

Background: Therapeutic antibodies such as adalimumab can elicit anti-drug antibodies.
Results: Monoclonal patient-derived antibodies were generated and found to compete for binding to adalimumab and are neutralizing, but they show markedly different fine-specificity.
Conclusion: Anti-adalimumab antibodies bind to overlapping but distinct epitopes on adalimumab.
Significance: Even for a fully human therapeutic antibody, there may be multiple determinants that contribute to immunogenicity.
Keywords: Antibody Engineering, Autoimmune Disease, Biotechnology, Humoral Response, Monoclonal Antibody, Tumor Necrosis Factor (TNF), Adalimumab, Immunogenicity
Abstract
The production of antibodies to adalimumab in autoimmune patients treated with adalimumab is shown to diminish treatment efficacy. We previously showed that these antibodies are almost exclusively neutralizing, indicating a restricted response. Here, we investigated the characteristics of a panel of patient-derived monoclonal antibodies for binding to adalimumab. Single B-cells were isolated from two patients, cultured, and screened for adalimumab specificity. Analysis of variable region sequences of 16 clones suggests that the immune response against adalimumab is broad, involving multiple B-cell clones each using different combinations of V(D)J segments. A strong bias for replacement mutations in the complementarity determining regions was found, indicating an antigen-driven response. We recombinantly expressed 11 different monoclonal antibodies and investigated their affinity and specificity. All clones except one are of high affinity (Kd between 0.6 and 233 pm) and compete with TNF as well as each other for binding to adalimumab. However, binding to a panel of single-point mutants of adalimumab indicates markedly different fine specificities that also result in a differential tendency of each clone to form dimeric and multimeric immune complexes. We conclude that although all anti-adalimumab antibodies compete for binding to TNF, the response is clonally diverse and involves multiple epitopes on adalimumab. These results are important for understanding the relationship between self and non-self or idiotypic determinants on therapeutic antibodies and their potential immunogenicity.
Introduction
The introduction of therapeutic monoclonal antibodies has given a major boost to the treatment of individuals suffering from autoimmune disorders, including rheumatoid arthritis (RA).2 However, some of the patients develop an immune response against the therapeutic, which results in the formation of anti-drug antibodies (ADA). The formation of ADA has been linked to lower serum drug levels and reduced clinical response (1–7). ADA formation can influence treatment efficiency via two possible mechanisms. First, the formation of ADA can lead to enhanced clearance of the drug. Second, the formation of neutralizing antibodies can prevent the therapeutic agent from binding to its target.
In the case of adalimumab, a fully human anti-TNF agent, recent data indicate that essentially all ADA are anti-idiotypic antibodies, directed against the TNF binding region of adalimumab and resulting in neutralization of the drug (8). This is confirmed by inhibition experiments showing that TNF completely prevents binding of ADA in patient sera (9). These observations suggest that the immune response against adalimumab is restricted, but it leaves unanswered the question of whether the immune system targets just a single epitope or multiple determinants that are spatially confined to the TNF-binding site on adalimumab. Although adalimumab is a fully human therapeutic antibody, it still carries unique stretches of amino acids that determine its specificity for TNF. Because its sequence is close to germ line, the number of unique foreign determinants, or idiotopes, is expected to be limited. Therefore, a likely explanation for the restricted anti-adalimumab response would be that there is only a single dominant B-cell epitope present on adalimumab. Alternatively, only limited numbers of B-cell clones participate in the ADA response leading to a restricted response. To investigate these hypotheses, we have cloned the antibody-specific sequences of 16 patient-derived adalimumab specific B-cells. We have expressed 11 of these fully human monoclonal antibodies in HEK293 cells and analyzed their neutralization capacity as well as their binding profiles to adalimumab and to a panel of single-point mutants of adalimumab.
MATERIALS AND METHODS
Patients
Monoclonal ADA were derived from two adalimumab (Abbott)-treated patients. Patient 1 was treated for ankylosing spondylitis, and patient 2 was treated for RA. Both patients had high titers of ADA as detected in the antigen binding test (2960 and 8570 arbitrary units/ml, respectively). Informed consent was obtained from both patients. The study was approved by the local ethics committee (1, 10).
Isolation, Proliferation, and Identification of Adalimumab-specific Single B-cells
Antigen-specific B-cells were isolated and cultured as described before (8). In short, PBMC's were isolated using a Percoll gradient, and B-cells were isolated using anti-CD19 beads. Antigen-specific memory cells were sorted using anti-CD27 and two differently labeled adalimumab Fab fragments and seeded 0.5 cells per well or sorted single cell per well. Cells were cultured in the presence of CD40 ligand-expressing cells and a cytokine mixture. After 10–14 days, the supernatants were tested for the presence of ADA using the bridging ELISA as described before (8). Furthermore, the isotype of each clone was determined by ELISA. Supernatants were incubated in plates coated with isotype-specific antibodies followed by detection using biotinylated F(ab′)2 fragments of adalimumab.
Production of Recombinant Human Antibodies
RNA was isolated from wells positive for anti-adalimumab in the bridging ELISA using TRIzol (Peqlab, Erlangen, Germany). cDNA synthesis and 5′-RACE PCR were performed using the SMART cDNA synthesis kit (Clontech, catalog no. 634914). RACE PCR products for VL and VH were sequenced, ordered, and cloned into pcDNA3.3 or pcDNA3.1 (Invitrogen) expression vectors together with the constant domains of the human κ, λ, and human IgG1 allotype f genes, essentially as described before (12). In some cases, an additional nested PCR was performed to obtain the VL and VH sequences. Antibodies were expressed by transient co-transfection of heavy and light chain containing vectors of HEK293F cells with 293fectin and OptiMEM (Invitrogen), using the Freestyle HEK293F expression system (Invitrogen) according to the instructions supplied by the manufacturer.
Nucleotide Sequence Analysis
Nucleotide analysis was performed using JOINSOLVER®. Replacement (R) and silent (S) mutation frequencies were calculated for framework regions and CDRs based on the average number of nucleotides in all analyzed sequences according to the definitions of IMGT/Joinsolver (13).
Generation of F(ab′)2 and Fab Fragments
F(ab′)2 and Fab fragments were generated using pepsin, and subsequent reduction and alkylation with dithioerythritol and N-ethylmaleimide, respectively, were as described previously (14).
Biotinylation
Antibodies and Fab fragments (100 μg) were biotinylated by incubation with 60 μg of Sulfo-NHS-LC-Biotin (Thermo Scientific, Rockford, IL) in 0.1 m NaHCO3. After 2 h of incubation in the dark at room temperature, biotinylated antibodies were dialyzed against PBS.
Affinity Measurement of Monoclonal Antibodies
Affinities were determined as described previously (15, 25). Briefly, serial 4-fold dilutions of anti-adalimumab monoclonal antibody (0.05–1000 ng/ml) were incubated with a fixed concentration of 63 pg/ml (1.25 pm) of adalimumab Fab-DyLight 488 (ada-Fab-488) (31 pg/ml (0.625 pm) for anti-adalimumab 2.2 and 2.12). Samples (1000 μl) were applied using a thermostatted autosampler (25 °C) to a Superdex 200 HR 10/300 column (GE Healthcare), which was connected to an ÄKTAexplorer HPLC system (GE Healthcare). Elution profiles of ada-Fab-488 were monitored by measuring the fluorescence (excitation/emission 488/525 nm) with a Prominence RF-20Axs on-line fluorescence detector (Shimadzu, Kyoto, Japan). To calculate dissociation constants, fluorescence at 13 or 17 ml, corresponding to the peak maxima of bound or free ada-Fab-488, respectively, was plotted against the concentration of anti-adalimumab antibody (molar concentration of the number of Fab arms), and a 1:1 binding model was fitted to the data using Microcal Origin software.
Competition ELISA with Recombinant Monoclonal Antibodies
Maxisorp ELISA plates were coated overnight with 0.5 μg/ml adalimumab in PBS. The binding of biotinylated monoclonal antibody was analyzed following incubation of the coat with increasing amounts of different unlabeled monoclonal antibodies or as a control using an irrelevant human monoclonal antibody, cetuximab (Erbitux, Merck). All incubations were performed in high performance ELISA buffer (Sanquin). Binding of biotinylated antibody was detected using streptavidin-poly-HRP as described before (8).
TNF Bioassay
104 TNF-responsive endothelial cells (ECRF-24 cells) (16) were seeded in a 96-well plate in IMDM containing 5% FCS (Bodinco), 100 units/ml penicillin, 100 μg/ml streptomycin, 50 μm 2-mercaptoethanol, 20 μg/ml human apo-transferrin (Sigma). After 24 h of incubation, the samples containing TNF, adalimumab, and monoclonal antibody were added. Supernatants were harvested after 24 h of incubation and tested for IL-8 concentration by ELISA (17).
Competition ELISA with Single-point Mutants of Adalimumab
A bridging ELISA for measurement of anti-adalimumab was carried out essentially as described before (8) but with an additional inhibition step as described below. In short, ELISA plates were coated with adalimumab, and after incubation with the anti-adalimumab-containing sample, 100 ng/ml Fab fragments of adalimumab or single-point mutants of adalimumab (18) were added followed by incubation for 1 h. Then, without washing, biotinylated adalimumab was added, and after 1 h of incubation and washing the binding was assessed with streptavidin-poly-HRP. The single-point mutants of anti-adalimumab were produced as described before (18).
Complex Formation of Adalimumab and Anti-adalimumab
To form immune complexes, adalimumab and anti-adalimumab were combined 1:1 at 50 μg/ml in PBS. Samples were analyzed for IgG monomer, IgG dimer, and IgG multimer content on a calibrated Superdex200 10/300 gel filtration column (30 cm, 24 ml, GE Healthcare) connected to an HPLC system at room temperature with a flow rate of 0.5 ml/min and PBS as running buffer. Elution profiles were obtained by recording the absorbance at 215 nm. A computer program (Unicorn version 5.11) was used to integrate the areas under the curve of the peaks shown in the chromatograms.
RESULTS
Gene Characteristics and Affinities of Monoclonal Anti-adalimumab Antibodies
To obtain the sequences of the VH and VL chain of various anti-adalimumab antibodies, antigen-specific memory B-cells isolated from the peripheral blood of two ADA-positive patients were cultured starting from a single cell per well for 10–14 days. Supernatants were screened for ADA production, and RNA was extracted from ADA-positive wells. IgG subclasses were determined as follows: 8/16 (50%) were IgG1; 7/16 (44%) were IgG4; and 1/16 (6%) was undetermined. After the production of cDNA, the IgG-specific sequence was amplified and sequenced, resulting in the sequences of three clones from the first patient and 13 clones from the second patient (summarized in Table 1). All the monoclonal antibodies used different combinations of V(D)J segments indicating that they originated from different precursor B-cells. IGHV1 and IGKV1 genes were the most frequently used heavy and light chain V segments (Fig. 1A). The majority of antibodies have κ light chains, but this probably reflects a selection bias in our system. The heavy chain CDR3 length varies substantially but within the normal range. The majority of the antibodies underwent extensive somatic hypermutation (Fig. 1B). The total median number of mutations was 23 (IQR(19–27)) and 12 (IQR(8–18)) for VH and VL, respectively. R and S mutation frequencies for the framework regions and CDR1 and CDR2 regions were analyzed (Fig. 1B) and indicate an antigen-driven affinity maturation (19), which was confirmed using the Focused test (20); mean R/S ratios are 1.7 and 1.5 for the framework regions of VH and VL, and 8.0 and 4.6 for CDRs of VH and VL, respectively. We were able to express 11 of the above-described monoclonal antibodies in HEK293 cells in large enough quantities to investigate their binding affinity and specificity. All recombinant monoclonal antibodies were expressed as IgG1 and bound specifically to adalimumab when tested by ELISA (data not shown). The affinities of these clones are generally high, ranging from 0.6 to 233 pm with one low affinity clone with a Kd of ∼50 nm (Fig. 1C). There was no correlation between the number of mutations in the sequence and the affinity of the antibodies (data not shown). Taken together, these results show that the ADA response in these patients is a very diverse response that is antigen-driven and the result of substantial affinity maturation.
TABLE 1.
All monoclonal antibodies are derived from different precursor B-cells
V(D)J usage of the different monoclonal antibodies, the CDR3 length, and the number of mutations leading to amino acid replacements (R) or silent mutations (S) is shown.
| Clone | Heavy chain |
Light chain |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Isotype | V gene | D gene | J gene | CDR3-IMGT | Length | R | S | κ/λ | V gene | J gene | CDR3-IMGT | Length | R | S | |
| 1.1 | IgG1 | 1–03 | 2–02 | 4 | ARDIVVVPVAMHPDY | 15 | 16 | 4 | κ | 1–33 | 4 | QHYDNLPS | 8 | 6 | 0 |
| 1.2 | IgG4 | 1–02 | 2–15 | 5 | ARDKWGPAAQYPDNWFDS | 18 | 9 | 7 | κ | 1–12 | 1 | QQANSFPLT | 9 | 6 | 2 |
| 1.3 | IgG1 | 1–18 | 1–14 | 4 | AREPYDYSGTADY | 13 | 15 | 4 | κ | 1–39 | 2 | QQSYTTPYT | 9 | 0 | 3 |
| 2.1 | IgG1 | 1–03 | 3–09 | 4 | ASEGLLTGFPLDY | 13 | 16 | 15 | κ | 3–20 | 4 | QQYASSPLT | 9 | 8 | 5 |
| 2.2 | IgG4 | 1–69 | 3–10 | 6 | ARLAIPWFGEAVFSYHYDMDV | 21 | 15 | 9 | κ | 1–33 | 4 | QQYDDVPLT | 9 | 10 | 11 |
| 2.3 | IgG4 | 4–31 | 6–13 | 3 | AREPAATGPSGDAFDI | 16 | 21 | 5 | κ | 1–39 | 2 | QQSYSEPYT | 9 | 7 | 8 |
| 2.4 | IgG1 | 1–03 | 3–16 | 3 | ARMGERGLDV | 10 | 19 | 7 | κ | 3–20 | 4 | QQYVSSPLT | 9 | 8 | 3 |
| 2.5 | IgG4 | 4–59 | 6–13 | 3 | ARQTLLMAADGDDAFDI | 17 | 16 | 11 | κ | 1–39 | 4 | QQSYSTPFT | 9 | 14 | 9 |
| 2.6 | IgG1 | 4–39 | 1–26 | 4 | ARRSVAAFDY | 10 | 14 | 6 | κ | 3–20 | 2 | QQYGSSET | 8 | 9 | 2 |
| 2.7 | IgG1 | 4–34 | 1–26 | 3 | AREGKNSGSYYVRLGDTFDI | 20 | 5 | 1 | κ | 4–1 | 2 | QQYYSAPPT | 9 | 2 | 2 |
| 2.8 | IgG4 | 1–69 | 6–19 | 5 | ARDQKGQWFDP | 11 | 21 | 2 | κ | 3–20 | 4 | HQSALPPRT | 9 | 9 | 5 |
| 2.9 | IgG1 | 3–48 | 2–21 | 6 | ARVKDDIVVPTGLGMDV | 17 | 23 | 9 | κ | 1–33 | 4 | QQYNDIPFT | 9 | 13 | 6 |
| 2.10 | IgG4 | 1–03 | 2–21 | 5 | AELASSGLFDP | 11 | 15 | 6 | κ | 3–20 | 4 | QQYGSSPLT | 9 | 9 | 4 |
| 2.11 | N.D. | 1–69 | 6–19 | 4 | ARLHSRGWSDFDY | 13 | 14 | 8 | κ | 1–33 | 4 | QEYDNLVVN | 9 | 5 | 5 |
| 2.12 | IgG4 | 1–18 | 2–21 | 6 | AREIAPGDMDE | 11 | 25 | 10 | λ | 3–25 | 1 | LSADSSASYV | 10 | 18 | 6 |
| 2.13 | IgG1 | 3–48 | 5–5 | 3 | ARTGGHSHGPGGFDI | 15 | 9 | 1 | λ | 3–25 | 3 | QSGDTSGAPIWV | 12 | 6 | 2 |
FIGURE 1.
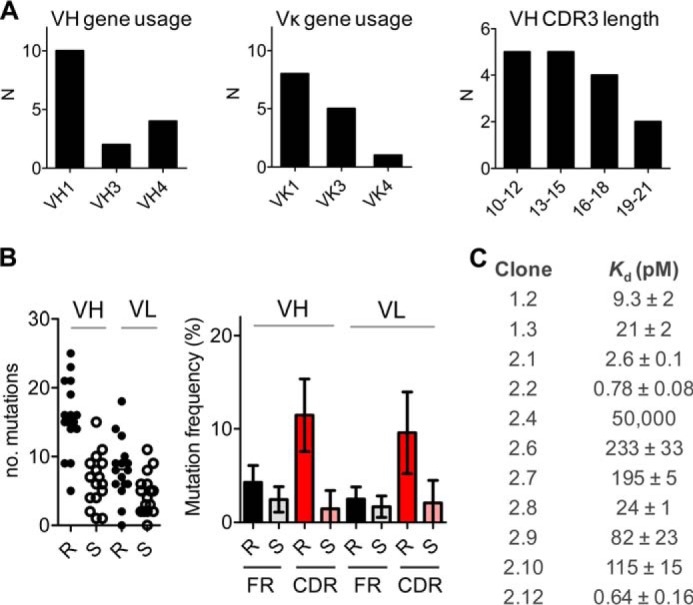
Gene characteristics and affinities of monoclonal anti-adalimumab antibodies. A, VH and Vκ gene usage and VH CDR3 length. B, number of R and S mutations (left panel) and mean mutation frequency for framework (FR) and CDR1 and CDR2 regions (right panel); error bars represent S.D. C, affinities of clones as measured with fluorescence-assisted HPLC (15).
Competition and Neutralization of Monoclonal Antibodies
Competition experiments were carried out to investigate the relative binding locations of the anti-adalimumab clones to adalimumab. To this end, binding of biotinylated anti-adalimumab to coated adalimumab was inhibited with all the other monoclonal antibodies. An example is shown in Fig. 2A, which shows that all antibodies can inhibit binding of anti-adalimumab 2.4 to adalimumab with some variation in the efficiency of inhibition. The variation in inhibition may be explained by differences in affinity between the monoclonal antibodies. In similar experiments, it was found that all monoclonal antibodies compete with each other for the binding of adalimumab (data not shown). Furthermore, it was found that all antibodies could prevent binding of adalimumab to TNF as tested with the ECRF cell line, which responds to TNF by producing IL-8 (Fig. 2B), demonstrating that all antibodies are neutralizing. None of the antibodies could prevent binding of infliximab to TNF (data not shown). The low affinity clone 2.4 appeared unable to neutralize adalimumab, but a higher concentration of anti-adalimumab 2.4 could restore IL-8 production (data not shown). In summary, despite the fact that the different anti-adalimumab antibodies are clonally unrelated, they target sites on adalimumab that overlap with the TNF-binding site.
FIGURE 2.

All monoclonal antibodies compete with anti-adalimumab 2.4 and TNF for the binding to adalimumab. A, binding of biotinylated anti-adalimumab 2.4 to adalimumab was inhibited by the addition of unlabeled monoclonal anti-adalimumab antibodies. The control monoclonal antibody cetuximab did not inhibit binding. B, in response to 1 ng of TNF, ECRF-24 cells produce IL-8 in the linear range of the titration curve. Adalimumab (30 ng) can neutralize TNF, thereby preventing IL-8 production. 0.1 μg of all antibodies (except anti-adalimumab 2.4) was sufficient to completely restore IL-8 production in the presence of adalimumab. Results represent mean and S.E. (error bars) from three experiments. Abbreviation used is as follows: ADL, adalimumab.
Specificity and Immune Complex Formation of Monoclonal Antibodies
Next, we investigated whether the clones bind to one specific epitope on adalimumab or to multiple epitopes that are spatially confined. We set up an inhibition assay in which binding of biotinylated adalimumab to an anti-adalimumab clone is assessed in the presence of Fab fragments of either wild-type or single-point variants of adalimumab (Fig. 3). All variants retained their capacity to bind TNF (18). The point mutations are all located in or closely to the CDR of adalimumab (Fig. 3A), the regions expected to be involved in the binding to TNF. The inhibitory capacity of the Fab fragments of adalimumab variants directly reflects their binding potential to the anti-adalimumab clone. For each anti-adalimumab clone, inhibition of binding to adalimumab was assessed for all adalimumab variants. A schematic overview of the assay is depicted in Fig. 3B, and a representative example of the results is shown for clone 2.8 in Fig. 3C. Although several adalimumab variants inhibit binding of biotinylated adalimumab similarly strongly as does adalimumab, other variants do not, indicating that these particular point mutations unfavorably affect the binding of anti-adalimumab 2.8 to adalimumab. These experiments were repeated for all anti-adalimumab clones, and a range of different inhibition profiles was obtained (Fig. 3D). In other words, the precise way in which each clone binds to adalimumab differs, effectively ruling out the possibility of a single dominant B-cell epitope on adalimumab. Interestingly, inhibition profiles of sera from four RA patients with an anti-adalimumab response are similar and appear to represent an “average” of the panel of anti-adalimumab clones, suggesting that similar epitopes are recognized by different individuals and that the panel of clones is representative for the overall anti-adalimumab response.
FIGURE 3.

Binding of monoclonal anti-adalimumab antibodies to single-point mutants of adalimumab. A, sequences of adalimumab VH and VL. CDRs are highlighted in different shades of green; amino acids that deviate from germ line V and J segments are underlined. B and C, representative inhibition experiment in which binding of biotinylated adalimumab to adalimumab-captured ADA (clone 2.8; 0.2–20 ng/ml) is inhibited by Fab fragments of single-point mutants of adalimumab (100 ng/ml). The positions of the single-point mutations are also indicated in A with carets. Whenever binding was observed, the point mutation was negatively affecting the binding of 2.8 to adalimumab. D, summary of inhibition experiments (n = 3) for the different anti-adalimumab clones. Residual binding is color-coded as % relative to uninhibited at an appropriate concentration of ADA (as indicated in C by the arrow). Inhibition experiments were also carried out for four sera from RA patients that developed an anti-adalimumab response (s1–s4). Clones 1.1–1.3 are derived from patient s1.
One consequence of the different fine specificities may be that the clones form different sizes of immune complexes (21). To investigate immune complex formation, the anti-adalimumab clones were mixed 1:1 with adalimumab to form immune complexes that were analyzed by high performance size exclusion chromatography (Fig. 4). The clones were found to differ in their propensity to form dimeric or tetrameric/multimeric complexes, additionally demonstrating that the clones differ in their precise mode of binding. Furthermore, it demonstrates that larger immune complexes may still be formed if anti-adalimumab antibodies develop. These findings complement a previous study where only small immune complexes could be detected in the sera of patients treated with adalimumab (8), and the findings suggest that if larger immune complexes are formed in vivo, they will probably be rapidly cleared.
FIGURE 4.

Formation of adalimumab-anti-adalimumab immune complexes. A, adalimumab and anti-adalimumab 2.10 were combined 1:1 at 50 μg/ml in PBS and analyzed by high performance size exclusion chromatography. Dimeric and multimeric complexes can be distinguished. B, contribution of dimeric complexes for the different anti-adalimumab clones. Means and S.E. of three experiments are shown.
DISCUSSION
Previously, we have shown that in ADA-positive patients all antibodies are directed against a small region on adalimumab, because the polyclonal antibody response in the sera of patients could be inhibited by a single monoclonal antibody against adalimumab. Here, we demonstrate that this restriction is the result of the presence of multiple B-cell epitopes on adalimumab. Furthermore, the results also suggest that multiple B-cell clones are involved in the anti-adalimumab response.
The sequences of the monoclonal antibodies are the result of different V(D)J rearrangements, showing that they derived from different precursor B-cells. This suggests that the antibody response against adalimumab results from the outgrowth of multiple B-cell clones all recognizing overlapping regions on the adalimumab molecule. From our data it is impossible to extract the number of different B-cells involved in the immune response against adalimumab, due to the limited number of sequences obtained. For other immune responses, such as the antibody response against tetanus toxoid or citrullinated autoantigens, it has been described that as many as 100 different B-cell clones can be involved (22, 23), but there also exist antibody responses such as anti-RhD that compose a more restricted repertoire (24).
The amino acid sequences of the variable domains of adalimumab are close to germ line, which suggests that only a limited number of potential epitopes are present on adalimumab. However, many patients develop anti-adalimumab antibodies. The experiments in this study strongly suggest that there are still multiple epitopes on adalimumab that can be recognized by the patients' immune system, which may in part explain the relatively high immunogenic potential of adalimumab. One might speculate about the possibilities to reduce the immunogenicity of adalimumab by eliminating one or more of these determinants; however, a more detailed analysis would be required to more precisely map these determinants. Furthermore, it may prove impossible to remove determinants without impairing TNF binding or introducing new immunogenic epitopes. The functional consequences of the different fine specificities vary. Because antibodies are relatively large molecules, they can inhibit binding of TNF even if they bind to a site that only partially overlaps with the TNF binding region. A large part of the unique determinants on adalimumab is located in or near the CDRs and spatially in close proximity. Therefore, it is not surprising that most if not all anti-adalimumab antibodies are neutralizing. However, in terms of immune complex formation, we did observe substantial differences between the different clones. Although side effects of adalimumab are relatively rare, it would be of interest to examine the anti-adalimumab repertoire of patients that do experience potential rare side effects such as a lupus-like syndrome. It may be that in these patients this repertoire is skewed in such a way that larger immune complexes are formed more easily.
The affinities of the anti-adalimumab clones were high on average, with a median Kd of 22 pm. This is in line with the high average number of mutations and the high R/S ratios in the CDRs, and it fits with a T-cell-dependent, antigen-driven antibody response. Furthermore, previous data from our group have shown that the immune response against adalimumab mainly consists of IgG1 and IgG4 antibodies (11), which is supported by the present data, because all, except one, monoclonal ADA found were originally identified as IgG1 and IgG4 antibodies. The IgG4 clones tended to have a higher affinity compared with the IgG1 clones, but the difference was not significant. We cannot fully exclude the possibility that our screening methods have introduced a bias toward high affinities. However, the low affinity clone 2.4 can still be detected in the bridging ELISA that was used for the screening in case the supernatants would contain ∼200 ng/ml, a level of antibody production that is easily reached in the culture system applied for the screening.3
In summary, here we present the analysis of 16 human monoclonal antibodies against adalimumab all obtained from different precursor B-cells. The results show that the anti-adalimumab response is clonally diverse and involves multiple adalimumab epitopes, all at least partially overlapping with the TNF binding region.
Acknowledgments
We thank Eric Mul and Floris van Alphen for their help with the FACS sorting, and Gestur Vidarsson for sharing expertise on the different cloning steps.
This article was selected as a Paper of the Week.
P. A. van Schouwenburg, L. Lighaam, and T. Rispens, unpublished observations.
- RA
- rheumatoid arthritis
- CDR
- complementarity determining region
- ADA
- anti-drug antibody
- R
- replacement
- S
- silent
- VH
- variable heavy chain
- VL
- variable light chain.
REFERENCES
- 1. Bartelds G. M., Krieckaert C. L., Nurmohamed M. T., van Schouwenburg P. A., Lems W. F., Twisk J. W., Dijkmans B. A., Aarden L., Wolbink G. J. (2011) Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA 305, 1460–1468 [DOI] [PubMed] [Google Scholar]
- 2. Wolbink G. J., Vis M., Lems W., Voskuyl A. E., de Groot E., Nurmohamed M. T., Stapel S., Tak P. P., Aarden L., Dijkmans B. (2006) Development of antiinfliximab antibodies and relationship to clinical response in patients with rheumatoid arthritis. Arthritis Rheum. 54, 711–715 [DOI] [PubMed] [Google Scholar]
- 3. Radstake T. R., Svenson M., Eijsbouts A. M., van den Hoogen F. H., Enevold C., van Riel P. L., Bendtzen K. (2009) Formation of antibodies against infliximab and adalimumab strongly correlates with functional drug levels and clinical responses in rheumatoid arthritis. Ann. Rheum. Dis. 68, 1739–1745 [DOI] [PubMed] [Google Scholar]
- 4. Weinblatt M. E., Keystone E., C., Furst D. E., Moreland L. W., Weisman M. H., Birbara C. A., Teoh L. A., Fischkoff S. A., Chartash E. K. (2003) Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate, the ARMADA trial. Arthritis Rheum. 48, 35–45 [DOI] [PubMed] [Google Scholar]
- 5. Pascual-Salcedo D., Plasencia C., Ramiro S., Nuño L., Bonilla G., Nagore D., Ruiz Del Agua A., Martinez A., Aarden L., Martín-Mola E., Balsa A. (2011) Influence of immunogenicity on the efficacy of long-term treatment with infliximab in rheumatoid arthritis. Rheumatology 50, 1445–1452 [DOI] [PubMed] [Google Scholar]
- 6. Bendtzen K., Geborek P, Svenson M., Larsson L., Kapetanovic M. C., Saxne T. (2006) Individualized monitoring of drug bioavailability and immunogenicity in rheumatoid arthritis patients treated with the tumor necrosis factor α inhibitor infliximab. Arthritis Rheum. 54, 3782–3789 [DOI] [PubMed] [Google Scholar]
- 7. Svenson M., Geborek P., Saxne T., Bendtzen K. (2007) Monitoring patients treated with anti-TNFα biopharmaceuticals, assessing serum infliximab and anti-infliximab antibodies. Rheumatology 46, 1828–1834 [DOI] [PubMed] [Google Scholar]
- 8. van Schouwenburg P. A., van de Stadt L. A., de Jong R. N., van Buren E. E., Kruithof S., de Groot E., Hart M., van Ham S. M., Rispens T., Aarden L., Wolbink G. J., Wouters D. (2013) Adalimumab elicits a restricted anti-idiotypic antibody response in autoimmune patients resulting in functional neutralisation. Ann. Rheum. Dis. 72, 104–109 [DOI] [PubMed] [Google Scholar]
- 9. van Schie K. A., Hart M. H., de Groot E. R., Kruithof S., Aarden L. A., Wolbink G. J., Rispens T. (2014) The antibody response against human and chimeric anti-TNF therapeutic antibodies primarily targets the TNF binding region. Ann. Rheum. Dis. 10.1136/annrheumdis-2014-206237 [DOI] [PubMed] [Google Scholar]
- 10. de Vries M. K., Brouwer E., van der Horst-Bruinsma I. E., Spoorenberg A., van Denderen J. C., Jamnitski A., Nurmohamed M. T., Dijkmans B. A., Aarden L. A., Wolbink G. J. (2009) Decreased clinical response to adalimumab in ankylosing spondylitis is associated with antibody formation. Ann. Rheum. Dis. 68, 1787–1788 [DOI] [PubMed] [Google Scholar]
- 11. van Schouwenburg P A., Krieckaert C. L., Nurmohamed M., Hart M., Rispens T., Aarden L., Wouters D., Wolbink G. J. (2012) IgG4 production against adalimumab during long term treatment of RA patients. J. Clin. Immunol. 32, 1000–1006 [DOI] [PubMed] [Google Scholar]
- 12. de Jong R. N., Daniëls M. A., Kaptein R., Folkers G. E. (2006) Enzyme free cloning for high throughput gene cloning and expression. J. Struct. Funct. Genomics 7, 109–118 [DOI] [PubMed] [Google Scholar]
- 13. Souto-Carneiro M. M., Longo N. S., Russ D. E., Sun H. W., Lipsky P. E. (2004) Characterization of the human Ig heavy chain antigen binding complementarity determining region 3 using a newly developed software algorithm, JOINSOLVER. J. Immunol. 172, 6790–6802 [DOI] [PubMed] [Google Scholar]
- 14. van Schouwenburg P. A., Bartelds G. M., Hart M. H., Aarden L., Wolbink G. J., Wouters D. (2010) A novel method for the detection of antibodies to adalimumab in the presence of drug reveals “hidden” immunogenicity in rheumatoid arthritis patients. J. Immunol. Methods 362, 82–88 [DOI] [PubMed] [Google Scholar]
- 15. Rispens T., Ooijevaar-de Heer P., Derksen N. I., Wolbink G., van Schouwenburg P. A., Kruithof S., Aalberse R. C. (2013) Nanomolar to sub-picomolar affinity measurements of antibody-antigen interactions and protein multimerizations, fluorescence-assisted high-performance liquid chromatography. Anal. Biochem. 437, 118–122 [DOI] [PubMed] [Google Scholar]
- 16. Fontijn R., Hop C., Brinkman H. J., Slater R., Westerveld A., van Mourik J. A., Pannekoek H. (1995) Maintenance of vascular endothelial cell-specific properties after immortalization with an amphitropic replication-deficient retrovirus containing human papilloma virus 16 E6/E7 DNA. Exp. Cell Res. 216, 199–207 [DOI] [PubMed] [Google Scholar]
- 17. Hack C. E., Hart M., van Schijndel R. J., Eerenberg A. J., Nuijens J. H., Thijs L. G., Aarden L. A. (1992) Interleukin-8 in sepsis: relation to shock and inflammatory mediators. Infect. Immun. 60, 2835–2842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Votsmeier C., Plittersdorf H., Hesse O., Scheidig A., Strerath M., Gritzan U., Pellengahr K., Scholz P., Eicker A., Myszka D., Coco W. M., Haupts U. (2012) Femtomolar Fab binding affinities to a protein target by alternative CDR residue co-optimization strategies without phage or cell surface display. MAbs 4, 341–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Insel R. A, Varade W. S. (1994) Bias in somatic hypermutation of human VH genes. Int. Immunol. 6, 1437–1443 [DOI] [PubMed] [Google Scholar]
- 20. Uduman M., Yaari G., Hershberg U., Stern J. A., Shlomchik M. J., Kleinstein S. H. (2011) Detecting selection in immunoglobulin sequences. Nucleic Acids Res. 39, W499–W504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roux K. H., Strelets L., Michaelsen T. E. (1997) Flexibility of human IgG subclasses. J. Immunol. 159, 3372–3382 [PubMed] [Google Scholar]
- 22. Poulsen T. R., Meijer P. J., Jensen A., Nielsen L. S., Andersen P. S. (2007) Kinetic, affinity, and diversity limits of human polyclonal antibody responses against tetanus toxoid. J. Immunol. 179, 3841–3850 [DOI] [PubMed] [Google Scholar]
- 23. Amara K., Steen J., Murray F., Morbach H., Fernandez-Rodriguez B. M., Joshua V., Engström M., Snir O., Israelsson L., Catrina A. I., Wardemann H., Corti D., Meffre E., Klareskog L., Malmström V. (2013) Monoclonal IgG antibodies generated from joint-derived B cells of RA patients have a strong bias toward citrullinated autoantigen recognition. J. Exp. Med. 210, 445–455 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24. Dohmen S. E., Verhagen O. J., Muit J., Ligthart P. C., van der Schoot C. E. (2006) The restricted use of IGHV3 superspecies genes in anti-Rh is not limited to hyperimmunized anti-D donors. Transfusion 46, 2162–2168 [DOI] [PubMed] [Google Scholar]
- 25. Rispens T., Davies A. M., Ooijevaar-de Heer P., Absalah S., Bende O., Sutton B. J., Vidarsson G., Aalberse R. C. (2014) Dynamics of inter-heavy chain interactions in human immunoglobulin G (IgG) subclasses studied by kinetic Fab arm exchange. J. Biol. Chem. 289, 6098–6109 [DOI] [PMC free article] [PubMed] [Google Scholar]