

Background: Localization and identification of interaction partners of two splice variants of the human 3-mercaptopyruvate sulfurtransferase TUM1.
Results: We show that TUM1 interacts with proteins involved in Moco and FeS cluster biosynthesis.
Conclusion: Human TUM1 is a dual localized protein in the cytosol and mitochondria with distinct roles in sulfur transfer and interaction partners.
Significance: The study contributes to the sulfur transfer pathway for the biosynthesis of sulfur-containing biofactors.
Keywords: Fluorescence, Mitochondria, Molybdenum, Sulfur, Transfer RNA (tRNA)
Abstract
The human tRNA thiouridine modification protein (TUM1), also designated as 3-mercaptopyruvate sulfurtransferase (MPST), has been implicated in a wide range of physiological processes in the cell. The roles range from an involvement in thiolation of cytosolic tRNAs to the generation of H2S as signaling molecule both in mitochondria and the cytosol. TUM1 is a member of the sulfurtransferase family and catalyzes the conversion of 3-mercaptopyruvate to pyruvate and protein-bound persulfide. Here, we purified and characterized two novel TUM1 splice variants, designated as TUM1-Iso1 and TUM1-Iso2. The purified proteins showed similar kinetic behavior and comparable pH and temperature dependence. Cellular localization studies, however, showed a different localization pattern between the isoforms. TUM1-Iso1 is exclusively localized in the cytosol, whereas TUM1-Iso2 showed a dual localization both in the cytosol and mitochondria. Interaction studies were performed with the isoforms both in vitro using the purified proteins and in vivo by fluorescence analysis in human cells, using the split-EGFP system. The studies showed that TUM1 interacts with the l-cysteine desulfurase NFS1 and the rhodanese-like protein MOCS3, suggesting a dual function of TUM1 both in sulfur transfer for the biosynthesis of the molybdenum cofactor, and for the thiolation of tRNA. Our studies point to distinct roles of each TUM1 isoform in the sulfur transfer processes in the cell, with different compartmentalization of the two splice variants of TUM1.
Introduction
The human 3-mercaptopyruvate sulfurtransferase (MPST)3 (EC 2.8.1.2), also designated as TUM1 (tRNA thiouridin modification protein 1), belongs to an enzyme superfamily of proteins that contain a rhodanese-like domain (RLD) (1). Representatives of this enzyme family consist of one or up to four RLDs, of which usually only the C-terminal domain harbors the catalytically active site loop. The function of the N-terminal domain is believed to be of regulatory importance and involved in signaling processes (1). Catalytically active or inactive single RLDs have been found associated with various domains of other enzyme families, such as adenylation domains, RNA-binding motifs, several phosphatase families, and ubiquitinating enzymes (1, 2). RLDs often share only low amino acid sequence homology among each other, but exhibit highly conserved structural criteria (3, 4). Substrate specificity of the RLDs with catalytic activity is dependent on the constitution of the active site center, with either a conserved CRXGX(T/R) motif in the case of thiosulfate sulfurtransferases (EC 2.8.1.1), or a more defined CG(S/T)GV(T/S) motif in the case of MPSTs (1, 5). Rhodanese-like enzymes bind sulfur in the form of persulfides, with a formal redox status of S0 at their conserved active site cysteine residue (6, 7). To date the physiological function of rhodanese-like proteins is not fully understood, but has been linked to a wide variety of biological processes, including the detoxification of cyanide, the homeostasis of cellular sulfur in general, the participation in the degradation of l-cysteine, mitochondrial production of hydrogen sulfide (H2S) as signaling molecule, in addition to the biosynthesis of enzymatic cofactors, vitamins, and sulfur-containing nucleic acids (8–13). Human TUM1 shares about 60% amino acid sequence identity to MPST homologues from various sources. The crystal structure of human and yeast TUM1, also in complex with 3-MP has been solved at 2.15-Å resolution (13, 14). TUM1 is expressed in kidney cells, liver cells, cardiac cells, and neurological cells (15). Human individuals lacking proper formation of TUM1 were shown to excrete elevated levels of mercaptolactate-cysteine disulfide in the urine, an inborn disorder referred to as mercaptolactate-cysteine disulfiduria (16, 17). TUM1 was described to be involved in H2S production and signaling, which is also produced by cystathione β-synthase and cystathionine γ-lyase in the cytosol (18). In the TUM1-dependent route for H2S generation, 3-MP originating from cysteine is converted to pyruvate and TUM1-bound persulfide, which can be released as H2S in the presence of reducing systems like GSH or thioredoxin (19).
Thio modification of uridine in the 2-position is known to ensure accurate deciphering of the genetic code and stabilization of tRNA structure. In Saccharomyces cerevisiae the wobble bases of tRNAs contain two thiouridines, 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U34) in cytoplasmic tRNAs and 5-carboxymethyl-2-thiouridine (cmnm5s2U34) in mitochondrial tRNAs. Previously, the yeast Tum1p protein was described to be involved in the sulfur transfer for the thiolation of tRNA. In a genetic screen for the identification of cytoplasmic tRNA modification for the formation of mcm5s2U at the wobble position of U34, the proteins Tum1p, Uba4p, Nfs1p, Urm1p, Ncs2p, and Nsc6p were identified to be involved in this process. Nfs1p is a l-cysteine desulfurase, which was described, not only to function as a direct supplier of persulfide to thionucleosides in the cytosol, but also to function as the main protein that supplies sulfur for FeS formation in mitochondria (20). An in vitro sulfur transfer experiment suggested that Tum1p stimulates the cysteine desulfurase activity, however, a direct interaction of the proteins was not shown. In contrast, Uba4p was shown to be capable of accepting sulfur from Nfs1p, but because these experimental assays contained DTT, which can be prone to unspecific sulfide release to the solution, the physiological significance of the results remain unclear. Urm1p is an ubiquitin-related modifier and Uba4p is an E1-like Urm1p activating enzyme that is involved in protein urmylation. The carboxyl terminus of Urm1p is first activated as an acyl adenylate intermediate (-COAMP) and then thiocarboxylated (-COSH) by Uba4p. The activated thiocarboxylate can be utilized in the subsequent reaction for 2-thiouridine formation, mediated by a heterodimer complex consisting of Ncs2p and Ncs6p.
In humans, a similar pathway for mcm5s2U34 thiolation exists in the cytosol, consisting of the proteins (yeast homologues are given in parentheses): NFS1 (Nfs1p), MOCS3 (Uba4), URM1 (Urm1p), CTU1 (Ncs2), and CTU2 (Ncs6). For mcm5s2U34 modification of cytosolic tRNAs, sulfur is transferred from the rhodanese-like protein MOCS3 to the C terminus of the ubiquitin-related modifier URM1 (20–25). This complex first activates URM1 by adenylation followed by the sulfur transfer step, which results in the formation of a thiocarboxylate group at the C-terminal Gly of URM1. By the interaction with the CTU1·CTU2 complex, which binds and activates the tRNAs for further sulfur transfer, thiocarboxylated URM1 transfers the sulfur to tRNALys, tRNAGln, and tRNAGlu (26). The sulfur for the persulfide group of MOCS3 was shown to originate from NFS1 in the cytosol. A specific interaction between both proteins was identified recently, confirming the additional cytosolic localization of NFS1 and its involvement in the pathways mediated by MOCS3. In contrast to yeast Uba4p, human MOCS3 is a dual function protein that is involved in two sulfur transfer pathways. MOCS3 was initially identified to be involved in molybdenum cofactor (Moco) biosynthesis in the cytosol (27). In this case, MOCS3 interacts with MOCS2A and forms a thiocarboxylate group at the C terminus of MOCS2A (24, 25, 27). MOCS2A subsequently assembles with MOCS2B to form the molybdopterin (MPT) synthase (28). The MPT synthase binds the first intermediate of Moco biosynthesis, cyclic pyranopterin monophosphate (cPMP) and generates MPT after the transfer of two sulfur atoms from two MOCS2A proteins (28). MOCS2B binds cPMP in this reaction. The two sulfur atoms of MPT coordinate the molybdenum atom in the final step of Moco biosynthesis. In humans, Moco is required for the activity of xanthine dehydrogenase, aldehyde oxidase, sulfite oxidase, and the mitochondrial amidoxime reducing components, mARC1 and mARC2 (29). The role of human MOCS3, thus, is different from yeast Uba4p, which only interacts with Urm1p in the cytosol, because proteins for the biosynthesis of the molybdenum cofactor like MOCS2A are not present in yeast.
By comparison to yeast Tum1p, the role of human TUM1 in tRNA thiolation or Moco biosynthesis, to date, is not well resolved. In addition, two splice variants exist for TUM1, which so far were neglected to be characterized. In turn, we purified for the first time the two splice variants of TUM1 and characterized the two proteins in terms of their kinetic behavior, localization, and interaction partners. Both TUM1 isoforms showed similar kinetic characteristics. However, their localization in human cells and the respective interaction partners proved to be different. TUM1-Iso1 and TUM1-Iso2 were identified both to be localized in the cytosol, whereas TUM1-Iso2 is additionally localized in the mitochondria. Furthermore, we identified by in vivo and in vitro methods that in the cytosol, TUM1-Iso1 interacted with both NFS1 and MOCS3, whereas TUM1-Iso2 exclusively interacted with MOCS3 in the cytosol and NFS1 in the mitochondria.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Plasmids, Media, and Growth Conditions
Escherichia coli cell strains containing expression plasmids were grown aerobically at 30 °C in LB medium containing either 150 μg/ml of ampicillin or 50 μg/ml of chloramphenicol. All E. coli strains, human cell lines, and plasmids used in this study are listed in Table 1. E. coli MoaE was expressed in BL21(DE3) cells from plasmid pMWaE15 and purified as described in Ref. 30, and human MOCS2A was expressed and purified as described in Ref. 21. Coexpression and co-purification of human NFS1Δ1–55 and ISD11 was achieved as described in Ref. 31, and human MOCS3 was expressed and purified as described in Ref. 24.
TABLE 1.
Plasmids, E. coli strains, and cell lines used in this study
| Plasmid, strain, or human cell line | Characteristics, genotype, or origin | Reference |
|---|---|---|
| Plasmids used in E. coli | ||
| pBF18 | TUM1 gene fragment encoding for TUM1-RLD2 cloned into NdeI/XhoI sites of pET24a, KanR | This study |
| pBF21 | TUM1 gene encoding for TUM1-Iso1 cloned into BamHI/XhoI sites of pGEX4T-1, AmpR | This study |
| pBF22 | TUM1 gene encoding for TUM1-Iso2 cloned into BamHI/XhoI sites of pGEX4T-1, AmpR | This study |
| pBF24 | Mutated TUM1 gene encoding for TUM1-Iso2C248A cloned into BamHI/XhoI sites of pGEX4T-1, AmpR | This study |
| pMMC30 | MOCS3 gene cloned into BamHI/XbaI sites of pFastBacDuet-1, AmpR | 24 |
| pAM21 | MOCS3-RLD gene fragment cloned into NdeI/BamHI sites of pET15b, AmpR | 27 |
| pPW9710 | sseA gene cloned into NdeI/XhoI sites of pET28a, KanR | 41 |
| pMWaE15 | moaE gene cloned into NcoI/BamHI sites of pET15b, AmpR | 28 |
| pSL174 | MOCS2A gene and promoter region cloned into XbaI/BamHI sites of pET15b, AmpR | 21 |
| pZM2 | NFS1Δ1–55 gene fragment cloned into XhoI/BamHI sites of pET15b, AmpR | 31 |
| pZM4 | ISD11 gene fragment cloned into NcoI/HindIII sites of pACYCDuet-1, CmR | 31 |
| Plasmids used in cell culture | ||
| pCR01 | TUM1 gene encoding for TUM1-Iso1 cloned into XhoI/BamHI sites of pEGFP(1–157)-N1 generating a TUM1-Iso1-EGFP(1–157) fusion | This study |
| pCR05 | TUM1 gene fragment encoding for TUM1-Iso2 cloned into XhoI/BamHI sites of pEGFP(1–157)-N1 generating a TUM1-Iso2-EGFP(1–157) fusion | This study |
| pZM144 | MOCS3 gene cloned into XhoI/BamHI of pEGFP(158–238) generating a MOCS3-EGFP(158–238) fusion | 53 |
| pZM146 | NFS1 gene cloned into XhoI/BamHI of pEGFP(158–238)-N1 generating a NFS1-EGFP(158–238) fusion | 53 |
| pZM148 | NFS1Δ1–55 gene fragment cloned into XhoI/BamHI of pEGFP(158–238) generating a NFS1-EGFP(158–238)-N1 fusion | 53 |
| Bacterial strains | ||
| BL21 (DE3) | F-, ompT, rB-mB- hsdS, gal (λclts857, ind1, San7, nin5, lac UV5-T7 gene1) (DE3) | 54 |
| Human cell lines | ||
| HeLa | Human cervix carcinoma cell line | 55 |
| HEK293a | Human embryonal kidney cell line | 56 |
Cloning, Expression, and Purification of Human TUM1
For heterologous expression of TUM1 in E. coli, the genes for TUM1-Iso1 and TUM1-Iso2 were amplified by PCR from a human cDNA library (Clontech). Primers were designed that allowed for cloning into the BamHI-XhoI restriction sites of the expression vector pGEX4T-1 (GE Healthcare), which expresses both TUM1 isoforms as N-terminal GST fusion proteins. The resulting plasmids were designated pBF15 (expressing TUM1-Iso1) and pBF16 (expressing TUM1-Iso2) (Table 1). E. coli BL21(DE3) expression cultures were grown at 37 °C in LB medium under aerobic conditions until A600 = 0.6 was reached. Expression was induced with 100 μm isopropyl β-d-1-thiogalactopyranoside and growth was continued at 16 °C for 16 h. Cells were harvested at 11,000 × g, resuspended in 50 mm Tris-HCl, 200 mm NaCl, pH 8.0, and stored at −20 °C. Cell lysis was achieved by sonification using a Heinemann HTU Soni130 (Heinemann HTU, Schwäbisch-Gmünd, Germany) in the presence of 1 μg/ml of DNase I. Cleared lysates were applied to 0.5 ml of glutathione-agarose (GSH-Matrix) (Macherey-Nagel, Düren, Germany) per liter of expression culture and incubated at 4 °C for 1 h on a rotary shaker. The GSH-Matrix was washed with 20 column volumes of 50 mm Tris-HCl, 200 mm NaCl, 5 mm DTT, pH 8.0, and 10 column volumes of 50 mm Tris-HCl, 200 mm NaCl, pH 8.0. The GST fusion proteins bound to GSH-Matrix were cleaved overnight with 5 mg/ml of thrombin at 4 °C, and the proteins were eluted with 1 column volume of 50 mm Tris-HCl, 200 mm NaCl, pH 8.0. For separation of the thrombin, the elution fraction was applied to 2 volumes of benzamidine-Sepharose (GE Healthcare), and TUM1-Iso1 and TUM1-Iso2 were eluted with 1 column volume of 50 mm Tris-HCl, 200 mm NaCl, pH 8.0, and stored at −80 °C until further use.
Size Exclusion Chromatography
Size exclusion chromatography was performed at 8 °C using a Superdex 200 column (GE Healthcare) with a bed volume of 25 ml equilibrated in 50 mm Tris-HCl, 200 mm NaCl, pH 8.0 (flow rate of 0.5 ml/min). Proteins were applied in a volume of 500 μl and elution fractions were analyzed by SDS-PAGE and Coomassie Brilliant Blue staining.
Fluorescence Spectroscopy of TUM1 Variants
Fluorescence measurements were performed using a HORIBA Jobin Yvon FluoroMax-4 Spectrofluorometer (Horiba, Kyoto, Japan) equipped with a temperature-controlled stirred cell holder. All measurements were performed at 37 °C using 5 μm enzyme in a total volume of 1 ml of 50 mm Tris-HCl, 200 mm NaCl, pH 8.0. The excitation wavelength was set to 280 nm with a slit width of 2 nm. Emission spectra were recorded from 300 to 400 nm with a slit width of 5 nm. As substrates, 10 mm KCN or 1 mm 3-MP were added and incubated for 1 min prior to recording.
3-Mercaptopyruvate:Dithiothreitol Sulfurtransferase Activity of TUM1
3-Mercaptopyruvate:dithiothreitol sulfurtransferase activities of TUM1 were quantified using the methylene blue method (32). The reaction mixtures were incubated for 3 min at 37 °C in a final volume of 400 μl of 50 mm Tris-HCl, 200 mm NaCl, pH 10.5. Sodium 3-MP was used at concentrations of 0.05–0.75 mm and DTT at 0.25–5 mm. Reactions were initiated by the addition of 0.1 μm TUM1 and terminated by the addition of 50 μl of 20 mm N,N-dimethyl-p-phenylenediamine (DMPD) in 7.2 m HCl. Color development was started by the addition of 50 μl of 30 mm FeCl3 in 1.2 m HCl, following an incubation time of 5 min at room temperature. Precipitated protein was removed by centrifugation at 15,000 × g for 3 min, and the produced methylene blue was quantified at 670 nm using a standard curve with known amounts of sodium sulfide.
3-Mercaptopyruvate:Cyanide Sulfurtransferase Activity
3-Mercaptopyruvate:cyanide sulfurtransferase activities of TUM1 were quantified by the method described by Sörbo (33). The reaction mixtures contained 3-MP at varying concentrations of 0.5–5 mm, and DTT at 2–20 mm in a final volume of 500 μl of 100 mm CAPS, pH 10.5. Following initiation of the reaction by the addition of 0.2 μm TUM1 the mixtures were incubated for 10 min at 37 °C. The reactions were terminated by the addition of 250 μl of 15% formaldehyde, and 750 μl of a coloring agent were added (containing 100 g of Fe(NO3)3·9 H2O, 200 ml of 65% HNO3 in a final volume of 1500 ml of H2O). Following centrifugation at 15,000 × g for 3 min to remove precipitated protein, produced thiocyanate was quantified at 460 nm against a standard curve of known amounts of sodium thiocyanate.
CD Spectroscopy
CD spectroscopy was performed with purified proteins in 40 mm NaPO4, 140 mm NaF, pH 6.7, buffer using protein concentrations of 0.15 mg/ml and 1.00-mm quartz cuvettes. All spectra were recorded on a Jasco-J715 CD-spectrometer (Jasco) equipped with a thermoelectric temperature-controlled cuvette holder at 0.5 nm/min scan speed with 5 repetitions. The CD spectra were baseline corrected by subtraction of the corresponding buffer spectrum. Ellipticity values in degrees cm2 dmol−1 were calculated on a mean residue molecular mass basis of 111.2 Da for TUM1-Iso1 and 111.7 Da for TUM1-Iso2.
Surface Plasmon Resonance (SPR) Measurements
All binding experiments were performed on an SPR-based BiacoreTM T200 instrument employing CM5 sensor chips at a temperature of 25 °C and a flow rate of 30 μl/min using the Biacore control T200 software and evaluation T200 software (GE Healthcare). The autosampler rack containing the samples was cooled throughout the entire measurements to 8 °C. Proteins were immobilized with the following recovery units: BSA, 500–1000; NFS1Δ1–55, 1581; NFS1Δ1–55·ISD11, 150–700; TUM1-Iso1, 626; TUM1-Iso2, 551; MOCS3, 400–1100; and MOCS3-RLD, 600–650. Proteins for each immobilization process resulted from independent purifications. As running buffer 20 mm phosphate, 150 mm NaCl, 0.005% (v/v) Tween 20, pH 7.4, was used. Analytes with concentrations of 0.16, 0.31, 0.63, 1.25, 2.5, 5, 10, and 20 μm were injected for 4.5 min at a flow rate of 30 μl/min followed by a 15-min dissociation and regeneration of the sensor surface with 50 mm HCl for 1 min. Bovine serum albumin (BSA) served as a control ligand. Binding curves were corrected by subtraction of buffer injection curves for both flow cells.
Cysteine Desulfurase Activity Measurements
To analyze the influence of TUM1 and/or MOCS3 on the l-cysteine desulfurase activity of the NFS1·ISD11 complex total sulfide production was measured employing the methylene blue method (32). 1 μm NFS1·ISD11 complex was preincubated with 0–6 μm TUM1 and MOCS3 variants in 50 mm Tris-HCl buffer, pH 8.0, for 30 min at 4 °C. After the addition of DTT to a final concentration of 1 mm, the reaction was started by addition of 1 mm l-cysteine. After incubation in a total volume of 400 μl at 37 °C for 15 min, the reaction was stopped by the addition of 50 μm N,N-dimethyl-4-phenylenediamine in 7.2 m HCl. Methylene blue was formed by addition of 50 μm of a FeCl3 solution in 1.2 m HCl and quantified at 670 nm against a sulfide standard curve.
In Vitro MPT Production
MPT synthase reactions were performed at room temperature in a total volume of 400 μl of 100 mm Tris-HCl, pH 8.0. The produced MPT was converted to the stable oxidation product Form A and quantified as described by published procedures (34, 35). For MPT synthesis 5 μm E. coli MoaE, 15 μm MOCS2A, 5 μm MOCS3, 2.5 mm Mg-ATP, 20 μm NFS1Δ1–55·ISD11, and 10 μm TUM1 were mixed. As sulfur source 0.4 mm sodium sulfide, sodium thiosulfate, 3-MP, or l-cysteine was used. The reaction was initiated by the addition of cPMP in excess, which was purified as described previously (36).
Cell Culture Maintenance
HEK293a and HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM, PAN-Biotech, Germany) supplemented with 10% fetal bovine serum (FBS, PAN-Biotech, Germany) and glutamine. Cell cultures were maintained at a temperature of 37 °C and 5% CO2 atmosphere. For localization and FRET analyses, HEK293a or HeLa cells were grown on poly-l-lysine-coated coverslips prior to transfection.
Immunodetection of TUM1 in Fractionated HEK295 Cell Lysates
The subcellular fractionation of HEK293a cells was performed as described previously (37). The two fractions were concentrated 10–20-fold by ultrafiltration using Centriprep devices with a cut-off of 3 kDa. The protein concentration of the extract was quantified by using the Bradford Reagent Coomassie PlusTM Protein Assay Reagent (Thermo) with bovine serum albumin as a standard. 75 μg of protein extracts were separated by 12% SDS-PAGE and transferred onto a PVDF membrane (Sigma). The membrane was blocked with 5% milk powder in TBS containing 0.1% Tween (TBST) for 1 h at room temperature. After washing in TBST the membranes were incubated with anti-MPST (Sigma, 1:2000) or anti-citrate synthase (Sigma, 1:5000) antibodies overnight at 4 °C, and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (Thermo, 1:1000) at room temperature for 1 h. The protein bands were visualized by the enhanced chemiluminescence (ECL) detection system (Super Signal; Thermo, Pierce).
Subcellular Interaction Studies Using the Split-EGFP System
Coding regions for TUM1-Iso1 and TUM1-Iso2 were amplified by PCR using HEK293a cDNA as a template and the obtained fragments were cloned into mammalian expression vectors pEGFP(1–157)-N1, resulting in plasmids pCR1 (TUM1-Iso1) and pCR5 (TUM1-Iso2). The cell culture plasmids used in this study are listed in Table 1. HEK293a cells were transiently transfected with the corresponding plasmids: pCR1 with pZM44, pZM146, or pZM148 or pCR5 with pZM44, pZM146, or pZM148 using Lipofectamine® (Invitrogen). For the staining of the mitochondria and nuclei MitoTracker DeepRed® (Invitrogen, 1:20,000) and DAPI (Sigma, 1:1,000) were used. 24 h after transfection, cells were fixed for 20 min at 4 °C using 3% paraformaldehyde in PBS. The cells were washed twice with PBS and mounted onto slides with Mowiol (Roth). Images for EGFP fluorescence were imaged with a confocal microscope LSM710 (Carl Zeiss Microscopy, Jena, Germany) using a PlanApo 1.4/×63 Oil or EC Plan-Neofluar 1.3/×40 Oil objective. DAPI, EGFP, and MitoTracker were excited sequentially (multi-track mode) at 405, 488, and 633 nm. Images were taken with a depth of 12 bits in the spectral range of the emission at 413–560 nm (for DAPI), 493–612 nm (for EGFP), and 637–735 nm (for MitoTracker). The imaging software ZEN2009 was used for operating the system and image acquisition. For processing, the ImageJ (MacBiophotonics) program was used.
Indirect Immunofluorescence
For indirect immunofluorescence studies in the HEK293a cell line cells were grown overnight on poly-l-lysine-coated coverslips. The cells were fixed for 15 min using ice-cold 3% paraformaldehyde in PBS, blocked in Dulbecco's modified Eagle's medium (DMEM, PAN-Biotech, Germany) supplemented with 10% fetal bovine serum (FBS, PAN-Biotech, Germany) and permeabilized with 0.1% Triton X-100 in PBS. The cells were washed with PBS and incubated with anti-MPST antibody (Sigma, 1:100) in PBS supplemented with 4% FBS overnight at 4 °C. The cells were washed with PBS and incubated with Alexa Fluor® 488-conjugated secondary antibody (Invitrogen, 1:400) supplemented with 4% FBS. Cells were then washed with PBS and mounted onto slides with Mowiol (Roth). Alexa Fluor 488 was excited sequentially at 488 nm and images were taken with a depth of 12 bits in the spectral range of the emission at 519 nm. ZEN2009 software was used for operating the system and image acquisition. For processing, the ImageJ (MacBiophotonics) program was used.
RESULTS
Expression and Purification of Human TUM1 Variants
The TUM1 gene encoding for 3-mercaptopyruvate sulfurtransferase is located on chromosome 22. It was predicted that two splice variants exist for TUM1, resulting in two discrete isoforms (38) designated as TUM1-Iso1 and TUM1-Iso2 in this work. However, the existence of the two TUM1 protein isoforms in human cells remained unproven and a biochemical characterization of the isolated proteins was not performed so far. For heterologous expression in E. coli, the two coding regions were amplified by PCR using cDNA from a human cDNA library and cloned into vector pGEX4T-1, resulting in N-terminal GST fusions of the two proteins. The major difference of the two proteins is a 20-amino acid extension at the N terminus of TUM1-Iso2, thereby flanking the 20-amino acid mitochondrial targeting sequence (Fig. 1A). Following affinity purification and cleavage of the GST tag, the two TUM1 isoforms were purified with a yield of ∼10 mg/liter of E. coli culture. Eluted proteins were analyzed on SDS-PAGE. After staining with Coomassie Brilliant Blue R two protein bands were visualized that corresponded to the calculated molecular masses of 35.394 kDa for TUM-Iso1 and 33.322 kDa for TUM-Iso2 (Fig. 1B). In addition, the C-terminal RLD of TUM1 (amino acids 167–297 with respect to TUM1-Iso2), referred to as TUM1-RLD2, was expressed as a fusion protein with a C-terminal His6 tag. Protein purification was performed by nickel-nitrilotriacetic acid chromatography with a yield of ∼15 mg/liter of E. coli culture (Fig. 1B). The three purified proteins were judged to be >90% pure based on SDS-PAGE analysis. Analytical gel filtration using a Superdex 200 column showed that the two TUM1 isoforms were purified as monomers (Fig. 1C), whereas the TUM1-RLD2 formed aggregates. Additionally, a TUM1-Iso2-C248A active site variant was generated, in which the catalytically important cysteine residue at the 6-amino acid active site loop of the rhodanese-like domain was substituted for an alanine.
FIGURE 1.

Analytical size-exclusion chromatography and domain structure of TUM1 variants. A, domain structure of TUM1 isoforms. MTS, mitochondrial targeting sequence. Numbers represent the position in the respective amino acid sequence. Identification of targeting sequences and enzyme domains was achieved by sequence analysis using the Pfam and SignalP online tools. B, SDS-PAGE of 12-μl samples of fractions collected at maximum peak heights: 1, TUM1-Iso1; 2, TUM1-Iso2 both separated on a 12% SDS acrylamide gel; 3, TUM1-RLD2 separated on 10% SDS acrylamide gel. C, analytical size-exclusion chromatography was performed with 20 μm of each TUM1 variant on a Superdex 200 column equilibrated with 50 mm Tris-HCl, 200 mm NaCl, pH 8.0, the elution signals were recorded at 280 nm: solid line, TUM1-Iso1; dashed line, TUM1-Iso2; dotted line, TUM1-RLD2. Inset, plot of the protein standard (Bio-Rad): thyroglobulin (670 kDa), γ-globulin (158 kDa), ovalbumin (44 kDa), myoglobin (17 kDa), and vitamin B12 (1.3 kDa).
Intrinsic Fluorescence of Human TUM1
A specific characteristic of several sulfurtransferases with a rhodanese-like domain is to exhibit quenched intrinsic fluorescence after addition of stoichiometric amounts of substrate, due to the location of a tryptophan residue close to the active site (39, 40). TUM1-Iso1 and TUM-Iso2 showed maximal fluorescence at λex = 336 nm, which is similar to the fluorescence maximum of SseA, a TUM1 homologue from E. coli (41). Cyanide addition to either purified protein resulted in no major changes in fluorescence, showing that both isoforms were purified in a non-persulfurated state. After the addition of stoichiometric amounts of 3-MP, the intrinsic fluorescence was decreased due to the persulfide formation on Cys-248 (Fig. 2). In comparison, fluorescence of the TUM1-Iso2-C248A variant was not quenched after substrate addition, showing that the protein is inactive. Additionally, the TUM1-RLD2 variant proved to be inactive, because no differences in fluorescence were recorded after substrate addition, showing that aggregation of the protein contributed to its inactivity.
FIGURE 2.
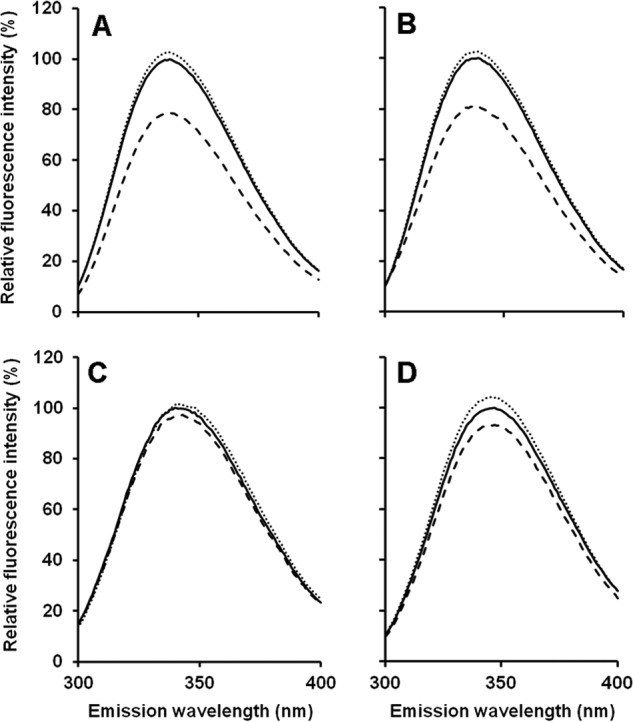
Intrinsic fluorescence of TUM1 variants. Tryptophane residues of purified proteins present in 50 mm Tris-HCl, 200 mm NaCl, pH 8.0, were excited photometrically at 280 nm and the intrinsic fluorescence spectra were recorded ranging from 300 to 400 nm. Solid lines, intrinsic fluorescence of proteins as purified; dashed lines, after addition of KCN; dotted lines, after the addition of 3-MP. A, TUM1-Iso1; B, TUM1-Iso2; C, TUM1-Iso2C248A; D, TUM1-RLD2.
Temperature and pH Dependence of MPST Activity of TUM1
To further characterize the two TUM1 isoforms and identify potential differences in their stability or kinetic behavior, the pH dependence of the activity of TUM1-Iso1 and TUM1-Iso2 was determined in a pH range of 7.0 to 11.0 with 3-MP as substrate. The results in Fig. 3 show a bell-shaped curve with a pH maximal of 10.5 for both TUM1 isoforms. The temperature dependence was analyzed in steps of 3 degrees from 22 to 61 °C. Maximum activity was detected at 49 °C for both TUM1 isoforms (Fig. 4).
FIGURE 3.
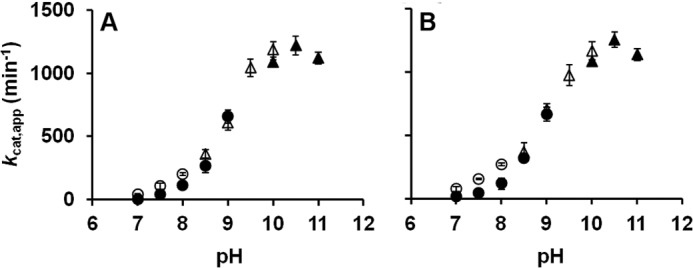
pH dependence of the 3-mercaptopyruvate sulfurtransferase activity of TUM1. The sulfurtransferase activity of purified TUM1 isoforms was recorded as the amount of produced sulfide per min at varying pH values in overlapping buffer systems. A, TUM1-Iso1; B, TUM1-Iso2; empty circles, HEPES; filled circles, Tris-HCl; empty triangles, CHES; filled triangles, CAPS.
FIGURE 4.
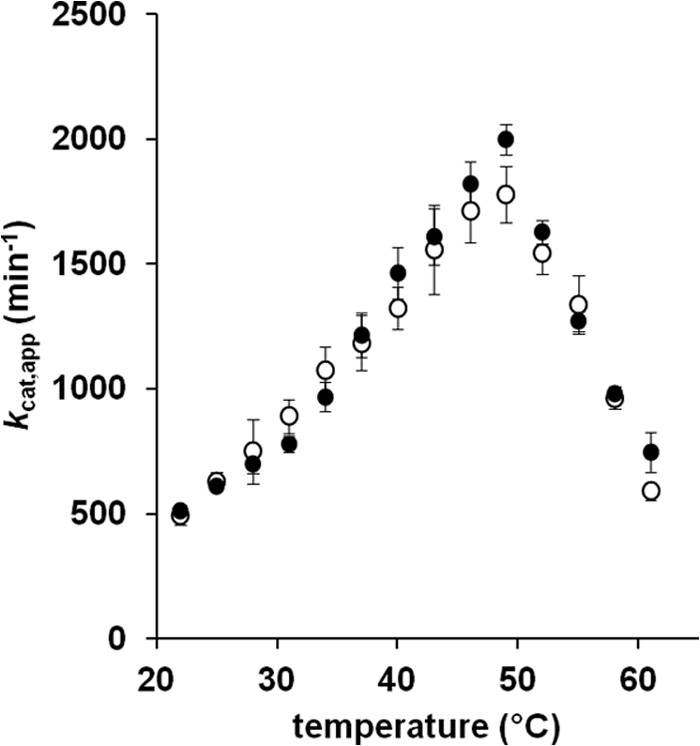
Temperature dependence of the 3-mercaptopyruvate sulfurtransferase activity of TUM1. The activity of TUM1 was measured in 100 mm CAPS, pH 10.5, over a range of 22–61 °C in steps of 3 degrees and quantified as the amount of produced sulfide (42). Filled circles, TUM1-Iso1; empty circles, TUM1-Iso2.
To analyze the influence of the 20-amino acid extension of TUM1-Iso1 on the overall folding of the respective protein, both isoforms were subjected to CD spectroscopy. The CD spectra in Fig. 5 show identical intensities between 185–200 and 210–230 nm for both proteins, showing that no differences in their α-helical and β-strand composition exist and that the overall folding of the protein is not altered by the amino acid extension at the N terminus of TUM1-Iso1.
FIGURE 5.

Circular dichroism (CD) spectra of TUM1-Iso1 and TUM1-Iso2. CD spectra on TUM1-Iso1 and TUM1-Iso2 were recorded in a 1.00-mm path-length Suprasil quartz cell with a Jasco-J715 CD spectrometer equipped with a thermoelectric temperature-controlled cuvette holder. Far-UV CD was recorded in a range of 190–260 nm using a step size of 0.5 nm with a signal averaging time of 4 s at each wavelength step. 300 μl of TUM1-Iso1 (black line) and TUM1-Iso2 (dashed line) at a concentration of 0.15 mg/ml was used and normalized to the mean residue weight of each TUM1 isoform.
Steady-state Kinetics of TUM1
The kinetic constants of the TUM1 variants were determined as bisubstrate kinetics by quantification of produced sulfide or thiocyanate (33, 42). As shown in Table 2, the kcat for sulfide production by TUM1-Iso1 was determined to be 1252 ± 16 min−1, with Km values of 0.55 ± 0.06 mm for 3-MP and 2.69 ± 0.49 mm for DTT. The corresponding catalytic efficiencies were calculated to be 2289 min−1 mm−1 for 3-MP and 466 min−1 mm−1 for DTT, respectively. In comparison, a kcat of 1211 ± 19 min−1, and Km values of 0.54 ± 0.03 mm for 3-MP and 2.59 ± 0.42 mm for DTT were determined for TUM1-Iso2, resulting in catalytic efficiencies of 2291 min−1 mm−1 for 3-MP and 460 min−1 mm−1 for DTT. Analysis of thiocyanate production by TUM1-Iso1 showed a kcat of 528 ± 8 min−1 and Km values of 1.33 ± 0.30 mm for 3-MP and 4.45 ± 0.72 mm for KCN, respectively. Calculation of catalytic efficiencies delivered values of 398 min−1 mm−1 for 3-MP, respectively, 118 min−1 mm−1 for KCN. For TUM1-Iso2 a kcat of 517 ± 14 min−1, and Km values of 1.29 ± 0.62 mm for 3-MP and 4.50 ± 1.22 mm for KCN were determined, resulting in the catalytic efficiencies of 402 min−1 mm−1 for 3-MP and 115 min−1 mm−1 for KCN. These results show that both TUM1 isoforms have similar kinetic constants under our assay conditions.
TABLE 2.
Kinetic constants of TUM1 variants
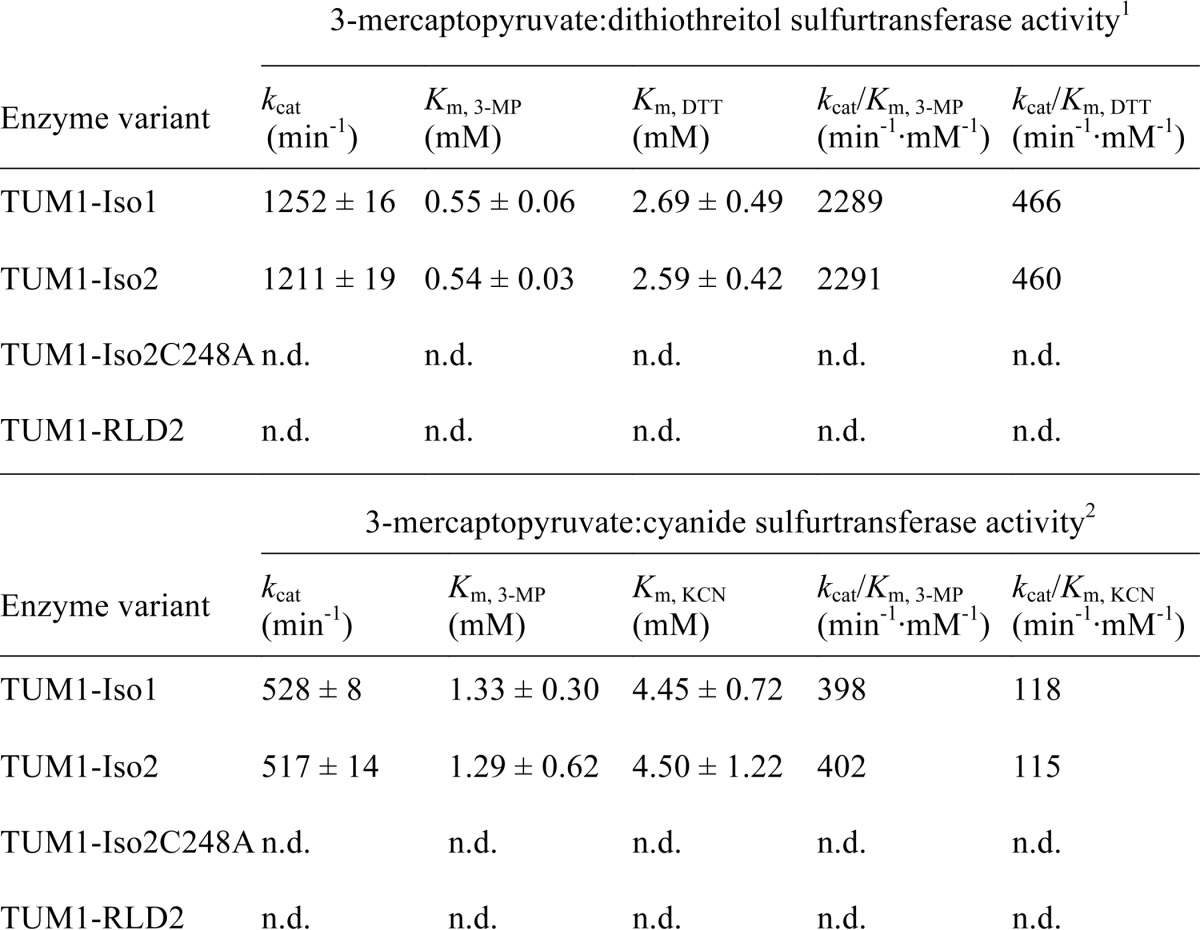
1 Determined in 50 mm Tris-HCl, 200 mm NaCl, pH 10.5, at 37 °C using the methylene blue assay (32).
2 Determined in 100 mM CAPS, pH 10.5, at 37 °C using the assay after Sörbo (33). n.d., = not detectable.
Analysis of Protein-Protein Interactions by Surface Plasmon Resonance Experiments
Because an involvement of TUM1 in sulfur transfer was postulated, we wanted to analyze the direct interaction of the two human TUM1-Iso1 and TUM1-Iso2 isoforms with the l-cysteine desulfurase NFS1 and the rhodanese-like protein MOCS3 from humans. For detection of in vitro interactions of TUM1-Iso1 and TUM1-Iso2, the stable forms of the proteins NFS1Δ1–55, NFS1Δ1–55·ISD11, and MOCS3 were purified as described previously. SPR experiments were performed in both directions with proteins either immobilized on a CM5 chip via amine coupling or used as analytes. Mean KD values obtained from at least three independent SPR measurements for the protein pairs are listed in Table 3. Immobilized TUM1-Iso1 and TUM-Iso2 were shown to interact with NFS1Δ1–55 with KD values of 0.18 ± 0.08 and 0.26 ± 0.1 μm, respectively. Also in the absence of ISD11, the stabilizing protein for NFS1, an interaction with both TUM1-Iso1 and TUM1-Iso2 was obtained, however, the KD values were about 10 times higher (Table 3). Similar results were obtained in the other direction, when NFS1Δ1–55·ISD11 or NFS1 were immobilized, with KD values of 2.92 ± 0.76 (TUM1-Iso1) and 2.91 ± 1.22 μm (TUM1-Iso2) for the interaction with NFS1·ISD11, and higher KD values of 4.06 ± 1.19 (TUM1-Iso1) and 2.45 ± 0.55 μm (TUM1-Iso2) for the interaction with NFS1. Additionally, an interaction of TUM1 with MOCS3 and its C-terminal rhodanese-like domain MOCS3-RLD was obtained. Measurements with immobilized MOCS3 and the TUM1 isoforms as analytes showed KD values of 3.07 ± 0.08 μm for TUM1-Iso1 and 3.58 ± 0.55 μm for TUM1-Iso2. Similar KD values were obtained with immobilized MOCS3-RLD, with a KD of 2.61 ± 0.33 μm for TUM1-Iso1 and a KD of 3.19 ± 1.80 for TUM1-Iso2. This shows that the interaction of both proteins is mainly mediated by the MOCS3-RLD, which contains an active-site cysteine for sulfur transfer. In comparison, with BSA or E. coli IscS no interaction was determined.
TABLE 3.
Results from SPR measurements using purified proteins
| Immobilized proteina | Protein partnerb | KDc |
|---|---|---|
| μm | ||
| TUM1-Iso1 | NFS1Δ1–55 | 1.63 ± 0.14 |
| NFS1Δ1–55·ISD11 | 0.18 ± 0.08 | |
| BSA | NDd | |
| E. coli IscS | ND | |
| TUM1-Iso2 | NFS1Δ1–55 | 1.11 ± 0.21 |
| NFS1Δ1–55·ISD11 | 0.26 ± 0.10 | |
| BSA | ND | |
| E. coli IscS | ND | |
| NFS1Δ1–55 | TUM1-Iso1 | 4.06 ± 1.19 |
| TUM1-Iso2 | 2.45 ± 0.55 | |
| BSA | ND | |
| E. coli IscS | ND | |
| NFS1Δ1–55·ISD11 | TUM1-Iso1 | 2.92 ± 0.76 |
| TUM1-Iso2 | 2.91 ± 1.22 | |
| BSA | ND | |
| E. coli IscS | ND | |
| MOCS3 | TUM1-Iso1 | 3.07 ± 0.08 |
| TUM1-Iso2 | 3.58 ± 0.55 | |
| BSA | ND | |
| E. coli IscS | ND | |
| MOCS3-RLD | TUM1-Iso1 | 2.61 ± 0.33 |
| TUM1-Iso2 | 3.19 ± 1.8 | |
| BSA | ND | |
| E. coli IscS | ND |
a Proteins were immobilized via amine coupling.
b Proteins were injected by application of the KINJECT protocol. Concentrations were in the range of 0.3–20 μm. Regeneration of the flow cell was achieved by injection of 20 mm HCl.
c KD values and standard deviations were obtained by global fitting to a 1:1 binding model from at least two independent measurements.
d ND, no binding detectable.
Analysis of the l-Cysteine Desulfurase Activity of NFS1 in the Presence of TUM1 and MOCS3
To analyze whether TUM1-Iso1 or TUM1-Iso2 stimulated l-cysteine desulfurase activity of human NFS1, each isoform was incubated with the stable NFS1Δ1–55·ISD11 complex and sulfide production was determined by methylene blue quantification (23, 32). The results in Fig. 6 show that neither TUM1 nor MOCS3 alone enhanced the l-cysteine desulfurase activity of NFS1Δ1–55·ISD11. However, when both proteins were present in 4-fold excess, the sulfide production was increased about 2-fold in comparison to the activity of NFS1Δ1–55·ISD11 alone. This shows that a ternary complex is likely formed with the four proteins, resulting in more efficient sulfide production by NFS1. By comparison, TUM1-Iso1, TUM1-Iso2, or MOCS3 alone were inactive with l-cysteine as sulfur source production (data not shown).
FIGURE 6.

l-cysteine desulfurase activity of the NFS1. The diagrams show the desulfurase activity of the NFS1·ISD11 complex alone and after the addition of varying ratios of: A, TUM1-Iso1 and MOCS3-RLD; B, TUM1-Iso1 and full-length MOCS3; C, TUM1-Iso2 and MOCS3-RLD; D, TUM1-Iso2 and full-length MOCS3. TUM1 isoforms and the respective MOCS3 variant were added at a 1:1 ratio when both components were included in the assay.
Influence of TUM1 on in Vitro MPT Production
Because an interaction of the TUM1 isoforms with MOCS3 was identified above, and because MOCS3 has an additional role in Moco biosynthesis, it was of interest to analyze whether TUM1 stimulated in vitro production of MPT. For in vitro MPT synthesis, a defined synthesis system was utilized that contained excess cPMP, Mg-ATP, varying combinations of the purified proteins NFS1Δ1–55·ISD11, TUM1-Iso1, or TUM1-Iso2, MOCS3, and MOCS2A and E. coli oaE, and 0.4 mm sodium sulfide, sodium thiosulfate, 3-MP or l-cysteine as sulfur sources. E. coli MoaE was used instead of the human homolog MOCS2B, because the former was shown to exhibit a higher activity with MOCS2A in vitro than the latter (28). The formed MPT was quantified after its conversion to the fluorescent oxidation product Form A by acidic iodine and alkaline phosphatase treatment (34). This assay does not contain DTT and, thus, the sulfur transfer reaction is specific from one protein to another.
Maximal MPT production was obtained for assays consisting of MOCS3, MOCS2A, and MoaE with sodium sulfide as direct sulfur source or sodium thiosulfate as substrate for MOCS3 (27, 43). The fluorescence intensity for the produced Form A in these assay mixtures was set to 100%, and all further assay mixtures were related to this value. To determine whether TUM1 is able to transfer its persulfide sulfur to MOCS3, the two TUM1 isoforms were included in the assay using 3-MP as sulfur source. In these assay mixtures a relative amount of Form A of 28.0% with TUM1-Iso1 and 30.0% with TUM1-Iso2 was obtained. In assay mixtures containing NFS1·ISD11 and l-cysteine as sulfur donor for MOCS3, the relative amount of detected Form A was 51%. In contrast, when TUM1-Iso1 or TUM1-Iso2 were additionally present using NFS1·ISD11 and l-cysteine as sulfur source, the relative amount of detected Form A increased to 82.7% in the presence of TUM1-Iso1 and 77.5% in the presence of TUM1-Iso2 (Fig. 7). These results show that NFS1·ISD11, MOCS3, and TUM1 are required to increase the l-cysteine desulfurase activity of NFS1, resulting in a better sulfur transfer to acceptor molecules (as measured by the amount of produced MPT).
FIGURE 7.
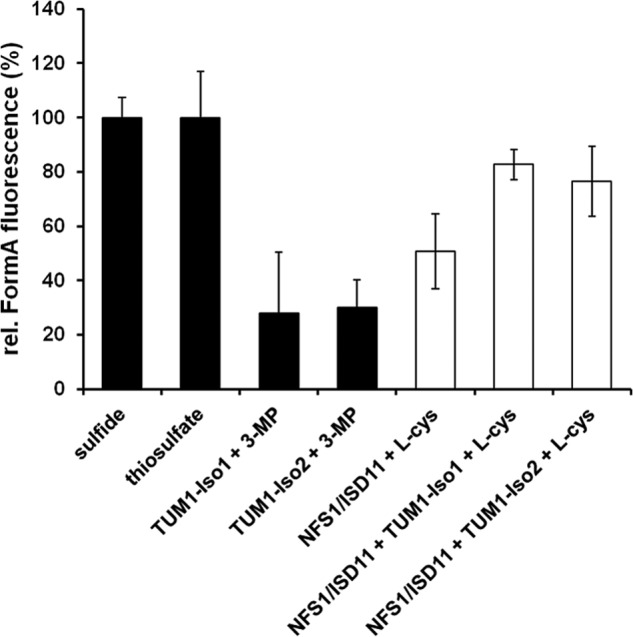
In vitro MPT production. MPT was produced using an in vitro system consisting of 5 μm MoaE, 15 μm MOCS2A, 5 μm MOCS3, 20 μm NFS1·ISD11, 10 μm TUM1-Iso1 or Iso2 and cPMP in excess. Reaction mixtures were set up in 100 mm Tris-HCl, pH 8.0, and incubated for 60 min at room temperature. Following oxidation of MPT, Form A fluorescence was quantified after separation on an HPLC C-18 reversed phase column.
Indirect Immunofluorescence of TUM1 in HEK293a Cells
To analyze the subcellular localization of the two isoforms of TUM1 in human cells, we first performed indirect immunofluorescence for total TUM1 using HEK293a cells. Using an Alexa Fluor 488-conjugated secondary antibody we were able to detect TUM1 in the cytosol and mitochondria (Fig. 8), which is consistent with the previously obtained results (15). As control, we used DAPI for visualization of the nuclei and MitoTracker DeepRed for visualization of the mitochondria. The yellow color in the merged picture (Fig. 8, right panel) reflects the colocalization of the MitoTracker DeepRed and the Alexa Fluor 488 fluorescence and shows that TUM1 is localized in the mitochondria. The same results were obtained with HeLa cells (data not shown).
FIGURE 8.
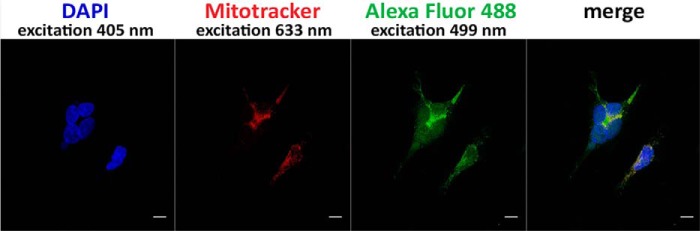
Cellular localization of TUM1 using indirect immunofluorescence in HEK293a cells. Dual localization of TUM1 in the cytosol and the mitochondria using anti-MPST antibody and a Alexa Fluor 488-conjugated secondary antibody. The nuclei were visualized with DAPI stain (blue) and the mitochondria were visualized with MitoTracker DeepRed (red). The yellow color shown in the merged pictures in the right panel indicate colocalization of MitoTracker DeepRed and Alexa Fluor 488 fluorescence. The scale bar represents a range of 10 μm.
Immunodetection of TUM1 in Fractionated Cell Lysates
The localization studies of TUM1 were unable to detect a different localization of TUM1-Iso1 and TUM1-Iso2. To individually analyze the localization of each TUM1 isoform separately, we performed subcellular fractionation of HEK293a cells with subsequent immunoblot detection. We detected two bands in the cytosolic fraction, which were detected by the anti-MPST antibody, in which the band with a molecular mass of 35 kDa corresponds to TUM1-Iso1 and the band with a molecular mass of 33 kDa corresponds to TUM1-Iso2 (Fig. 9A). The lower band with a size of 33 kDa corresponding to TUM1-Iso2 was additionally detected in the mitochondrial fraction. For the analysis of the quality of the subcellular fractionation an anti-citrate synthase antibody was used as a mitochondrial marker (Fig. 9A). We obtained the same result for fractionated HeLa cells (Fig. 9B). Furthermore, we analyzed the cross-reactivity of the anti-MPST antibody with purified thiosulfate sulfurtransferase, which is present only in the mitochondria (5). Fig. 9C shows that the anti-MPST antibody did not cross-react with the purified thiosulfate sulfurtransferase protein, but indeed with purified TUM1-Iso1 and TUM1-Iso2. These data show that the two TUM1 isoforms have a distinct localization pattern with TUM1-Iso1 localized in the cytosol and TUM1-Iso2 localized both in the cytosol and mitochondria.
FIGURE 9.

Immunodetection of TUM1-Iso1 and TUM1-Iso2. A, cytosol (1) and mitochondria (2) fractions were obtained from HEK293a cells. Proteins of each fraction were analyzed by immunoblotting using anti-MPST antibody for the localization of TUM1-Iso1 and TUM1-Iso2. Anti-citrate synthase antibody was used as a marker protein for the mitochondrial matrix. B, cytosol (1) and mitochondria (2) fractions were obtained from HeLa cells. C, Western blot analysis of recombinant TUM1-Iso1 (1), TUM1-Iso2 (2), and thiosulfate sulfurtransferase (3) with anti-MPST antibody. Proteins of each fraction were analyzed by immunodetection using anti-MPST antibody for the localization of TUM1-Iso1 and TUM1-Iso2.
Detection of in Vivo Protein-Protein Interactions with the Split-EGFP System
To directly analyze the cellular interactions of TUM1-Iso1 and TUM1-Iso2 with MOCS3 and NFS1 in human cells, we used the split enhanced green fluorescence protein (split-EGFP) complementation assay. For this the N-terminal 1–157 amino acids of EGFP or the C-terminal 158–238 amino acids of EGFP were fused to TUM1-Iso1, TUM1-Iso2, NFS1, NFS1Δ1–55, or MOCS3 and the two plasmids containing the fusion proteins were co-transfected in HEK392 cells. By a specific interaction of two proteins, the two EGFP parts reassemble, resulting in EGFP fluorescence. As shown in Figs. 10 and 11, we were only able to detect EGFP fluorescence with TUM1-Iso2-EGFP(1–157) and NFS1-EGFP(158–238) in the mitochondria (Fig. 11A) and not with TUM1-Iso1-EGFP(1–157) and NFS1-EGFP(158–238) (Fig. 10A). When the cytosolic form of NFS1 was used without the mitochondrial targeting sequence (NFS1Δ1–55-EGFP(158–238)) fluorescence was only detected in the cytosol with TUM1-Iso1-EGFP(1–157) (Fig. 10B), but not with TUM1-Iso2-EGFP(1–157) (Fig. 11B). Additionally, fluorescence in the cytosol was detected for both TUM1-Iso1-EGFP(1–157) and TUM1-Iso2-EGFP-(1–157) with MOCS3-EGFP-(158–238) (Figs. 10C and 11C). Using HeLa cells the same results were obtained (data not shown). These results show that TUM1-Iso1 interacts with NFS1 and MOCS3 in the cytosol and TUM1-Iso2 interacts with NFS1 in the mitochondria and MOCS3 in the cytosol.
FIGURE 10.

Analysis of TUM1-Iso1 interactions with NFS1, NFS1Δ1–55, and MOCS3 using the split-EGFP system in HEK293a cells. Subcellular EGFP assembly of different split-EGFP fusion proteins was analyzed in HEK293a cells by confocal fluorescent microscopy. The following proteins were expressed after cotransfection (assembly of EGFP(1–157) and EGFP(158–238) resulting in a green pseudocolor): A, TUM1-Iso1-EGFP(1–157) and NFS1-EGFP(158–238); B, TUM1-Iso1-EGFP(1–157) and NFS1Δ1–55-EGFP(158–238); C, TUM1-Iso1-EGFP(1–157) and MOCS3-EGFP(158–238). Mitochondria of HEK293a cells were visualized with MitoTracker DeepRed (red). The nuclei were visualized with DAPI stain (blue). Merged pictures are shown in the right panel and the yellow color indicates colocalization of MitoTracker DeepRed and EGFP fluorescence. The scale bar represents a range of 10 μm.
FIGURE 11.

Analysis of TUM-Iso2 interactions with NFS1, NFS1Δ1–55, and MOCS3 using the split-EGFP system in HEK293a cells. Subcellular EGFP assembly of different split-EGFP fusion proteins was analyzed in HEK293a cells by confocal fluorescent microscopy. The following proteins were expressed after cotransfection (assembly of EGFP(1–157) and EGFP(158–238) resulting in a green pseudocolor): A, TUM1-Iso2-EGFP(1–157) and NFS1-EGFP(158–238); B, TUM1-Iso2-EGFP(1–157) and NFS1Δ1–55-EGFP(158–238); C, TUM1-Iso2-EGFP(1–157) and MOCS3-EGFP(158–238). Mitochondria of HEK293a cells were visualized with MitoTracker DeepRed (red). The nuclei were visualized with DAPI stain (blue). Merged pictures are shown in the right panel and the yellow color indicates colocalization of MitoTracker DeepRed and EGFP fluorescence. The scale bar represents a range of 10 μm.
DISCUSSION
Initially, the main role of TUM1 was described to be involved in cysteine degradation in mitochondria. Here, cysteine is converted by the action of aspartate (or cysteine) aminotransferase to 3-MP, which is converted by TUM1 to pyruvate and enzyme-bound persulfide (Fig. 12) (5, 8). The TUM1-bound persulfide can be transferred to acceptors such as cyanide to generate the less toxic thiocyanate (44). Alternatively, H2S can be released from TUM1 in the presence of reducing systems like thioredoxin or glutathione (45, 46). Inhibition of TUM1 conserves cysteine and contributes to an increase in the cysteine pool (47, 48). An increase in the cysteine content in the cell results in an increase in the content of cellular reductants such as thioredoxin or glutathione. Thus, TUM1 has a major role to maintain the overall redox homeostasis in the cell (49).
FIGURE 12.
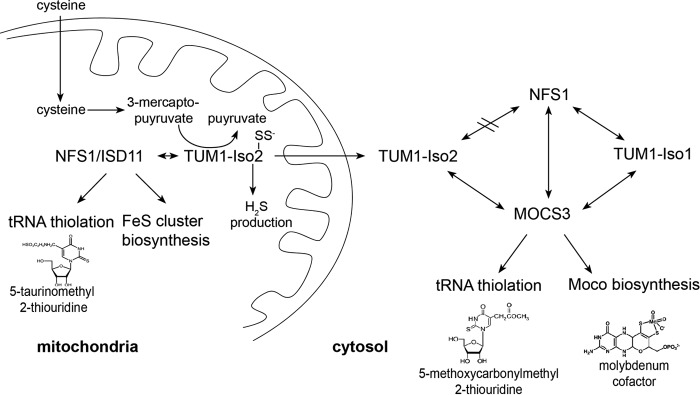
Model of the interaction network of TUM1-Iso1, TUM1-Iso2, NFS1·ISD11, and MOCS3. For TUM1-Iso1 and TUM1-Iso2 a different localization pattern was identified. TUM-Iso1 is exclusively localized in the cytosol where it interacts with MOCS3 and NFS1 (with or without ISD11) for sulfur transfer for the biosynthesis of Moco and the thiolation of tRNA. In contrast, TUM1-Iso2 has a dual localization in both mitochondria and the cytosol. In mitochondria, TUM1-Iso2 interacts with NFS1·ISD11, whereas in the cytosol TUM1-Iso2 interacts with MOCS3 but not with NFS1·ISD11.
In this study we characterized for the first time two isoforms of TUM1, which are derived from two different splice variants. So far, all previous characterizations of TUM1 did not consider that TUM1 exists in two different isoforms in the cell.
The two TUM1 isoforms are distinguished by a 20-amino acid extension at the N terminus of TUM1-Iso1, which results in capping of the mitochondrial targeting sequence of the protein. Mitochondrial TUM1-Iso2 is consequently about 45 amino acids shorter after cleavage of the mitochondrial localization signal as compared with cytosolic TUM1-Iso1.
We first characterized the purified TUM1 isoforms after heterologous expression and purification from E. coli, to differentiate their stability and kinetic behavior. Both proteins showed similar properties in activity, catalytic constants, protein stability, pH dependence, and temperature optimum, showing that the general characteristics of the protein are not influenced by the 20-amino acid extension on TUM1-Iso1. To exclude the fact that the protein purified from E. coli is not correctly folded, we additionally purified both TUM1 variants from SF9 insect cells as a N-terminal His6 tag fusion. The purified protein from the eukaryotic host showed the same pH dependence, excluding the fact that the His6 tag present on the protein contributes to the difference in the pH-dependent catalytic reaction (data not shown). However, because the protein purified from the SF9 cells was about 10-fold less active, all further experiments were performed with the protein expressed and purified from E. coli.
Our results in human cell lines unambiguously prove the existence of the two splice variants TUM1-Iso1 and TUM1-Iso2. However, surprisingly we identified a different cellular localization of both proteins. Our results show that TUM1-Iso1 is exclusively localized in the cytosol, whereas TUM1-Iso2 shows a dual localization both in the cytosol and mitochondria (Fig. 12). The mechanism of how TUM1-Iso2 remains in the cytosol or whether it is targeted back from mitochondria to the cytosol is unknown and has to be elucidated in future studies. Several mechanisms for the eclipsed localization of proteins in the cell have been discussed previously (50–52), including the reverse transport to the cytosol, the incorrect folding in the cytosol or the binding of protein partners, which results in an inhibition of the mitochondrial transport, e.g. by blocking the targeting sequence. However, in our studies TUM1-Iso2 is processed and is localized in the cytosol without the mitochondrial targeting sequence. Thus, in future studies, the enigma of dual localized proteins has to be resolved in detail.
For yeast Tum1p, a role in the sulfur transfer reaction for the thiolation of cytosolic tRNAs was proposed, because in a reverse genetic approach Tum1p was identified to be essential for the formation of mcm5s2U at the wobble position of U34 in tRNAs for Gln, Glu, and Lys (25). This mechanism involves the initial sulfur mobilization by the l-cysteine desulfurase Nfs1p and further activation of Urm1p by Uba4p, ending in the thiocarboxylation of Urm1p. In contrast to humans, these proteins exclusively perform the sulfur transfer reaction for the thiolation of uridine in tRNAs. The second interaction partner of Uba4p, the MOCS2A homologue, is not present in yeast, because yeast cells lost the ability to synthesize the molybdenum cofactor.
Human NFS1 is also a dual localized protein, because its major function is FeS cluster biosynthesis in mitochondria. The function of NFS1 in the cytosol was described to be a sulfur donor for MOCS3, a dual function protein that interacts with URM1 for tRNA thiolation and with MOCS2A for the biosynthesis of Moco (22, 53). Thus, to characterize the role of TUM1 in humans, we analyzed the interaction of both isoforms with MOCS3 and NFS1. In vitro interaction studies using the purified proteins showed that TUM1-Iso1 and TUM1-Iso2 both interacted with MOCS3 and NFS1, and with the NFS1·ISD11 complex with only slight variations in the dissociation constants. Studies on the enzyme activities showed, that TUM1, MOCS3, and NFS1·ISD11 likely form a ternary complex for effective sulfur transfer from the l-cysteine desulfurase NFS1. This was proven by analyzing the NFS1 activity for effective sulfide production in the methylene blue assay, or by the detection of the effective MPT production in a complex in vitro system containing all the components required for the conversion of cPMP to MPT in Moco biosynthesis. In both cases, the highest activity was obtained when all four proteins (TUM1, MOCS3, and NFS1·ISD11) were present.
For the detection of protein-protein interactions in human cells, the split-EGFP system was used. In vivo in HEK293a and HeLa cells, an interaction of TUM1-Iso1 with both MOCS3 and NFS1 in the cytosol was determined, consistent with the interaction studies determined in vitro. However, for TUM1-Iso2 a different pattern was obtained. Here, TUM1-Iso2 interacted with NFS1 only in mitochondria and not in the cytosol, where an interaction of TUM1-Iso2 with MOCS3 was readily determined. This observation might point to different roles of TUM1-Iso1 and TUM1-Iso2 in both compartments. In mitochondria, TUM1-Iso2 interacts with NFS1 for sulfur transfer for taurine-modified tRNAs. Here, NFS1 also has a dual role, because on the one hand it mobilizes the sulfur for the biosynthesis of FeS clusters, and on the other hand an involvement of NFS1 for the synthesis of thiolated mitochondrial tRNAs was described. Thus, TUM1-Iso2 might be involved in the interaction of NFS1 to direct sulfur transfer to the tRNA thiolation, e.g. by blocking the interaction site for ISCU on NFS1.
By comparison, the functional roles for each of the TUM1 isoforms can be expected as different in the cytosol. To date, the principal function of NFS1 in the cytosol has been to perform sulfur transfer to MOCS3 for Moco biosynthesis and the thiolation of cytoplasmic tRNAs (53). In this compartment, TUM1-Iso1 interacts with both MOCS3 and NFS1. The additional presence of TUM1-Iso2 and its ability to interact only with MOCS3, and not with NFS1, might enable an additional route for sulfur transfer from alternative substrates like 3-MP or by other, so far not identified interaction partners (Fig. 12). Here, the difference to yeast cells is obvious, where TUM1 isoforms are not present, which might not be necessary for effective sulfur transfer, because only the route for tRNA thiolation exists, and not the additional route for sulfur transfer for Moco formation. Thus in humans, the additional splice variant of TUM1 might ensure an additional sulfur transfer route to the interaction partner MOCS3, which might be necessary due to its additional involvement in Moco biosynthesis. In total, TUM1 seems to be a multifunctional protein for which different splice variants exist, which contribute to different localization patterns and multifunctional roles both in the cytosol and mitochondria of human cells (Fig. 12).
Acknowledgments
We thank Angelika Lehmann for technical assistance, Klaus Gast for help with CD spectroscopy, Otto Baumann for help with cellular localization studies, and Benjamin Duffus for critical reading of the manuscript and helpful comments.
This work was supported by Deutsche Forschungsgemeinschaft Grant LE1171/9-1.
- MPST
- 3-mercaptopyruvate sulfurtransferase
- TUM1
- tRNA thiouridine modification protein 1
- NFS1
- nitrogen fixation homolog 1
- MOCS3
- molybdenum cofactor synthesis protein 3
- RLD
- rhodanese-like domain
- URM1
- ubiquitin-related modifier 1
- Uba4
- ubiquitin-like protein activator 4
- MPT
- molybdopterin
- cPMP
- cyclic pyranopterin monophosphate
- Moco
- molybdenum cofactor
- MOCS2A
- molybdenum cofactor synthesis protein 2A
- 3-MP
- 3-mercaptopyruvate
- SPR
- surface plasmon resonance
- EGFP
- enhanced green fluorescent protein
- CAPS
- 3-(cyclohexylamino)propanesulfonic acid
- CHES
- 2-(cyclohexylamino)ethanesulfonic acid.
REFERENCES
- 1. Bordo D., Bork P. (2002) The rhodanese/Cdc25 phosphatase superfamily. Sequence-structure-function relations. EMBO Rep. 3, 741–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Keyse S. M., Ginsburg M. (1993) Amino acid sequence similarity between CL100, a dual-specificity MAP kinase phosphatase and cdc25. Trends Biochem. Sci 18, 377–378 [DOI] [PubMed] [Google Scholar]
- 3. Ploegman J. H., Drent G., Kalk K. H., Hol W. G. (1978) Structure of bovine liver rhodanese: I. structure determination at 2.5-Å resolution and a comparison of the conformation and sequence of its two domains. J. Mol. Biol. 123, 557–594 [PubMed] [Google Scholar]
- 4. Ploegman J. H., Drent G., Kalk K. H., Hol W. G., Heinrikson R. L., Keim P., Weng L., Russell J. (1978) The covalent and tertiary structure of bovine liver rhodanese. Nature 273, 124–129 [DOI] [PubMed] [Google Scholar]
- 5. Nagahara N., Okazaki T., Nishino T. (1995) Cytosolic mercaptopyruvate sulfurtransferase is evolutionarily related to mitochondrial rhodanese: striking similarity in active site amino acid sequence and the increase in the mercaptopyruvate sulfurtransferase activity of rhodanese by site-directed mutagenesis. J. Biol. Chem. 270, 16230–16235 [DOI] [PubMed] [Google Scholar]
- 6. Mueller E. G. (2006) Trafficking in persulfides: delivering sulfur in biosynthetic pathways. Nat. Chem. Biol. 2, 185–194 [DOI] [PubMed] [Google Scholar]
- 7. Toohey J. I. (2011) Sulfur signaling: is the agent sulfide or sulfane? Anal. Biochem. 413, 1–7 [DOI] [PubMed] [Google Scholar]
- 8. Shibuya N., Tanaka M., Yoshida M., Ogasawara Y., Togawa T., Ishii K., Kimura H. (2009) 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid. Redox Signal. 11, 703–714 [DOI] [PubMed] [Google Scholar]
- 9. Ubuka T., Ohta J., Yao W. B., Abe T., Teraoka T., Kurozumi Y. (1992) l-Cysteine metabolism via 3-mercaptopyruvate pathway and sulfate formation in rat liver mitochondria. Amino Acids 2, 143–155 [DOI] [PubMed] [Google Scholar]
- 10. Westley J. (1989) Depletion of the sulphane pool: toxicological implications. in Sulphur-Containing Drugs and Related Organic Compounds. pp. 87–99, Wiley & Sons, New York [Google Scholar]
- 11. Dahl J. U., Urban A., Bolte A., Sriyabhaya P., Donahue J. L., Nimtz M., Larson T. J., Leimkühler S. (2011) The identification of a novel protein involved in molybdenum cofactor biosynthesis in Escherichia coli. J. Biol. Chem. 286, 35801–35812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Palenchar P. M., Buck C. J., Cheng H., Larson T. J., Mueller E. G. (2000) Evidence that ThiI, an enzyme shared between thiamin and 4-thiouridine biosynthesis, may be a sulfurtransferase that proceeds through a persulfide intermediate. J. Biol. Chem. 275, 8283–8286 [DOI] [PubMed] [Google Scholar]
- 13. Yadav P. K., Yamada K., Chiku T., Koutmos M., Banerjee R. (2013) Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J. Biol. Chem. 288, 20002–20013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Qiu R., Wang F., Liu M., Lou T., Ji C. (2012) Crystal structure of the Tum1 protein from the yeast Saccharomyces cerevisiae. Protein Pept. Lett. 19, 1139–1143 [DOI] [PubMed] [Google Scholar]
- 15. Taniguchi T., Kimura T. (1974) Role of 3-mercaptopyruvate sulfurtransferase in the formation of the iron-sulfur chromophore of adrenal ferredoxin. Biochim. Biophys. Acta 364, 284–295 [DOI] [PubMed] [Google Scholar]
- 16. Hannestad U., Mårtensson J., Sjödahl R., Sörbo B. (1981) 3-Mercaptolactate cysteine disulfiduria: biochemical studies on affected and unaffected members of a family. Biochem. Med. 26, 106–114 [DOI] [PubMed] [Google Scholar]
- 17. Billaut-Laden I., Rat E., Allorge D., Crunelle-Thibaut A., Cauffiez C., Chevalier D., Lo-Guidice J. M., Broly F. (2006) Evidence for a functional genetic polymorphism of the human mercaptopyruvate sulfurtransferase (MPST), a cyanide detoxification enzyme. Toxicol. Lett. 165, 101–111 [DOI] [PubMed] [Google Scholar]
- 18. Hildebrandt T. M., Grieshaber M. K. (2008) Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. Febs J. 275, 3352–3361 [DOI] [PubMed] [Google Scholar]
- 19. Mikami Y., Shibuya N., Kimura Y., Nagahara N., Ogasawara Y., Kimura H. (2011) Thioredoxin and dihydrolipoic acid are required for 3-mercaptopyruvate sulfurtransferase to produce hydrogen sulfide. Biochem. J. 439, 479–485 [DOI] [PubMed] [Google Scholar]
- 20. Nakai Y., Nakai M., Lill R., Suzuki T., Hayashi H. (2007) Thio modification of yeast cytosolic tRNA is an iron-sulfur protein-dependent pathway. Mol. Cell. Biol. 27, 2841–2847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schmitz J., Chowdhury M. M., Hänzelmann P., Nimtz M., Lee E. Y., Schindelin H., Leimkühler S. (2008) The sulfurtransferase activity of Uba4 presents a link between ubiquitin-like protein conjugation and activation of sulfur carrier proteins. Biochemistry 47, 6479–6489 [DOI] [PubMed] [Google Scholar]
- 22. Leidel S., Pedrioli P. G., Bucher T., Brost R., Costanzo M., Schmidt A., Aebersold R., Boone C., Hofmann K., Peter M. (2009) Ubiquitin-related modifier Urm1 acts as a sulphur carrier in thiolation of eukaryotic transfer RNA. Nature 458, 228–232 [DOI] [PubMed] [Google Scholar]
- 23. Noma A., Sakaguchi Y., Suzuki T. (2009) Mechanistic characterization of the sulfur-relay system for eukaryotic 2-thiouridine biogenesis at tRNA wobble positions. Nucleic Acids Res. 37, 1335–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chowdhury M. M., Dosche C., Löhmannsröben H. G., Leimkühler S. (2012) Dual role of the molybdenum cofactor biosynthesis protein MOCS3 in tRNA thiolation and molybdenum cofactor biosynthesis in humans. J. Biol. Chem. 287, 17297–17307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nakai Y., Nakai M., Hayashi H. (2008) Thio-modification of yeast cytosolic tRNA requires a ubiquitin-related system that resembles bacterial sulfur transfer systems. J. Biol. Chem. 283, 27469–27476 [DOI] [PubMed] [Google Scholar]
- 26. Van der Veen A. G., Schorpp K., Schlieker C., Buti L., Damon J. R., Spooner E., Ploegh H. L., Jentsch S. (2011) Role of the ubiquitin-like protein Urm1 as a noncanonical lysine-directed protein modifier. Proc. Natl. Acad. Sci. U.S.A. 108, 1763–1770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Matthies A., Rajagopalan K. V., Mendel R. R., Leimkühler S. (2004) Evidence for the physiological role of a rhodanese-like protein for the biosynthesis of the molybdenum cofactor in humans. Proc. Natl. Acad. Sci. U.S.A. 101, 5946–5951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leimkuhler S., Freuer A., Araujo J. A., Rajagopalan K. V., Mendel R. R. (2003) Mechanistic studies of human molybdopterin synthase reaction and characterization of mutants identified in group B patients of molybdenum cofactor deficiency. J. Biol. Chem. 278, 26127–26134 [DOI] [PubMed] [Google Scholar]
- 29. Hille R., Nishino T., Bittner F. (2011) Molybdenum enzymes in higher organisms. Coord. Chem. Rev. 255, 1179–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wuebbens M. M., Rajagopalan K. V. (2003) Mechanistic and mutational studies of Escherichia coli molybdopterin synthase clarify the final step of molybdopterin biosynthesis. J. Biol. Chem. 278, 14523–14532 [DOI] [PubMed] [Google Scholar]
- 31. Marelja Z., Stöcklein W., Nimtz M., Leimkühler S. (2008) A novel role for human Nfs1 in the cytoplasm: Nfs1 acts as a sulfur donor for MOCS3, a protein involved in molybdenum cofactor biosynthesis. J. Biol. Chem. 283, 25178–25185 [DOI] [PubMed] [Google Scholar]
- 32. Fogo J. K., Popowsky M. (1949) Spectrophotometric determination of hydrogen sulfide. Anal. Chem. 21, 732–734 [Google Scholar]
- 33. Sorbo B. (1957) A colorimetric method for the determination of thiosulfate. Biochim. Biophys. Acta 23, 412–416 [DOI] [PubMed] [Google Scholar]
- 34. Johnson J. L., Hainline B. E., Rajagopalan K. V., Arison B. H. (1984) The pterin component of the molybdenum cofactor. Structural characterization of two fluorescent derivatives. J. Biol. Chem. 259, 5414–5422 [PubMed] [Google Scholar]
- 35. Schmitz J., Wuebbens M. M., Rajagopalan K. V., Leimkühler S. (2007) Role of the C-terminal Gly-Gly motif of Escherichia coli MoaD, a molybdenum cofactor biosynthesis protein with a ubiquitin fold. Biochemistry 46, 909–916 [DOI] [PubMed] [Google Scholar]
- 36. Wuebbens M. M., Rajagopalan K. V. (1995) Investigation of the early steps of molybdopterin biosynthesis in Escherichia coli through the use of in vivo labeling studies. J. Biol. Chem. 270, 1082–1087 [DOI] [PubMed] [Google Scholar]
- 37. Miyamoto R., Otsuguro K., Yamaguchi S., Ito S. (2014) Contribution of cysteine aminotransferase and mercaptopyruvate sulfurtransferase to hydrogen sulfide production in peripheral neurons. J. Neurochem. 130, 29–40 [DOI] [PubMed] [Google Scholar]
- 38. Dunham I., Shimizu N., Roe B. A., Chissoe S., Hunt A. R., Collins J. E., Bruskiewich R., Beare D. M., Clamp M., Smink L. J., Ainscough R., Almeida J. P., Babbage A., Bagguley C., Bailey J., Barlow K., Bates K. N., Beasley O., Bird C. P., Blakey S., Bridgeman A. M., Buck D., Burgess J., Burrill W. D., O'Brien K. P. (1999) The DNA sequence of human chromosome 22. Nature 402, 489–495 [DOI] [PubMed] [Google Scholar]
- 39. Horowitz P., Criscimagna N. L. (1983) The use of intrinsic protein fluorescence to quantitate enzyme-bound persulfide and to measure equilibria between intermediates in rhodanese catalysis. J. Biol. Chem. 258, 7894–7896 [PubMed] [Google Scholar]
- 40. Cannella C., Berni R., Rosato N., Finazzi-Agrò A. (1986) Active site modifications quench intrinsic fluorescence of rhodanese by different mechanisms. Biochemistry 25, 7319–7323 [DOI] [PubMed] [Google Scholar]
- 41. Colnaghi R., Cassinelli G., Drummond M., Forlani F., Pagani S. (2001) Properties of the Escherichia coli rhodanese-like protein SseA: contribution of the active-site residue Ser-240 to sulfur donor recognition. FEBS Lett. 500, 153–156 [DOI] [PubMed] [Google Scholar]
- 42. Pandurangappa M., Samrat D. (2010) Micellar-mediated extractive spectrophotometric determination of hydrogen sulfide/sulfide through Prussian Blue reaction: application to environmental samples. Anal. Sci. 26, 83–87 [DOI] [PubMed] [Google Scholar]
- 43. Matthies A., Nimtz M., Leimkühler S. (2005) Molybdenum cofactor biosynthesis in humans: identification of a persulfide group in the rhodanese-like domain of MOCS3 by mass spectrometry. Biochemistry 44, 7912–7920 [DOI] [PubMed] [Google Scholar]
- 44. Nagahara N., Ito T., Minami M. (1999) Mercaptopyruvate sulfurtransferase as a defense against cyanide toxication: molecular properties and mode of detoxification. Histol. Histopathol. 14, 1277–1286 [DOI] [PubMed] [Google Scholar]
- 45. Westrop G. D., Georg I., Coombs G. H. (2009) The mercaptopyruvate sulfurtransferase of Trichomonas vaginalis links cysteine catabolism to the production of thioredoxin persulfide. J. Biol. Chem. 284, 33485–33494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Williams R. A., Kelly S. M., Mottram J. C., Coombs G. H. (2003) 3-Mercaptopyruvate sulfurtransferase of Leishmania contains an unusual C-terminal extension and is involved in thioredoxin and antioxidant metabolism. J. Biol. Chem. 278, 1480–1486 [DOI] [PubMed] [Google Scholar]
- 47. Crawhall J. C., Parker R., Sneddon W., Young E. P., Ampola M. G., Efron M. L., Bixby E. M. (1968) β-Mercaptolactate-cysteine disulfide: analog of cystine in the urine of a mentally retarded patient. Science 160, 419–420 [DOI] [PubMed] [Google Scholar]
- 48. Nagahara N., Ito T., Kitamura H., Nishino T. (1998) Tissue and subcellular distribution of mercaptopyruvate sulfurtransferase in the rat: confocal laser fluorescence and immunoelectron microscopic studies combined with biochemical analysis. Histochem. Cell Biol. 110, 243–250 [DOI] [PubMed] [Google Scholar]
- 49. Nagahara N., Katayama A. (2005) Post-translational regulation of mercaptopyruvate sulfurtransferase via a low redox potential cysteine-sulfenate in the maintenance of redox homeostasis. J. Biol. Chem. 280, 34569–34576 [DOI] [PubMed] [Google Scholar]
- 50. Regev-Rudzki N., Karniely S., Ben-Haim N. N., Pines O. (2005) Yeast aconitase in two locations and two metabolic pathways: seeing small amounts is believing. Mol. Biol. Cell 16, 4163–4171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sass E., Karniely S., Pines O. (2003) Folding of fumarase during mitochondrial import determines its dual targeting in yeast. J. Biol. Chem. 278, 45109–45116 [DOI] [PubMed] [Google Scholar]
- 52. Yogev O., Naamati A., Pines O. (2011) Fumarase: a paradigm of dual targeting and dual localized functions. FEBS J. 278, 4230–4242 [DOI] [PubMed] [Google Scholar]
- 53. Marelja Z., Mullick Chowdhury M., Dosche C., Hille C., Baumann O., Löhmannsröben H. G., Leimkühler S. (2013) The l-cysteine desulfurase NFS1 is localized in the cytosol where it provides the sulfur for molybdenum cofactor biosynthesis in humans. PLoS One 8, e60869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Studier F. W., Moffatt B. A. (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189, 113–130 [DOI] [PubMed] [Google Scholar]
- 55. Scherer W. F., Syverton J. T., Gey G. O. (1953) Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix. J. Exp. Med. 97, 695–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Graham F. L., Smiley J., Russell W. C., Nairn R. (1977) Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36, 59–74 [DOI] [PubMed] [Google Scholar]