images; the hypointensity is more pronounced on gradient-echo than on fast-spin-echo images, and on images with longer echo times (117,122,130). These nodules usually appear hypointense on T1-weighted images but, depending on the image weighting and the iron concentration, may appear slightly hyperintense (63). Some dysplastic nodules, especially high-grade dysplastic nodules, may contain intracellular fat in higher concentration than background liver (42). Such dysplastic nodules appear hyperintense on T1-weighted in-phase images and show signal loss on out-of-phase images (131) (Fig 7). Intralesional steatosis is more frequent in early HCC than in dysplastic nodules and also may occur in progressed HCCs, however, so the detection of intracellular lipid is not diagnostic of dysplastic nodule (132).
Figure 7a:
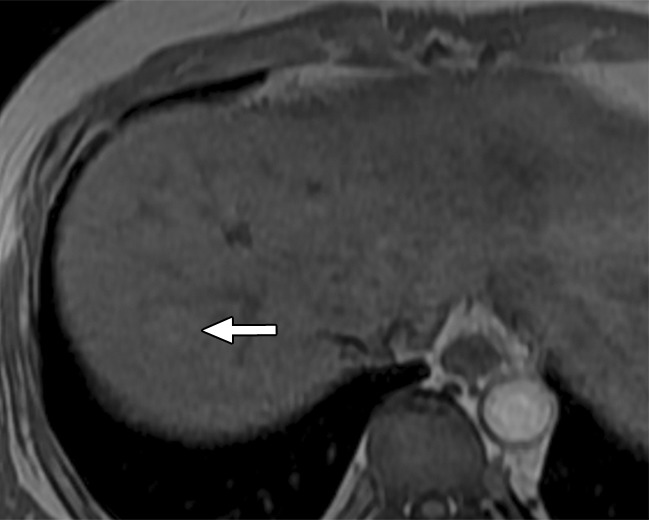
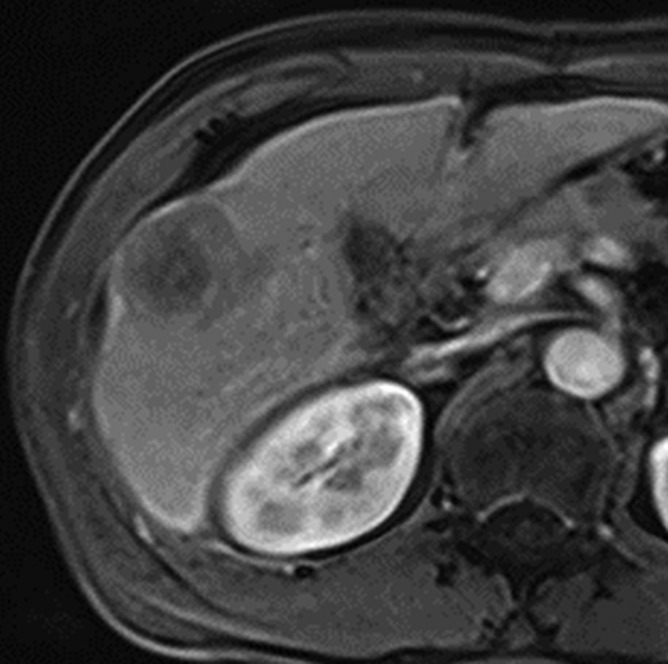
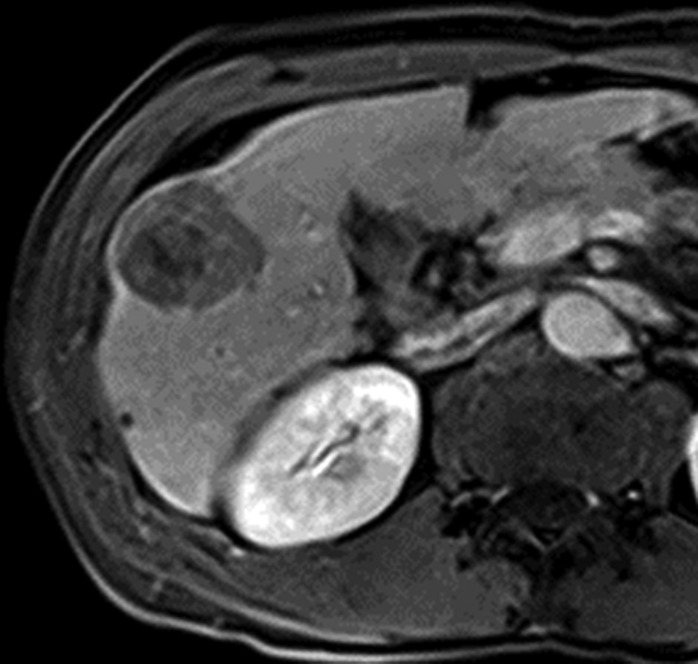
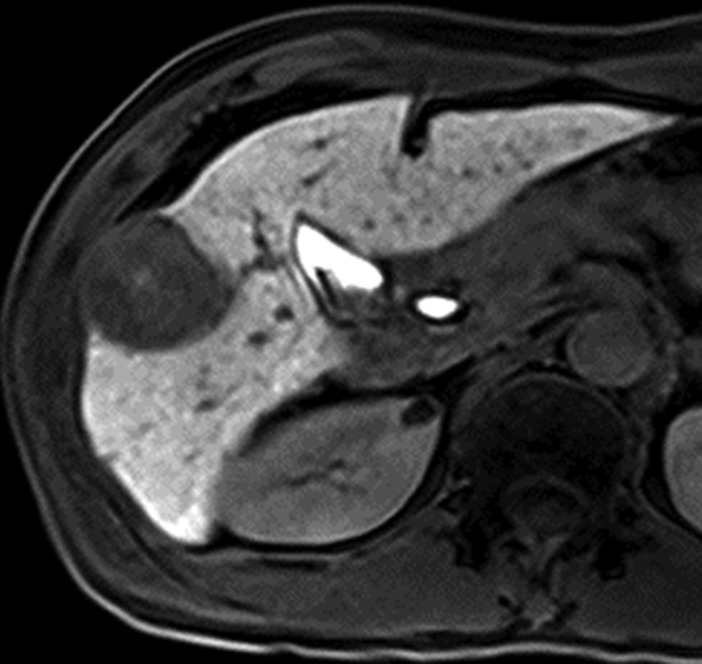
MR images in a 58-year-old man with fat-containing high-grade dysplastic nodule and hepatitis B–related cirrhosis. (a, b) Axial dual-echo GRE images show a nodule in segment VII of liver. Nodule loses signal intensity on (b) out-of-phase (7.8/2.3) image compared with (a) in-phase (7.8/5.1) image, indicating intralesional fat. (c) Nodule is isointense on T2-weighted fast-recovery fast spin-echo (3500/101) image. (d) Nodule is hypointense on hepatobiliary phase image, which favors a diagnosis of high-grade dysplastic nodule or early HCC over low-grade dysplastic nodule or cirrhotic nodule. Notice nodule-in-nodule architecture on hepatobiliary phase image, which raises concern for an incipient HCC subnodule developing within a parent dysplastic nodule. Percutaneous needle biopsy with histologic analysis was consistent with high-grade dysplastic nodule with fatty change. Sampling errors may occur with biopsy, and it is possible that an HCC subnodule was missed.
Figure 7b:
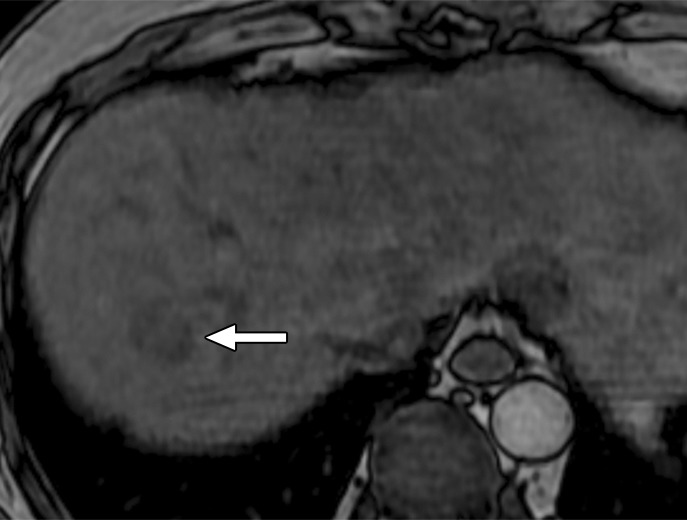
MR images in a 58-year-old man with fat-containing high-grade dysplastic nodule and hepatitis B–related cirrhosis. (a, b) Axial dual-echo GRE images show a nodule in segment VII of liver. Nodule loses signal intensity on (b) out-of-phase (7.8/2.3) image compared with (a) in-phase (7.8/5.1) image, indicating intralesional fat. (c) Nodule is isointense on T2-weighted fast-recovery fast spin-echo (3500/101) image. (d) Nodule is hypointense on hepatobiliary phase image, which favors a diagnosis of high-grade dysplastic nodule or early HCC over low-grade dysplastic nodule or cirrhotic nodule. Notice nodule-in-nodule architecture on hepatobiliary phase image, which raises concern for an incipient HCC subnodule developing within a parent dysplastic nodule. Percutaneous needle biopsy with histologic analysis was consistent with high-grade dysplastic nodule with fatty change. Sampling errors may occur with biopsy, and it is possible that an HCC subnodule was missed.
Figure 7c:
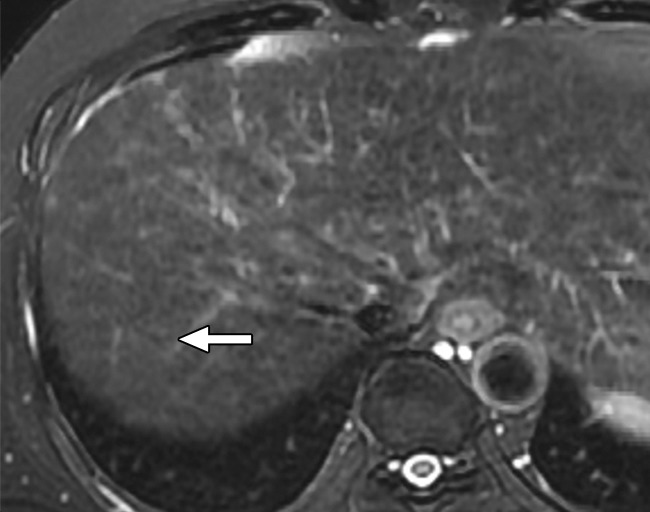
MR images in a 58-year-old man with fat-containing high-grade dysplastic nodule and hepatitis B–related cirrhosis. (a, b) Axial dual-echo GRE images show a nodule in segment VII of liver. Nodule loses signal intensity on (b) out-of-phase (7.8/2.3) image compared with (a) in-phase (7.8/5.1) image, indicating intralesional fat. (c) Nodule is isointense on T2-weighted fast-recovery fast spin-echo (3500/101) image. (d) Nodule is hypointense on hepatobiliary phase image, which favors a diagnosis of high-grade dysplastic nodule or early HCC over low-grade dysplastic nodule or cirrhotic nodule. Notice nodule-in-nodule architecture on hepatobiliary phase image, which raises concern for an incipient HCC subnodule developing within a parent dysplastic nodule. Percutaneous needle biopsy with histologic analysis was consistent with high-grade dysplastic nodule with fatty change. Sampling errors may occur with biopsy, and it is possible that an HCC subnodule was missed.
Figure 7d:
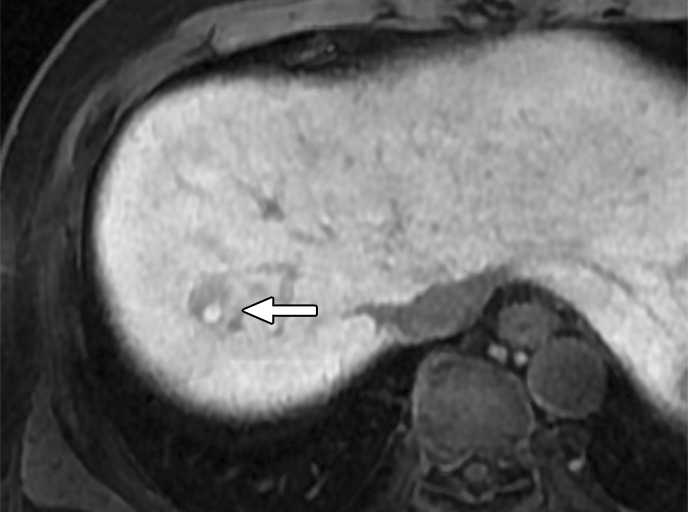
MR images in a 58-year-old man with fat-containing high-grade dysplastic nodule and hepatitis B–related cirrhosis. (a, b) Axial dual-echo GRE images show a nodule in segment VII of liver. Nodule loses signal intensity on (b) out-of-phase (7.8/2.3) image compared with (a) in-phase (7.8/5.1) image, indicating intralesional fat. (c) Nodule is isointense on T2-weighted fast-recovery fast spin-echo (3500/101) image. (d) Nodule is hypointense on hepatobiliary phase image, which favors a diagnosis of high-grade dysplastic nodule or early HCC over low-grade dysplastic nodule or cirrhotic nodule. Notice nodule-in-nodule architecture on hepatobiliary phase image, which raises concern for an incipient HCC subnodule developing within a parent dysplastic nodule. Percutaneous needle biopsy with histologic analysis was consistent with high-grade dysplastic nodule with fatty change. Sampling errors may occur with biopsy, and it is possible that an HCC subnodule was missed.
Differentiation of low-grade dysplastic nodules, high-grade dysplastic nodules, early HCC, and small progressed HCCs may be impossible at unenhanced MR imaging, as all four nodule types may be isointense or hyperintense on T1-weighted images, may be isointense or hypointense on T2-weighted images, and contain intralesional fat (32,133) (Figs 5–7). Some imaging features may help in the differential diagnosis, however. Dysplastic nodules almost never are hyperintense on T2-weighted images (32,133) or show restricted diffusion (134). Thus, in the differential diagnosis of dysplastic nodule and HCC, the presence of mild-moderate T2 hyperintensity or restricted diffusion strongly favors the diagnosis of HCC. These signal intensity characteristics can also be observed in ICCs, however, so they do not clinch the diagnosis of HCC. Exceptionally, both cirrhotic nodules and dysplastic nodules may infarct due to ischemic necrosis, usually after episodes of hypotension with hepatic hypoperfusion, and appear hyperintense on T2-weighted images (135); at MR imaging, such nodules may be difficult to differentiate from hypovascular HCC. On the other hand, the detection of diffuse iron deposition within a nodule, as indicated by marked hypointensity on T2-weighted and T2*-weighted MR images, favors a nonmalignant etiology (low-grade dysplastic nodule or high-grade dysplastic nodule) over HCC, because malignant hepatocytes typically are iron resistant (136). Rarely, HCC may develop within an iron-rich nodule, however, and the emergence within an iron-rich nodule of an iron-poor (nonsiderotic) subnodule (ie, nodule-in-nodule architecture) raises concern for an incident HCC.
Low-grade dysplastic nodules and most high-grade dysplastic nodules have relatively preserved arterial blood supply and so usually are isoattenuating or isointense to liver in the late hepatic arterial phase at CT or MR imaging (137). Some high-grade dysplastic nodules have elevated arterial supply due to neo-arterialization and enhance more than liver in the late hepatic arterial phase, potentially being mistaken for hypervascular, progressed HCC (138). Analysis of portal venous and delayed phase images is helpful for interpreting arterial-phase hyperenhanced nodules. As discussed further in part II, hypervascular high-grade dysplastic nodules rarely show washout or capsule appearance in the portal venous or delayed phases, while these features are characteristic of progressed HCC (139). Thus, the combination of arterial phase hyperenhancement and either washout or capsule appearance is considered diagnostic of HCC.
Cirrhosis-associated nodules without arterial-phase hyperenhancement include cirrhotic nodules, low-grade dysplastic nodules, high-grade dysplastic nodules, and early HCCs. CT and MR imaging with extracellular agents are unreliable in the differentiation of such nodules, as each nodule type may show isoenhancement or hypoenhancement in all postcontrast phases (140). Hepatobiliary contrast agents show promise for differentiating early HCCs and premalignant nodules (high-grade dysplastic nodules) from lower-risk nodules (low-grade dysplastic nodules and cirrhotic nodules). Since OATP expression declines during hepatocarcinogenesis, hepatobiliary phase hypointensity is a strong predictor of premalignancy or malignancy, and its presence favors high-grade dysplastic nodule or early HCC over low-grade dysplastic nodule or cirrhotic nodule (141) (Fig 7). A potential pitfall is that iron-rich low-grade dysplastic nodules may appear hypointense in the hepatobiliary phase due to the T2* shortening effects of iron. Thus, in nodules with imaging evidence of iron accumulation, hepatobiliary phase hypointensity is nonspecific.
Conclusion
HCC is an increasingly common cause of cancer death in Western nations. Hepatocarcinogenesis is driven by the progressive accumulation of epigenetic and genetic alterations potentially involving many different genes and regulatory pathways. The process evolves over years or decades in parallel with the progression of fibrosis in the context of chronic liver disease. Most HCCs emerge after cirrhosis has been established. Growing evidence suggests that many HCCs arise from hepatic stem cells, which in part may explain the predisposition of patients with cirrhosis and chronic hepatitis to develop tumors spanning a spectrum of hepatocellular and cholangiocellular differentiation phenotypes. Histologically, hepatocarcinogenesis is characterized by the repeated emergence and expansion of successively less differentiated precursor nodules. Key alterations during hepatocarcinogenesis include elevation of arterial flow, reduction in portal venous flow, and reduction in OATP expression. Changes in fat and iron content also may occur. Multiphasic CT and MR imaging with extracellular agents permit diagnosis of HCC based mainly on assessment of vascularity. Critical contrast-enhanced phases include the late arterial, portal venous, and delayed phases. MR imaging with the hepatobiliary agents, gadobenate dimeglumine and gadoxetate disodium, provides information on hepatocellular function in addition to vascularity; the delay required for the hepatobiliary phase depends on the agent. Due to its rapid uptake by hepatocytes, gadoxetate disodium does not provide a conventional delayed phase but instead provides a transitional phase in which the intracellular and extracellular pools both contribute to parenchymal enhancement. The ability to use CT and MR imaging with extracellular agents to identify and differentiate cirrhotic nodules, low-grade dysplastic nodules, high-grade dysplastic nodules, and early HCCs is limited; hepatobiliary phase MR imaging shows promise for characterization of precursor lesions and for identifying high-grade dysplastic nodules and early HCCs prior to neo-arterialization and progression to overt HCC.
Essentials
■ Hepatocarcinogenesis, the gradual transformation of nonmalignant liver cells into hepatocellular carcinoma (HCC), is a complex, multistep process characterized at the molecular and cellular level by the progressive accumulation of epigenetic and genetic alterations and at the histologic level by the emergence and progression of successively more advanced precancerous, early cancerous, and overtly malignantly lesions.
■ Early HCC is an incipient stage of HCC development, analogous to “carcinoma in situ” or “microinvasive carcinoma” of other organs, whereas progressed HCC is an overtly malignant neoplasm with ability to invade vessels and metastasize.
■ Key alterations during hepatocarcinogenesis include elevation of arterial flow, reduction in portal venous flow, and reduction in OATP expression.
■ The ability to use CT and MR imaging with extracellular agents to identify and differentiate cirrhotic nodules, low-grade dysplastic nodules, high-grade dysplastic nodules, and early HCCs is limited.
■ Hepatobiliary phase MR imaging shows promise for characterization of precursor lesions and for identifying high-grade dysplastic nodules and early HCCs prior to neoarterialization and progression to overt HCC.
Received October 9, 2013; revision requested November 25; revision received February 6, 2014; final version accepted March 4.
Funding: Supported by the National Institutes of Health (grant DK088925).
Disclosures of Conflicts of Interest: J.Y.C. disclosed no relevant relationships. J.M.L. Activities related to the present article: disclosed no relevant relationships. Activities not related to the present article: grants and personal fees from Bayer Healthcare; grants from GE Healthcare, Dongseo Medical, Central Medical Service, Acuzen, and Starmed; and personal fees from Siemens Healthcare. Other relationships: disclosed no relevant relationships. C.B.S. Activities related to the present article: disclosed no relevant relationships. Activities not related to the present article: grants from GE and grants and personal fees from Bayer. Other relationships: disclosed no relevant relationships.
Abbreviations:
- GRE
- gradient echo
- HCC
- hepatocellular carcinoma
- ICC
- intrahepatic cholangiocarcinoma
- MRP
- multidrug resistance–associated protein
- OATP
- organic anionic transporting polypeptides
- 3D
- three dimensional
References
- 1.van Malenstein H, van Pelt J, Verslype C. Molecular classification of hepatocellular carcinoma anno 2011. Eur J Cancer 2011;47(12):1789–1797. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag HB, Davila JA, Petersen NJ, McGlynn KA. The continuing increase in the incidence of hepatocellular carcinoma in the United States: an update. Ann Intern Med 2003;139(10):817–823. [DOI] [PubMed] [Google Scholar]
- 3.McGlynn KA, London WT. Epidemiology and natural history of hepatocellular carcinoma. Best Pract Res Clin Gastroenterol 2005;19(1):3–23. [DOI] [PubMed] [Google Scholar]
- 4.Kudo M. Real practice of hepatocellular carcinoma in Japan: conclusions of the Japan Society of Hepatology 2009 Kobe Congress. Oncology 2010;78(Suppl 1):180–188. [DOI] [PubMed] [Google Scholar]
- 5.Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan. Lancet 1981;2(8256):1129–1133. [DOI] [PubMed] [Google Scholar]
- 6.Theise ND, Curado MP, Franceschi S, et al. Hepatocellular carcinoma. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, eds. WHO Cassification of Tumours of the Digestive System. 4th ed. Lyon, France: IARC, 2010; 205–216. [Google Scholar]
- 7.El-Serag HB. Hepatocellular carcinoma. N Engl J Med 2011;365(12):1118–1127. [DOI] [PubMed] [Google Scholar]
- 8.Baffy G, Brunt EM, Caldwell SH. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J Hepatol 2012;56(6):1384–1391. [DOI] [PubMed] [Google Scholar]
- 9.McGlynn KA, London WT. The global epidemiology of hepatocellular carcinoma: present and future. Clin Liver Dis 2011;15(2):223–243, vii–x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tyson GL, El-Serag HB. Risk factors for cholangiocarcinoma. Hepatology 2011;54(1):173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trevisani F, Cantarini MC, Wands JR, Bernardi M. Recent advances in the natural history of hepatocellular carcinoma. Carcinogenesis 2008;29(7):1299–1305. [DOI] [PubMed] [Google Scholar]
- 12.Bruix J, Sherman M; American Association for the Study of Liver Diseases. Management of hepatocellular carcinoma: an update. Hepatology 2011;53(3):1020–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.European Association For The Study Of The Liver; European Organisation For Research And Treatment Of Cancer . EASL-EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol 2012;56(4):908–943. [DOI] [PubMed] [Google Scholar]
- 14.Omata M, Lesmana LA, Tateishi R, et al. Asian Pacific Association for the Study of the Liver consensus recommendations on hepatocellular carcinoma. Hepatol Int 2010;4(2):439–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Padhya KT, Marrero JA, Singal AG. Recent advances in the treatment of hepatocellular carcinoma. Curr Opin Gastroenterol 2013;29(3):285–292. [DOI] [PubMed] [Google Scholar]
- 16.Marrero JA, Welling T. Modern diagnosis and management of hepatocellular carcinoma. Clin Liver Dis 2009;13(2):233–247. [DOI] [PubMed] [Google Scholar]
- 17.Nishida N, Goel A. Genetic and epigenetic signatures in human hepatocellular carcinoma: a systematic review. Curr Genomics 2011;12(2):130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Libbrecht L, Craninx M, Nevens F, Desmet V, Roskams T. Predictive value of liver cell dysplasia for development of hepatocellular carcinoma in patients with non-cirrhotic and cirrhotic chronic viral hepatitis. Histopathology 2001;39(1):66–73. [DOI] [PubMed] [Google Scholar]
- 19.Park YN. Update on precursor and early lesions of hepatocellular carcinomas. Arch Pathol Lab Med 2011;135(6):704–715. [DOI] [PubMed] [Google Scholar]
- 20.Brody RI, Theise ND. An inflammatory proposal for hepatocarcinogenesis. Hepatology 2012;56(1):382–384. [DOI] [PubMed] [Google Scholar]
- 21.Aravalli RN, Cressman EN, Steer CJ. Cellular and molecular mechanisms of hepatocellular carcinoma: an update. Arch Toxicol 2013;87(2):227–247. [DOI] [PubMed] [Google Scholar]
- 22.Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet 2002;31(4):339–346. [DOI] [PubMed] [Google Scholar]
- 23.Theise ND. Macroregenerative (dysplastic) nodules and hepatocarcinogenesis: theoretical and clinical considerations. Semin Liver Dis 1995;15(4):360–371. [DOI] [PubMed] [Google Scholar]
- 24.Theise ND. Cirrhosis and hepatocellular neoplasia: more like cousins than like parent and child. Gastroenterology 1996;111(2):526–528. [DOI] [PubMed] [Google Scholar]
- 25.Sun M, Eshleman JR, Ferrell LD, et al. An early lesion in hepatic carcinogenesis: loss of heterozygosity in human cirrhotic livers and dysplastic nodules at the 1p36-p34 region. Hepatology 2001;33(6):1415–1424. [DOI] [PubMed] [Google Scholar]
- 26.Aihara T, Noguchi S, Sasaki Y, Nakano H, Imaoka S. Clonal analysis of regenerative nodules in hepatitis C virus-induced liver cirrhosis. Gastroenterology 1994;107(6):1805–1811. [DOI] [PubMed] [Google Scholar]
- 27.Frenette C, Gish RG. Hepatocellular carcinoma: molecular and genomic guideline for the clinician. Clin Liver Dis 2011;15(2):307–321, vii–x. [DOI] [PubMed] [Google Scholar]
- 28.Palmer WC, Patel T. Are common factors involved in the pathogenesis of primary liver cancers? A meta-analysis of risk factors for intrahepatic cholangiocarcinoma. J Hepatol 2012;57(1):69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coleman WB. Mechanisms of human hepatocarcinogenesis. Curr Mol Med 2003;3(6):573–588. [DOI] [PubMed] [Google Scholar]
- 30.International Working Party . Terminology of nodular hepatocellular lesions. Hepatology 1995;22(3):983–993. [DOI] [PubMed] [Google Scholar]
- 31.Taguchi K, Asayama Y, Aishima S, et al. Morphologic approach to hepatocellular carcinoma development in man: de novo or the so-called ‘dysplastic nodule-carcinoma’ sequence? Oncol Rep 2002;9(4):737–743. [PubMed] [Google Scholar]
- 32.Park YN, Kim MJ. Hepatocarcinogenesis: imaging-pathologic correlation. Abdom Imaging 2011;36(3):232–243. [DOI] [PubMed] [Google Scholar]
- 33.International Consensus Group for Hepatocellular Neoplasia . Pathologic diagnosis of early hepatocellular carcinoma: a report of the international consensus group for hepatocellular neoplasia. Hepatology 2009;49(2):658–664. [DOI] [PubMed] [Google Scholar]
- 34.Bruix J, Sherman M; Practice Guidelines Committee, American Association for the Study of Liver Diseases. Management of hepatocellular carcinoma. Hepatology 2005;42(5):1208–1236. [DOI] [PubMed] [Google Scholar]
- 35.Kobayashi M, Ikeda K, Hosaka T, et al. Dysplastic nodules frequently develop into hepatocellular carcinoma in patients with chronic viral hepatitis and cirrhosis. Cancer 2006;106(3):636–647. [DOI] [PubMed] [Google Scholar]
- 36.Roskams T, Kojiro M. Pathology of early hepatocellular carcinoma: conventional and molecular diagnosis. Semin Liver Dis 2010;30(1):17–25. [DOI] [PubMed] [Google Scholar]
- 37.Sakamoto M. Pathology of early hepatocellular carcinoma. Hepatol Res 2007;37(Suppl 2):S135–S138. [DOI] [PubMed] [Google Scholar]
- 38.Nakashima O, Sugihara S, Kage M, Kojiro M. Pathomorphologic characteristics of small hepatocellular carcinoma: a special reference to small hepatocellular carcinoma with indistinct margins. Hepatology 1995;22(1):101–105. [PubMed] [Google Scholar]
- 39.Khalili K, Kim TK, Jang HJ, Yazdi LK, Guindi M, Sherman M. Indeterminate 1-2-cm nodules found on hepatocellular carcinoma surveillance: biopsy for all, some, or none? Hepatology 2011;54(6):2048–2054. [DOI] [PubMed] [Google Scholar]
- 40.Stevens WR, Gulino SP, Batts KP, Stephens DH, Johnson CD. Mosaic pattern of hepatocellular carcinoma: histologic basis for a characteristic CT appearance. J Comput Assist Tomogr 1996;20(3):337–342. [DOI] [PubMed] [Google Scholar]
- 41.Villanueva A, Newell P, Chiang DY, Friedman SL, Llovet JM. Genomics and signaling pathways in hepatocellular carcinoma. Semin Liver Dis 2007;27(1):55–76. [DOI] [PubMed] [Google Scholar]
- 42.Kojiro M. Histopathology of liver cancers. Best Pract Res Clin Gastroenterol 2005;19(1):39–62. [DOI] [PubMed] [Google Scholar]
- 43.Yano H, Iemura A, Fukuda K, Mizoguchi A, Haramaki M, Kojiro M. Establishment of two distinct human hepatocellular carcinoma cell lines from a single nodule showing clonal dedifferentiation of cancer cells. Hepatology 1993;18(2):320–327. [PubMed] [Google Scholar]
- 44.Okusaka T, Okada S, Ueno H, et al. Satellite lesions in patients with small hepatocellular carcinoma with reference to clinicopathologic features. Cancer 2002;95(9):1931–1937. [DOI] [PubMed] [Google Scholar]
- 45.Wang J, Li Q, Sun Y, et al. Clinicopathologic features between multicentric occurence and intrahepatic metastasis of multiple hepatocellular carcinomas related to HBV. Surg Oncol 2009;18(1):25–30. [DOI] [PubMed] [Google Scholar]
- 46.Park YN, Yang CP, Fernandez GJ, Cubukcu O, Thung SN, Theise ND. Neoangiogenesis and sinusoidal “capillarization” in dysplastic nodules of the liver. Am J Surg Pathol 1998;22(6):656–662. [DOI] [PubMed] [Google Scholar]
- 47.Kojiro M, Roskams T. Early hepatocellular carcinoma and dysplastic nodules. Semin Liver Dis 2005;25(2):133–142. [DOI] [PubMed] [Google Scholar]
- 48.Asayama Y, Yoshimitsu K, Irie H, et al. Poorly versus moderately differentiated hepatocellular carcinoma: vascularity assessment by computed tomographic hepatic angiography in correlation with histologically counted number of unpaired arteries. J Comput Assist Tomogr 2007;31(2):188–192. [DOI] [PubMed] [Google Scholar]
- 49.El-Assal ON, Yamanoi A, Soda Y, et al. Clinical significance of microvessel density and vascular endothelial growth factor expression in hepatocellular carcinoma and surrounding liver: possible involvement of vascular endothelial growth factor in the angiogenesis of cirrhotic liver. Hepatology 1998;27(6):1554–1562. [DOI] [PubMed] [Google Scholar]
- 50.Ueda K, Matsui O, Kawamori Y, et al. Hypervascular hepatocellular carcinoma: evaluation of hemodynamics with dynamic CT during hepatic arteriography. Radiology 1998;206(1):161–166. [DOI] [PubMed] [Google Scholar]
- 51.Kitao A, Zen Y, Matsui O, Gabata T, Nakanuma Y. Hepatocarcinogenesis: multistep changes of drainage vessels at CT during arterial portography and hepatic arteriography—radiologic-pathologic correlation. Radiology 2009;252(2):605–614. [DOI] [PubMed] [Google Scholar]
- 52.Matsui O, Kobayashi S, Sanada J, et al. Hepatocelluar nodules in liver cirrhosis: hemodynamic evaluation (angiography-assisted CT) with special reference to multi-step hepatocarcinogenesis. Abdom Imaging 2011;36(3):264–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kadoya M, Matsui O, Takashima T, Nonomura A. Hepatocellular carcinoma: correlation of MR imaging and histopathologic findings. Radiology 1992;183(3):819–825. [DOI] [PubMed] [Google Scholar]
- 54.Ishigami K, Yoshimitsu K, Nishihara Y, et al. Hepatocellular carcinoma with a pseudocapsule on gadolinium-enhanced MR images: correlation with histopathologic findings. Radiology 2009;250(2):435–443. [DOI] [PubMed] [Google Scholar]
- 55.Ishizaki M, Ashida K, Higashi T, et al. The formation of capsule and septum in human hepatocellular carcinoma. Virchows Arch 2001;438(6):574–580. [DOI] [PubMed] [Google Scholar]
- 56.Imaeda T, Kanematsu M, Mochizuki R, Goto H, Saji S, Shimokawa K. Extracapsular invasion of small hepatocellular carcinoma: MR and CT findings. J Comput Assist Tomogr 1994;18(5):755–760. [DOI] [PubMed] [Google Scholar]
- 57.Ng IO, Lai EC, Ng MM, Fan ST. Tumor encapsulation in hepatocellular carcinoma. A pathologic study of 189 cases. Cancer 1992;70(1):45–49. [DOI] [PubMed] [Google Scholar]
- 58.Lim JH, Choi D, Park CK, Lee WJ, Lim HK. Encapsulated hepatocellular carcinoma: CT-pathologic correlations. Eur Radiol 2006;16(10):2326–2333. [DOI] [PubMed] [Google Scholar]
- 59.Iguchi T, Aishima S, Sanefuji K, et al. Both fibrous capsule formation and extracapsular penetration are powerful predictors of poor survival in human hepatocellular carcinoma: a histological assessment of 365 patients in Japan. Ann Surg Oncol 2009;16(9):2539–2546. [DOI] [PubMed] [Google Scholar]
- 60.Takayama T, Makuuchi M, Hirohashi S, et al. Malignant transformation of adenomatous hyperplasia to hepatocellular carcinoma. Lancet 1990;336(8724):1150–1153. [DOI] [PubMed] [Google Scholar]
- 61.Kutami R, Nakashima Y, Nakashima O, Shiota K, Kojiro M. Pathomorphologic study on the mechanism of fatty change in small hepatocellular carcinoma of humans. J Hepatol 2000;33(2):282–289. [DOI] [PubMed] [Google Scholar]
- 62.Salomao M, Remotti H, Vaughan R, Siegel AB, Lefkowitch JH, Moreira RK. The steatohepatitic variant of hepatocellular carcinoma and its association with underlying steatohepatitis. Hum Pathol 2012;43(5):737–746. [DOI] [PubMed] [Google Scholar]
- 63.Zhang J, Krinsky GA. Iron-containing nodules of cirrhosis. NMR Biomed 2004;17(7):459–464. [DOI] [PubMed] [Google Scholar]
- 64.Terada T, Kadoya M, Nakanuma Y, Matsui O. Iron-accumulating adenomatous hyperplastic nodule with malignant foci in the cirrhotic liver. Histopathologic, quantitative iron, and magnetic resonance imaging in vitro studies. Cancer 1990;65(9):1994–2000. [DOI] [PubMed] [Google Scholar]
- 65.Gurusamy K. Trace element concentration in primary liver cancers—a systematic review. Biol Trace Elem Res 2007;118(3):191–206. [DOI] [PubMed] [Google Scholar]
- 66.Nassif A, Jia J, Keiser M, et al. Visualization of hepatic uptake transporter function in healthy subjects by using gadoxetic acid-enhanced MR imaging. Radiology 2012;264(3):741–750. [DOI] [PubMed] [Google Scholar]
- 67.Kitao A, Matsui O, Yoneda N, et al. The uptake transporter OATP8 expression decreases during multistep hepatocarcinogenesis: correlation with gadoxetic acid enhanced MR imaging. Eur Radiol 2011;21(10):2056–2066. [DOI] [PubMed] [Google Scholar]
- 68.Kogita S, Imai Y, Okada M, et al. Gd-EOB-DTPA-enhanced magnetic resonance images of hepatocellular carcinoma: correlation with histological grading and portal blood flow. Eur Radiol 2010;20(10):2405–2413. [DOI] [PubMed] [Google Scholar]
- 69.Tsuboyama T, Onishi H, Kim T, et al. Hepatocellular carcinoma: hepatocyte-selective enhancement at gadoxetic acid-enhanced MR imaging—correlation with expression of sinusoidal and canalicular transporters and bile accumulation. Radiology 2010;255(3):824–833. [DOI] [PubMed] [Google Scholar]
- 70.Vasuri F, Golfieri R, Fiorentino M, et al. OATP 1B1/1B3 expression in hepatocellular carcinomas treated with orthotopic liver transplantation. Virchows Arch 2011;459(2):141–146. [DOI] [PubMed] [Google Scholar]
- 71.Durnez A, Verslype C, Nevens F, et al. The clinicopathological and prognostic relevance of cytokeratin 7 and 19 expression in hepatocellular carcinoma. A possible progenitor cell origin. Histopathology 2006;49(2):138–151. [DOI] [PubMed] [Google Scholar]
- 72.Tsuda N, Matsui O. Cirrhotic rat liver: reference to transporter activity and morphologic changes in bile canaliculi—gadoxetic acid-enhanced MR imaging. Radiology 2010;256(3):767–773. [DOI] [PubMed] [Google Scholar]
- 73.Kitao A, Zen Y, Matsui O, et al. Hepatocellular carcinoma: signal intensity at gadoxetic acid-enhanced MR Imaging—correlation with molecular transporters and histopathologic features. Radiology 2010;256(3):817–826. [DOI] [PubMed] [Google Scholar]
- 74.Nakashima Y, Nakashima O, Tanaka M, Okuda K, Nakashima M, Kojiro M. Portal vein invasion and intrahepatic micrometastasis in small hepatocellular carcinoma by gross type. Hepatol Res 2003;26(2):142–147. [DOI] [PubMed] [Google Scholar]
- 75.Sakon M, Nagano H, Nakamori S, et al. Intrahepatic recurrences of hepatocellular carcinoma after hepatectomy: analysis based on tumor hemodynamics. Arch Surg 2002;137(1):94–99. [DOI] [PubMed] [Google Scholar]
- 76.Okuda K. Hepatocellular carcinoma: clinicopathological aspects. J Gastroenterol Hepatol 1997;12(9-10):S314–S318. [DOI] [PubMed] [Google Scholar]
- 77.Edmondson HA, Steiner PE. Primary carcinoma of the liver: a study of 100 cases among 48,900 necropsies. Cancer 1954;7(3):462–503. [DOI] [PubMed] [Google Scholar]
- 78.Pomfret EA, Washburn K, Wald C, et al. Report of a national conference on liver allocation in patients with hepatocellular carcinoma in the United States. Liver Transpl 2010;16(3):262–278. [DOI] [PubMed] [Google Scholar]
- 79.Shin WY, Suh KS, Lee HW, et al. Prognostic factors affecting survival after recurrence in adult living donor liver transplantation for hepatocellular carcinoma. Liver Transpl 2010;16(5):678–684. [DOI] [PubMed] [Google Scholar]
- 80.Cha C, Fong Y, Jarnagin WR, Blumgart LH, DeMatteo RP. Predictors and patterns of recurrence after resection of hepatocellular carcinoma. J Am Coll Surg 2003;197(5):753–758. [DOI] [PubMed] [Google Scholar]
- 81.Llovet JM, Fuster J, Bruix J; Barcelona-Clínic Liver Cancer Group. The Barcelona approach: diagnosis, staging, and treatment of hepatocellular carcinoma. Liver Transpl 2004;10(2 Suppl 1):S115–S120. [DOI] [PubMed] [Google Scholar]
- 82.Kojiro M, Kawabata K, Kawano Y, Shirai F, Takemoto N, Nakashima T. Hepatocellular carcinoma presenting as intrabile duct tumor growth: a clinicopathologic study of 24 cases. Cancer 1982;49(10):2144–2147. [DOI] [PubMed] [Google Scholar]
- 83.Iguchi T, Aishima S, Taketomi A, et al. Extracapsular penetration is a new prognostic factor in human hepatocellular carcinoma. Am J Surg Pathol 2008;32(11):1675–1682. [DOI] [PubMed] [Google Scholar]
- 84.Shirabe K, Kanematsu T, Matsumata T, Adachi E, Akazawa K, Sugimachi K. Factors linked to early recurrence of small hepatocellular carcinoma after hepatectomy: univariate and multivariate analyses. Hepatology 1991;14(5):802–805. [DOI] [PubMed] [Google Scholar]
- 85.Schima W, Kulinna C, Ba-Ssalamah A, Grünberger T. Multidetector computed tomography of the liver [in German]. Radiologe 2005;45(1):15–23. [DOI] [PubMed] [Google Scholar]
- 86.Frydrychowicz A, Lubner MG, Brown JJ, et al. Hepatobiliary MR imaging with gadolinium-based contrast agents. J Magn Reson Imaging 2012;35(3):492–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miraglia R, Pietrosi G, Maruzzelli L, et al. Predictive factors of tumor response to trans-catheter treatment in cirrhotic patients with hepatocellular carcinoma: a multivariate analysis of pre-treatment findings. World J Gastroenterol 2007;13(45):6022–6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kim MJ, Choi JY, Lim JS, et al. Optimal scan window for detection of hypervascular hepatocellular carcinomas during MDCT examination. AJR Am J Roentgenol 2006;187(1):198–206. [DOI] [PubMed] [Google Scholar]
- 89.Ichikawa T, Kitamura T, Nakajima H, et al. Hypervascular hepatocellular carcinoma: can double arterial phase imaging with multidetector CT improve tumor depiction in the cirrhotic liver? AJR Am J Roentgenol 2002;179(3):751–758. [DOI] [PubMed] [Google Scholar]
- 90.Earls JP, Rofsky NM, DeCorato DR, Krinsky GA, Weinreb JC. Hepatic arterial-phase dynamic gadolinium-enhanced MR imaging: optimization with a test examination and a power injector. Radiology 1997;202(1):268–273. [DOI] [PubMed] [Google Scholar]
- 91.Hong HS, Kim HS, Kim MJ, De Becker J, Mitchell DG, Kanematsu M. Single breath-hold multiarterial dynamic MRI of the liver at 3T using a 3D fat-suppressed keyhole technique. J Magn Reson Imaging 2008;28(2):396–402. [DOI] [PubMed] [Google Scholar]
- 92.Murakami T, Hori M, Kim T, Kawata S, Abe H, Nakamura H. Multidetector row CT and MR imaging in diagnosing hepatocellular carcinoma. Intervirology 2004;47(3-5):209–226. [DOI] [PubMed] [Google Scholar]
- 93.Iannaccone R, Laghi A, Catalano C, et al. Hepatocellular carcinoma: role of unenhanced and delayed phase multi-detector row helical CT in patients with cirrhosis. Radiology 2005;234(2):460–467. [DOI] [PubMed] [Google Scholar]
- 94.Rimola J, Forner A, Reig M, et al. Cholangiocarcinoma in cirrhosis: absence of contrast washout in delayed phases by magnetic resonance imaging avoids misdiagnosis of hepatocellular carcinoma. Hepatology 2009;50(3):791–798. [DOI] [PubMed] [Google Scholar]
- 95.Lim JH, Choi D, Kim SH, et al. Detection of hepatocellular carcinoma: value of adding delayed phase imaging to dual-phase helical CT. AJR Am J Roentgenol 2002;179(1):67–73. [DOI] [PubMed] [Google Scholar]
- 96.Takayasu K, Furukawa H, Wakao F, et al. CT diagnosis of early hepatocellular carcinoma: sensitivity, findings, and CT-pathologic correlation. AJR Am J Roentgenol 1995;164(4):885–890. [DOI] [PubMed] [Google Scholar]
- 97.Doyle DJ, O’Malley ME, Jang HJ, Jhaveri K. Value of the unenhanced phase for detection of hepatocellular carcinomas 3 cm or less when performing multiphase computed tomography in patients with cirrhosis. J Comput Assist Tomogr 2007;31(1):86–92. [DOI] [PubMed] [Google Scholar]
- 98.Davies BE, Kirchin MA, Bensel K, et al. Pharmacokinetics and safety of gadobenate dimeglumine (multihance) in subjects with impaired liver function. Invest Radiol 2002;37(5):299–308. [DOI] [PubMed] [Google Scholar]
- 99.Swan SK, Lambrecht LJ, Townsend R, et al. Safety and pharmacokinetic profile of gadobenate dimeglumine in subjects with renal impairment. Invest Radiol 1999;34(7):443–448. [DOI] [PubMed] [Google Scholar]
- 100.Ringe KI, Husarik DB, Sirlin CB, Merkle EM. Gadoxetate disodium-enhanced MRI of the liver: part 1, protocol optimization and lesion appearance in the noncirrhotic liver. AJR Am J Roentgenol 2010;195(1):13–28. [DOI] [PubMed] [Google Scholar]
- 101.Frericks BB, Loddenkemper C, Huppertz A, et al. Qualitative and quantitative evaluation of hepatocellular carcinoma and cirrhotic liver enhancement using Gd-EOB-DTPA. AJR Am J Roentgenol 2009;193(4):1053–1060. [DOI] [PubMed] [Google Scholar]
- 102.Huppertz A, Haraida S, Kraus A, et al. Enhancement of focal liver lesions at gadoxetic acid-enhanced MR imaging: correlation with histopathologic findings and spiral CT—initial observations. Radiology 2005;234(2):468–478. [DOI] [PubMed] [Google Scholar]
- 103.Tsuda N, Harada K, Matsui O. Effect of change in transporter expression on gadolinium-ethoxybenzyl-diethylenetriamine pentaacetic acid-enhanced magnetic resonance imaging during hepatocarcinogenesis in rats. J Gastroenterol Hepatol 2011;26(3):568–576. [DOI] [PubMed] [Google Scholar]
- 104.Tanimoto A, Kuwatsuru R, Kadoya M, et al. Evaluation of gadobenate dimeglumine in hepatocellular carcinoma: results from phase II and phase III clinical trials in Japan. J Magn Reson Imaging 1999;10(3):450–460. [DOI] [PubMed] [Google Scholar]
- 105.Libra A, Fernetti C, Lorusso V, et al. Molecular determinants in the transport of a bile acid-derived diagnostic agent in tumoral and nontumoral cell lines of human liver. J Pharmacol Exp Ther 2006;319(2):809–817. [DOI] [PubMed] [Google Scholar]
- 106.Bartolozzi C, Battaglia V, Bozzi E. Hepatocellular nodules in liver cirrhosis: contrast-enhanced MR. Abdom Imaging 2011;36(3):290–299. [DOI] [PubMed] [Google Scholar]
- 107.Rohrer M, Bauer H, Mintorovitch J, Requardt M, Weinmann HJ. Comparison of magnetic properties of MRI contrast media solutions at different magnetic field strengths. Invest Radiol 2005;40(11):715–724. [DOI] [PubMed] [Google Scholar]
- 108.Filippone A, Blakeborough A, Breuer J, et al. Enhancement of liver parenchyma after injection of hepatocyte-specific MRI contrast media: a comparison of gadoxetic acid and gadobenate dimeglumine. J Magn Reson Imaging 2010;31(2):356–364. [DOI] [PubMed] [Google Scholar]
- 109.Tanimoto A, Higuchi N, Ueno A. Reduction of ringing artifacts in the arterial phase of gadoxetic acid-enhanced dynamic MR imaging. Magn Reson Med Sci 2012;11(2):91–97. [DOI] [PubMed] [Google Scholar]
- 110.Svensson J, Petersson JS, Ståhlberg F, Larsson EM, Leander P, Olsson LE. Image artifacts due to a time-varying contrast medium concentration in 3D contrast-enhanced MRA. J Magn Reson Imaging 1999;10(6):919–928. [DOI] [PubMed] [Google Scholar]
- 111.Schmid-Tannwald C, Herrmann K, Oto A, Panteleon A, Reiser M, Zech C. Optimization of the dynamic, Gd-EOB-DTPA-enhanced MRI of the liver: the effect of the injection rate. Acta Radiol 2012;53(9):961–965. [DOI] [PubMed] [Google Scholar]
- 112.Tamada T, Ito K, Yoshida K, et al. Comparison of three different injection methods for arterial phase of Gd-EOB-DTPA enhanced MR imaging of the liver. Eur J Radiol 2011;80(3):e284–e288. [DOI] [PubMed] [Google Scholar]
- 113.Davenport MS, Viglianti BL, Al-Hawary MM, et al. Comparison of acute transient dyspnea after intravenous administration of gadoxetate disodium and gadobenate dimeglumine: effect on arterial phase image quality. Radiology 2013;266(2):452–461. [DOI] [PubMed] [Google Scholar]
- 114.Kühn JP, Hegenscheid K, Siegmund W, Froehlich CP, Hosten N, Puls R. Normal dynamic MRI enhancement patterns of the upper abdominal organs: gadoxetic acid compared with gadobutrol. AJR Am J Roentgenol 2009;193(5):1318–1323. [DOI] [PubMed] [Google Scholar]
- 115.Tanimoto A, Lee JM, Murakami T, Huppertz A, Kudo M, Grazioli L. Consensus report of the 2nd International Forum for Liver MRI. Eur Radiol 2009;19(Suppl 5):S975–S989. [DOI] [PubMed] [Google Scholar]
- 116.Nakamura Y, Toyota N, Date S, et al. Clinical significance of the transitional phase at gadoxetate disodium-enhanced hepatic MRI for the diagnosis of hepatocellular carcinoma: preliminary results. J Comput Assist Tomogr 2011;35(6):723–727. [DOI] [PubMed] [Google Scholar]
- 117.Krinsky GA, Lee VS, Nguyen MT, et al. Siderotic nodules at MR imaging: regenerative or dysplastic? J Comput Assist Tomogr 2000;24(5):773–776. [DOI] [PubMed] [Google Scholar]
- 118.Xu PJ, Yan FH, Wang JH, Shan Y, Ji Y, Chen CZ. Contribution of diffusion-weighted magnetic resonance imaging in the characterization of hepatocellular carcinomas and dysplastic nodules in cirrhotic liver. J Comput Assist Tomogr 2010;34(4):506–512. [DOI] [PubMed] [Google Scholar]
- 119.Krinsky GA, Israel G. Nondysplastic nodules that are hyperintense on T1-weighted gradient-echo MR imaging: frequency in cirrhotic patients undergoing transplantation. AJR Am J Roentgenol 2003;180(4):1023–1027. [DOI] [PubMed] [Google Scholar]
- 120.Hamilton SR, Aaltonen LA. Tumours of the liver and intrahepatic bile ducts. In: World Health Organization Classification of Tumours. Lyon, France: IARC, 2000; 159–172. [Google Scholar]
- 121.Murakami T, Nakamura H, Hori S, et al. CT and MRI of siderotic regenerating nodules in hepatic cirrhosis. J Comput Assist Tomogr 1992;16(4):578–582. [DOI] [PubMed] [Google Scholar]
- 122.Hanna RF, Aguirre DA, Kased N, Emery SC, Peterson MR, Sirlin CB. Cirrhosis-associated hepatocellular nodules: correlation of histopathologic and MR imaging features. RadioGraphics 2008;28(3):747–769. [DOI] [PubMed] [Google Scholar]
- 123.Freeny PC, Grossholz M, Kaakaji K, Schmiedl UP. Significance of hyperattenuating and contrast-enhancing hepatic nodules detected in the cirrhotic liver during arterial phase helical CT in pre-liver transplant patients: radiologic-histopathologic correlation of explanted livers. Abdom Imaging 2003;28(3):333–346. [DOI] [PubMed] [Google Scholar]
- 124.Tsuda N, Matsui O. Signal profile on Gd-EOB-DTPA-enhanced MR imaging in non-alcoholic steatohepatitis and liver cirrhosis induced in rats: correlation with transporter expression. Eur Radiol 2011;21(12):2542–2550. [DOI] [PubMed] [Google Scholar]
- 125.Manfredi R, Maresca G, Baron RL, et al. Gadobenate dimeglumine (BOPTA) enhanced MR imaging: patterns of enhancement in normal liver and cirrhosis. J Magn Reson Imaging 1998;8(4):862–867. [DOI] [PubMed] [Google Scholar]
- 126.Lim JH, Choi BI. Dysplastic nodules in liver cirrhosis: imaging. Abdom Imaging 2002;27(2):117–128. [DOI] [PubMed] [Google Scholar]
- 127.Baron RL, Peterson MS. From the RSNA refresher courses: screening the cirrhotic liver for hepatocellular carcinoma with CT and MR imaging: opportunities and pitfalls. RadioGraphics 2001;21(Spec No):S117–S132. [DOI] [PubMed] [Google Scholar]
- 128.Martín J, Puig J, Darnell A, Donoso L. Magnetic resonance of focal liver lesions in hepatic cirrhosis and chronic hepatitis. Semin Ultrasound CT MR 2002;23(1):62–78. [DOI] [PubMed] [Google Scholar]
- 129.Earls JP, Theise ND, Weinreb JC, et al. Dysplastic nodules and hepatocellular carcinoma: thin-section MR imaging of explanted cirrhotic livers with pathologic correlation. Radiology 1996;201(1):207–214. [DOI] [PubMed] [Google Scholar]
- 130.Ito K, Mitchell DG, Gabata T, et al. Hepatocellular carcinoma: association with increased iron deposition in the cirrhotic liver at MR imaging. Radiology 1999;212(1):235–240. [DOI] [PubMed] [Google Scholar]
- 131.Martín J, Sentís M, Zidan A, et al. Fatty metamorphosis of hepatocellular carcinoma: detection with chemical shift gradient-echo MR imaging. Radiology 1995;195(1):125–130. [DOI] [PubMed] [Google Scholar]
- 132.Rimola J, Forner A, Tremosini S, et al. Non-invasive diagnosis of hepatocellular carcinoma ≤ 2 cm in cirrhosis. Diagnostic accuracy assessing fat, capsule and signal intensity at dynamic MRI. J Hepatol 2012;56(6):1317–1323. [DOI] [PubMed] [Google Scholar]
- 133.Willatt JM, Hussain HK, Adusumilli S, Marrero JA. MR. Imaging of hepatocellular carcinoma in the cirrhotic liver: challenges and controversies. Radiology 2008;247(2):311–330. [DOI] [PubMed] [Google Scholar]
- 134.Park MJ, Kim YK, Lee MH, Lee JH. Validation of diagnostic criteria using gadoxetic acid-enhanced and diffusion-weighted MR imaging for small hepatocellular carcinoma (<= 2.0 cm) in patients with hepatitis-induced liver cirrhosis. Acta Radiol 2013;54(2):127–136. [DOI] [PubMed] [Google Scholar]
- 135.Kim T, Baron RL, Nalesnik MA. Infarcted regenerative nodules in cirrhosis: CT and MR imaging findings with pathologic correlation. AJR Am J Roentgenol 2000;175(4):1121–1125. [DOI] [PubMed] [Google Scholar]
- 136.Takeshita K, Nagashima I, Frui S, et al. Effect of superparamagnetic iron oxide-enhanced MRI of the liver with hepatocellular carcinoma and hyperplastic nodule. J Comput Assist Tomogr 2002;26(3):451–455. [DOI] [PubMed] [Google Scholar]
- 137.Honda H, Tajima T, Kajiyama K, et al. Vascular changes in hepatocellular carcinoma: correlation of radiologic and pathologic findings. AJR Am J Roentgenol 1999;173(5):1213–1217. [DOI] [PubMed] [Google Scholar]
- 138.Hayashi M, Matsui O, Ueda K, Kawamori Y, Gabata T, Kadoya M. Progression to hypervascular hepatocellular carcinoma: correlation with intranodular blood supply evaluated with CT during intraarterial injection of contrast material. Radiology 2002;225(1):143–149. [DOI] [PubMed] [Google Scholar]
- 139.Forner A, Vilana R, Ayuso C, et al. Diagnosis of hepatic nodules 20 mm or smaller in cirrhosis: Prospective validation of the noninvasive diagnostic criteria for hepatocellular carcinoma. Hepatology 2008;47(1):97–104. [DOI] [PubMed] [Google Scholar]
- 140.Ito K. Hepatocellular carcinoma: conventional MRI findings including gadolinium-enhanced dynamic imaging. Eur J Radiol 2006;58(2):186–199. [DOI] [PubMed] [Google Scholar]
- 141.Golfieri R, Grazioli L, Orlando E, et al. Which is the best MRI marker of malignancy for atypical cirrhotic nodules: hypointensity in hepatobiliary phase alone or combined with other features? Classification after Gd-EOB-DTPA administration. J Magn Reson Imaging 2012;36(3):648–657. [DOI] [PubMed] [Google Scholar]