

Abstract
The Saccharomyces cerevisiae genome encodes two sequence related acetyl-CoA carboxylases, the cytosolic Acc1p and the mitochondrial Hfa1p, required for respiratory function. Several aspects of expression of the HFA1 gene and its evolutionary origin have remained unclear. Here, we determined the HFA1 transcription initiation sites by 5′ RACE analysis. Using a novel “Stop codon scanning” approach, we mapped the location of the HFA1 translation initiation site to an upstream AUU codon at position −372 relative to the annotated start codon. This upstream initiation leads to production of a mitochondrial targeting sequence preceding the ACC domains of the protein. In silico analyses of fungal ACC genes revealed conserved “cryptic” upstream mitochondrial targeting sequences in yeast species that have not undergone a whole genome duplication. Our Δhfa1 baker's yeast mutant phenotype rescue studies using the protoploid Kluyveromyces lactis ACC confirmed functionality of the cryptic upstream mitochondrial targeting signal. These results lend strong experimental support to the hypothesis that the mitochondrial and cytosolic acetyl-CoA carboxylases in S. cerevisiae have evolved from a single gene encoding both the mitochondrial and cytosolic isoforms. Leaning on a cursory survey of a group of genes of our interest, we propose that cryptic 5′ upstream mitochondrial targeting sequences may be more abundant in eukaryotes than anticipated thus far.
Introduction
A fairly recently recognized feature of mitochondria, conserved in all eukaryotes, is their ability to synthesize fatty acids in an acyl carrier protein dependent manner. The mitochondrial fatty acid synthesis (mtFAS) pathway is best described in the yeast Saccharomyces cerevisiae, where the disruption of the pathway results in a respiratory deficient phenotype, lack of cytochromes, and loss of or decrease in lipoic acid. There is overwhelming evidence that lipoic acid is one of the downstream products of mtFAS, an enzyme cofactor essential for the function of several mitochondrial enzyme complexes [1], [2]. The mtFAS process has been proposed to act as a regulatory circuit, controlling mitochondrial biogenesis in response to acetyl-CoA availability, and the presented evidence suggests a physiological role for fatty acids products of mtFAS longer than the octanoic acid precursor of lipoic acid in this regulatory circuit [3]. Acetyl-CoA carboxylase (ACC) catalyses the ATP-dependent carboxylation of acetyl-CoA to form malonyl-CoA, the first committed step in FAS. This reaction product is an intermediate in the de novo synthesis of long-chain fatty acids both in mitochondria and cytosol. It is also a substrate for distinct fatty acyl-CoA elongation enzymes in the endoplasmic reticulum. In mammals and probably also in other metazoans, malonyl-CoA is a transmitter of a signaling cascade sensing nutrition status of the body and tissue and controls the transport of long-chain fatty acids into mitochondria for β-oxidation by inhibiting carnitine palmitoyltransferaseI (CPT1) [4]. In key tissues, the regulation of the rate of β-oxidation plays a major role in orchestrating whole body metabolic adaptations to changes in nutrient availability and to fuel traffic between organelles. There are still several intriguing questions surrounding the origin and expression of mitochondrial matrix ACC in yeast (HFA1) as well as higher eukaryotes.
In S. cerevisiae, Acc1p is found to be the cytosolic version of acetyl-CoA carboxylase [5] while Hfa1p represents the mitochondrial counterpart [6]. Translation initiated from the first ATG start codon of the HFA1 ORF does not produce a functional enzyme, and homology to ACC1 extends far towards the 5′ direction of the annotated ATG of HFA1 [6]. Further analysis for this upstream region revealed a putative mitochondrial import sequence required for mitochondrial function. The only additional, far upstream in-frame ATGs are almost immediately followed by stop codons, requiring exotic mechanisms like mRNA editing or ribosomal frame-shifting to allow translation of HFA1. It has been recognized already in the 1980s that yeast is capable to initiate gene product translation from non AUG start codons [7], and a physiological role of translation from non-canonical initiation sites in the production of mitochondrially localized protein isoforms of several tRNA synthetases in S. cerevisiae [8]–[11] has been well documented. A perhaps more feasible solution to the problem of HFA1 expression would therefore be a non AUG start codon in HFA1 translation as has been previously proposed [6]. To date, there are large discrepancies in the existing reports for the transcription and translation start sites of HFA1.
In addition to the question of HFA1 expression, the evolutionary origin of this gene and its ACC1 counterpart has remained unresolved. Kellis et al. used the ACC1/HFA1 gene pair as an example for gene specialization after the genome duplication that led to the speciation of S. cerevisiae. The authors argued for the gain of a mitochondrial import sequence by the gene that was to become HFA1 after duplication of the proto-fungal ACC [12]. The discussion on the evolution of HFA1 by Hoja et al. can also be interpreted in a similar manner. In contrast, Turunen et al. suggested the common ancestor of both genes to be a one-gene variant encoding both the cytosolic and mitochondrial proteins, the former initiated from a canonical ATG translation start codon and the latter from a non-AUG triplet [13], but in vivo experimental support for this hypothesis is lacking.
In this study, we identified the transcription and translation initiation sites to provide insight into how the expression of HFA1 is achieved. We have used Kluyveromyces lactis, a protoploid yeast strain closely related to the degenerate diploid S. cerevisiae and commonly used for comparative genetic studies, to address the question of the evolutionary origin of yeast acetyl-CoA carboxylases. The presence of cryptic 5′ untranslated regions (UTRs) containing mitochondrial targeting sequences in yeast raises the intriguing question about the possibility of the presence of a hidden cache of low abundance eukaryotic mitochondrial proteins that have not been recognized due to a primary function in another compartment.
Materials and Methods
Yeast Strains, media and genetic methods
Wild type and mutant strains of S. cerevisiae and sources are summarized in Table 1. Media used are as follows: YPD [1% yeast extract (DIFCO, NJ, USA), 2% Bacto- peptone (DIFCO), 2% glucose], SCD/-Uracil [0.67% Yeast Nitrogen Base without amino acids (DIFCO), 0.19% Synthetic complete drop out mix without uracil (SIGMA-Aldrich, St. Louis, MO, USA), 2% glucose], SCD[0.67% Yeast Nitrogen Base without amino acids, 0.19% Synthetic complete drop out mix without uracil, 0.008% Uracil, 2% glucose], SCG[0.67% Yeast Nitrogen Base without amino acids, 0.19% Synthetic complete drop out mix without uracil, 0.008% Uracil, 3% glycerol], SCL[0.67% Yeast Nitrogen Base without amino acids, 0.19% Synthetic complete drop out mix without uracil, 0.008% Uracil, 1% D/L lactic acid]. SCD -Leucine [0.67% Yeast Nitrogen Base without amino acids, 0.16% Synthetic complete drop out mix without leucine, 2% glucose]. Solid media was prepared with 2%agar.
Table 1. Yeast strains used for this study.
| Strain | Genotype | Reference |
| W1536 5B | MAT a ; Δade2; Δade3; his3-11; his3-12; leu2-3; trp1-1; ura3-1 | H. Zou and R. Rothstein [27] [43] |
| W1536 8B | MATα; Δade2; Δade3; his3-11; his3-12; leu2-3; trp1-1; ura3-1 | H. Zou and R. Rothstein [27] [43] |
| BY4741 Δhfa1 | MAT a ; Δhis3-1; Δleu2-0; Δ met15-0; Δura3-0; YMR207c::kanM4 | Euroscarf |
| BY4742 Δhfa1 | MATα; Δ his3-1; Δleu2-0; Δlys2-0; Δura3-0; YMR207c::KanMX | Euroscarf |
| W1536 8B Δhfa1 pTSV30 HFA1 | MATα; Δade2; Δade3; his3-11; his3-12; leu2-3; trp1-1; ura3-1; YMR207c::kanMX4 pTSV30 HFA1 | This study |
| W1536 8B Δhfa1 | MATα; Δade2; Δade3; his3-11; his3-12; leu2-3; trp1-1; ura3-1 | This study |
| W1536 8B Δetr1 | MATα; Δade2; Δade3; his3-11; his3-12; leu2-3; trp1-1; ura3-1; mrf::kanMX | This study |
Construction of plasmids
Plasmids used for this study are listed in Table 2. Because HFA1 is a very large gene, a PstI site was introduced in position +2449 (relative to the annotated ATG) to allow easier handling. The PstI site does not change the encoded amino acid sequence. To generate this construct, the sequence encoding the C-terminal end of HFA1 was PCR amplified from wild type yeast genomic DNA using oligonucleotides HFA1 +2449 5′PstI and HFA1 clon/KO 3′SmaI and cloned into the PstI and SmaI sites of YCplac33 to generate YCp33HFA1 C-term. The N-terminal end of HFA1 was PCR amplified using oligonucleotids HFA1 -980 5′HindIII and HFA1 +2449 3′PstI and ligated into YCp33HFA1 C-term to generate YCp33HFA1 with HFA1 gene under its own regulatory sequences. The YCp33hfa1-1 was generated like YCp33HFA1, but the sequence encoding the N-terminal part of hfa1-1 gene was PCR amplified from a synthetic petite mutant [3] and contained the C273T mutation, leading to the premature stop codon Q91STOP. The bluescript vector pBS KS (+) was digested with HindIII and PstI (New England Biolabs 20U/µl). The N-terminal part of HFA1 was cut out from YCp33 HFA1 with HindIII and PstI (New England Biolabs 20U/µl), inserted to the digested bluescript vector pBS KS (+). This plasmid was used for the site directed mutagenesis manipulations.YCp33 HFA1 -273, YCp33 HFA1 -282, YCp33 HFA1-312, YCp33 HFA1 -360, YCp33 HFA1 -363, YCp33 HFA1 -372, YCp33 HFA1 -375, YCp33 HFA1 -381 and YCp33 HFA1 -378 were generated by site-directed mutagenesis (QuikChange XL Site-Directed Mutagenesis Kit, Stratagene, La Jolla, California, USA) of pBS KS N-HFA1 (Table 2). HFA1 with point mutation was cloned into the expression vector YCp33 HFA1 (Table 2) using the PstI and HindIII restriction sites.
Table 2. Plasmids used for the acetyl-CoA carboxylases study in yeasts.
| Plasmid | Description | Reference |
| YCplac33 | Single copy vector, empty | [15] |
| YCp33 HFA1 | Full length HFA1with and engineered Pst1 site in position +2449, cloned into HindIII and SmaI restriction sites | This study |
| pET23a+ | Novagen (Merck Chemicals, Nottingham, United Kingdom) | |
| pET23a+ HFA1 tx start | Promoter region of HFA1 cloned within BamH1 and XbaI restriction sites (see description in text) | This study |
| YEplac181 | Single copy vector, empty | [15] |
| pBS KS + | Fermentas (Helsinki, Finland) | |
| pBS KS N-HFA1 | This study | |
| pTSV30A | Multi copy vector, empty | [27] |
| pTSV30HFA1 | Full length HFA1 cloned into XmaI and XbaI restriction sites | This study [44] |
| YCp33 HFA1 −381 | Generated through site directed mutagenesis from YCp33 HFA1 or pBS N-HFA1 using the appropriate primers | This study |
| YCp33 HFA1 −378 | Generated through site directed mutagenesis from YCp33 HFA1 or pBS N-HFA1 using the appropriate primers | This study |
| YCp33 HFA1 −375 | Generated through site directed mutagenesis from YCp33 HFA1 or pBS N-HFA1 using the appropriate primers | This study |
| YCp33 HFA1 −372 | Generated through site directed mutagenesis from YCp33 HFA1 or pBS N-HFA1 using the appropriate primers | This study |
| YCp33 HFA1 −363 | Generated through site directed mutagenesis from YCp33 HFA1 or pBS N-HFA1 using the appropriate primers | This study |
| YCp33 HFA1 −360 | Generated through site directed mutagenesis from YCp33 HFA1 or pBS N-HFA1 using the appropriate primers | This study |
| YCp33 HFA1 −312 | Generated through site directed mutagenesis from YCp33 HFA1 or pBS N-HFA1 using the appropriate primers | This study |
| YCp33 HFA1 −282 | Generated through site directed mutagenesis from YCp33 HFA1 or pBS N-HFA1 using the appropriate primers | This study |
| YCp33 HFA1 −273 | Full length HFA1 cloned into HindIII and SmaI restriction sites PCR amplified from genomic DNA of HFA1 point mutant (Kursu et al. 2013) | This study |
| Ycp33 ADH1 promoter | ADH promoter cloned into HindIII and XbaI restriction sites | This study |
| YCp33 K.lactis HFA1 * | Full length K.lactis HFA1 with upstream sequence predicted to be MTS cloned into SpeI and EcoRI restriction sites | This study |
| YCp33 K.lactis HFA1 * noMTS | Full length K.lactis HFA1 cloned into SpeI and EcoRI restriction sites | This study |
*these plasmids are unstable in E. coli and have to be maintained in S. cerevisiae frozen stocks
Plasmid YCp33pADH1 was constructed by amplification of the −720 to −12 region of the ADH1 promoter sequence (numbers are location upstream relative to the initiation ATG of the ADH1 ORF) from yeast genomic DNA using primers HindIIIADH1prom 5′ and ADH1 prom XbaI 3′ and ligation of this product digested with HindIII and XbaI into the corresponding sites of YCplac33.
The bluescript vector pBS KS (+) was digested with HindIII and PstI (New England Biolabs 20U/µl). The N-terminal part of HFA1 was cut out from YCp33 HFA1 with HindIII and PstI (New England Biolabs 20U/µl), inserted to the digested bluescript vector pBS KS (+).
Oligonucleotides
The DNA oligonucleotides designed for this study and their purpose are listed in Table 3.
Table 3. Oligonucleotides used for the acetyl-CoA carboxylases study in yeasts.
| Name | Sequence |
| HFA1 +2449 5′PstI | AAACTGCAGCCACTTCTTAAAATTAGTGAA |
| HFA1 clon/KO 3′SmaI | CTCTCAACCAATGTCACCAA |
| HFA1 −980 5′HindIII | CCCAAGCTTGTTGTACACAGCAGAGTG |
| HFA1 +2449 3′PstI | AAACTGCAGGGCAACATAAACTACATGAT |
| BamH1 Hfa1p transcr. Start 5′ | TTATGGATCCGCATTCTGAAAGTGAGATAG |
| Xba1 Hfa1p transcr. Start 3′ | TTATTCTAGAAGAGAGGTTCCTACTACTTC |
| Hfa1p transcr. Start 3′ seq | AGAGGTTCCTACTACTTC |
| Hfa1p 5′RACE Sp1 | ATCATTGAACGTCTCGTACGC |
| Hfa1p 5′ RACE Sp2 | CTTTAGATCTCTTTCTTTCACCGCAGCA |
| Hfa1p 5′ RACE Sp3 | CGAGATGAATGCCTATACG |
| Xba1 Hfa1p transcr. Start 3′ new | TTATTCTAGACGCGAGCTTGTACTGTCCTATTCG |
| HA-His tag coding Sph1 | GTACCCATACGACGTCCCAGACTACGCTCATCATCATCATCATCATT AGGCATG |
| HA-His tag complementing Pst1 | CTAATGATGATGATGATGATGAGCGTAGTCTGGGACGTCGTATGGGTACTGCA |
| Hfa1-Ile127 Pst1 | TTATCTGCAGGATACACGGGGATTTTAGCGC |
| Hfa1-Arg135 Pst1 | TTATCTGGCAGTCTATATGTGAACCAGACCAAG |
| Hfa1-prom-5′ EcoR1 | TTATGATATCCGTTCTTCGATACCCTGTCGAA |
| HFA1 Stop −381 5′-3′ | CCGGATACATTCTCACTTTTACATTACCTAATTCACAATTACTTG |
| HFA1 Stop −381 5′-3′ | CAAGTAATTGTGAATTAGGTAATGTAAAAGTGAGAATGTATCCGG |
| HFA1 Stop −378 5′-3′ | CTCGCCGGATACATTCTCACTTTTACATTCCCATATTCACAATTA |
| HFA1 Stop −378 3′-5′ | TAATTGTGAATATGGGAATGTAAAAGTGAGAATGTATCCGGCGAG |
| HFA1 Stop −375 5′-3′ | GGATACATTCTCACTTTTACATTACCATATAGACAATTACTTGCATTCGAATAGG |
| HFA1 Stop −375 3′-5′ | CCTATTCGAATGCAAGTAATTGTCTATATGGTAATGTAAAAGTGAGAATGTATCC |
| HFA1 Stop −372 5′-3′ | CATTACCATATTCACATAAACTTGCATTCGAATAG |
| HFA1 Stop −372 3′-5′ | TAAAAGTGAGAATGTATCCGG |
| HFA1 Stop −363 5′-3′ | TTCACAATTACTTGCTAACGAATAGGACAGTAC |
| HFA1 Stop −363 3′-5′ | TATGGTAATGTAAAAGTGTGAGAATGTACCG |
| HFA1 Stop −360 5′-3′ | ATATTCACAATTACTTGCATTTAGATAGGACAGTAC |
| HFA1 Stop −360 3′-5′ | GTACTGTCCTATCTAAATGCAAGTAATTGTGAATAT |
| HFA1 Stop −312- 5′-3″ | GGATCCTTATCGATTCTAGAATATAACGGGCAGCCAGATAGTACGG |
| HFA1 Stop −312 3′-5′ | CCGTACTATCTGGCTGCCCGTTATATTCTAGAATCGATAAGGATCC |
| HFA1 Stop −282 5′-3″ | GCAGCCAGATAGTACGGTAGAAAGGACAGCGCC |
| HFA1 Stop −282 3′-5′ | GGCGCTGTCCTTTCTACCGTACTATCTGGCTGC |
| HFA1-prom-5′ Sma1/Xma1 | TTATCCCGGGCGTTCTTCGATACCCTGTCG |
| HFA1-transcr_Start seq. 3′ new | CGCGAGCTTGTACTGTCCTATTCG |
| HFA1-980 5′ HindIII | CCCAAGCTTGTTGTACACAGCAGAGTG |
| HFA1+2450 3′ PstI | AAACTGCAGGGCAACATAAACTACATGAT |
| HFA1+2450 5′ PstI | AAACTGCAGCCCACTTCTTAAAATTAGTGAA |
| HFA1+350 5′seq | GGGTGACAAGATTTCTTCC |
| HFA1+950 5′ seq | CGAATACTTATATTCACCAAAAG |
| HFA1+1450 5′ seq | CGACTGGTTGGTTAGATG |
| HFA1+2000 5′ seq | GAAAAGTGATGGTGTAATTG |
| HFA1+3000 5′ seq | GATACAACTTCAGGATTTGTTC |
| HFA1+3500 5′ seq | CGTATTACATTTGCATTTATC |
| HFA1+4000 5′ seq | GAGTGCCTAGAAACAAAGAAG |
| HFA1+4500 5′ seq | CGAGAGAAGATTTGTTTTTTG |
| HFA1+5000 5′ seq | CTAACCTAACAAATTGGCG |
| HFA1+5500 5′ seq | CGTTCAAAACAGCTCAAAC |
| HFA1+6000 5′ seq | GATAGATCGACTAGGATGC |
| HFA1+200 3′ seeq | GACCGCATCCACATCC |
| HindIIIADH1prom 5′ | GATCAAGCTTGATATCCTTTTGTTGTTTCC |
| ADH1 prom XbaI 3′ | GATCTCTAGAAGTTGATTGTATGCTTGGTATAGC |
| ADH promoter klac ACC SpeI-F | CTTTTTCTGCACAATATTTCAAGCTATACCAAGCATACAATCAACT ACTAGTAGGTTAAAGAAAGTTTTG |
| ADH promoter klac-minusMTS ACC SpeI- F | CTTTTTCTGCACAATATTTCAAGCTATACCAAGCATACAATCAACTACTAGTATGAGTGAGGAAAATCTTTC |
| YCp33-EcoRI-Klac ACC R | ACGACGGCCAGTGAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGAATTCCAATCATATCTTTATT |
| ADH1promseq −90 → −68 | GTCATTGTTCTCGTTCCCTTTC |
| Klac pcr R check2 | GCCTTATCCACAGTGTTCAG |
Isolation of yeast genomic DNA
Genomic DNA was extracted following the standard protocol [14].
Disruption of the HFA1 gene
Initial attempts to generate the hfa1 deletion in the W1536 8B background consistently resulted in rho− derivatives. Subsequently, a strain carrying a wild type copy of HFA1 on a plasmid was used to prevent generation of a rho− mutant in the absence of HFA1. The W1536 8B Δhfa1 pTSV30 HFA1 strain was generated from W1536 8B carrying pTSV30 HFA1 by transformation with a PCR product containing the hfa1::kanMX4 cassette amplified from the genomic DNA of BY4741 Δhfa1.The transformation was carried out using a high efficiency transformation protocol [15] followed by selection for geneticin resistance. The W1536 8B Δhfa1 strain was obtained by allowing loss of the pTSV30 HFA1 plasmid on glucose. Colonies lacking the plasmid were identified by white colony color. In absence of the plasmid, the strain is geneticin resistant and can be complemented by plasmid-borne wild type HFA1. Similar to the previous report [6] the Δhfa1 strain shows differences in growth behavior depending on which non-fermentable carbon source is used. The W1536 8B Δhfa1 strain we generated is able to weakly grow on glycerol but unable to grow on lactate at elevated temperature (33°C) (data not shown).
Identification of the translation start site, “Stop codon scanning”
The idea of “stop codon scanning” is to pinpoint the exact translation start site by introducing in-frame stop codons in the promoter region and by monitoring the phenotype for the respiratory deficiency. An in-frame stop codon will lead to a growth deficient mutant phenotype on lactate medium if introduced downstream of the translation start site and should have no effect on the phenotype if located upstream of the translation start site. The stop codons were introduced from the region close to the location of the respiratory deficient stop codon mutation site towards upstream. We took care to design the changes in a manner so that in most cases a single base pair mutation led to an in-frame stop codon. The purpose was to make the smallest change necessary to minimize the possibility of disrupting a regulatory RNA structure or protein binding sites. Site directed mutagenesis was employed. The deletion strain W1536 8B Δhfa1 was transformed using the One Step Transformation protocol [16] with STOP-codon-mutagenized or control plasmids and incubated on selective media at 30°C. Transformants carrying the plasmids were then streaked for single colonies on SCL plates as well as SCD control plates and incubated at 33°C for four to seven days to test whether the plasmids were able to rescue the respiratory deficient phenotype of the deletion strain. The W1536 8B Δhfa1 YCp33 HFA1 and W1536 8B strains were used as positive control on the non-fermentable carbon source, while the respiratory deficient W1536 8B Δetr1 strain, lacking the mitochondrial thioester reductase of mtFAS [17], served as a negative control. For yeast serial dilution/spotting assays, the transformants carrying the relevant constructs were first grown over night on 2 ml SCD-Ura liquid medium. The next day, the cultures were re-inoculated in 5 ml fresh SCD-Ura medium and grown for five hours to reach and OD600 of approximately 0.5. The OD600 of the individual cultures was measured, and cell concentrations were all normalized to OD600 = 0.5. These 1x cell suspensions were diluted to 1/10, 1/100 and 1/1000 in sterile water and spotted on SCD and SCLactate plates. The plates were then incubated for two (SCD) or three days (SCL), when the growth was then documented.
Identification of the transcription start site with 5′RACE study
The 5′RACE experiment was done according to the protocol of the manufacturer (5′/3′ RACE Kit, 2nd Generation, Roche Applied Science, Mannheim, Germany). Total RNA isolated from W1536 8B was used as template for cDNA synthesis primed by the HFA1 specific primer SP1 (Table 3). The ss cDNA was treated with a terminal transferase that introduced a polyA tail at the 3′-end of the ss cDNA. A kit-specific polyT PCR anchor primer and a HFA1 specific SP2 primer were used to amplify the ss cDNA by using the DyNAzymeII polymerase (Fermentas, Helsinki, Finland).
Analysis of the K. lactis ACC nucleotide sequence
The in silico translation analysis of the 5′-upstream of the K. lactis ACC was performed to test for the possible presence of a mitochondrial targeting signal. We have used the MitoProtII [18] and Target P mitochondrial localization prediction programs [19] for the analysis.
Rescue of the respiratory deficiency of the Δhfa1 strain with a K.lactis ACC complementation construct
The translated amino acid sequence of the K. lactis ACC open reading frame (KLLA0F06072g) including the 5′ sequence preceding the designated start codon was obtained with the Expasy translate tool (http://web.expasy.org). The 5′ region upstream of the starting AUG of the sequence was translated up to the first in-frame STOP codon. This putative 83 amino acid sequence was analyzed with the MitoProtII program [18] and calculated for the mitochondrial targeting signal (MTS) probability. No colony developed from Escherichia coli cells transformed with plasmids ligated to K. lactis ACC1, indicating that this ORF was toxic to the bacteria. To overcome this problem, we took a yeast gap repair approach to produce the constructs directly in the yeast cells [20]. S. cerevisiae is able to repair gapped plasmids to circles if it is provided with a “patch” sequence that carries homology to the free plasmid arms on each end. This feature can be used to insert DNA molecules, carrying plasmid homology introduced to the ends by PCR, into linearized plasmids by co-transformation. Repaired plasmids can be identified by the plasmid nutritional marker. To control against transformation due to incompletely gapped plasmids, parallel mock transformations were carried out using only the gapped plasmid, but no insert with homologous ends. These control transformations should yield in no or much lower numbers of transformant colonies on the selective plates. The construct containing the predicted non-AUG initiated MTS together with the K.lactis ACC ORF was amplified with the primers ADH promoter klacACC SpeI-F and YCp33-EcoRI-klacACC-R. The K.lactis ACC ORF was amplified with the primer ADH promoter klac-minusMTS ACC SpeI-F and YCp33-EcoRI-klactisACC-R using genomic DNA of K.lactis as template (provided by Dr. Ana Rodriguez-Torres from Universidade da Coruña, Spain) and introducing 40-45 bp of homology to the ADH1 promoter at the 5′ end and the YCplac33 multiple cloning site. Plasmid YCp33 ADH promoter was digested with XbaI and EcoRI. The PCR amplified fragments and the digested vector were transformed into the W1536 8B Δhfa1 strain using the high frequency transformation method [15]. Candidate colonies carrying the repaired plasmids were picked up from SCD-Ura plates and then streaked for single colonies on SCL plates and incubated at 33°C for four to seven days to test whether the plasmids were able to rescue the respiratory deficient phenotype of the deletion strain. A growth control on SCD-Ura media was performed as well. As positive control on the non-fermentable carbon source the strains W1536 8B Δhfa1 YCp33 HFA1 and W1536 8B were used. The strain W1536 8B Δetr1 served as a negative, respiratory deficient control with an mtFAS defect. After the positive growth on SCL plate at 33°C was confirmed, the genomic DNA and the plasmid DNA of the candidate colonies were isolated with the yeast DNA miniprep method. PCR was performed with the ADH1promseq −90→ −68 primer and Klac pcr R check primer to confirm that the K. lactis ORF was present in these cells and fused to the ADH1 promoter. After the correct size product was produced, the PCR product encompassing the 3′ end of the ADH1 promoter, the fusion junction and the 5′ end of K.lactis ACC1 was sequenced and confirmed.
In silico analysis of fungal ACC evolution
Amino acid sequences of the fungal acetyl-CoA carboxylases are obtained from the KEGG database (http://www.kegg.jp/). The phylogenetic tree shown in Fig. 1 was rooted by using aligned amino acid sequences by ClustalW2-Phylogeny [21] or ETH Phylogenetic Tree (http://www.cbrg.ethz.ch/services/PhylogeneticTree). The probability of the MTS of each amino acid sequences was analyzed with MitoProtII [18]. The 5′end of the nucleotide sequence was obtained from KEGG (http://www.kegg.jp/), Génolevures (http://www.genolevures.org/index.html), and NCBI (http://www.ncbi.nlm.nih.gov/).
Figure 1. Identification of the HFA1 transcription initiation site by 5′RACE: The HFA1 specific primer SP1 (Table 4) was used for the reverse transcription reaction to synthesize a first strand cDNA.
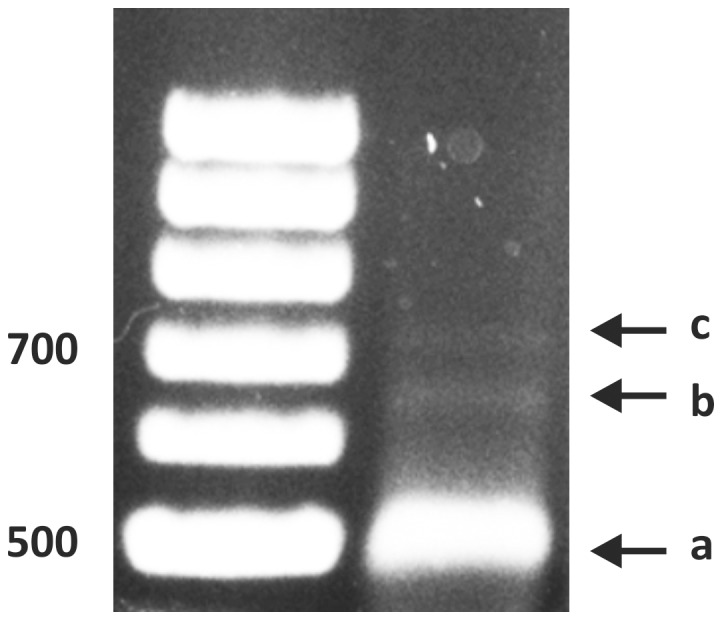
a) Major 5′ RACE product (500 bp), b) Minor 5′RACE product (650 bp) and c) third band produced only occasionally.
Each obtained nucleotide sequence was translated with the ExPASy translate tool (http://web.expasy.org/translate/) and the matching frame was chosen for the further analysis for the MTS. After the upstream amino acid sequence from each fungal species was obtained, the MTS was analyzed up to the first in-frame stop codon with the addition of the methionine for the convenience for the analysis with MitoProtII [18].
Results
Transcription initiation sites of HFA1
We performed 5′RACE studies of S. cerevisiae mRNA to unequivocally determine the HFA1 transcription initiation site(s). Two amplified products were consistently detected by agarose gel electrophoresis; a prominent band of just below 500 bp and a faint band of about 650 bp (Fig. 1). Occasionally, we also observed a third band somewhat larger than 700 bp. The two main bands were isolated, re-amplified by PCR, subcloned into the plasmid YCplac33 and then sequenced. The 5′ ends of three clones out of four from the plasmid containing the 650 bp terminated at nucleotide -587 bp upstream of the designated start codon. Another clone was found to harbor an insert with the 5′ end at −618 bp upstream. The majority of the 5′ ends sequenced from these plasmids harboring the more prominent PCR product contained the nucleotide sequence between bases −441 to −424 relative to the annotated start codon. All the products obtained from the major 5′ RACE PCR band were found to be further downstream of the transcription initiation site identified by Hoja et al. [6]. Changes in the mRNA sequence by hypothetical RNA editing, resulting in elimination of the stop codons or the generation of an in-frame start codon, were not observed. There was also no evidence for alternative splicing.
HFA1 translation initiates from a non-AUG start codon (Stop codon scanning assay)
A mutation located upstream of the annotated start codon of HFA1 was obtained during our study on the mtFAS in yeast seeking for respiratory deficient synthetic petite mutants [3]. This mutation is located upstream at −273 in frame with the designated AUG (Fig. 2). This finding further supported the hypothesis that the translation of HFA1 mRNA does not start at the designated codon but further upstream. W1536 8B Δhfa1 proved to be respiratory deficient when grown on lactate as the only carbon source at a growth temperature of 33°C. None of the transformants carrying plasmids with stop codon mutations downstream of the −372 site were able to grow like the wild type strain or the Δhfa1 strains carrying a wild type copy of HFA1 on the YCp33 HFA1 plasmid. However, stop codon mutations inserted upstream of the −375 position did not impede the rescue of the respiratory deficiency of the W1536 8B Δhfa1 strain. The only triplet in between the mutated codons that matches the sequence of previously reported non-AUG initiation codons is ATT at position −372. The sequence context is a good match for a Kozak consensus sequence [22] (Fig. 3). The mutation of the codon in at location −372 resulted in unchanged lactate deficiency of Δhfa1 strains transformed with the mutagenized rescue plasmid (Fig. 3). This is not the case when the preceding likely non-AUG translation initiation codon is (−381, ATA encoding Ile) is mutated to a stop codon (Fig. 3). We therefore suggest that the native translation initiation codon of HFA1 is ATT at −372, encoding isoleucine.
Figure 2. Schematic depiction of location of the introduced stop codons for stop –codon scanning assay and RNAse protection assay results.

All nucleotide numbers are given respective to the annotated start codon at +1. Position −450 is the predicted transcribed but not translated region of HFA1. The underlined region up to position −216 region shows the putative minimum mitochondrial import sequence and upstream position −141 shows the end of the sequence similarity to ACC1. The ORFof HFA1 annotated in the Saccharomyces Genome Database starts from +1. The stop codon found to lead to a respiratory deficient phenotype in the screen performed by Kursu et al. 2013 is located at −273 and 8 more stop codons at −282, −312, −360, −363, −372 −375 −378 and −381 were introduced upstream in the promoter region of HFA1.
Figure 3. The W1536 8B Δhfa1 strain carrying plasmids with inserted stop codon mutation at position downstream of −372 resulted in unchanged lactate deficiency.

Only stop codon mutations relevant to define the putative translation initiation site and controls are shown. The yeast cells were grown on media containing Glucose (SCD) or lactate (Lactate) as the sole carbon source at 33°C. Strains used for this study are W1536 8B, W1536 8B Δhfa1 or W1536 8B Δhtd2 (respiratory deficient control) and the plasmids carried by the strains are indicated at the left side of the panels. YCp33: empty plasmid; HFA1: YCp33 HFA1; −381: YCp33 HFA1 −381; −372: YCp33 HFA1 −372; −363: YCp33HFA1 −363. Only stop codon mutations relevant to define the putative translation initiation site and controls are shown. The results for other mutants shown in Fig. 2 can be found as supplementary data.
The region 5′ to the designated starting codon of K. lactis ACC is required for functional complementation of the hfa1 mutant
Turunen et al. have suggested that HFA1 and ACC1 originated following a whole genome duplication event from a single ACC gene encoding a dually localized ACC [13]. Our database research confirms that K.lactis is predicted to have only one ACC (KLLA0F06072g). We analyzed the translation of the 5′-upstream region of the K. lactis ACC with two different subcellular localization prediction programs, MitoProtII and Target P [18], [19]. MitoProtII predicted that the region upstream to the canonical translation initiation codon encodes an in-frame protein sequence with a probability of 0.9999 to be a mitochondrial import signal, while Target P produces a mTP (mitochondrial targeting peptide) score of 0.586 for mitochondrial targeting potential. In order to add experimental evidence to the hypothesis that the dually localized, one-gene encoded variant of ACC is the original state of ACC in yeasts, we cloned and expressed the K. lactis ACC with and without the 5′ upstream region predicted to encode a MTS, in the S. cerevisiae W1536 8B Δhfa1 strain. Because K. lactis ACC was apparently toxic to E. coli even when expressed in low plasmid copy number, we resorted to a gap repair cloning approach in yeast (see material and methods). Functional mitochondrial localization of the K. lactis ACC carrying the extra 5′ sequence was successfully demonstrated by the complementation of the respiratory deficiency of the mutant strain (12 out of 18 Ura+ candidates were respiratory competent) at 33°C. All 18 isolates from the W1536 8B Δhfa1 strain transformed with the K. lactis ACC without MTS were tested and failed to grown on the lactate plate at 33°C, suggesting that the hypothetical MTS is required for the complementation (Fig. 4).
Figure 4. K.lactis ACC complemented the respiratory deficiency when appended with the putative 5′ non-AUG initiated MTS, but not without this sequence.

Strains were grown on media containing glucose (SCD-URA), glycerol (SCG, non-fermentable) or lactate (SCL, non-fermentable) as the sole carbon source at 30°C and 33°C. 1) W1536 8B Δhfa1 +YCp33 HFA1. 2) W1536 8B Δhfa1+YCp33 3) W1536 8B Δhfa1. 4) W1536 8B (wild type). 5) W1536 8B Δhfa1+K.lactis ACC without 5′- encoded putative cryptic MTS sequence. 6) W1536 8B Δhfa1+K.lactis ACC with 5′ -encoded putative cryptic 5′ MTS.
MTS of the ACCs are highly conserved in fungi
Querying databases on the amino acid sequences of fungal ACCs, we found that the MTS is highly conserved among fungal species especially in the group of Saccharomycotina (Fig. 5). The alignment shows the homologous sequence of ACCs through all the fungal species, except for the N-terminal part. Our results are presented in a distance tree, displaying in the division of the fungal ACCs in two large groups (Fig. 5). One group (group 1) includes Candida albicans, Candida dubliniensis, Candida tropicalis, and Debaryomyces hansenii, and is restricted to organisms which translate CTG as serine instead of leucine. Group 2 contains the other Saccharomyces species, Ashbya gossypii, Kluyveromyces waltii, K. lactis, and two post-whole genomic duplication species, Candida glabrata and S. cerevisiae. This grouping matches with the fungal phylogeny data reported by Fitzpatrick et al. [23] [24]. S. cerevisiae Hfa1p is distantly located from the other ACCs from fungi, indicating that this protein has undergone a faster evolution compared to the other species.
Figure 5. Acetyl-CoA carboxylases from yeast split into two subgroups with the exception of the S. cerevisiae HFA1.

The phylogenetic tree of representative members of the acetyl-CoA carboxylases from fungi species was constructed using Clustal W2 Phylogeny and NJPlot as described in Material and methods. Numbers at branch points are bootstrap values and were calculated with Clustal X2.
The K. waltii ACC Kwal_6157 (KEGG entry) does not contain a MTS within the annotated ORF sequence. However, when the nucleotide sequence upstream of the ORF was investigated by a translation program, an in-frame amino acid sequence with a high probability of being a MTS (0.9923) (Table 4) was revealed.
Table 4. Acetyl-CoA carboxylases in yeasts and MTS probabilities.
| Organism | Accession number | Group number | MTS probability(MitoProtII)/cleavage site/AA sequence lengtha | Upstream MTS probability (MitoProtII)/cleavage site/AA sequence lengthb |
| C. albicans | EEQ43196.1 | 1 | 0.8764/NP/2271 | NC |
| C. dubliniensis | XP_002421671.1 | 1 | 0.0403/NP/2228 | NC |
| C. tropicalis | XP_002546225.1 | 1 | 0.9935/40/2275 | NC |
| D. hansenii | XP_457211.1 | 1 | 0.9693/NP/2297 | NC |
| P. pastoris | FN392319 | 1 | 0.0793/NP/2215 | 0.9050/31/2245 |
| Y. lipolytica | XP_501721.1 | 1 | 0.981621/2266 | NC |
| S. cerevisiae HFA1 | BK006946 | 2 | 0.4993/NP/2123 | 0.7297/80/2248c |
| K. waltii | Kwal_6157 (KEGG entry) | 2 | 0.0195/NP/2230 | 0.9923/19/2281 |
| K. lactis | XP_455355 | 2 | 0.0201/NP/2231 | 0.9999/90/2317 |
| A. gossypii | NP_982612 | 2 | 0.0321/NP/2231 | 0.9902/52/2298 |
| C. glabrata | CR380958 | 2 | 0.0090/NP/2233 | 0.5589/NP/2292 |
| S. cerevisiae ACC1 | ACC1(Locus Tag) | 2 | 0.0279/NP/2233 | 0.0452/NP/2252 |
: The whole amino acid sequence of each protein was analyzed with MitoProt II. b: The whole sequence with the additional 5′ upstream region of the ORF of each protein was analyzed up to the first in-frame stop codon. c: The whole amino acid sequence from the 5′ end upstream start codon identified in this study was analyzed. NP: not predicted. NC: not calculated
The analysis of the MTS of these fungal ACCs by MitoProtII revealed that 6 species of group 1 show high probability of MTS (<0.70) with the exception of C. dubliniensis and Pichia pastoris. Group 2 did not reveal a high probability of the MTS with their annotated ORF (Table 4). However, the translated 5′ upstream region of these ORFs showed a high probability prediction of an in-frame MTS (<0.98). Yarrowia lipolytica, a fungal genus in the Saccharomycotina, has branched before the division of the Candida CTG species, is also annotated to have one ACC (YALI0C11407p). The predicted protein is found to be maintaining a MTS at the N-terminus of the ORF, while P. pastoris, which diverged before the formation of the CTG clade [25], has only one ACC (FN392319) and does not have an MTS at the N-terminal region (MitoProtII: 0.0793). However, also this gene displays a nucleotide sequence encoding an in-frame putative MTS in the region 5′ to the annotated start codon (MitoProtII: 0.9050).
Cryptic non-AUG inititated MTSs of mtFAS-related proteins in yeast
A cursory survey of proteins related to mtFAS revealed at least three more likely candidates for cryptic mitochondrial import sequences (see Table 5). The BPL1 gene encodes a biotin protein ligase/holocarboxylase required to activate both Hfa1p and Acc1p and annotated to be localized in the cytosol. Analysis of the amino acid sequence 5′ to the annotated start codon of BPL1 revealed a high probability of functioning as an MTS. The FAA2 gene was also found to harbor an upstream sequence encoding a putative in-frame MTS, consistent with reports that a point mutation in this region which generates a new in-frame start codon can act as a suppressor of mtFAS mutations [26]–[28]. Poignantly, the wild type Faa2 protein has also recently been detected in low abundance in mitochondria [29]. Lastly, the sequence of the SAM2 gene, coding for isoform 2 of S-adenosylmethionine (SAM) synthase and induced during the diauxic shift encodes a putative 5′ MTS.
Table 5. Mitochondrial targeting prediction of selected proteins in S. cerevisiae.
| Gene name | MTS probability annotated ORF(MitoProt II)/cleavage site/sequence length | Upstream MTS probability/cleavage site/sequence length | Upstream MTS prediction/Target P | |||
| mTP | SP | Loc | RC | |||
| BPL1 | 0.0101/not (690) | 0.9329/51 (760) | 0.190 | 0.447 | S | 4 |
| FAA2 | 0.2275/not (744) | 0.8722/21 (764) | 0.283 | 0.074 | - | 4 |
| SAM2 | 0.0373/not (384) | 0.6337/not (431) | 0.363 | 0.516 | S | 5 |
| HFA1 | 0.4993/not (2123) | 0.7297/80 (2248) this study | 0.305 | 0.129 | - | 4 |
| ACP1 | 0.9996/37 (122) | NC | 0.960 | 0.009 | M | 1 |
The whole amino acid sequence and the whole amino acid sequence with the 5′ upstream region of the ORF of each protein was analyzed up to the first in-frame stop codon with MitoProt II. In case of the Hfa1p, the identified upstream start codon with the addition of the methionine for the convenience of the calculation with MitoProt II was used. The whole amino acid sequence with the 5′ upstream region of the ORF of each protein was analyzed up to the first in-frame stop codon with Target P. In case of Acp1p, the whole amino acid sequence was used for the calculation with Target P. mTP, SP: Final NN scores on which the final prediction is based. Note that the scores are not really probabilities, and they do not necessarily add to one. However, the location with the highest score is the most likely according to TargetP, and the relationship between the scores (the reliability class, see below) may be an indication of how certain the prediction is. Loc: S: Secretory pathway, i.e. the sequence contains SP, a signal peptide M: Mitochondrion, i.e. the sequence contains mTP, a mitochondrial targeting peptide; -: Any other location; RC: Reliability class, from 1 to 5, where 1 indicates the strongest prediction. RC is a measure of the size of the difference ('diff') between the highest (winning) and the second highest output scores. There are 5 reliability classes, defined as follows: 1: diff>0.800, 2: 0.800>diff>0.600, 3: 0.600> diff>0.400, 4: 0.400> diff>0.200, 5: 0.200> diff HFA1 and ACP1 are listed as controls for the prediction calculation of MitoProt II and target P.
The proteins required for or interacting with the mtFAS pathway are listed at the KEGG as the orthologs of S. cerevisiae BPL1, FAA2 and SAM2 showed highly conserved putative 5′ MTS (Table 6). What was found in common in the protein sequences in between S. cerevisiae, K. lactis and C. dubliniensis is the significantly higher probability of a 5′ upstream encoded MTS in comparison to the MTS calculated from the annotated start codon. Other fungi species shown on Table 6 were also found to carry 5′ upstream sequences predicted to be MTSs with high probability, but not all species showed the exact match in all these three proteins, indicating diversification during the molecular evolution. For example, C. glabrata carries two genes encoding hypothetical proteins with similarity to SAM synthases, one with low probability of harboring an MTS, the other one with higher probability if the 5′ upstream region is included in the analysis. The region 5′ to the annotated AUG and translated in frame with the SAM ORF revealed a putative MTS with a predicted cleavage site. C. glabrata is one of the fungal species which has undergone the whole genomic duplication. This could be another example of the molecular evolution of one protein gave rise to divergence of localization of paralogs after the genomic duplication. Other examples supporting this hypothesis are found in the pre-genomic duplication species such as K. lactis, A. gossypii and C. dubliniensis. These species carry a single gene encoded variants of these proteins with putative MTSs either or both in the 5′ sequence and annotated sequence.
Table 6. Mitochondrial targeting prediction of fungal homologues of proteins in Table 5.
| Gene name | Organism | Entry | Definition | MTS probability/cleavage site/AA sequence lengtha | Upstream MTS probability/cleavage site/AA sequence lengthb |
| BPL1 | S. cerevisiae | YDL141W | Biotin:apoprotein ligase | 0.0101/NP/690 | 0.9329/51/760 |
| K. lactis | KLLA0F05049g | hypothetical protein | 0.0477/NP/693 | 0.9870/30/732 | |
| A. gossypii | AGOS_ADR085W | hypothetical protein | 0.0314/NP/679 | 0.6205/NP/720 | |
| C. dubliniensis | CD36_35420 | biotin: apoprotein ligase | 0.0108/NP/665 | 0.6348/NP/689 | |
| Z. rouxii | ZYRO0F05940g | hypothetical protein | 0.0219/NP/690 | 0.8872/30/722 | |
| L.thermotolerans | KLTH0H02024g | biotin−protein ligase | 0.042/NP/660 | 0.4203/53/7110 | |
| K. africana | KAFR_0B01050 | hypothetical protein | 0.0219/NP/690 | 0.8872/30/722 | |
| FAA2 | S. cerevisiae | YER015W | medium-chain fatty acid-CoA ligase FAA2 | 0.1550/NP/744 | 0.8807/21/765 |
| K. lactis | KLLA0B11572g | long-chain fatty acyl-CoA synthetase | 0.1983/NP/745 | 0.2603/NP/749 | |
| C. glabrata | CAGL0H09460g | hypothetical protein | 0.3174/NP/741 | 0.4203/NP/752 | |
| T. delbureckii | TDEL_0H02790 | hypothetical protein | 0.2323/NP/741 | 0.8615/NP/750 | |
| SAM2 | S. cerevisiae | YDR502c | S-adenosylmethionine synthetase | 0.0373/NP/384 | 0.6377/NP/430 |
| K. lactis | KLLA0C01782g | hypothetical protein | 0.0159/NP/384 | 0.1565/NP/392 | |
| A. gossypii | AGOS_AFR692C | AFR692Cp | 0.3994/14/382 | 0.1639/NP/387 | |
| Z. rouxii | ZYRO0F17248g | hypothetical protein | 0.0769/NP/382 | 0.3633/NP/412 | |
| P. pastoris | PAS_chr3_0876 | S-adenosylmethionine synthetase | 0.0139/NP/384 | 0.5093/NP/423*1 | |
| C. glabrata | CAGL0J08415g | hypothetical protein | 0.0634/15/383 | 0.2737/29/397*2 | |
| C. glabrata | CAGL0B01122g | hypothetical protein | 0.0727/NP/382 | 0.0308/NP/394 | |
| C. dubliniensis | CD36_10740 | S-adenosylmethionine synthetase, putative | 0.0280/NP/385 | 0.9093/58/447 |
: The whole amino acid sequence of each protein was analyzed with MitoProt II. b: The 5′ upstream region of the ORF of each protein was analyzed up to the first in-frame stop codon. *1The P. pastoris SAM homolog is preceded by a 39 aa sequence with high probability of being an MTS and starting with a bona fide ATG that apparently has been omitted in the annotation, probably because this sequence does not show homology with the S. cerevisiae SAM1 or SAM2 genes. *2 Translation from the first likely initiation codon (ATA, Ile) results in a probability of 0.4938.
Discussion
HFA1 clearly does not produce a functional protein from the annotated ATG in the yeast database, and the in vivo translation initiation site is not known [6]. In addition, the existing reports on the transcription initiation site on the HFA1 mRNA showed disagreement in the 5′ extent of the transcript, placing it 633 to 607 base pairs [6], or 534 base pairs for the poly(a)RNA array and 430 base pairs for the total RNA array 5′ [30] relative to the annotated translation initiation AUG codon. The transcript reported by Hoja et al. would contain an AUG start codon in frame with the HFA1 coding region, which is soon followed by two stop codons. The variants identified by Fitzpatrick et al. would not contain any upstream in-frame AUG at all [24]. In this work, we have demonstrated that at least two HFA1 transcripts exist, with the major mRNA species being the shorter form initiated between nucleotides −442 → −435. The sequence analysis of the transcription products confirmed that there is no RNA editing. The existence of this major shorter transcript also makes a frame-shifting explanation unnecessary. In essence, both existing reports on the transcription start sites of HFA1 are correct [6], [30], but our results indicated that the most proximal to the annotated AUG is likely to be the physiologically most relevant one.
In this study, we have pinpointed the exact location of the translation initiation site using a “stop codon scanning” approach, which reveals the likely non-AUG start codon usage of this gene. This expression principle resembles the previously reported situation of tRNA synthetases, where the mitochondrial versions of dually localized proteins were described to be translated from non-AUG codons [10], [11]. In yeast, a redundancy of non-AUG initiation codons were reported for ALA1 [31] and GRS1 [32]. The non-AUG codons for these genes act as alternative translation initiation sites with downstream in-frame AUG initiation codons, which can be observed in our K.lactis model described in the paragraph below. Our stop codon scanning results also argue against a major role of the upstream ATGs in translation.
We chose the K. lactis ACC gene as a model to investigate the evolutionary history of S. cerevisiae ACC isoforms. In this study, we showed that the only ACC encoded by the K. lactis genome was capable of complementing the respiratory deficiency of an Δhfa1 strain only if the construct included the 5′ upstream region of the K. lactis gene, encoding a putative MTS. This result supports the hypothesis that the single K. lactis ACC gene encodes both the cytosolic and mitochondrial isoforms of the enzyme.
Using a combination of bioinformatics methodologies and a genetic approach, our results illustrate an important aspect of the evolution of one mitochondrial protein instigated by a whole genome duplication (WGD). A recent study on the comparative genomics of yeast revealed how the genomic evolution has been affected by the WGD [33]. Genomic duplications are considered to be an important source of evolutional novelty, providing extra genetic material that is free to diverge without compromising the organism, as a copy retaining the original function exists [34]. Not only fungi but also other higher eukaryotic genomes show a high degree of redundancy [35], [36] [33]. Wolfe and Shields proposed a model of massive gene deletion in the wake of the gene duplication in yeast [37]. By systematic study of the genomic data from K. lactis and S. cerevisiae targeting the duplicated segments of the S. cerevisiae genome, they have shown that most of the duplicated genes were lost. The remaining small fractions of the genes were rearranged by many reciprocal translocations between chromosomes. A preceding study has shown that duplicated genes are almost as likely to acquire a new and essential function as to be lost through acquisition of mutations that compromise protein function [35], [38]. This was also confirmed by Kellis et al. [12] in their study describing how the genome duplication of S. cerevisiae was followed by random local deletion of genes during the course of evolution. The authors propose that the paralogs derived from the genome duplication have specialized in their cellular localization or temporal expression. They take ACC1 and HFA1as one example pair and describe that the former gave rise to the latter one, suggesting a model that the ancestral cytoplasmic form acquired the MTS after the gene duplication event. The conclusions of Hoja et al. [6], who published their results on the analysis of HFA1 almost simultaneously, do not state the same hypothesis explicitly, but echo a similar conclusion. In contrast, an in silico study on functional specialization events after WGD in yeast reports that the new localization patterns have evolved in the duplicated genes [13]. They show ACC1 as one example that has lost the MTS, whereas HFA1 retained it through evolution. Our results support the in silico data, and suggest that the MTS of fungal ACC was gained long before the gene duplication event that generated S. cerevisiae. The views of Kellis et al. and Hoja et al. on the origin of present-day baker's yeast ACCs are not supported by our data. Our experimental data is much more consistent with the in silico results by Turunen et al., indicating that both ACC forms are derived from a dually localized ancestral gene and adapted to serve their more specialized roles in different compartments.
Our phylogenetic study on the amino acid composition of the ACCs in Saccharomycotina clarified that the ACCs are categorized in to two large groups, one is the CTG clade (group1) and the other one contains the WGD species and Saccharomycotina (group2) which have not undergone WGD. This study based on the protein sequence of the fungal ACCs matches to the fungal phylogeny based on the complete genome analysis [23]. Additional data that we obtained from the analysis of the MTS probability of the ACCs supports this grouping. All the group1 species carry ACCs with high MTS probability (<0.7) with the exception of the C. dubliniensis and P. pastoris and all the group 2 species except S. cerevisiae carry a putative MTS (<0.9) in the upstream of the start codon. This phylogenetic analysis and MTS conservation in fungi also suggests that the ancestral gene encoding acetyl-CoA carboxylase allowed the production of two proteins, one with a MTS which is almost always initiated from a non-AUG translation start codon, and a cytosolic variant of which translation starts from a canonical AUG. Hfa1p is distantly localized in the phylogenetic tree in this study, indicating that the evolution of this protein is faster than the other ACCs in fungi, possibly to allow specialization for the mitochondrial compartment. The favorable MTS of the ACC upstream of the start codon is also conserved in fungi species diverged before the formation of the CTG clade, supporting our hypothesis that the Hfa1p, carrying MTS is the original form and Acc1p arose after WGD by deleting the MTS.
A recent report on the GAL1/GAL3 gene duplication showed that specialization of proteins encoded by gene pairs can increase the evolutionary fitness of an organism, as the dual use of a product encoded by a single gene can imply compromise in function [39]. Our limited analysis of a small subset of genes of our interest resulted in a few candidates with a possible concealed mitochondrial localization. The obvious requirement for Bpl1p in mitochondria to activate Hfa1p is probably the clearest support for a physiological role of this putatively mitochondrial isoform. Results on the mitochondrial localization of human or mouse biotin protein ligase/holocarboxylase are ambiguous [40], [41] and deserve a more thorough investigation. The mitochondrial roles of the other two candidates are less clear cut. There is support in the literature on mitochondrial localization of Faa2p [29]. The most controversial of our candidates may be SAM2, as mitochondria are clearly dependent on import of SAM under normal growth conditions [42]. Intriguingly, the same publication demonstrated that, with exception of SAM synthase itself, the entire SAM recycling machinery is present in mitochondria. It may not be impossible to find a condition where cytosolic SAM is of too short supply to support the mitochondrial demand of this cofactor. Poignantly, one of each of the two versions of duplicated SAM synthase genes (MAT2A/Mat2a) in both the mouse and human genomes also potentially encodes a putative 5′-encoded non-AUG initiated MTSs (unpublished).
Supporting Information
Stop codon scanning mutant phenotype. The W1536 8B Δhfa1 strain carrying plasmids with inserted stop codon mutation at position −282, −312, −360, and −378 were plated onto the indicated media under the indicated growth temperatures to show the complementation or lack thereof of the different stop codon mutant constructs: The carbon sources of the media are Glucose (SCD, fermentable, left panel) or lactate (SCL, non-fermentable, middle and right panel) as the sole carbon source and cultured at 30°C and 33°C. The strains/transformants plated are as follows: 1. W1536 8B wild type, 2. W1536 8B Δhfa1+pTSV30 HFA1 (multicopy plasmid), 3. W1536 8B Δetr1 (respiratory deficient control), 4. W1536 8B Δhfa1, 5. W1536 8B Δhfa1+YCp33 (empty plasmid), 6. W1536 8B Δhfa1+YCp33 HFA1, 7–10. W1536 8B Δhfa1 carrying the indicated stop codon plasmids (four independent transformants). a. W1536 8B wild type, b. W1536 8B Δhfa1, c. W1536 8B Δetr1 (respiratory deficient control), d. W1536 8B Δhfa1+YCp33 HFA1, e. W1536 8B Δhfa1+YCp33 (empty plasmid), f-h. W1536 8B Δhfa1 colonies carrying the indicated stop codon plasmids (three independent transformants).
(TIF)
Acknowledgments
We thank Dr. Kalervo Hiltunen for support of the research work, stimulating discussions and critical reading of this manuscript, as well as Dr. Ana Rodriguez-Torres for providing us with K. lactis genomic DNA. This work was supported by the Academy of Finland.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the Academy of Finland Decision Number 263776 (AJK). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Schonauer MS, Kastaniotis AJ, Hiltunen JK, Dieckmann CL (2008) Intersection of RNA processing and the type II fatty acid synthesis pathway in yeast mitochondria. Mol Cell Biol 28:6646–6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hiltunen JK, Autio KJ, Schonauer MS, Kursu VA, Dieckmann CL, et al. (2010) Mitochondrial fatty acid synthesis and respiration. Biochim Biophys Acta 1797:1195–1202. [DOI] [PubMed] [Google Scholar]
- 3. Kursu VA, Pietikainen LP, Fontanesi F, Aaltonen MJ, Suomi F, et al. (2013) Defects in mitochondrial fatty acid synthesis result in failure of multiple aspects of mitochondrial biogenesis in saccharomyces cerevisiae. Mol Microbiol 90:824–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Saggerson D (2008) Malonyl-CoA, a key signaling molecule in mammalian cells. Annu Rev Nutr 28:253–272. [DOI] [PubMed] [Google Scholar]
- 5. Hasslacher M, Ivessa AS, Paltauf F, Kohlwein SD (1993) Acetyl-CoA carboxylase from yeast is an essential enzyme and is regulated by factors that control phospholipid metabolism. J Biol Chem 268:10946–10952. [PubMed] [Google Scholar]
- 6. Hoja U, Marthol S, Hofmann J, Stegner S, Schulz R, et al. (2004) HFA1 encoding an organelle-specific acetyl-CoA carboxylase controls mitochondrial fatty acid synthesis in saccharomyces cerevisiae. J Biol Chem 279:21779–21786. [DOI] [PubMed] [Google Scholar]
- 7. Zitomer RS, Walthall DA, Rymond BC, Hollenberg CP (1984) Saccharomyces cerevisiae ribosomes recognize non-AUG initiation codons. Mol Cell Biol 4:1191–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Natsoulis G, Hilger F, Fink GR (1986) The HTS1 gene encodes both the cytoplasmic and mitochondrial histidine tRNA synthetases of S. cerevisiae. Cell 46:235–243. [DOI] [PubMed] [Google Scholar]
- 9. Chatton B, Walter P, Ebel JP, Lacroute F, Fasiolo F (1988) The yeast VAS1 gene encodes both mitochondrial and cytoplasmic valyl-tRNA synthetases. J Biol Chem 263:52–57. [PubMed] [Google Scholar]
- 10. Tang HL, Yeh LS, Chen NK, Ripmaster T, Schimmel P, et al. (2004) Translation of a yeast mitochondrial tRNA synthetase initiated at redundant non-AUG codons. J Biol Chem 279:49656–49663. [DOI] [PubMed] [Google Scholar]
- 11. Chang KJ, Wang CC (2004) Translation initiation from a naturally occurring non-AUG codon in saccharomyces cerevisiae. J Biol Chem 279:13778–13785. [DOI] [PubMed] [Google Scholar]
- 12. Kellis M, Birren BW, Lander ES (2004) Proof and evolutionary analysis of ancient genome duplication in the yeast saccharomyces cerevisiae. Nature 428:617–624. [DOI] [PubMed] [Google Scholar]
- 13. Turunen O, Seelke R, Macosko J (2009) In silico evidence for functional specialization after genome duplication in yeast. FEMS Yeast Res 9:16–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaiser C, Michaelis S, Mitchell A (1994) Methods in yeast genetics: A cold spring harbor laboratory course manual. New York: Cold Spring Harbor Laboratory Press.
- 15. Gietz RD, Woods RA (2002) Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol 350:87–96. [DOI] [PubMed] [Google Scholar]
- 16. Chen DC, Yang BC, Kuo TT (1992) One-step transformation of yeast in stationary phase. Curr Genet 21:83–84. [DOI] [PubMed] [Google Scholar]
- 17. Torkko JM, Koivuranta KT, Miinalainen IJ, Yagi AI, Schmitz W, et al. (2001) Candida tropicalis Etr1p and saccharomyces cerevisiae Ybr026p (Mrf1'p), 2-enoyl thioester reductases essential for mitochondrial respiratory competence. Mol Cell Biol 21:6243–6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Claros MG, Vincens P (1996) Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem 241:779–786. [DOI] [PubMed] [Google Scholar]
- 19. Emanuelsson O, Brunak S, von Heijne G, Nielsen H (2007) Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc 2:953–971. [DOI] [PubMed] [Google Scholar]
- 20. Kostrub CF, Lei EP, Enoch T (1998) Use of gap repair in fission yeast to obtain novel alleles of specific genes. Nucleic Acids Res 26:4783–4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, et al. (2007) Clustal W and clustal X version 2.0. Bioinformatics 23:2947–2948. [DOI] [PubMed] [Google Scholar]
- 22. Kozak M (1984) Point mutations close to the AUG initiator codon affect the efficiency of translation of rat preproinsulin in vivo. Nature 308:241–246. [DOI] [PubMed] [Google Scholar]
- 23. Fitzpatrick DA, Logue ME, Stajich JE, Butler G (2006) A fungal phylogeny based on 42 complete genomes derived from supertree and combined gene analysis. BMC Evol Biol 6:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fitzpatrick DA, Creevey CJ, McInerney JO (2006) Genome phylogenies indicate a meaningful alpha-proteobacterial phylogeny and support a grouping of the mitochondria with the rickettsiales. Mol Biol Evol 23:74–85. [DOI] [PubMed] [Google Scholar]
- 25. De Schutter K, Lin YC, Tiels P, Van Hecke A, Glinka S, et al. (2009) Genome sequence of the recombinant protein production host pichia pastoris. Nat Biotechnol 27:561–566. [DOI] [PubMed] [Google Scholar]
- 26. Harington A, Herbert CJ, Tung B, Getz GS, Slonimski PP (1993) Identification of a new nuclear gene (CEM1) encoding a protein homologous to a beta-keto-acyl synthase which is essential for mitochondrial respiration in saccharomyces cerevisiae. Mol Microbiol 9:545–555. [DOI] [PubMed] [Google Scholar]
- 27. Kastaniotis AJ, Autio KJ, Sormunen RT, Hiltunen JK (2004) Htd2p/Yhr067p is a yeast 3-hydroxyacyl-ACP dehydratase essential for mitochondrial function and morphology. Mol Microbiol 53:1407–1421. [DOI] [PubMed] [Google Scholar]
- 28. Autio KJ, Kastaniotis AJ, Pospiech H, Miinalainen IJ, Schonauer MS, et al. (2008) An ancient genetic link between vertebrate mitochondrial fatty acid synthesis and RNA processing. FASEB J 22:569–578. [DOI] [PubMed] [Google Scholar]
- 29. Vogtle FN, Wortelkamp S, Zahedi RP, Becker D, Leidhold C, et al. (2009) Global analysis of the mitochondrial N-proteome identifies a processing peptidase critical for protein stability. Cell 139:428–439. [DOI] [PubMed] [Google Scholar]
- 30. David L, Huber W, Granovskaia M, Toedling J, Palm CJ, et al. (2006) A high-resolution map of transcription in the yeast genome. Proc Natl Acad Sci U S A 103:5320–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chang KJ, Lin G, Men LC, Wang CC (2006) Redundancy of non-AUG initiators. A clever mechanism to enhance the efficiency of translation in yeast. J Biol Chem 281:7775–7783. [DOI] [PubMed] [Google Scholar]
- 32. Chen SJ, Lin G, Chang KJ, Yeh LS, Wang CC (2008) Translational efficiency of a non-AUG initiation codon is significantly affected by its sequence context in yeast. J Biol Chem 283:3173–3180. [DOI] [PubMed] [Google Scholar]
- 33. Piskur J, Langkjaer RB (2004) Yeast genome sequencing: The power of comparative genomics. Mol Microbiol 53:381–389. [DOI] [PubMed] [Google Scholar]
- 34.Ohno S (1970) Evolution by gene duplication. Berlin, New York: Springer-Verlag. 160 p. [Google Scholar]
- 35. Nadeau JH, Taylor BA (1984) Lengths of chromosomal segments conserved since divergence of man and mouse. Proc Natl Acad Sci U S A 81:814–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schughart K, Kappen C, Ruddle FH (1989) Duplication of large genomic regions during the evolution of vertebrate homeobox genes. Proc Natl Acad Sci U S A 86:7067–7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wolfe KH, Shields DC (1997) Molecular evidence for an ancient duplication of the entire yeast genome. Nature 387:708–713. [DOI] [PubMed] [Google Scholar]
- 38. Nadeau JH, Sankoff D (1997) Comparable rates of gene loss and functional divergence after genome duplications early in vertebrate evolution. Genetics 147:1259–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hittinger CT, Carroll SB (2007) Gene duplication and the adaptive evolution of a classic genetic switch. Nature 449:677–681. [DOI] [PubMed] [Google Scholar]
- 40. Aoki Y, Suzuki Y, Sakamoto O, Li X, Takahashi K, et al. (1995) Molecular analysis of holocarboxylase synthetase deficiency: A missense mutation and a single base deletion are predominant in japanese patients. Biochim Biophys Acta 1272:168–174. [DOI] [PubMed] [Google Scholar]
- 41. Bailey LM, Wallace JC, Polyak SW (2010) Holocarboxylase synthetase: Correlation of protein localisation with biological function. Arch Biochem Biophys 496:45–52. [DOI] [PubMed] [Google Scholar]
- 42. Cherest H, Surdin-Kerjan Y (1978) S-adenosyl methionine requiring mutants in saccharomyces cerevisiae: Evidences for the existence of two methionine adenosyl transferases. Mol Gen Genet 163:153–167. [DOI] [PubMed] [Google Scholar]
- 43. Johnson ES, Gupta AA (2001) An E3-like factor that promotes SUMO conjugation to the yeast septins. Cell 106:735–744. [DOI] [PubMed] [Google Scholar]
- 44.Menger K (2008) Study of the expression of the yeast mitochondrial acetyl-CoA carboxylase HFA1 gene. Oulu: Menger K 72, 6 p. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Stop codon scanning mutant phenotype. The W1536 8B Δhfa1 strain carrying plasmids with inserted stop codon mutation at position −282, −312, −360, and −378 were plated onto the indicated media under the indicated growth temperatures to show the complementation or lack thereof of the different stop codon mutant constructs: The carbon sources of the media are Glucose (SCD, fermentable, left panel) or lactate (SCL, non-fermentable, middle and right panel) as the sole carbon source and cultured at 30°C and 33°C. The strains/transformants plated are as follows: 1. W1536 8B wild type, 2. W1536 8B Δhfa1+pTSV30 HFA1 (multicopy plasmid), 3. W1536 8B Δetr1 (respiratory deficient control), 4. W1536 8B Δhfa1, 5. W1536 8B Δhfa1+YCp33 (empty plasmid), 6. W1536 8B Δhfa1+YCp33 HFA1, 7–10. W1536 8B Δhfa1 carrying the indicated stop codon plasmids (four independent transformants). a. W1536 8B wild type, b. W1536 8B Δhfa1, c. W1536 8B Δetr1 (respiratory deficient control), d. W1536 8B Δhfa1+YCp33 HFA1, e. W1536 8B Δhfa1+YCp33 (empty plasmid), f-h. W1536 8B Δhfa1 colonies carrying the indicated stop codon plasmids (three independent transformants).
(TIF)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.