

Background: Aminopeptidase N (APN) mediates tumor cell motility and is a receptor for tumor-homing peptides containing NGR motifs.
Results: Using crystallographic and biochemical methods, we investigated how APN interacts with NGR motifs in tumor-homing peptides and extracellular matrix proteins.
Conclusion: APN forms specific and stable interactions with these NGR motifs, accounting for APN's functions.
Significance: This study facilitates development of APN-targeting cancer therapies.
Keywords: Cancer Biology, Cancer Therapy, Cell Motility, Crystallography, Extracellular Matrix Protein, Tumor Marker, Tumor Metastases
Abstract
Tumor cell surface aminopeptidase N (APN or CD13) has two puzzling functions unrelated to its enzymatic activity: mediating tumor cell motility and serving as a receptor for tumor-homing peptides (peptides that bring anti-cancer drugs to tumor cells). To investigate APN-based tumor-homing therapy, we determined the crystal structure of APN complexed with a tumor-homing peptide containing a representative Asn-Gly-Arg (NGR) motif. The tumor-homing peptide binds to the APN enzymatic active site, but it resists APN degradation due to a distorted scissile peptide bond. To explore APN-based tumor cell motility, we examined the interactions between APN and extracellular matrix (ECM) proteins. APN binds to, but does not degrade, NGR motifs in ECM proteins that share similar conformations with the NGR motif in the APN-bound tumor-homing peptide. Therefore, APN-based tumor cell motility and tumor-homing therapy rely on a unified mechanism in which both functions are driven by the specific and stable interactions between APN and the NGR motifs in ECM proteins and tumor-homing peptides. This study further implicates APN as an integrin-like molecule that functions broadly in cell motility and adhesion by interacting with its signature NGR motifs in the extracellular environment.
Introduction
Mammalian aminopeptidase N (APN),3 also called CD13, is a tumor marker that is overly expressed on the cell surface of almost all major tumor forms, including skin, ovary, lung, stomach, colon, kidney, bone, prostate, renal, pancreatic, thyroid, and breast cancers (1). As a zinc-dependent aminopeptidase, APN cleaves the N-terminal neutral residue off physiological peptides and functions ubiquitously in various peptide metabolism pathways (2). Consequently, tumor cell surface APN is required for tumor growth and development by cleaving and activating angiogenic peptides that are essential for tumor angiogenesis (3). However, tumor cell surface APN also mediates tumor cell motility and serves as a receptor for tumor-homing peptides that guide anti-cancer drugs to tumor cells; these functions appear to be unrelated to each other or to APN's enzymatic activity (3–8). It is puzzling how APN, a zinc-dependent aminopeptidase in essence, can function as a cell motility molecule and as a tumor-homing receptor. Solving these puzzles can lead to a better understanding of cancer biology and more effective cancer treatment.
APN is the most extensively studied member of the large M1 family of zinc-dependent aminopeptidases (2). APN is a cell surface-anchored ectoenzyme, with a small N-terminal intracellular domain, a single-pass transmembrane anchor, a small extracellular stalk, and a large C-terminal ectodomain (Fig. 1A). We recently determined the crystal structures of porcine APN (pAPN) ectodomain alone and in complex with a cleaved polyalanine peptide as well as the crystal structure of a catalytically inactive mutant of pAPN ectodomain in complex with an uncleaved polyalanine peptide (9). The pAPN ectodomain has a seahorse-like shape, with four distinct domains: head, side, body, and tail. pAPN forms a homodimer through interactions between the head domains. The zinc-dependent catalytic site is located in a cavity surrounded by the head, side, and body domains. The catalytic site contains a tightly chelated zinc, a zinc-activated catalytic water that attacks and breaks the scissile peptide bond of peptide substrates, and a number of pAPN residues that either participate directly in catalysis or anchor peptide substrates in position for catalysis. In addition to binding peptides, pAPN also binds the exposed N terminus of proteins by undergoing a closed-to-open conformational change, which opens up its active site cavity. These results have elucidated the enzymatic activity of APN, but the puzzling roles of APN in tumor cell motility and tumor-homing therapy are still unclear.
FIGURE 1.

Crystal structure of porcine APN in complex with a tumor-homing peptide CNGRCG. A, overall structure of the pAPN-CNGRCG complex. pAPN contains an ectodomain, a stalk, a transmembrane anchor (TM), and an intracellular tail (IC). The ectodomain contains four domains: head (cyan), side (brown), body (magenta), and tail (yellow). CNGRCG is shown in green as balls and sticks. Zinc is shown as a blue ball. Only one monomer of the dimeric pAPN is shown. B, electron density map of CNGRCG. The electron density map corresponds to Fo − Fc omit map calculated in the absence of CNGRCG and contoured at 2.2 σ. C, another view of the pAPN-CNGRCG complex. The view of the complex is obtained by rotating the view in A first by 90° along a vertical axis and then by 45° along a horizontal axis, in such a way that the active site cavity of pAPN is facing the reader. D, enlarged view of CNGRCG in the active site of pAPN.
APN plays a critical role in tumor cell migration and metastasis. It was previously shown that increased expression of APN on tumor cell surfaces greatly enhanced the migratory capacity of these tumor cells (10, 11). Moreover, decreasing the expression of APN on tumor cell surfaces or use of anti-APN antibodies or APN inhibitors to treat tumor cells blocked tumor cell migration and metastasis (8, 12, 13). It was suggested that APN degrades extracellular matrix (ECM) proteins for tumor cell migration and metastasis (14, 15). However, tumor cells overexpressing enzymatically inactive APN also demonstrate enhanced migration and metastasis (13, 16). Thus, APN-mediated tumor cell motility and metastasis reside on some unknown activity of APN that is independent from its zinc-dependent aminopeptidase activity. In addition to mediating tumor cell motility, APN also functions broadly in other cell motility and adhesion processes such as immune cell chemotaxis, sperm motility, and monocytic cell adhesion (17–23). These functions of APN are reminiscent of those of integrins, which mediate cell motility and adhesion via specifically interacting with a three-residue motif, arginine-glycine-aspartate (RGD), in ECM proteins or on the surface of other cells (24–28). Whereas the structures and functions of integrins have been extensively studied and well characterized, little is known about how APN mediates tumor cell motility or other cell motility and adhesion processes.
Tumor-homing therapy, also called targeted drug therapy, has recently emerged as one of the most promising approaches for cancer treatment (6, 29, 30). The concept of tumor-homing therapy is to link anti-tumor drugs to a tumor-homing peptide; the latter specifically recognizes a receptor that is uniquely or overly expressed on tumor cell surfaces and thereby actively guides the drugs to tumor cell surfaces. APN and integrins are two of the most promising tumor-homing receptors; APN- and integrin-based tumor-homing therapies are the only ones that are currently in clinical trials (29, 31–35). Consistent with serving as receptors for RGD motifs in ECM proteins, integrins are also receptors for tumor-homing peptides containing an RGD motif (6, 29, 30). The detailed interactions between integrins and the tumor-homing RGD peptide have been delineated by structural studies (24–26). However, APN is a zinc-dependent aminopeptidase that is not known to recognize any specific peptide motifs, and thus it was totally unexpected when phage display identified APN as the receptor for tumor-homing peptides containing an Asn-Gly-Arg (NGR) motif (4–6). Nevertheless, the APN-based tumor-homing NGR peptides have been rapidly developed and are now in phase I and II clinical trials to treat advanced solid tumors (31–33). Despite the great promises that APN-based tumor-homing therapy holds, the basic knowledge about this therapy is still lacking. It is not known where the binding site in APN is for the tumor-homing NGR peptides or what the detailed interactions are between APN and these peptides. The above knowledge would be essential for rational design and development of tumor-homing peptides with improved affinity and specificity for APN.
Here, we investigated the interactions between APN and ECM proteins and between APN and a tumor-homing NGR peptide. Using x-ray crystallographic and biochemical methods, we have identified a unified mechanism governing both APN-based tumor cell motility and tumor-homing therapy. These results not only solve the puzzles surrounding these APN-related functions, but also lay the foundation for future development of better APN-based cancer treatments. Furthermore, our study establishes APN as an integrin-like cell motility and adhesion molecule that should be investigated in depth and exploited therapeutically.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Porcine APN (pAPN) (GenBankTM accession number CAA82641.1) ectodomain (residues 62–963) was expressed and purified as described previously (9). Briefly, pAPN containing an N-terminal honeybee melittin signal peptide and a C-terminal His6 tag was cloned into pFastbac1 vector, expressed in Sf9 insect cells using Bac-to-Bac expression system (Invitrogen), and secreted to cell culture medium. The His6-tagged pAPN was harvested and purified sequentially on HiTrap nickel-chelating HP column and Superdex 200 gel filtration column (GE Healthcare). Fc-tagged pAPN was obtained by fusion of the human IgG4 Fc region to the C terminus of the pAPN ectodomain and was purified sequentially on HiTrap Protein G HP column and Superdex 200 gel filtration column. pAPN containing the E384Q mutation was constructed using site-directed mutagenesis, and it was expressed and purified in the same way as the wild type pAPN.
The human fibronectin gene that encodes residues 1–275 (including the free N terminus and domains 1–5) (GenBankTM accession number BAD52437.1) was synthesized commercially (Genscript). Constructs that encode different parts of the above fibronectin region along with a C-terminal His6 tag were cloned into vector pET42b(+) and expressed in Escherichia coli BL21 cells (Invitrogen) through induction with isopropyl β-d-1-thiogalactopyranoside (Sigma). Different fibronectin domains with a C-terminal His6 tag were purified subsequently on a HiTrap nickel-chelating HP column and Superdex 200 gel filtration column.
CNGRCG peptide and its mutants were fused to a C-terminal GST tag using a SGSGSGSG peptide linker. Constructs were cloned into vector pET42b(+) and expressed as described previously for fibronectin domains. Recombinant proteins were purified on GSTrap 4B column (GE Healthcare) and Superdex 200 gel filtration column sequentially. All the above recombinant proteins used in this study were stored in buffer containing 20 mm Tris-HCl, pH 7.4, and 200 mm NaCl.
Crystallization and Structural Determination
Crystallization of His6-tagged pAPN ectodomain was carried out as described previously (9). Briefly, crystallization was set up in sitting drops at 4 °C by adding 2 μl of protein solution to 2 μl of well solution containing 18% (v/v) PEG3350, 200 mm Li2SO4, and 100 mm HEPES, pH 7.2. Crystals appeared in 2 days and were allowed to grow for another 2 weeks. The crystals were then transferred to buffer containing 5 mm CNGRCG (AnaSpec), 20% (v/v) ethylene glycol, 25% (v/v) PEG3350, 200 mm Li2SO4, and 100 mm HEPES, pH 7.2. After 2 days, the crystals were flash-frozen in liquid nitrogen. X-ray diffraction data were collected at ALS beamline 4.2.2 and processed using software HKL2000 (36). The structure of the pAPN-CNGRCG complex was determined by molecular replacement using the pAPN structure as the search template (PDB code 4FKE). The Fo − Fc omit electron density map was calculated in the absence of CNGRCG and showed strong density for CNGRCG. Based on the Fo − Fc omit electron density map, the model of CNGRCG was built. The structure of the complex was subsequently refined at 1.95 Å resolution using software CNS and RefMac (37, 38).
APN Catalysis Inhibition Assay
The APN catalysis inhibition assay was carried out as described previously (39). Briefly, 2 nm His6-tagged pAPN and 0.5 mm l-alanine-p-nitroanilide (Sigma) were incubated in 100 μl of 60 mm KH2PO4, pH 7.2, in the presence of gradient concentrations of CNGRCG or GNGRG peptide (Genscript). The reactions were incubated at 37 °C for 30 min. Formation of product p-nitroanilide was measured every 10 min using an absorbance plate reader (BioTek) at 405 nm. The IC50 value was defined as the concentration of each peptide that led to 50% of maximal pAPN catalytic activity. Ki values for each peptide were calculated from the IC50 value using the Cheng-Prusoff equation, Ki = IC50/(1 + [S]/Km), in which Km was determined previously (9).
Wound Healing Assay
Tumor cell wound healing assay was performed as described previously (40). Briefly, 8 × 104 human fibrosarcoma HT-1080 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Invitrogen) and seeded onto 24-well plates. After serum starvation for 16 h, the scratch wounds were created on the confluent monolayers with a pipette tip. Each well was washed twice with serum-free media to remove cell debris. Then the cells in triplicates were treated with 10 μg/ml anti-APN antibody WM15 (Pharmingen) or 10 μg/ml anti-integrin αV/β3 antibody (Santa Cruz Biotechnology) for 8 h. Digital images at different time points were captured using an inverted contrasting microscope (Leica Microsystems). The wound healing effect was calculated as the distance of cells migrating into cell-free spaces compared with the initial wound. The relative migration was standardized against the control group without any antibody treatment.
Transwell Migration Assay
Transwell migration assay was conducted as described previously (41). Briefly, Transwell inserts (Corning Glass) with 6.5-mm diameter and 8-μm pore size were coated with 10 μg/cm2 fibronectin (Sigma) and air-dried. HT-1080 cells were seeded at a density of 8 × 104 and treated with 10 μg/ml anti-APN antibody WM15 (Pharmingen). 4 h after treatment, cells in the upper compartment were scraped off with a cotton swab. Cells passing through the membrane were fixed with 5% glutaraldehyde (Sigma) and stained with 0.5% crystal violet (Sigma). For quantification, cells that had migrated to the lower surface were counted under a microscope in three fields for triplicate experiments. The relative migration was standardized against the control group without antibody treatment.
Dot-blot Hybridization Assay
Dot-blot hybridization assay was carried out as described previously (42). Briefly, 10 μg of fibronectin, laminin, or type IV collagen (Sigma) was each dotted onto a nitrocellulose membrane. The membranes were dried completely and blocked with BSA at 4 °C overnight. The membranes were then incubated at 37 °C for 2 h with 50 μg/ml His6-tagged pAPN, which had been preincubated alone or with 20 μg/ml fibronectin domains 4–5, anti-APN antibody WM15 (Pharmingen), or anti-integrin αV/β3 antibody (Santa Cruz Biotechnology). The membranes were then washed five times with phosphate-buffered saline with Tween 20 (PBST), incubated with anti-His6 mouse monoclonal IgG1 antibody (Santa Cruz Biotechnology) at 37 °C for 2 h, washed five times with buffer PBST again, incubated with HRP-conjugated goat anti-mouse IgG antibody (Santa Cruz Biotechnology) at 37 °C for 1 h, and washed five times with buffer PBST. Finally, the bound proteins were detected using ECL Plus (GE Healthcare).
ELISA
Binding of pAPN to different ECM proteins was carried out using ELISA as described previously (43). Briefly, ELISA plates were coated overnight at 4 °C with 10 μg/ml fibronectin, laminin, type IV collagen, or PBS. After blocking at 37 °C for 2 h, the plates were incubated at 37 °C for 2 h with 50 μg/ml His6-tagged pAPN, which had been preincubated alone or with 20 μg/ml fibronectin domains 4–5 or anti-APN antibody WM15 (Pharmingen). The plate was then treated the same way as in the dot-blot hybridization assay. Finally, the bound proteins were detected using HRP substrate (G-Bioscience), and the color reaction was quantified using an absorbance plate reader (BioTek) at 630 nm.
AlphaScreen Protein-Protein Binding Assay
Binding of His6-tagged pAPN to GST-tagged proteins or peptides (e.g. fibronectin domains 4–5, NGR peptide, or its mutants) was carried out using AlphaScreen assay as described previously (43). Briefly, each of the GST-tagged proteins or peptides at a final concentration of 30 nm was mixed with His6-tagged pAPN also at a final concentration of 30 nm in ½ AreaPlate (PerkinElmer Life Sciences) for 1 h at room temperature. AlphaScreen anti-GST acceptor beads and nickel chelate donor beads (PerkinElmer Life Sciences) were added to the mixture at final concentrations of 10 μg/ml each. The mixture was incubated at room temperature for 1 h and protected from light. The assay plates were read in an EnSpire plate reader (PerkinElmer Life Sciences).
Binding of Fc-tagged pAPN to His6-tagged fibronectin domains was carried out in the same way as above, except that fibronectin domains had a final concentration of 9 nm and that AlphaScreen protein A acceptor beads (PerkinElmer Life Sciences) and AlphaScreen nickel chelate donor beads were added to the mixture. To block the binding interaction between pAPN and fibronectin domains 4–5, bestatin, methionine, or CNGRCG peptide at various concentrations was incubated with the mixture for 1 h before donor and acceptor beads were added.
SDS-PAGE
Fibronectin domains 4–5 at a final concentration of 0.5 μg/μl was incubated alone or with pAPN at a final concentration of 0.5 μg/μl at 37 °C for 2 h. Subsequently, the mixture was subjected to SDS-PAGE. The gel was stained by Brilliant Blue G (Sigma).
Mass Spectrometry
Fibronectin domains 4–5 at a final concentration of 100 μm was incubated alone or with pAPN at a final concentration of 10 μm at 37 °C for 2 h. Subsequently, the mixture was subjected to mass spectrometry at the Center for Mass Spectrometry and Proteomics at the University of Minnesota (Minneapolis, MN).
RESULTS
Crystal Structure of Porcine APN in Complex with a Tumor-homing NGR Peptide
To investigate the structural basis for the interactions between APN and tumor-homing NGR peptides, we determined the crystal structure of porcine APN (pAPN) in complex with peptide CNGRCG. CNGRCG was chosen for the study because it is the most commonly used tumor-homing NGR peptide and targets tumor cells more effectively than other NGR peptides such as GNGRG (6, 44). pAPN was chosen for this study because it was previously crystallized in a closed and catalytically active conformation under physiologically relevant pH, and the crystals diffracted to high resolution (∼2.0 Å) (9). In addition, pAPN and human APN share high sequence homology, with catalytic residues 100% conserved between the two proteins (9). To crystallize the pAPN-CNGRCG complex, pAPN was expressed, purified, and crystallized as described previously, and CNGRCG was soaked into pAPN crystals as described previously for other APN-binding ligands (9). The structure of the complex was determined by molecular replacement using the unliganded pAPN structure as the search template (Fig. 1A). The Fo − Fc omit electron density map calculated in the absence of CNGRCG showed strong density for this peptide, allowing the model to be built (Fig. 1B). Subsequently, the structure of the complex was refined at 1.95 Å resolution (Table 1).
TABLE 1.
Data collection and refinement statistics
| pAPN-CNGRCG complex | |
|---|---|
| Data Collection | |
| Space group | C2 |
| Cell dimensions | |
| a, b, c (Å) | 260.3, 62.9, 82.0 |
| α, β, γ (°) | 90, 100.6, 90 |
| Resolution (Å) | 50-1.92 (1.96-1.92)a |
| Rsym or Rmerge | 0.079 (0.615) |
| I/σI | 20.7 (1.7) |
| Completeness (%) | 97.9 (97.0) |
| Redundancy | 3.8 (3.9) |
| Refinement | |
| Resolution (Å) | 47.73-1.95 |
| No. of reflections | 88,780 |
| Rwork/Rfree | 0.140/0.189 |
| No. of atoms | 8498 |
| Protein | 7259 |
| CNGRCG | 40 |
| B-factors (Å2) | 48.9 |
| Protein | 47.3 |
| CNGRCG | 44.0 |
| R.M.S.b derivations | |
| Bond lengths (Å) | 0.012 |
| Bond angles (°) | 1.614 |
| Ramachandran plot | |
| Favored (%) | 97 |
| Allowed (%) | 2.6 |
| Disallowed (%) | 0.4 |
a Values in parentheses are for highest resolution shell.
b R.M.S. is root mean square.
The CNGRCG tumor-homing peptide binds to the catalytic site of pAPN (Fig. 1, C and D). The two cysteines in CNGRCG form a disulfide bond (Fig. 1B). Consequently, the NGR region forms a short loop with a sharp turn, facilitated by the presence of a flexible glycine in the middle of the loop. The NGR loop makes sequence-specific interactions with the pAPN active site through the side chains of asparagine and arginine (Fig. 2, A and B). Specifically, the side chain of asparagine in the NGR loop is parallel to the side chain of pAPN residue His-383; it also forms five water-mediated hydrogen bonds with the side chains of pAPN residues Glu-413, Ser-410, and Glu-384, and one water-mediated interaction with the carbonyl oxygen of pAPN residue Val-380 (Fig. 2A). Moreover, the side chain of the arginine in the NGR loop forms a hydrophobic interaction with Cα of pAPN residue Ala-346, one hydrogen bond with the side chain of pAPN residue Asn-345, two hydrogen bonds with the carbonyl oxygen of pAPN residue Asn-345, and a water-mediated hydrogen bond with the side chain of pAPN residue Arg-358 (Fig. 2B). Although glycine in the NGR loop has no direct contact with pAPN, its conformational flexibility is critical for the formation of the NGR loop (Fig. 2C). CNGRCG also makes sequence nonspecific interactions with the pAPN active site through the main chain groups, as described previously for pAPN-bound polyalanine peptide (Fig. 2D) (9). Because all of the APN residues involved in binding CNGRCG are completely conserved between human and porcine APNs (Fig. 2, A, B, and D) (9), the CNGRCG peptide is expected to interact with both APNs in the same way.
FIGURE 2.
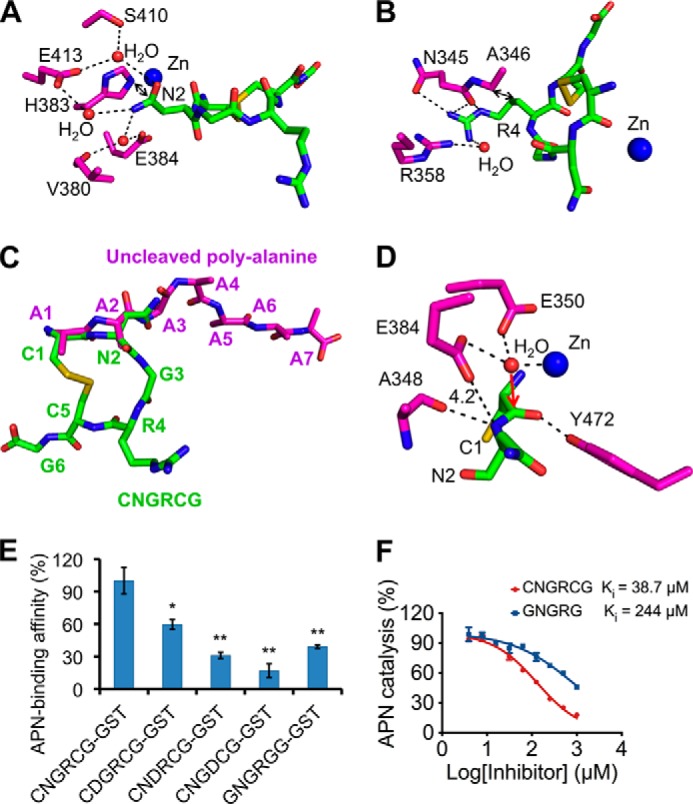
Detailed interactions between pAPN and CNGRCG. A, detailed interactions between pAPN and the side chain of the asparagine in CNGRCG. pAPN residues are in magenta, and CNGRCG is in green. B, detailed interactions between pAPN and the side chain of the arginine in CNGRCG. C, comparison of the conformations of CNGRCG in the crystal of the wild type pAPN-CNGRCG complex and the uncleaved polyalanine peptide in the crystal of mutant pAPN-polyalanine complex (PDB code 4NAQ). D, active site geometry of the pAPN-CNGRCG complex. The presumable scissile peptide bond of CNGRCG has a catalytically inactive conformation, resulting in its leaving nitrogen group being too far away from the proton-transferring pAPN residue Glu-384. Red arrow indicates the potential attack of the scissile peptide bond by the catalytic water at the pAPN active site. Unit of distance is in angstroms. E, pAPN binding by CNGRCG and its mutants. CNGRCG and the mutant peptides were fused to a C-terminal GST tag. The binding affinities of these fusion proteins with pAPN were measured using AlphaScreen protein-protein binding assay. The binding affinity of GST-tagged CNGRCG with pAPN was used as the standard and taken as 100%. Error bars indicate S.E. (compared with the standard two-tailed t test; *, p < 0.05; **, p < 0.01; n = 3). F, inhibition of APN catalytic activity by CNGRCG and GNGRG peptides. The catalytic activity of pAPN on Ala-p-nitroanilide in the absence of any inhibitor was taken as 100%. Error bars indicate S.E. (n = 3).
To further understand the APN/CNGRCG interactions, we investigated the roles of both the NGR motif and the disulfide bond-fortified loop conformation in APN binding using biochemical methods. To this end, we introduced mutations into CNGRCG, and fused CNGRCG and each of the mutant peptides to a C-terminal GST tag. We then measured the binding affinity between APN and the GST-tagged peptides using AlphaScreen protein-protein binding assay. Mutations of each of the residues in the NGR motif to an aspartate significantly weakened APN binding and so did mutations of each of the two flanking cysteines in CNGRCG to a glycine (Fig. 2E). Furthermore, we evaluated the contribution of the disulfide bond in CNGRCG to APN binding using APN catalysis inhibition assay. CNGRCG inhibits APN catalysis more effectively than GNGRG (Fig. 2F), again suggesting that the loop formation in CNGRCG contributes critically to APN binding. Indeed, soaking the GNGRG peptide into pAPN crystals did not yield any clear electron density of the peptide, consistent with the weak APN binding by GNGRG. Taken together, both structural and biochemical data revealed that the interactions between APN and CNGRCG depend on the specific sequence of the NGR motif as well as the loop conformation of the peptide. These sequence-specific and conformation-dependent interactions between APN and CNGRCG explain the high affinity and specificity of APN binding to the tumor-homing peptide; they also define APN as a functional receptor for the NGR motif with a loop conformation in tumor-homing peptides or other biological settings.
The CNGRCG tumor-homing peptide resists enzymatic degradation by pAPN. Our previous study demonstrated that the crystallized pAPN is catalytically active, and when peptide substrate polyalanine was soaked into the pAPN crystals, electron density showed a broken scissile peptide bond and hence a degraded polyalanine (Fig. 3A). In contrast, when soaked into crystals of a catalytically inactive mutant of pAPN, polyalanine remained uncleaved (Fig. 3B). In this study, CNGRCG was soaked into catalytically active pAPN crystals under the same soaking condition as for polyalanine. Electron density revealed that CNGRCG remained uncleaved in the crystals (Fig. 1B), suggesting that CNGRCG is a much poorer substrate than polyalanine for APN. To investigate why CNGRCG resists APN degradation, we compared the active site geometry of the pAPN-CNGRCG complex with that of the pAPN-polyalanine complex. At the active site of the pAPN-polyalanine complex (Fig. 3C), a zinc-activated catalytic water attacks and breaks the N-terminal scissile peptide bond of the peptide substrate; simultaneously, the catalytic water transfers a proton through the pAPN residue Glu-384 to the leaving nitrogen group of the peptide substrate, which subsequently becomes the N terminus of the newly formed peptide product (9). At the active site of the pAPN-CNGRCG complex, however, the scissile peptide bond of CNGRCG deviates from the optimal geometry required for peptide bond hydrolysis due to the sharp turn of the NGR loop (Fig. 2C). The outcome is that the leaving nitrogen group of CNGRCG is too far away from Glu-384 to accept the transferring proton, leading to a disconnected proton transfer pathway and an intact CNGRCG (Fig. 2D). Consequently, CNGRCG can serve as an efficient and stable tumor-homing peptide by targeting tumor cell surface APN without being turned over immediately during the tumor-homing process.
FIGURE 3.
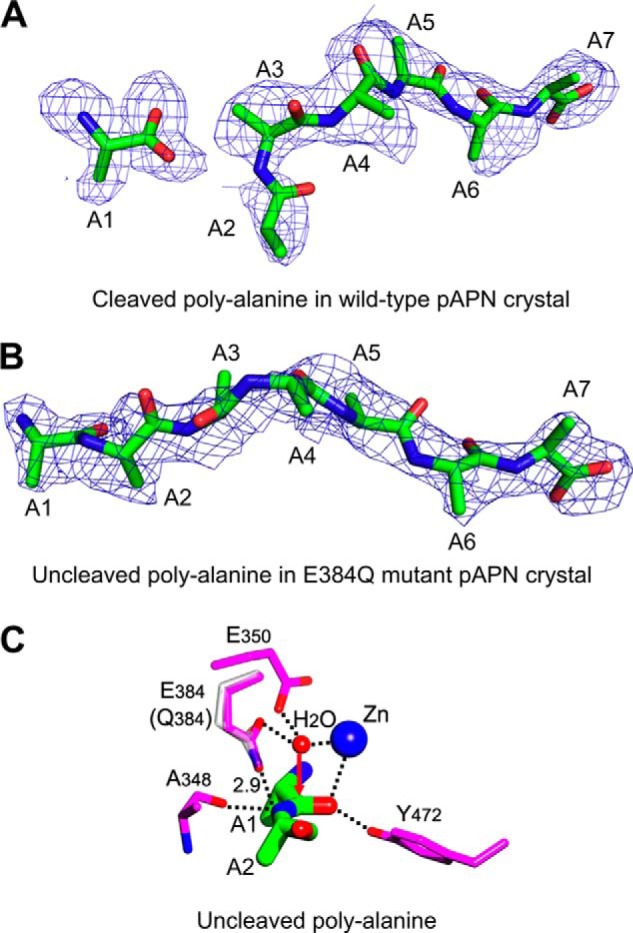
Previously published catalytic mechanism of APN (9). A, cleaved polyalanine peptide bound to wild type pAPN where the peptide was degraded in the crystal (PDB code 4NZ8). The electron density map corresponds to Fo − Fc omit map calculated in the absence of the cleaved polyalanine and contoured at 1.5 σ. B, uncleaved polyalanine peptide bound to E384Q mutant pAPN, which is catalytically inactive (PDB code 4NAQ). The electron density map corresponds to the Fo − Fc omit map calculated in the absence of the uncleaved polyalanine and contoured at 2.0 σ. C, interactions between catalytic residues of pAPN (magenta) and scissile peptide bond of polyalanine (green). Although Gln-384 (white) was introduced to pAPN to generate a catalytically incompetent enzyme for crystallographic studies (PDB code 4NAQ), Glu-384 (magenta) from wild type pAPN (PDB code 4FKE) was grafted here to illustrate the catalytic mechanism of pAPN. Red arrow indicates the potential attack of the scissile peptide bond by the catalytic water at the pAPN-active site. Zinc is shown as a blue ball and catalytic water as a red ball. Unit of distance is in angstroms.
Interactions between APN and Extracellular Matrix Proteins
The above structural study has established APN as a functional receptor for the NGR motif in tumor-homing peptides developed in vitro. Our structural finding raises the possibility that the NGR motifs may exist in vivo and that APN may interact with in vivo NGR motifs to perform its physiological functions. Indeed, it was previously observed that fibronectin, one type of the ECM proteins, contains four NGR motifs (44). In this study, we looked into the sequences of other types of ECM proteins and found that laminin contains three NGR motifs, whereas type IV collagen contains none. Then we investigated whether APN mediates tumor cell motility by interacting with ECM proteins containing NGR motifs. To this end, we examined the role of APN in tumor cell motility using both the tumor cell wound healing assay and transwell migration assay. The used cell line was HT-1080 (human fibrosarcoma). In the wound healing assay, tumor cells secrete a mixture of ECM proteins into the extracellular environment such that tumor cells can move by interacting with these secreted ECM proteins. The results showed that both anti-APN antibody and anti-integrin antibody significantly decreased tumor cell motility (Fig. 4, A and B). In the transwell migration assay, tumor cell motility was measured in a fibronectin-coated transwell chamber. The motility of HT-1080 was significantly reduced by anti-APN antibody (Fig. 4C). Both of these tumor cell motility assays demonstrated that APN plays an important role in tumor cell motility by interacting with ECM proteins (e.g. fibronectin) in the extracellular environment. Next, we analyzed the interactions between pAPN and individual ECM proteins using both dot-blot hybridization and ELISA assays (Fig. 5, A and B). pAPN specifically binds fibronectin and laminin, both of which contain NGR motifs, but not type IV collagen that does not contain any NGR motif. In addition, these interactions were significantly inhibited by anti-APN antibody (Fig. 5, A and B). These results suggest that APN specifically interacts with ECM proteins containing NGR motifs, contributing to the APN-mediated cell motility.
FIGURE 4.
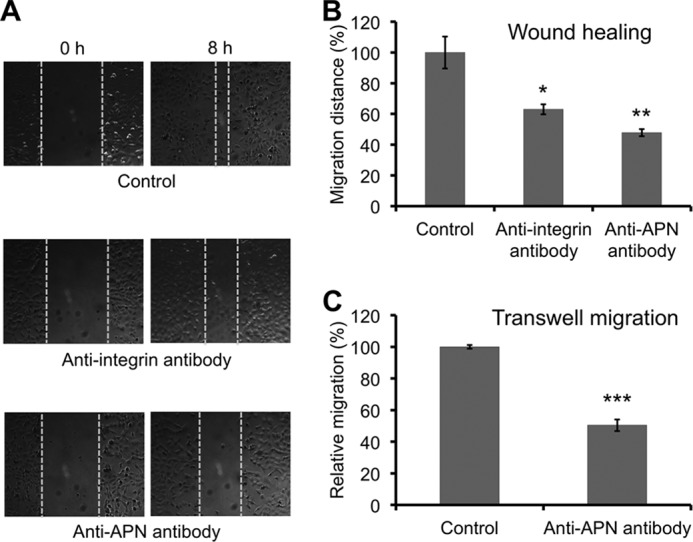
Tumor cell motility assays. A, microscopic photos showing the inhibition of HT-1080 cell motility by anti-APN or anti-integrin antibody in tumor cell wound healing assay. B, quantification of HT-1080 cell motility in wound healing assay. The migration distance in the control group without any antibody treatment was taken as 100%. C, transwell migration assay showing the inhibition of HT-1080 cell motility by anti-APN antibody. The relative migration was standardized against the control group without antibody treatment. Error bars indicate S.E. (two-tailed t test; *, p < 0.05; **, p < 0.01; ***, p < 0.001; n = 3).
FIGURE 5.
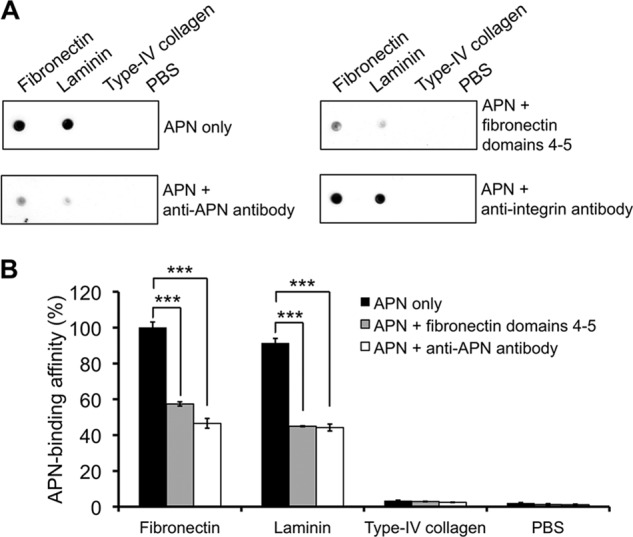
Interactions between APN and extracellular matrix proteins. Both dot blot hybridization assay (A) and ELISA (B) were performed to detect the interactions between pAPN and individual ECM proteins with or without NGR motifs. These assays also measured competitive inhibition by anti-APN antibody or fibronectin NGR domains, with anti-integrin αV/β3 antibody as the negative control. His6-tagged pAPN was detected by anti-His6 antibody. Error bars indicate S.E. (two-tailed t test; ***, p < 0.001; n = 3).
We further investigated the detailed interactions between APN and ECM proteins containing NGR motifs. First, we examined the interactions between APN and fibronectin using AlphaScreen protein-protein binding assay. Because APN is capable of interacting with the exposed N terminus of proteins, we investigated whether APN binds fibronectin at its exposed N terminus or a specific domain containing the NGR motif (i.e. NGR domain) (Fig. 6A). To this end, we expressed different parts of fibronectin with or without the exposed N terminus or the NGR domain, and analyzed their binding interactions with pAPN (Fig. 6B). Although domain 5 contains the NGR motif, it needed to be expressed together with domain 4 due to the interactions between the two domains (45). The results from the AlphaScreen assay showed that pAPN has significantly higher affinity for the NGR domain than for the exposed N terminus of fibronectin. Moreover, this NGR domain competitively blocked the interactions between pAPN and fibronectin or laminin (Fig. 5, A and B). In addition, when this NGR domain was fused to a C-terminal GST tag, the GST-tagged NGR domain has significantly higher pAPN-binding affinity than GST alone (Fig. 6C). Thus, APN primarily binds fibronectin in the NGR domain. Second, we analyzed the interactions between APN and the fibronectin NGR domain containing mutations in the NGR motif (Fig. 6D). The results from AlphaScreen assay showed that mutations in the NGR motif significantly decreased the binding affinity between pAPN and the fibronectin NGR domain. Thus, APN specifically interacts with the NGR motif in the fibronectin NGR domain. Finally, we probed the binding site of the fibronectin NGR motif in APN using three active-site inhibitors of APN, methionine, bestatin, and the CNGRCG peptide (Fig. 6E). The results from the AlphaScreen assay showed that active-site inhibitors of APN inhibited the binding of the fibronectin NGR motif to pAPN, supporting direct binding of the fibronectin NGR motif to the APN active site. Moreover, a catalytically inactive mutant pAPN containing an E384Q mutation in the catalytic site bound the fibronectin NGR domain in the same way as the wild type pAPN did, implying that the catalytic activity of APN is not a prerequisite for binding the fibronectin NGR domain (Fig. 6E). Here, the E384Q mutation at the pAPN active site did not have a significant effect on the binding of pAPN to the fibronectin NGR domain or any of the active site inhibitors due to the numerous other interactions between pAPN and its ligands (9). Collectively, these results suggest that APN specifically binds the NGR motif of ECM proteins and that the binding site for the NGR motif of ECM proteins is located at the APN active site.
FIGURE 6.

Interactions between APN and the NGR motif in fibronectin. A, schematic structure of the free N terminus and five N-terminal domains of fibronectin. The free N terminus is 18 residues long. The NGR motif is located in domain 5. B, interactions between pAPN and different fibronectin domains. AlphaScreen assay was performed. The AlphaScreen signal measured between pAPN and fibronectin domains 1–5 with the N terminus was taken as 100%. C, interactions between pAPN and GST or fibronectin domains 4–5 fused to the N terminus of GST. AlphaScreen signal measured between pAPN and GST-tagged fibronectin domains 4–5 was taken as 100%. D, interactions between pAPN and fibronectin NGR domain (domains 4–5) containing mutations in the NGR motif. AlphaScreen signal measured between pAPN and fibronectin NGR domain (domains 4–5) without any mutation was taken as 100%. E, inhibition of the interactions between pAPN (wild type or E384Q mutant) and the fibronectin NGR domain using APN active site inhibitors methionine, bestatin, or CNGRCG peptide. The AlphaScreen signal measured between wild-type pAPN and fibronectin NGR domain in the absence of any inhibitor was used as the standard and taken as 100%. Error bars indicate S.E. (compared with the standard; two-tailed t test; not significant (N.S.), p > 0.05; *, p < 0.05; **, p < 0.01; ***, p < 0.001; n = 3).
The NGR motif of fibronectin shares similar structural conformations with the NGR motif in the APN-bound tumor-homing peptide. As revealed by the NMR solution structure of the fibronectin NGR domain (i.e. domain 5), the NGR motif is located on an exposed and extruding loop that is connected to a stem region (Fig. 7A) (45). The NGR loop has a sharp turn, facilitated by the glycine residue in the middle of the NGR motif and two other glycine residues flanking the NGR motif. The conformation of the NGR loop depends on the tertiary structure of fibronectin, including the inter-domain interactions between domain 5 and domain 4. As a result, efforts to prepare the NGR loop outside the context of the fibronectin domains have been unsuccessful in yielding the crystal structure of APN in complex with the fibronectin NGR loop. Nevertheless, comparison of the structural conformation of the fibronectin NGR loop to that of APN-bound CNGRCG shows that the two NGR loops share similar structural conformations, suggesting that APN interacts with the fibronectin NGR motif and the tumor-homing NGR peptide using similar structural mechanisms (Fig. 7A). Moreover, as demonstrated by SDS-PAGE and mass spectrometry, incubation of the fibronectin NGR domain with APN in solution did not lead to APN cleavage of the NGR motif of the fibronectin NGR domain, indicating that APN binding to the NGR motifs in ECM proteins is stable and nondamaging (Fig. 7, B and C).
FIGURE 7.

Stable interactions between APN and fibronectin NGR domain. A, structure of fibronectin NGR domain (domain 5; in magenta), which is stabilized by inter-domain interactions with its neighboring domain (domain 4; in yellow) (PDB code 1FBR) (45). Also shown is the superposition of the NGR motifs in fibronectin domain 5 (in magenta) and in the tumor-homing peptide CNGRCG (in green). B, SDS-PAGE showing that pAPN did not cleave the NGR motif in the fibronectin NGR domain after they were incubated together. If the NGR motif in fibronectin domains 4–5 (molecular mass of 11 kDa) was cleaved by pAPN, two cleavage products (molecular mass of 9 and 2 kDa) would be detected on SDS-PAGE of the reaction mixture. This was not supported by SDS-PAGE. C, mass spectrometry also showing that pAPN did not cleave the NGR motif in the fibronectin NGR domain.
DISCUSSION
It is puzzling how APN, a zinc-dependent aminopeptidase in essence, can mediate tumor cell motility and serve as a receptor for tumor-homing peptides. Here, we systematically investigated the underlying molecular and structural mechanisms for these functions of APN that are seemingly unrelated to each other or to APN's enzymatic activity. Our study has identified a unified mechanism for APN-based tumor cell motility and tumor-homing therapy where APN carries out these functions by specifically interacting with the NGR motif in ECM proteins and in tumor-homing peptides, respectively (Fig. 8). The NGR motifs in ECM proteins and in tumor-homing peptides share similar structural conformations by both forming a short loop with a sharp turn. The structural conformations of the NGR motifs are stabilized either by local tertiary structure as in ECM proteins or by a flanking disulfide bond as in tumor-homing peptides. The binding site of the NGR motifs in APN is located at the zinc-aminopeptidase active site. APN recognizes the NGR motifs in a sequence-specific and conformation-dependent manner. Despite binding to the APN active site, the NGR motifs resist APN degradation because the presumed scissile peptide bonds are in catalytically inactive conformations. Therefore, the interactions between APN and the NGR motifs are specific and stable, allowing APN to provide traction for tumor cell motility and to also serve as a receptor for tumor-homing peptides. The structural information provided in this study on the detailed interactions between APN and the NGR motif can guide the design and development of tumor-homing peptides that efficiently target tumor cell surface APN and inhibitors that effectively block APN-mediated tumor cell motility and metastasis.
FIGURE 8.
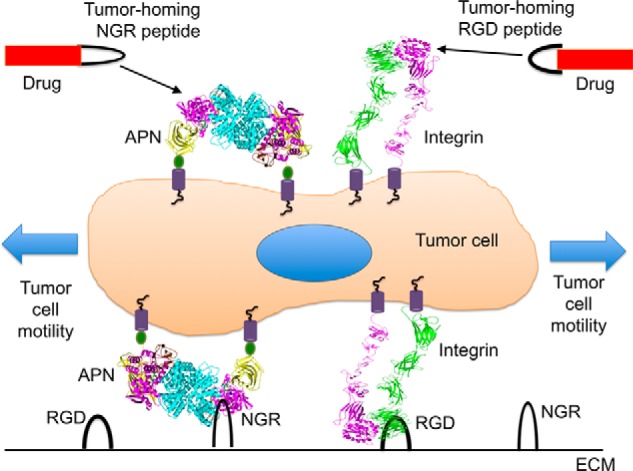
A unified mechanism for APN-based tumor cell motility and tumor-homing therapy. ECM is shown as a black line on the bottom. Black loops in two different shapes indicate the signature NGR motif recognized by APN and the signature RGD motif recognized by integrins, respectively. APN and integrins interact with their respective signature motifs in ECM proteins to provide tractions for tumor cell motility. Anti-tumor drugs (in red) are connected either to a tumor-homing NGR peptide that targets tumor cell surface APN or to a tumor-homing RGD peptide that targets tumor cell surface integrins.
The implications of our findings go beyond APN-based tumor cell motility and tumor-homing therapy; this study has established APN as an integrin-like cell motility and adhesion molecule. Indeed, despite sharing neither sequence similarity nor catalytic activity, APN and integrins resemble each other in several important ways (Fig. 8). First, integrins mediate cell motility and adhesion via binding to their signature RGD motifs in ECM proteins and on the surface of other cells. Like integrins, APN functions not only in tumor cell motility but also in other cell motility and adhesion processes such as immune cell chemotaxis, sperm motility, and monocytic cell adhesion (17–23). Here, we propose that APN mediates these other cell motility and adhesion processes by binding to its signature NGR motifs in ECM proteins and on the surface of other cells. Second, both APN and integrins relay signal transduction between cells and the extracellular environment (2). The closed-to-open conformational changes of APN may contribute to APN-mediated signal transduction (9). Third, both APN and integrins serve as functional receptors for tumor-homing peptides that contain NGR and RGD motifs, respectively. It is worth noting that although the above functions represent the main function of integrins, they are only secondary functions for APN, whose main role is to regulate the metabolism of peptides. Whereas integrins have been extensively studied and therapeutically targeted, our knowledge about APN has been rather limited. This study has laid the foundation for a better understanding of the physiological functions and therapeutic implications of APN.
Acknowledgments
We thank Carrie Wilmot and Erik Yukl for discussion and comments and the staff at ALS beamline 4.2.2 for assistance in data collection. Computer resources were provided by the Basic Sciences Computing Laboratory at the University of Minnesota Supercomputing Institute.
The atomic coordinates and structure factors (code 4OU3) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- APN
- aminopeptidase N
- pAPN
- porcine APN
- ECM
- extracellular matrix.
REFERENCES
- 1. Wickström M., Larsson R., Nygren P., Gullbo J. (2011) Aminopeptidase N (CD13) as a target for cancer chemotherapy. Cancer Sci. 102, 501–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mina-Osorio P. (2008) The moonlighting enzyme CD13: old and new functions to target. Trends Mol. Med. 14, 361–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bauvois B. (2004) Transmembrane proteases in cell growth and invasion: new contributors to angiogenesis? Oncogene 23, 317–329 [DOI] [PubMed] [Google Scholar]
- 4. Pasqualini R., Koivunen E., Kain R., Lahdenranta J., Sakamoto M., Stryhn A., Ashmun R. A., Shapiro L. H., Arap W., Ruoslahti E. (2000) Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 60, 722–727 [PMC free article] [PubMed] [Google Scholar]
- 5. Curnis F., Sacchi A., Borgna L., Magni F., Gasparri A., Corti A. (2000) Enhancement of tumor necrosis factor α antitumor immunotherapeutic properties by targeted delivery to aminopeptidase N (CD13). Nat. Biotechnol. 18, 1185–1190 [DOI] [PubMed] [Google Scholar]
- 6. Arap W., Pasqualini R., Ruoslahti E. (1998) Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 279, 377–380 [DOI] [PubMed] [Google Scholar]
- 7. Petrovic N., Schacke W., Gahagan J. R., O'Conor C. A., Winnicka B., Conway R. E., Mina-Osorio P., Shapiro L. H. (2007) CD13/APN regulates endothelial invasion and filopodia fonnation. Blood 110, 142–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carl-McGrath S., Lendeckel U., Ebert M., Röcken C. (2006) Ectopeptidases in tumour biology: a review. Histol. Histopathol. 21, 1339–1353 [DOI] [PubMed] [Google Scholar]
- 9. Chen L., Lin Y. L., Peng G., Li F. (2012) Structural basis for multifunctional roles of mammalian aminopeptidase N. Proc. Natl. Acad. Sci. U.S.A. 109, 17966–17971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fujii H., Nakajima M., Saiki I., Yoneda J., Azuma I., Tsuruo T. (1995) Human melanoma invasion and metastasis enhancement by high expression of aminopeptidase N/CD13. Clin. Exp. Metastasis 13, 337–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kehlen A., Lendeckel U., Dralle H., Langner J., Hoang-Vu C. (2003) Biological significance of aminopeptidase N/CD13 in thyroid carcinomas. Cancer Res. 63, 8500–8506 [PubMed] [Google Scholar]
- 12. Hashida H., Takabayashi A., Kanai M., Adachi M., Kondo K., Kohno N., Yamaoka Y., Miyake M. (2002) Aminopeptidase N is involved in cell motility and angiogenesis: its clinical significance in human colon cancer. Gastroenterology 122, 376–386 [DOI] [PubMed] [Google Scholar]
- 13. Chang Y. W., Chen S. C., Cheng E. C., Ko Y. P., Lin Y. C., Kao Y. R., Tsay Y. G., Yang P. C., Wu C. W., Roffler S. R. (2005) CD13 (aminopeptidase N) can associate with tumor-associated antigen L6 and enhance the motility of human lung cancer cells. Int. J. Cancer 116, 243–252 [DOI] [PubMed] [Google Scholar]
- 14. Menrad A., Speicher D., Wacker J., Herlyn M. (1993) Biochemical and functional characterization of aminopeptidase N expressed by human melanoma cells. Cancer Res. 53, 1450–1455 [PubMed] [Google Scholar]
- 15. Saiki I., Fujii H., Yoneda J., Abe F., Nakajima M., Tsuruo T., Azuma I. (1993) Role of aminopeptidase N (CD13) in tumor-cell invasion and extracellular matrix degradation. Int. J. Cancer 54, 137–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fukasawa K., Fujii H., Saitoh Y., Koizumi K., Aozuka Y., Sekine K., Yamada M., Saiki I., Nishikawa K. (2006) Aminopeptidase N (APN/CD13) is selectively expressed in vascular endothelial cells and plays multiple roles in angiogenesis. Cancer Lett. 243, 135–143 [DOI] [PubMed] [Google Scholar]
- 17. Mina-Osorio P., Winnicka B., O'Conor C., Grant C. L., Vogel L. K., Rodriguez-Pinto D., Holmes K. V., Ortega E., Shapiro L. H. (2008) CD13 is a novel mediator of monocytic/endothelial cell adhesion. J. Leukocyte Biol. 84, 448–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carlsson L., Ronquist G., Eliasson R., Egberg N., Larsson A. (2006) Flow cytometric technique for determination of prostasomal quantity, size and expression of CD10, CD13, CD26 and CD59 in human seminal plasma. Int. J. Androl. 29, 331–338 [DOI] [PubMed] [Google Scholar]
- 19. Irazusta J., Valdivia A., Fernández D., Agirregoitia E., Ochoa C., Casis L. (2004) Enkephalin-degrading enzymes in normal and subfertile human semen. J. Androl. 25, 733–739 [DOI] [PubMed] [Google Scholar]
- 20. González Buitrago J. M., Navajo J. A., García Diez L. C., Herruzo A. (1985) Seminal plasma leucine aminopeptidase in male fertility. Andrologia 17, 139–142 [DOI] [PubMed] [Google Scholar]
- 21. Tani K., Ogushi F., Huang L., Kawano T., Tada H., Hariguchi N., Sone S. (2000) CD13/aminopeptidase N, a novel chemoattractant for T lymphocytes in pulmonary sarcoidosis. Am. J. Respir. Crit. Care Med. 161, 1636–1642 [DOI] [PubMed] [Google Scholar]
- 22. Shimizu T., Tani K., Hase K., Ogawa H., Huang L., Shinomiya F., Sone S. (2002) CD13/aminopeptidase N-induced lymphocyte involvement in inflamed joints of patients with rheumatoid arthritis. Arthritis Rheum. 46, 2330–2338 [DOI] [PubMed] [Google Scholar]
- 23. Mina-Osorio P., Shapiro L. H., Ortega E. (2006) CD13 in cell adhesion: aminopeptidase N (CD13) mediates homotypic aggregation of monocytic cells. J. Leukocyte Biol. 79, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Arnaout M. A., Goodman S. L., Xiong J. P. (2002) Coming to grips with integrin binding to ligands. Curr. Opin. Cell Biol. 14, 641–651 [DOI] [PubMed] [Google Scholar]
- 25. Xiong J. P., Stehle T., Diefenbach B., Zhang R., Dunker R., Scott D. L., Joachimiak A., Goodman S. L., Arnaout M. A. (2001) Crystal structure of the extracellular segment of integrin αVβ3. Science 294, 339–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xiong J. P., Stehle T., Zhang R., Joachimiak A., Frech M., Goodman S. L., Arnaout M. A. (2002) Crystal structure of the extracellular segment of integrin αVβ3 in complex with an Arg-Gly-Asp ligand. Science 296, 151–155 [DOI] [PubMed] [Google Scholar]
- 27. Desgrosellier J. S., Cheresh D. A. (2010) Integrins in cancer: biological implications and therapeutic opportunities. Nat. Rev. Cancer 10, 9–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guo W., Giancotti F. G. (2004) Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 5, 816–826 [DOI] [PubMed] [Google Scholar]
- 29. Svensen N., Walton J. G., Bradley M. (2012) Peptides for cell-selective drug delivery. Trends Pharmacol. Sci. 33, 186–192 [DOI] [PubMed] [Google Scholar]
- 30. Sugahara K. N., Teesalu T., Karmali P. P., Kotamraju V. R., Agemy L., Greenwald D. R., Ruoslahti E. (2010) Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science 328, 1031–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gregorc V., Santoro A., Bennicelli E., Punt C. J., Citterio G., Timmer-Bonte J. N., Caligaris Cappio F., Lambiase A., Bordignon C., van Herpen C. M. (2009) Phase Ib study of NGR-hTNF, a selective vascular targeting agent, administered at low doses in combination with doxorubicin to patients with advanced solid tumours. Br. J. Cancer 101, 219–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gregorc V., Citterio G., Vitali G., Spreafico A., Scifo P., Borri A., Donadoni G., Rossoni G., Corti A., Caligaris-Cappio F., Del Maschio A., Esposito A., De Cobelli F., Dell'Acqua F., Troysi A., Bruzzi P., Lambiase A., Bordignon C. (2010) Defining the optimal biological dose of NGR-hTNF, a selective vascular targeting agent, in advanced solid tumours. Eur. J. Cancer 46, 198–206 [DOI] [PubMed] [Google Scholar]
- 33. Gregorc V., Zucali P. A., Santoro A., Ceresoli G. L., Citterio G., De Pas T. M., Zilembo N., De Vincenzo F., Simonelli M., Rossoni G., Spreafico A., Grazia Viganò M., Fontana F., De Braud F. G., Bajetta E., Caligaris-Cappio F., Bruzzi P., Lambiase A., Bordignon C. (2010) Phase II study of asparagine-glycine-arginine-human tumor necrosis factor α, a selective vascular targeting agent, in previously treated patients with malignant pleural mesothelioma. J. Clin. Oncol. 28, 2604–2611 [DOI] [PubMed] [Google Scholar]
- 34. Reardon D. A., Fink K. L., Mikkelsen T., Cloughesy T. F., O'Neill A., Plotkin S., Glantz M., Ravin P., Raizer J. J., Rich K. M., Schiff D., Shapiro W. R., Burdette-Radoux S., Dropcho E. J., Wittemer S. M., Nippgen J., Picard M., Nabors L. B. (2008) Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J. Clin. Oncol. 26, 5610–5617 [DOI] [PubMed] [Google Scholar]
- 35. Eskens F. A., Dumez H., Hoekstra R., Perschl A., Brindley C., Böttcher S., Wynendaele W., Drevs J., Verweij J., van Oosterom A. T. (2003) Phase I and pharmacokinetic study of continuous twice weekly intravenous administration of Cilengitide (EMD 121974), a novel inhibitor of the integrins αvβ3 and αvβ5 in patients with advanced solid tumours. Eur. J. Cancer 39, 917–926 [DOI] [PubMed] [Google Scholar]
- 36. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 37. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 38. Murshudov G. N., Vagin A. A., Lebedev A., Wilson K. S., Dodson E. J. (1999) Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr. D Biol. Crystallogr. 55, 247–255 [DOI] [PubMed] [Google Scholar]
- 39. Yang Y., Liu C., Lin Y. L., Li F. (2013) Structural insights into central hypertension regulation by human aminopeptidase A. J. Biol. Chem. 288, 25638–25645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shor A. C., Keschman E. A., Lee F. Y., Muro-Cacho C., Letson G. D., Trent J. C., Pledger W. J., Jove R. (2007) Dasatinib inhibits migration and invasion in diverse human sarcoma cell lines and induces apoptosis in bone sarcoma cells dependent on SRC kinase for survival. Cancer Res. 67, 2800–2808 [DOI] [PubMed] [Google Scholar]
- 41. Chen Y., Lu B., Yang Q., Fearns C., Yates J. R., 3rd, Lee J. D. (2009) Combined integrin phosphoproteomic analyses and small interfering RNA-based functional screening identify key regulators for cancer cell adhesion and migration. Cancer Res. 69, 3713–3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Peng G., Sun D., Rajashankar K. R., Qian Z., Holmes K. V., Li F. (2011) Crystal structure of mouse coronavirus receptor-binding domain complexed with its murine receptor. Proc. Natl. Acad. Sci. U.S.A. 108, 10696–10701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Du L., Zhao G., Yang Y., Qiu H., Wang L., Kou Z., Tao X., Yu H., Sun S., Tseng C. T., Jiang S., Li F., Zhou Y. (2014) A conformation-dependent neutralizing monoclonal antibody specifically targeting receptor-binding domain in Middle East respiratory syndrome coronavirus Spike protein. J. Virol. 88, 7045–7053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Di Matteo P., Curnis F., Longhi R., Colombo G., Sacchi A., Crippa L., Protti M. P., Ponzoni M., Toma S., Corti A. (2006) Immunogenic and structural properties of the Asn-Gly-Arg (NGR) tumor neovasculature-homing motif. Mol. Immunol. 43, 1509–1518 [DOI] [PubMed] [Google Scholar]
- 45. Williams M. J., Phan I., Harvey T. S., Rostagno A., Gold L. I., Campbell I. D. (1994) Solution structure of a pair of fibronectin type 1 modules with fibrin binding activity. J. Mol. Biol. 235, 1302–1311 [DOI] [PubMed] [Google Scholar]