

Abstract
Regional glucose hypometabolism is a defining feature of Alzheimer disease (AD). One emerging link between glucose hypometabolism and progression of AD is the nutrient-responsive post-translational O-GlcNAcylation of nucleocytoplasmic proteins. O-GlcNAc is abundant in neurons and occurs on both tau and amyloid precursor protein. Increased brain O-GlcNAcylation protects against tau and amyloid-β peptide toxicity. Decreased O-GlcNAcylation occurs in AD, suggesting that glucose hypometabolism may impair the protective roles of O-GlcNAc within neurons and enable neurodegeneration. Here, we review how O-GlcNAc may link cerebral glucose hypometabolism to progression of AD and summarize data regarding the protective role of O-GlcNAc in AD models.
Keywords: Alzheimer Disease, Amyloid-β (Aβ), Brain Metabolism, Glucose Metabolism, Glycobiology, Glycoprotein, Glycosylation, O-GlcNAcylation, O-Linked N-Acetylglucosamine (O-GlcNAc), Tau Protein (Tau)
Introduction
Alzheimer disease (AD)4 is the most common neurodegenerative disease, currently afflicting over 35 million worldwide. Age is the greatest risk factor for AD, and accordingly, the aging demographic in many countries is leading to a dramatic increase in the number of AD patients. By 2050, the number of patients is expected to nearly triple in the United States to reach 16 million. This increasing incidence represents a major emerging socioeconomic challenge for governments around the world. Although the root causes of AD remain obscure, the progression of AD is recognized as starting many years before cognitive impairments manifest (1). Various groups, including the Alzheimer's Disease Neuroimaging Initiative, are working to improve methods to identify AD patients, identify clinically useful biomarkers, and define the pathological progression of AD. Longitudinal monitoring of groups of patients, ranging from those who are cognitively normal to those suffering from mild cognitive impairment and those exhibiting full-blown AD, using various imaging modalities and biomarkers in cerebrospinal fluid and blood have led to major advances in defining the pathological cascade of AD (Fig. 1) (1–3).
FIGURE 1.
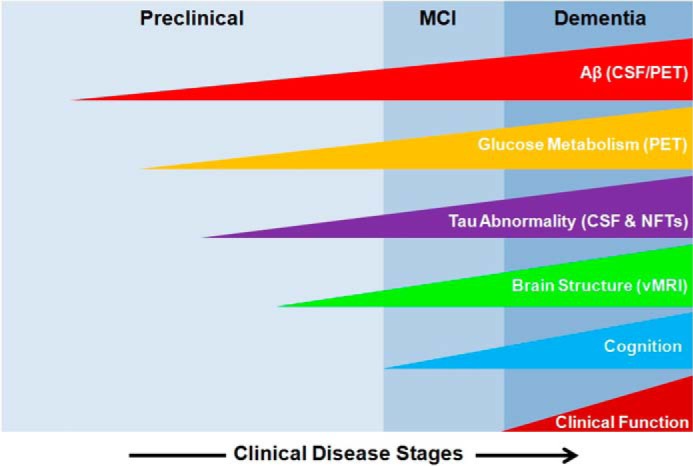
Hypothetical model of the change in biomarkers and their association with AD progression. Aβ in cerebrospinal fluid (CSF) or in the brain assessed by PET amyloid imaging is the earliest known biomarker that changes during the course of the disease, with changes beginning to develop during the preclinical phase of the disease. Subsequent to changes in Aβ, glucose metabolism becomes impaired, and increased tau in cerebrospinal fluid results from pronounced tau abnormalities. As the disease begins to advance to the symptomatic phase (the mild cognitive impairment stage (MCI)), differences in brain structure measured by volumetric MRI (vMRI) and in cognitive and clinical function begin to deteriorate steadily until the disease produces pronounced dementia. Biomarkers increase from null to maximum magnitude as a function of disease stage. This figure is adapted from a model proposed in Ref. 3.
Collectively, these biomarker studies have revealed that heightened production of amyloid-β peptides (Aβ), which are derived by proteolytic processing of the amyloid precursor protein (APP) (Fig. 2), is the first observed change among the studied biomarkers. Levels of Aβ rise in cerebrospinal fluid many years before other disease features manifest and then decline as amyloid plaques (protein deposits composed of Aβ that are one defining pathological feature of AD) develop within the AD brain. Notably, the Aβ species that are toxic appear to be oligomers, and these impair neuronal function to enable progression of AD. Neither cerebrospinal fluid levels of Aβ nor the extent of plaques in the brain correlates closely with neurodegeneration (7). Brain amyloid is therefore generally accepted as being essential for disease progression but not sufficient on its own to drive disease. The next observable change in the brain is impaired glucose metabolism within the AD brain, assessed using positron emission tomography (PET) to monitor 2-deoxy-2-[18F]fluoroglucose (FDG) uptake within the brain. Nearly parallel to this change yet lagging behind is the significant accumulation of abnormally hyperphosphorylated microtubule-associated protein tau. This hyperphosphorylation of tau within neurons is generally accepted as being a driver of tau toxicity (Fig. 2), which manifests as increased cerebrospinal fluid tau and phosphorylated tau, as well as aggregation of hyperphosphorylated tau into paired helical filaments (PHFs), which then aggregate together to form neurofibrillary tangles (NFTs). These NFTs are the second defining pathological hallmark of AD. FDG PET imaging studies have shown that brain regions exhibiting impaired glucose metabolism are often coincident with regions showing downstream neurodegeneration. Indeed, the development of glucose hypometabolism, as measured by FDG PET, and the deposition of NFTs precede measurable neurodegeneration and brain atrophy, which can be monitored by volumetric MRI (4–6). It is important to note, however, that this proposed sequence of events is based on challenging clinical studies, and a definitive series of events has not been unambiguously defined. Notwithstanding this limitation, it is clear that although aberrant volumetric MRI findings correlate most closely with severity of cognitive impairment, glucose hypometabolism and the extent of NFTs in the brain also correlate well with cognitive deficits. Based on these imaging and biomarker studies, it is emerging that brain glucose hypometabolism and tau toxicity likely reflect central events in the progression of AD (1, 2, 8).
FIGURE 2.

Molecular events giving rise to Aβ and tau pathologies. On the outside of the cell, the combined action of β- and γ-secretase cleavage of the transmembrane APP gives rise to soluble amyloid precursor protein β (sAPPβ) and secreted Aβ, which can then self-associate to form Aβ oligomers. These Aβ oligomers then combine to form higher order amyloid plaques. The APP intracellular domain (AICD) is also liberated by this secretase pathway. On the inside of the cell, phosphorylation of the tau protein promotes its dissociation from microtubules, enabling its aggregation to form competent nuclei or tau oligomers, which can then grow to form larger PHFs. These PHFs aggregate to form the higher order NFTs that are clinically observed in the AD brain.
Impaired Glucose Metabolism as a Contributing Factor to AD
The clear observation that brain amyloid is essential but not sufficient for progression of AD suggests that various disease modifiers are operative. Thus, although impaired glucose metabolism in the brain is generally thought to stem from synaptic dysfunction driven by Aβ toxicity, another hypothesis that has emerged is that changes in metabolism within neurons are instead factors contributing to disease progression. This hypothesis gains some support from the well established link with type 2 diabetes mellitus (T2DM) (9, 10), which is well characterized as having associated defects in brain glucose metabolism and insulin signaling, being a major risk factor for the development of AD.
Various mechanistic proposals linking T2DM to AD have been made (11–13). From a metabolic standpoint, it is notable that insulin resistance is commonly observed in AD, and the levels of insulin receptors are higher in concentration than in age-matched controls (14), which may reflect an effort to maintain glucose homeostasis within the brain. Furthermore, GLUT1 and GLUT3, which are key glucose transporters in neurons, are decreased in the AD brain (15). Because GLUT3 localization to the plasma membrane depends on insulin signaling, it is possible that the insulin resistance that is a risk factor for AD may lead to decreased cell-surface GLUT3 localization and cause downstream impairments in glucose utilization.
Studies using transgenic mouse models of AD show that Aβ toxicity can drive impairments in glucose metabolism within the brain (16–18), which is notable because this suggests that impaired glucose metabolism is not simply a confounding feature in AD patients. Some imaging studies in humans have also suggested that certain regions showing high amyloid deposition also show glucose hypometabolism (19, 20), which supports amyloid having toxic effects that influence glucose metabolism. Because insulin resistance decreases the levels of cell-surface glucose transporters, T2DM on its own causes impaired brain glucose metabolism. Accordingly, within humans, high blood glucose correlates with glucose hypometabolism in brain regions that are susceptible to neurodegeneration in AD (21, 22). In animal models, it has been shown that insulin resistance (23) induces tau and Aβ pathologies and, in transgenic AD models (24–26), greatly exacerbates AD progression. Collectively, these observations reveal that AD pathologies both drive impaired brain glucose metabolism and exacerbate the advance of AD. Overall, these emerging data from both human and animal studies reveal that impairments in glucose metabolism are a central feature of AD. Mechanistically, however, the molecular processes by which impaired glucose utilization drives these pathologies in the brain remain unclear.
Several proposals as to how impaired glucose metabolism in the brain might impact the progression of AD have been advanced, but one intriguing molecular link is a nutrient-sensing pathway that involves the post-translational modification of nuclear and cytoplasmic proteins with O-linked N-acetylglucosamine (O-GlcNAc). This hypothesis suggests that impaired glucose metabolism in the brain induced by Aβ toxicity and T2DM leads to decreased brain O-GlcNAc levels. Such a decrease in O-GlcNAc levels reflects a failure of the protective mechanism of O-GlcNAc in the brain and thereby enables progression of AD. Although still speculative, this hypothesis is appealing because O-GlcNAc is well established as being a protective stress response (27) and because it is fairly well accommodated within the proposed pathological cascade and fits with biochemical changes within the AD brain. Furthermore, the glucose responsiveness of O-GlcNAc offers an explanation as to how T2DM can operate at the molecular level as a major risk factor in AD. Although this hypothesis is still speculative, the prospect of such a link has stimulated growing interest in this research area. Here, we summarize the research in this area, with a focus on studies showing that perturbation of O-GlcNAc processing within animal models provides a protective benefit in transgenic models of AD.
O-GlcNAc Is a Common Post-translational Modification of Nucleocytoplasmic Proteins
The post-translational glycosylation of nuclear and cytoplasmic proteins by O-GlcNAc was discovered 30 years ago by Torres and Hart (28) and has emerged as a conserved modification found in all multicellular eukaryotes. Mass spectrometry-based proteomics studies of mouse and human brain tissues (29–31) have revealed O-GlcNAc on over 1000 proteins, ranging from highly abundant cytoskeletal proteins such as tau to membrane-associated proteins such as APP, as well as low-abundance proteins, including transcription factors. O-GlcNAcylated proteins play diverse cellular roles (32). Remarkably, despite its abundance, O-GlcNAcylation is regulated primarily by the action of only two enzymes (Fig. 3), each of which has various splice forms (for review, see Ref. 33).
FIGURE 3.

Potential protective role of O-GlcNAcylation within the healthy brain, which may become deficient in the AD brain. Impairments in glucose utilization (perhaps by decreased cell-surface glucose transporters) as shown here would result in decreased flux down the hexosamine biosynthetic pathway (HBSP) and ultimately lead to decreased O-GlcNAc on tau, APP, and nicastrin (a non-catalytic component of the γ-secretase complex), which could result in the increased production of NFTs and amyloid plaques, the hallmark features of the AD brain. This figure is adapted from Ref. 100.
O-GlcNAc is installed onto serine or threonine hydroxyl groups by the glycosyltransferase O-GlcNAc transferase (OGT). This enzyme uses UDP-GlcNAc as the source of the GlcNAc residue (34, 35). There is no known consensus sequence governing which residues are O-GlcNAcylated, although most sites of glycosylation are in regions that are intrinsically disordered (30). O-GlcNAc is removed from proteins by the glycoside hydrolase O-GlcNAcase (OGA) (36) (Fig. 3). Together, these two enzymes make O-GlcNAc a reversible post-translational modification. Studies on the half-life of O-GlcNAc are few, but available data indicate that the half-life of O-GlcNAc is in the range of several hours (37, 38).
The presence of O-GlcNAc on serine and threonine residues raises the potential for interplay with protein phosphorylation. In some cases, O-GlcNAc has been found to occur in a reciprocal manner to serine and threonine phosphorylation (39, 40), whereas in others, O-GlcNAc has been found close to known phosphorylation sites (41, 42). Using inhibitors of OGA to increase O-GlcNAc levels in an acute manner, it has been shown that phosphorylation at some phosphorylation sites increases, whereas at others, it decreases (43). More recent studies have found that O-GlcNAc and phosphorylation are not more coincident than by chance (30), arguing against a widespread role for O-GlcNAc in regulating protein phosphorylation. Nevertheless, the influence of O-GlcNAc on phosphorylation in specific instances such as for calcium/calmodulin-dependent kinase IV (42) and CK2 (44) has been demonstrated. Notably, both targeted and shotgun proteomics studies have found that acute administration of OGA inhibitors can decrease protein phosphorylation (43, 45–47), but longer term OGA inhibition does not affect phosphorylation on proteins such as tau (47–49). These observations suggest that O-GlcNAc can impact the activity of kinases and phosphatases but that these effects are attenuated with time, perhaps because cells adapt to sustained perturbations in overall protein O-GlcNAcylation.
O-GlcNAc and O-GlcNAc-processing Enzymes Are Particularly Abundant in Neurons
O-GlcNAc is widely distributed in cells and has been found in every tissue studied to date, but the focus here is on the levels of O-GlcNAc and its processing enzymes, OGA and OGT, in the central nervous system. mRNAs encoding OGT and OGA are widely distributed, but the highest levels encoding OGA are found in the brain (36) and those encoding OGT are found in the pancreas and brain (34, 35). Consistent with these observations, OGT activity in the brain has been found to be 10-fold higher than in peripheral tissues (50). Various studies have monitored O-GlcNAc levels as a function of age in rat and mouse brains. In mice, one study found higher brain O-GlcNAc at 5 months compared with 3 or 13 months (51). The most rigorous study (52), in which O-GlcNAc, OGA, and OGT levels in rat brain were monitored from embryos to 2 years of age, showed that O-GlcNAc levels were most abundant in prenatal rats, and soon after birth, levels became stable. This study also showed variation in the levels of OGT splice variants and the activities of both OGT and OGA. OGA activity decreased after birth, whereas OGT activity increased after birth such that both reached stable levels postnatally. Immunohistochemical staining for O-GlcNAc, as well as OGA and OGT, revealed developmental changes in staining, but in postnatal mice, these markers all showed a similar distribution in the brain, with the greatest immunoreactivity being present within all classes of neurons throughout all regions of the brain (52). Other studies reported particularly high levels of OGT expression and O-GlcNAc in the cerebellar cortex and hippocampus (53, 54). Subcellular localization studies of O-GlcNAc and OGT have focused on neurons, where it has been shown O-GlcNAc is present in both the cytoplasm and nucleus. More detailed studies have shown that the termini of neurons (55) and the nodes of Ranvier (56) are particularly rich in O-GlcNAc. These studies collectively suggest that O-GlcNAc plays particularly important roles in neurons, an idea that is consistent with neuron-specific deletion of OGT leading to neuronal apoptosis (57) and recent data indicating that OGT plays a role in mitochondrial transport within neurons (58).
Nutrient-mediated Regulation of O-GlcNAc
As noted above, glucose hypometabolism in AD occurs in brain regions prone to neurodegeneration. The ability of O-GlcNAc levels to change in response to variations in cellular glucose availability is therefore of interest and makes O-GlcNAc a possible contributor to neurodegeneration. This nutrient responsiveness occurs both in cultured cells and in vivo, where decreased glucose levels induced by fasting correlate with decreased brain O-GlcNAcylation (59). This nutrient responsiveness stems from the levels of UDP-GlcNAc being dependent on flux through the hexosamine biosynthetic pathway, the end product of which is UDP-GlcNAc. Approximately 2–5% of all glucose that is taken up by cellular glucose transporters is assimilated by the hexosamine biosynthetic pathway to generate UDP-GlcNAc (60). The hypothesis that impaired O-GlcNAc levels might contribute to the progression of AD has been bolstered by studies showing that brain tissue from AD patients has lower O-GlcNAc levels when considering post-mortem delay (46) and, more recently, in a different patient population in which decreased overall cytosolic O-GlcNAc levels were observed in the frontal cortex but not in the cerebellum (61). However, other studies using different analytic tools have suggested that O-GlcNAc levels in fact increase either generally (62) or within the detergent-insoluble fraction of AD brain tissue from regions other than the cerebellum (63). More rigorous studies of human tissues, ideally coupled with immunohistochemical analysis of O-GlcNAc within neurons, are needed to unambiguously clarify this issue.
Tau O-GlcNAcylation in AD
Tau protein exists as six splice variants that are all highly soluble and intrinsically disordered proteins. A factor generally accepted as driving aggregation of tau is its pathological hyperphosphorylation, which increases the stoichiometry of phosphorylation on tau by 6–8-fold (64). Dysfunction of many different kinases and phosphatases has been implicated in aberrant tau phosphorylation across >45 serine and threonine residues (Fig. 4). A major consequence of increased phosphorylation of tau is that it impairs its ability to bind to microtubules and thereby promote microtubule polymerization (65–67), leading to increased levels of free tau within the cytoplasm. Hyperphosphorylation of tau also leads to its compaction and enables its aggregation in vitro (64, 68) to assemble into the hyperphosphorylated PHFs found in vivo (69, 70). Data have increasingly indicated that tau pathology spreads prion-like through the brain (71), with the toxic species being small oligomers of tau (72–74) and the large aggregates being non-toxic. Studies in transgenic mice using kinase inhibitors have supported the importance of tau hyperphosphorylation as a primary factor in tau toxicity (75, 76).
FIGURE 4.

Domain map of the largest isoform of human tau (Tau441), with annotation of the locations of three different tau post-translational modifications. G, O-GlcNAcylation; Ac, acetylation; P, phosphorylation; MTBRs, microtubule-binding repeats.
O-GlcNAc on tau was first observed in bovine samples and was proposed to have a stoichiometry of four O-GlcNAc units per tau molecule (77). Since that time, several studies using both genetic methods and chemical approaches have shown that phosphorylation and O-GlcNAc on tau show some reciprocity. Gong and co-workers (46) showed that O-GlcNAc can antagonize phosphorylation of tau at various sites in tissue culture cells and rat brain slices. They also showed that human tau is O-GlcNAcylated and observed that aggregates of tau from human AD patients bear no O-GlcNAc. Support for this reciprocity comes from studies of fasted mice, which exhibited increased tau phosphorylation (59), as well as from Lefebvre et al. (45), who noted that inducing phosphorylation of tau by blocking protein phosphatase action resulted in less tau O-GlcNAcylation. Although less direct, mouse genetic studies also support this view. The neuron-specific deletion of the gene encoding OGT in mice causes increased tau phosphorylation (57). Using the selective OGA inhibitor Thiamet-G, which increases brain O-GlcNAcylation (47), Yuzwa et al. (78) found that acute OGA inhibition decreases tau phosphorylation at several pathologically relevant residues, an observation recently replicated by researchers at EMD Serono (48).
These studies have stimulated efforts to map O-GlcNAcylation sites on tau. Four O-GlcNAc sites on human tau have been mapped to Thr-123, Ser-208, Ser-400, and Ser-409/Ser-412/Ser-413 (Fig. 4) (78, 79). Ser-400 was also detected on human tau expressed in mice (78) and on rat tau (80). NMR studies using in vitro O-GlcNAcylated tau also found the Ser-208 and Ser-400 modification sites (41). Smet-Nocca et al. (41) went on to demonstrate how Ser-400 O-GlcNAcylation blocks phosphorylation at Ser-400 and subsequent sequential phosphorylation at Ser-396, providing a rationale as to how increased O-GlcNAcylation can antagonize phosphorylation at Ser-396, as seen in mice treated with the OGA inhibitor (47).
Effects of Pharmacological Inhibition of OGA on Tau Toxicity
The potential for increased O-GlcNAc to antagonize tau phosphorylation motivated research into the effect of OGA inhibitors on tau toxicity. Various mouse models of tauopathy have been generated by transgenic expression of mutant forms of tau associated with frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17), a progressive neurodegenerative disease that exhibits only tau pathology (81). JNPL3 mice (82) express the 0N4R isoform of P301L human tau under the control of the prion promoter. Due to the expression being driven by this promoter, tau hyperphosphorylation and neurofibrillary pathology are therefore seen mostly in the hindbrain, brainstem, and spinal cord. Gradual motor impairments develop, leading to muscle atrophy and consequent weight loss. The Tau.P301L model expresses the 2N4R isoform under the control of the thy-1 promoter and exhibits tau pathology primarily in the hindbrain and cervical spinal cord and, to a lesser extent, in the midbrain and cortex (83). Tg4510 mice, which express the P301L mutant tau 0N4R isoform under the control of a tetracycline-responsive operator (tetO) (72), exhibit tau pathology first in the neocortex, followed by spreading to the hippocampus. These mice show brain atrophy and a clear behavioral phenotype.
Treatment of JNPL3 mice with Thiamet-G over a period of months (78) decreased loss of motor neurons in the cervical spinal cord, reduced the extent of Sarkosyl-insoluble tau and the number of NFTs in the brain, and reduced weight loss (78). OGA inhibition was shown to increase O-GlcNAcylation of human tau at Ser-400 using a polyclonal antibody against tau O-GlcNAcylated at this site (79) and by mass spectrometry. Surprisingly, however, sustained OGA inhibition over a period of months did not lead to a reduction in phosphorylation of soluble tau (78), causing the authors to speculate that O-GlcNAc may itself hinder tau aggregation independent of phosphorylation. Analysis of various aggregation-prone tau fragments (78) and, more recently, full-length tau (84) showed that O-GlcNAc at Ser-400 hinders aggregation of tau in vitro without affecting the conformation of tau.
Using Thiamet-G, researchers at EMD Serono treated Tg4510 mice for 4 months and made observations (48) largely consistent with those made previously by Yuzwa et al. (78). EMD Serono also generated a monoclonal antibody against Ser-400 O-GlcNAcylation (85) and, using this tool, noted that OGA inhibition induced a 9-fold increase in tau O-GlcNAcylation at Ser-400. Short-term OGA inhibition reduced phosphorylation of soluble tau, but again, long-term administration had no influence on its phosphorylation. Decreases in NFT burden were also seen, but the authors also noted that the levels of pathological hyperphosphorylated tau species were markedly decreased. These species also did not appear to be O-GlcNAcylated, which is consistent with observations made in human AD tissues (46).
Using Thiamet-G in the Tau.P301L model, Van Leuven and co-workers (86) found that 2.5 months of treatment decreased the number of neurons showing tau pathology and decreased behavioral defects and weight loss while also increasing survival at 9.5 months. Interestingly, the investigators proposed that, within this model, tau is not O-GlcNAcylated, which varies from observations made by other groups in transgenic tauopathy models and human brain.
On balance, with the exception of the absence of tau glycosylation in the Tau.P301L studies, these independent studies, as well as data from H. Lundbeck A/S presented at the Alzheimer Association International Conference 2014, are generally in close agreement. The difference between the effects of short- and long-term administration of OGA inhibitors on tau phosphorylation is intriguing. Perhaps, acute OGA inhibition affects the activity of kinases and phosphatases, but the cell adapts to these changes over time. Alternatively, stoichiometric blockade of phosphorylation by O-GlcNAc may be overcome by sustained action of kinases over time to return phosphorylation to normal levels. Regardless, these studies reveal that chronic OGA inhibition leads to increases in tau O-GlcNAc modification at Ser-400 and probably at other residues, which likely lowers the aggregation propensity of tau and limits tau pathology and neurodegeneration. Currently, it is unclear whether OGA inhibition blocks pathological hyperphosphorylation of tau by direct blockade of tau phosphorylation at specific sites or indirectly by O-GlcNAc acting on other cellular processes; perhaps both direct and indirect effects are operative.
O-GlcNAc and Aβ in AD
APP is cleaved by α-secretase (87) and β-secretase (88), as well as by the γ-secretase complex (89) (Fig. 2). γ-Secretase is composed of the catalytic subunit presenilin and several non-catalytic components, including nicastrin, which aids in the maturation and trafficking of the γ-secretase complex and in the binding of substrates of the γ-secretase complex. Cleavage of APP can occur via a non-amyloidogenic pathway, which yields non-toxic peptide fragments, or via the amyloidogenic pathway, which results in the formation of Aβ40 or Aβ42 (90). As noted above, there is strong support for the amyloid cascade hypothesis, in which aberrant Aβ processing is the earliest recognized feature of AD that is required to trigger AD, but is not sufficient on its own.
Although impaired brain glucose metabolism may be a downstream consequence of Aβ toxicity, it has also been noted that T2DM can exacerbate Aβ pathology in AD models. Thus, impaired O-GlcNAcylation in the brain could, in principle, contribute to Aβ toxicity. Less research has focused on this area, although APP was shown to be O-GlcNAc-modified almost 20 years ago (91). Following this work, Jacobsen and Iverfeldt (92) demonstrated that increased O-GlcNAcylation leads to decreased production of Aβ40 in cultured SH-SY5Y cells; however, this studied relied on the nonselective OGA inhibitor O-(2-acetamido-2-deoxy-d-glucopyranosylidene)amino N-phenylcarbamate (93). An exciting recent study by Mook-Jung and co-workers (94) using the selective OGA inhibitor 1,2-dideoxy-2′-propyl-α-d-glucopyranoso-[2,1-D]-Δ2′-thiazoline in the 5xFAD Aβ mouse model showed that long-term OGA inhibition leads to reductions in the levels of Aβ40 and Aβ42 in the brain, decreased plaque formation and neuroinflammation, and improved cognition. These investigators found that 1,2-dideoxy-2′-propyl-α-d-glucopyranoso-[2,1-D]-Δ2′-thiazoline treatment of CHO cells expressing the Swedish mutation in APP resulted in decreased levels of the C-terminal fragment of APP (APP-CTF). These effects were proposed to stem from decreased γ-secretase activity induced by O-GlcNAcylation (94). Aβ40 and Aβ42 levels were not assessed, making it challenging to conclude whether the in vitro effects account for the in vivo observations. With regard to specific O-GlcNAcylation sites on APP or APP-processing enzymes, thus far, O-GlcNAc has been mapped only to Ser-708 of nicastrin (Fig. 3) by Mook-Jung and co-workers (94) .
A recent study using bigenic TAPP mice, which manifest both amyloid and tau pathologies, reported the effects of long-term OGA inhibition using Thiamet-G (95). OGA inhibition blocked cognitive decline in these mice in parallel to decreases in Aβ levels and amyloid plaques in the brain and showed a clear trend toward decreased Sarkosyl-insoluble tau. Notably, the authors reported no effect of OGA inhibition on the amount of Aβ42 released from HEK cells stably overexpressing the Swedish mutant form of APP or on the cellular levels of APP-CTF. Furthermore, in primary hippocampal neurons, no changes were observed in APP processing as evaluated using these same measures. Accordingly, OGA inhibition does not appear to influence Aβ42 release from cells or APP-CTF levels, suggesting that other processes may protect against Aβ42 toxicity in these mice. Regardless, further studies should help clarify how increased O-GlcNAc levels protect against Aβ42 and reconcile these recent reports regarding the effects of OGA inhibitors on APP processing.
Acute Effects of O-GlcNAc and Other Model Systems
In addition to the above discussion, of interest are observations that speak to the acute effects of increased O-GlcNAcylation. OGA inhibition has been found to decrease the numbers of axonal filopodia (96), enhance long-term potentiation (97), and also to correct breathing defects that occur in Tau.P301L mice (86). These data suggest that OGA inhibition may have beneficial effects in AD that are independent of blocking tau and Aβ toxicity. Further research into the processes by which O-GlcNAc induces these acute effects and also protects against both tau and Aβ toxicity will be important. In this regard, genetic studies on Caenorhabditis elegans overexpressing the 1N4R isoform of V337M FTDP-17 mutant tau showed that deletion of OGA had no effect on tau toxicity, whereas deletion of OGT was protective in an O-GlcNAc-dependent manner (98). Perhaps this discrepancy between mammalian models and C. elegans stems from differences caused by complete ablation of these genes as opposed to the use of OGA inhibitors or simply from the fact that C. elegans does not recapitulate mammalian systems in this regard. Further study using this model is, however, likely to cast light on important aspects of how O-GlcNAc affects tau toxicity.
Conclusion
The central role of glucose hypometabolism in the AD brain, coupled with the recognized impairments in insulin signaling that occur, has prompted some to define AD as a form of type 3 diabetes (99). Although many processes are disrupted in the brain by these changes, we have focused here on the role of O-GlcNAc, which has emerged as an important nutrient-sensitive modification controlling multiple cellular functions. The combined evidence discussed herein has begun to suggest that the O-GlcNAc modification may play a protective role in the brain. Deficiencies in this protective mechanism, brought on perhaps by impairments in glucose utilization, may be an important driver in AD pathobiology. These impairments in glucose utilization may result in impairments in O-GlcNAcylation of tau, APP, or other factors having an influence on their downstream toxicity. Accordingly, decreased O-GlcNAc may thus contribute to the propagation of toxic species in the brain and enable the spread of these hallmark pathologies within the AD brain. Nevertheless, regardless of the precise mechanisms that are operative, the consistent lack of apparent toxicity of OGA inhibitors (48, 78, 86, 94, 95, 100), coupled with the clear protective effects of perturbing O-GlcNAc cycling, suggests that OGA inhibitors are a promising potential disease-modifying monotherapy for AD and other tauopathies. Such compounds may represent an opportunity to positively influence the toxicity associated with both tau and Aβ and accordingly alter the course of both hallmark pathologies of AD.
This work was supported in part by the Natural Sciences and Engineering Research Council of Canada, Canadian Institutes of Health Research Grant MOP 275394, and the Alzheimer Society of Canada. S. A. Y., X. S., and D. J. V. may receive royalties from SFU for commercialization of technology relating to OGA inhibitors. D. J. V. is a cofounder, holds equity in, and serves as CSO of Alectos Therapeutics. This is the seventh article in the Minireview Series on the Thirtieth Anniversary of Research on O-GlcNAcylation of Nuclear and Cytoplasmic Proteins: Nutrient Regulation of Cellular Metabolism and Physiology by O-GlcNAcylation.
- AD
- Alzheimer disease
- Aβ
- amyloid-β peptide
- APP
- amyloid precursor protein
- PET
- positron emission tomography
- FDG
- 2-deoxy-2-[18F]fluoroglucose
- PHF
- paired helical filament
- NFT
- neurofibrillary tangle
- T2DM
- type 2 diabetes mellitus
- OGT
- O-GlcNAc transferase
- OGA
- O-GlcNAcase
- FTDP-17
- frontotemporal dementia and parkinsonism linked to chromosome 17
- CTF
- C-terminal fragment.
REFERENCES
- 1. Lo R. Y., Hubbard A. E., Shaw L. M., Trojanowski J. Q., Petersen R. C., Aisen P. S., Weiner M. W., Jagust W. J. (2011) Longitudinal change of biomarkers in cognitive decline. Arch. Neurol. 68, 1257–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beckett L. A., Harvey D. J., Gamst A., Donohue M., Kornak J., Zhang H., Kuo J. H. (2010) The Alzheimer's Disease Neuroimaging Initiative: annual change in biomarkers and clinical outcomes. Alzheimers Dement. 6, 257–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sperling R. A., Aisen P. S., Beckett L. A., Bennett D. A., Craft S., Fagan A. M., Iwatsubo T., Jack C. R., Jr., Kaye J., Montine T. J., Park D. C., Reiman E. M., Rowe C. C., Siemers E., Stern Y., Yaffe K., Carrillo M. C., Thies B., Morrison-Bogorad M., Wagster M. V., Phelps C. H. (2011) Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 7, 280–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ibáñez V., Pietrini P., Alexander G. E., Furey M. L., Teichberg D., Rajapakse J. C., Rapoport S. I., Schapiro M. B., Horwitz B. (1998) Regional glucose metabolic abnormalities are not the result of atrophy in Alzheimer's disease. Neurology 50, 1585–1593 [DOI] [PubMed] [Google Scholar]
- 5. de Leon M. J., Convit A., Wolf O. T., Tarshish C. Y., DeSanti S., Rusinek H., Tsui W., Kandil E., Scherer A. J., Roche A., Imossi A., Thorn E., Bobinski M., Caraos C., Lesbre P., Schlyer D., Poirier J., Reisberg B., Fowler J. (2001) Prediction of cognitive decline in normal elderly subjects with 2-[18F]fluoro-2-deoxy-d-glucose/poitron-emission tomography (FDG/PET). Proc. Natl. Acad. Sci. U.S.A. 98, 10966–10971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. De Santi S., de Leon M. J., Rusinek H., Convit A., Tarshish C. Y., Roche A., Tsui W. H., Kandil E., Boppana M., Daisley K., Wang G. J., Schlyer D., Fowler J. (2001) Hippocampal formation glucose metabolism and volume losses in MCI and AD. Neurobiol. Aging 22, 529–539 [DOI] [PubMed] [Google Scholar]
- 7. McLean C. A., Cherny R. A., Fraser F. W., Fuller S. J., Smith M. J., Beyreuther K., Bush A. I., Masters C. L. (1999) Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann. Neurol. 46, 860–866 [DOI] [PubMed] [Google Scholar]
- 8. Mosconi L., Pupi A., De Leon M. J. (2008) Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer's disease. Ann. N.Y. Acad. Sci. 1147, 180–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ott A., Stolk R. P., van Harskamp F., Pols H. A., Hofman A., Breteler M. M. (1999) Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 53, 1937–1942 [DOI] [PubMed] [Google Scholar]
- 10. Peila R., Rodriguez B. L., Launer L. J., Honolulu-Asia Aging S. (2002) Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes 51, 1256–1262 [DOI] [PubMed] [Google Scholar]
- 11. Calvo-Ochoa E., Arias C. (2014) Cellular and metabolic alterations in the hippocampus caused by insulin signaling dysfunction and its association with cognitive impairment during aging and Alzheimer's disease. Animal models of study. Diabetes Metab. Res. Rev. 10.1002/dmrr.2531 [DOI] [PubMed] [Google Scholar]
- 12. Correia S. C., Santos R. X., Carvalho C., Cardoso S., Candeias E., Santos M. S., Oliveira C. R., Moreira P. I. (2012) Insulin signaling, glucose metabolism and mitochondria: major players in Alzheimer's disease and diabetes interrelation. Brain Res. 1441, 64–78 [DOI] [PubMed] [Google Scholar]
- 13. de la Monte S. M. (2009) Insulin resistance and Alzheimer's disease. BMB Rep. 42, 475–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Frölich L., Blum-Degen D., Bernstein H. G., Engelsberger S., Humrich J., Laufer S., Muschner D., Thalheimer A., Türk A., Hoyer S., Zöchling R., Boissl K. W., Jellinger K., Riederer P. (1998) Brain insulin and insulin receptors in aging and sporadic Alzheimer's disease. J. Neural. Transm. 105, 423–438 [DOI] [PubMed] [Google Scholar]
- 15. Liu Y., Liu F., Iqbal K., Grundke-Iqbal I., Gong C. X. (2008) Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 582, 359–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dodart J. C., Mathis C., Bales K. R., Paul S. M., Ungerer A. (1999) Early regional cerebral glucose hypometabolism in transgenic mice overexpressing the V717F β-amyloid precursor protein. Neurosci. Lett. 277, 49–52 [DOI] [PubMed] [Google Scholar]
- 17. Nicholson R. M., Kusne Y., Nowak L. A., LaFerla F. M., Reiman E. M., Valla J. (2010) Regional cerebral glucose uptake in the 3xTG model of Alzheimer's disease highlights common regional vulnerability across AD mouse models. Brain Res. 1347, 179–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sadowski M., Pankiewicz J., Scholtzova H., Ji Y., Quartermain D., Jensen C. H., Duff K., Nixon R. A., Gruen R. J., Wisniewski T. (2004) Amyloid-β deposition is associated with decreased hippocampal glucose metabolism and spatial memory impairment in APP/PS1 mice. J. Neuropathol. Exp. Neurol. 63, 418–428 [DOI] [PubMed] [Google Scholar]
- 19. Edison P., Archer H. A., Hinz R., Hammers A., Pavese N., Tai Y. F., Hotton G., Cutler D., Fox N., Kennedy A., Rossor M., Brooks D. J. (2007) Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology 68, 501–508 [DOI] [PubMed] [Google Scholar]
- 20. Cohen A. D., Price J. C., Weissfeld L. A., James J., Rosario B. L., Bi W., Nebes R. D., Saxton J. A., Snitz B. E., Aizenstein H. A., Wolk D. A., Dekosky S. T., Mathis C. A., Klunk W. E. (2009) Basal cerebral metabolism may modulate the cognitive effects of Aβ in mild cognitive impairment: an example of brain reserve. J. Neurosci. 29, 14770–14778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baker L. D., Cross D. J., Minoshima S., Belongia D., Watson G. S., Craft S. (2011) Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch. Neurol. 68, 51–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burns C. M., Chen K., Kaszniak A. W., Lee W., Alexander G. E., Bandy D., Fleisher A. S., Caselli R. J., Reiman E. M. (2013) Higher serum glucose levels are associated with cerebral hypometabolism in Alzheimer regions. Neurology 80, 1557–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bitel C. L., Kasinathan C., Kaswala R. H., Klein W. L., Frederikse P. H. (2012) Amyloid-β and tau pathology of Alzheimer's disease induced by diabetes in a rabbit animal model. J. Alzheimers Dis. 32, 291–305 [DOI] [PubMed] [Google Scholar]
- 24. Julien C., Tremblay C., Phivilay A., Berthiaume L., Emond V., Julien P., Calon F. (2010) High-fat diet aggravates amyloid-β and tau pathologies in the 3xTg-AD mouse model. Neurobiol. Aging 31, 1516–1531 [DOI] [PubMed] [Google Scholar]
- 25. Ke Y. D., Delerue F., Gladbach A., Götz J., Ittner L. M. (2009) Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer's disease. PLoS ONE 4, e7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leboucher A., Laurent C., Fernandez-Gomez F. J., Burnouf S., Troquier L., Eddarkaoui S., Demeyer D., Caillierez R., Zommer N., Vallez E., Bantubungi K., Breton C., Pigny P., Buée-Scherrer V., Staels B., Hamdane M., Tailleux A., Buée L., Blum D. (2013) Detrimental effects of diet-induced obesity on tau pathology are independent of insulin resistance in tau transgenic mice. Diabetes 62, 1681–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zachara N. E., O'Donnell N., Cheung W. D., Mercer J. J., Marth J. D., Hart G. W. (2004) Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J. Biol. Chem. 279, 30133–30142 [DOI] [PubMed] [Google Scholar]
- 28. Torres C. R., Hart G. W. (1984) Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem. 259, 3308–3317 [PubMed] [Google Scholar]
- 29. Vosseller K., Trinidad J. C., Chalkley R. J., Specht C. G., Thalhammer A., Lynn A. J., Snedecor J. O., Guan S., Medzihradszky K. F., Maltby D. A., Schoepfer R., Burlingame A. L. (2006) O-Linked N-acetylglucosamine proteomics of postsynaptic density preparations using lectin weak affinity chromatography and mass spectrometry. Mol. Cell. Proteomics 5, 923–934 [DOI] [PubMed] [Google Scholar]
- 30. Trinidad J. C., Barkan D. T., Gulledge B. F., Thalhammer A., Sali A., Schoepfer R., Burlingame A. L. (2012) Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol. Cell. Proteomics 11, 215–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Skorobogatko Y. V., Deuso J., Adolf-Bryfogle J., Nowak M. G., Gong Y., Lippa C. F., Vosseller K. (2011) Human Alzheimer's disease synaptic O-GlcNAc site mapping and iTRAQ expression proteomics with ion trap mass spectrometry. Amino Acids 40, 765–779 [DOI] [PubMed] [Google Scholar]
- 32. Hart G. W., Housley M. P., Slawson C. (2007) Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 446, 1017–1022 [DOI] [PubMed] [Google Scholar]
- 33. Vocadlo D. J. (2012) O-GlcNAc processing enzymes: catalytic mechanisms, substrate specificity, and enzyme regulation. Curr. Opin. Chem. Biol. 16, 488–497 [DOI] [PubMed] [Google Scholar]
- 34. Kreppel L. K., Blomberg M. A., Hart G. W. (1997) Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J. Biol. Chem. 272, 9308–9315 [DOI] [PubMed] [Google Scholar]
- 35. Lubas W. A., Frank D. W., Krause M., Hanover J. A. (1997) O-Linked GlcNAc transferase is a conserved nucleocytoplasmic protein containing tetratricopeptide repeats. J. Biol. Chem. 272, 9316–9324 [DOI] [PubMed] [Google Scholar]
- 36. Gao Y., Wells L., Comer F. I., Parker G. J., Hart G. W. (2001) Dynamic O-glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a neutral, cytosolic β-N-acetylglucosaminidase from human brain. J. Biol. Chem. 276, 9838–9845 [DOI] [PubMed] [Google Scholar]
- 37. Chou C. F., Smith A. J., Omary M. B. (1992) Characterization and dynamics of O-linked glycosylation of human cytokeratin 8 and 18. J. Biol. Chem. 267, 3901–3906 [PubMed] [Google Scholar]
- 38. Roquemore E. P., Chevrier M. R., Cotter R. J., Hart G. W. (1996) Dynamic O-GlcNAcylation of the small heat shock protein αB-crystallin. Biochemistry 35, 3578–3586 [DOI] [PubMed] [Google Scholar]
- 39. Cheng X., Hart G. W. (2001) Alternative O-glycosylation/O-phosphorylation of serine-16 in murine estrogen receptor β. Post-translational regulation of turnover and transactivation activity. J. Biol. Chem. 276, 10570–10575 [DOI] [PubMed] [Google Scholar]
- 40. Chou T. Y., Hart G. W., Dang C. V. (1995) c-Myc is glycosylated at threonine 58, a known phosphorylation site and a mutational hot spot in lymphomas. J. Biol. Chem. 270, 18961–18965 [DOI] [PubMed] [Google Scholar]
- 41. Smet-Nocca C., Broncel M., Wieruszeski J. M., Tokarski C., Hanoulle X., Leroy A., Landrieu I., Rolando C., Lippens G., Hackenberger C. P. (2011) Identification of O-GlcNAc sites within peptides of the tau protein and their impact on phosphorylation. Mol. Biosyst. 7, 1420–1429 [DOI] [PubMed] [Google Scholar]
- 42. Dias W. B., Cheung W. D., Wang Z., Hart G. W. (2009) Regulation of calcium/calmodulin-dependent kinase IV by O-GlcNAc modification. J. Biol. Chem. 284, 21327–21337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Z., Gucek M., Hart G. W. (2008) Cross-talk between GlcNAcylation and phosphorylation: site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc. Natl. Acad. Sci. U.S.A. 105, 13793–13798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tarrant M. K., Rho H. S., Xie Z., Jiang Y. L., Gross C., Culhane J. C., Yan G., Qian J., Ichikawa Y., Matsuoka T., Zachara N., Etzkorn F. A., Hart G. W., Jeong J. S., Blackshaw S., Zhu H., Cole P. A. (2012) Regulation of CK2 by phosphorylation and O-GlcNAcylation revealed by semisynthesis. Nat. Chem. Biol. 8, 262–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lefebvre T., Ferreira S., Dupont-Wallois L., Bussière T., Dupire M. J., Delacourte A., Michalski J. C., Caillet-Boudin M. L. (2003) Evidence of a balance between phosphorylation and O-GlcNAc glycosylation of tau proteins–a role in nuclear localization. Biochim. Biophys. Acta 1619, 167–176 [DOI] [PubMed] [Google Scholar]
- 46. Liu F., Iqbal K., Grundke-Iqbal I., Hart G. W., Gong C. X. (2004) O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 101, 10804–10809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yuzwa S. A., Macauley M. S., Heinonen J. E., Shan X., Dennis R. J., He Y., Whitworth G. E., Stubbs K. A., McEachern E. J., Davies G. J., Vocadlo D. J. (2008) A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat. Chem. Biol. 4, 483–490 [DOI] [PubMed] [Google Scholar]
- 48. Graham D. L., Gray A. J., Joyce J. A., Yu D., O'Moore J., Carlson G. A., Shearman M. S., Dellovade T. L., Hering H. (2014) Increased O-GlcNAcylation reduces pathological tau without affecting its normal phosphorylation in a mouse model of tauopathy. Neuropharmacology 79, 307–313 [DOI] [PubMed] [Google Scholar]
- 49. Yu Y., Zhang L., Li X., Run X., Liang Z., Li Y., Liu Y., Lee M. H., Grundke-Iqbal I., Iqbal K., Vocadlo D. J., Liu F., Gong C. X. (2012) Differential effects of an O-GlcNAcase inhibitor on tau phosphorylation. PLoS ONE 7, e35277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Okuyama R., Marshall S. (2003) UDP-N-acetylglucosaminyltransferase (OGT) in brain tissue: temperature sensitivity and subcellular distribution of cytosolic and nuclear enzyme. J. Neurochem. 86, 1271–1280 [DOI] [PubMed] [Google Scholar]
- 51. Rex-Mathes M., Werner S., Strutas D., Griffith L. S., Viebahn C., Thelen K., Schmitz B. (2001) O-GlcNAc expression in developing and ageing mouse brain. Biochimie 83, 583–590 [DOI] [PubMed] [Google Scholar]
- 52. Liu Y., Li X., Yu Y., Shi J., Liang Z., Run X., Li Y., Dai C. L., Grundke-Iqbal I., Iqbal K., Liu F., Gong C. X. (2012) Developmental regulation of protein O-GlcNAcylation, O-GlcNAc transferase, and O-GlcNAcase in mammalian brain. PLoS ONE 7, e43724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Akimoto Y., Comer F. I., Cole R. N., Kudo A., Kawakami H., Hirano H., Hart G. W. (2003) Localization of the O-GlcNAc transferase and O-GlcNAc-modified proteins in rat cerebellar cortex. Brain Res. 966, 194–205 [DOI] [PubMed] [Google Scholar]
- 54. Liu K., Paterson A. J., Zhang F., McAndrew J., Fukuchi K., Wyss J. M., Peng L., Hu Y., Kudlow J. E. (2004) Accumulation of protein O-GlcNAc modification inhibits proteasomes in the brain and coincides with neuronal apoptosis in brain areas with high O-GlcNAc metabolism. J. Neurochem. 89, 1044–1055 [DOI] [PubMed] [Google Scholar]
- 55. Cole R. N., Hart G. W. (2001) Cytosolic O-glycosylation is abundant in nerve terminals. J. Neurochem. 79, 1080–1089 [DOI] [PubMed] [Google Scholar]
- 56. Zhang X., Bennett V. (1996) Identification of O-linked N-acetylglucosamine modification of ankyrin G isoforms targeted to nodes of Ranvier. J. Biol. Chem. 271, 31391–31398 [DOI] [PubMed] [Google Scholar]
- 57. O'Donnell N., Zachara N. E., Hart G. W., Marth J. D. (2004) Ogt-dependent X-chromosome-linked protein glycosylation is a requisite modification in somatic cell function and embryo viability. Mol. Cell. Biol. 24, 1680–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pekkurnaz G., Trinidad J. C., Wang X., Kong D., Schwarz T. L. (2014) Glucose regulates mitochondrial motility via milton modification by O-GlcNAc transferase. Cell 158, 54–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li X., Lu F., Wang J. Z., Gong C. X. (2006) Concurrent alterations of O-GlcNAcylation and phosphorylation of tau in mouse brains during fasting. Eur. J. Neurosci. 23, 2078–2086 [DOI] [PubMed] [Google Scholar]
- 60. Marshall S., Bacote V., Traxinger R. R. (1991) Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem. 266, 4706–4712 [PubMed] [Google Scholar]
- 61. Liu F., Shi J., Tanimukai H., Gu J., Gu J., Grundke-Iqbal I., Iqbal K., Gong C. X. (2009) Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer's disease. Brain 132, 1820–1832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Förster S., Welleford A. S., Triplett J. C., Sultana R., Schmitz B., Butterfield D. A. (2014) Increased O-GlcNAc levels correlate with decreased O-GlcNAcase levels in Alzheimer disease brain. Biochim. Biophys. Acta 1842, 1333–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Griffith L. S., Schmitz B. (1995) O-linked N-acetylglucosamine is upregulated in Alzheimer brains. Biochem. Biophys. Res. Commun. 213, 424–431 [DOI] [PubMed] [Google Scholar]
- 64. Ksiezak-Reding H., Liu W. K., Yen S. H. (1992) Phosphate analysis and dephosphorylation of modified tau associated with paired helical filaments. Brain Res. 597, 209–219 [DOI] [PubMed] [Google Scholar]
- 65. Cho J. H., Johnson G. V. (2004) Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3β (GSK3β) plays a critical role in regulating tau's ability to bind and stabilize microtubules. J. Neurochem. 88, 349–358 [DOI] [PubMed] [Google Scholar]
- 66. Lindwall G., Cole R. D. (1984) Phosphorylation affects the ability of tau protein to promote microtubule assembly. J. Biol. Chem. 259, 5301–5305 [PubMed] [Google Scholar]
- 67. Sengupta A., Kabat J., Novak M., Wu Q., Grundke-Iqbal I., Iqbal K. (1998) Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch. Biochem. Biophys. 357, 299–309 [DOI] [PubMed] [Google Scholar]
- 68. Alonso A., Zaidi T., Novak M., Grundke-Iqbal I., Iqbal K. (2001) Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. U.S.A. 98, 6923–6928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Grundke-Iqbal I., Iqbal K., Quinlan M., Tung Y. C., Zaidi M. S., Wisniewski H. M. (1986) Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 261, 6084–6089 [PubMed] [Google Scholar]
- 70. Nukina N., Kosik K. S., Selkoe D. J. (1987) Recognition of Alzheimer paired helical filaments by monoclonal neurofilament antibodies is due to crossreaction with tau protein. Proc. Natl. Acad. Sci. U.S.A. 84, 3415–3419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. de Calignon A., Polydoro M., Suárez-Calvet M., William C., Adamowicz D. H., Kopeikina K. J., Pitstick R., Sahara N., Ashe K. H., Carlson G. A., Spires-Jones T. L., Hyman B. T. (2012) Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 73, 685–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Santacruz K., Lewis J., Spires T., Paulson J., Kotilinek L., Ingelsson M., Guimaraes A., DeTure M., Ramsden M., McGowan E., Forster C., Yue M., Orne J., Janus C., Mariash A., Kuskowski M., Hyman B., Hutton M., Ashe K. H. (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Frost B., Jacks R. L., Diamond M. I. (2009) Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 284, 12845–12852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kfoury N., Holmes B. B., Jiang H., Holtzman D. M., Diamond M. I. (2012) Trans-cellular propagation of tau aggregation by fibrillar species. J. Biol. Chem. 287, 19440–19451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Le Corre S., Klafki H. W., Plesnila N., Hübinger G., Obermeier A., Sahagún H., Monse B., Seneci P., Lewis J., Eriksen J., Zehr C., Yue M., McGowan E., Dickson D. W., Hutton M., Roder H. M. (2006) An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 103, 9673–9678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Uno Y., Iwashita H., Tsukamoto T., Uchiyama N., Kawamoto T., Kori M., Nakanishi A. (2009) Efficacy of a novel, orally active GSK-3 inhibitor 6-methyl-N-[3-[[3-(1-methylethoxy)propyl]carbamoyl]-1H-pyrazol-4-yl]pyridine-3-carboxamide in tau transgenic mice. Brain Res. 1296, 148–163 [DOI] [PubMed] [Google Scholar]
- 77. Arnold C. S., Johnson G. V., Cole R. N., Dong D. L., Lee M., Hart G. W. (1996) The microtubule-associated protein tau is extensively modified with O-linked N-acetylglucosamine. J. Biol. Chem. 271, 28741–28744 [DOI] [PubMed] [Google Scholar]
- 78. Yuzwa S. A., Shan X., Macauley M. S., Clark T., Skorobogatko Y., Vosseller K., Vocadlo D. J. (2012) Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat. Chem. Biol. 8, 393–399 [DOI] [PubMed] [Google Scholar]
- 79. Yuzwa S. A., Yadav A. K., Skorobogatko Y., Clark T., Vosseller K., Vocadlo D. J. (2011) Mapping O-GlcNAc modification sites on tau and generation of a site-specific O-GlcNAc tau antibody. Amino Acids 40, 857–868 [DOI] [PubMed] [Google Scholar]
- 80. Wang Z., Udeshi N. D., O'Malley M., Shabanowitz J., Hunt D. F., Hart G. W. (2010) Enrichment and site mapping of O-linked N-acetylglucosamine by a combination of chemical/enzymatic tagging, photochemical cleavage, and electron transfer dissociation mass spectrometry. Mol. Cell. Proteomics 9, 153–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Baker M., Kwok J. B., Kucera S., Crook R., Farrer M., Houlden H., Isaacs A., Lincoln S., Onstead L., Hardy J., Wittenberg L., Dodd P., Webb S., Hayward N., Tannenberg T., Andreadis A., Hallupp M., Schofield P., Dark F., Hutton M. (1997) Localization of frontotemporal dementia with parkinsonism in an Australian kindred to chromosome 17q21-22. Ann. Neurol. 42, 794–798 [DOI] [PubMed] [Google Scholar]
- 82. Lewis J., McGowan E., Rockwood J., Melrose H., Nacharaju P., Van Slegtenhorst M., Gwinn-Hardy K., Paul Murphy M., Baker M., Yu X., Duff K., Hardy J., Corral A., Lin W. L., Yen S. H., Dickson D. W., Davies P., Hutton M. (2000) Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 25, 402–405 [DOI] [PubMed] [Google Scholar]
- 83. Terwel D., Lasrado R., Snauwaert J., Vandeweert E., Van Haesendonck C., Borghgraef P., Van Leuven F. (2005) Changed conformation of mutant tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of tau-4R/2N transgenic mice. J. Biol. Chem. 280, 3963–3973 [DOI] [PubMed] [Google Scholar]
- 84. Yuzwa S. A., Cheung A. H., Okon M., McIntosh L. P., Vocadlo D. J. (2014) O-GlcNAc modification of tau directly inhibits its aggregation without perturbing the conformational properties of tau monomers. J. Mol. Biol. 426, 1736–1752 [DOI] [PubMed] [Google Scholar]
- 85. Cameron A., Giacomozzi B., Joyce J., Gray A., Graham D., Ousson S., Neny M., Beher D., Carlson G., O'Moore J., Shearman M., Hering H. (2013) Generation and characterization of a rabbit monoclonal antibody site-specific for tau O-GlcNAcylated at serine 400. FEBS Lett. 587, 3722–3728 [DOI] [PubMed] [Google Scholar]
- 86. Borghgraef P., Menuet C., Theunis C., Louis J. V., Devijver H., Maurin H., Smet-Nocca C., Lippens G., Hilaire G., Gijsen H., Moechars D., Van Leuven F. (2013) Increasing brain protein O-GlcNAcylation mitigates breathing defects and mortality of Tau.P301L mice. PLoS ONE 8, e84442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lammich S., Kojro E., Postina R., Gilbert S., Pfeiffer R., Jasionowski M., Haass C., Fahrenholz F. (1999) Constitutive and regulated α-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. U.S.A. 96, 3922–3927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Vassar R., Bennett B. D., Babu-Khan S., Kahn S., Mendiaz E. A., Denis P., Teplow D. B., Ross S., Amarante P., Loeloff R., Luo Y., Fisher S., Fuller J., Edenson S., Lile J., Jarosinski M. A., Biere A. L., Curran E., Burgess T., Louis J. C., Collins F., Treanor J., Rogers G., Citron M. (1999) β-Secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741 [DOI] [PubMed] [Google Scholar]
- 89. De Strooper B., Saftig P., Craessaerts K., Vanderstichele H., Guhde G., Annaert W., Von Figura K., Van Leuven F. (1998) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391, 387–390 [DOI] [PubMed] [Google Scholar]
- 90. Estus S., Golde T. E., Kunishita T., Blades D., Lowery D., Eisen M., Usiak M., Qu X. M., Tabira T., Greenberg B. D. (1992) Potentially amyloidogenic, carboxyl-terminal derivatives of the amyloid protein precursor. Science 255, 726–728 [DOI] [PubMed] [Google Scholar]
- 91. Griffith L. S., Mathes M., Schmitz B. (1995) β-Amyloid precursor protein is modified with O-linked N-acetylglucosamine. J. Neurosci. Res. 41, 270–278 [DOI] [PubMed] [Google Scholar]
- 92. Jacobsen K. T., Iverfeldt K. (2011) O-GlcNAcylation increases non-amyloidogenic processing of the amyloid-β precursor protein (APP). Biochem. Biophys. Res. Commun. 404, 882–886 [DOI] [PubMed] [Google Scholar]
- 93. Macauley M. S., Vocadlo D. J. (2010) Increasing O-GlcNAc levels: an overview of small-molecule inhibitors of O-GlcNAcase. Biochim. Biophys. Acta 1800, 107–121 [DOI] [PubMed] [Google Scholar]
- 94. Kim C., Nam D. W., Park S. Y., Song H., Hong H. S., Boo J. H., Jung E. S., Kim Y., Baek J. Y., Kim K. S., Cho J. W., Mook-Jung I. (2013) O-Linked β-N-acetylglucosaminidase inhibitor attenuates β-amyloid plaque and rescues memory impairment. Neurobiol. Aging 34, 275–285 [DOI] [PubMed] [Google Scholar]
- 95. Yuzwa S. A., Shan X., Jones B. A., Zhao G., Woodward M. L., Li X., Zhu Y., McEachern E. J., Silverman M. A., Watson N. V., Gong C. X., Vocadlo D. J. (2014) Pharmacological Inhibition of O-GlcNAcase (OGA) prevents cognitive decline and amyloid plaque formation in bigenic tau/APP mutant mice. Mol. Neurodegener. 9, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Francisco H., Kollins K., Varghis N., Vocadlo D., Vosseller K., Gallo G. (2009) O-GlcNAc post-translational modifications regulate the entry of neurons into an axon branching program. Dev. Neurobiol. 69, 162–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Tallent M. K., Varghis N., Skorobogatko Y., Hernandez-Cuebas L., Whelan K., Vocadlo D. J., Vosseller K. (2009) In vivo modulation of O-GlcNAc levels regulates hippocampal synaptic plasticity through interplay with phosphorylation. J. Biol. Chem. 284, 174–181 [DOI] [PubMed] [Google Scholar]
- 98. Wang P., Lazarus B. D., Forsythe M. E., Love D. C., Krause M. W., Hanover J. A. (2012) O-GlcNAc cycling mutants modulate proteotoxicity in Caenorhabditis elegans models of human neurodegenerative diseases. Proc. Natl. Acad. Sci. U.S.A. 109, 17669–17674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. de la Monte S. M., Tong M. (2014) Brain metabolic dysfunction at the core of Alzheimer's disease. Biochem. Pharmacol. 88, 548–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Macauley M. S., Shan X., Yuzwa S. A., Gloster T. M., Vocadlo D. J. (2010) Elevation of global O-GlcNAc in rodents using a selective O-GlcNAcase inhibitor does not cause insulin resistance or perturb glucohomeostasis. Chem. Biol. 17, 949–958 [DOI] [PMC free article] [PubMed] [Google Scholar]