

Background: Galbonolide (GAL) was proposed to be synthesized by a modular type I polyketide synthase (PKS) with GalA-E serving a supporting role.
Results: GalA-C constitute a sole type I PKS that is involved in GAL biosynthesis in S. galbus.
Conclusion: GalA-C catalyze the de novo formation of GAL macrolactone.
Significance: GalA-C constitute a novel iterative PKS that incorporates methylmalonate units with highly programmed β-keto group modifications.
Keywords: Actinobacteria, Bacterial Genetics, Gene Knockout, Natural Product Biosynthesis, Polyketide, Streptomyces galbus, Galbonolide, Highly Reducing Type I Polyketide Synthase
Abstract
Galbonolide (GAL) A and B are antifungal macrolactone polyketides produced by Streptomyces galbus. During their polyketide chain assembly, GAL-A and -B incorporate methoxymalonate and methylmalonate, respectively, in the fourth chain extension step. The methoxymalonyl-acyl carrier protein biosynthesis locus (galG to K) is specifically involved in GAL-A biosynthesis, and this locus is neighbored by a gene cluster composed of galA-E. GalA-C constitute a single module, highly reducing type I polyketide synthase (PKS). GalD and GalE are cytochrome P450 and Rieske domain protein, respectively. Gene knock-out experiments verified that galB, -C, and -D are essential for GAL biosynthesis. A galD mutant accumulated a GAL-C that lacked two hydroxyl groups and a double bond when compared with GAL-B. A [U-13C]propionate feeding experiment indicated that no rare precursor other than methoxymalonate was incorporated during GAL biogenesis. A search of the S. galbus genome for a modular type I PKS system, the type that was expected to direct GAL biosynthesis, resulted in the identification of only one modular type I PKS gene cluster. Homology analysis indicated that this PKS gene cluster is the locus for vicenistatin biosynthesis. This cluster was previously reported in Streptomyces halstedii. A gene deletion of the vinP2 ortholog clearly demonstrated that this modular type I PKS system is not involved in GAL biosynthesis. Therefore, we propose that GalA-C direct macrolactone polyketide formation for GAL. Our studies provide a glimpse into a novel biochemical strategy used for polyketide synthesis; that is, the iterative assembly of propionates with highly programmed β-keto group modifications.
Introduction
Galbonolide (GAL)2 A and B were first isolated from Streptomyces galbus subsp. eurythermus Tü 2253 due to their antifungal activities (1). When tested against several fungal strains, including Rhizoctonia solani and Pichia farinose, GAL-A exhibited a greater potency than GAL-B. GAL-A and -B were also reported in Micromonospora narashinoensis and Micromonospora chalcea and were named as rustmicin and neorustmicin A, respectively (2, 3). GAL-A is potent against wheat stem rust fungus Puccinia graminis, Botrytis cinerea, and other phytopathogenic fungi. GAL-A also exhibits remarkable potency against several human pathogens, including one of the most common opportunistic fungal pathogens, Cryptococcus neoformans. It was later found that a selective inhibition of fungal sphingolipid biosynthesis at the level of inositol phosphoceramide synthase is responsible for the antifungal activity of GAL-A (4).
Many clinically important drugs, including erythromycin, daunorubicin, and lovastatin, are synthesized by polyketide pathways. Polyketide pathways generate structurally diverse natural products but are common in their biochemical strategy for C-C bond formation. Polyketide synthases (PKSs) all catalyze C-C bond formation through decarboxylative Claisen condensation with their catalytic triad (Cys-His-His/Asn). This condensation is carried out between an acylthioester attached to the catalytic Cys residue of β-ketoacyl-thioester synthase (KS) and an incoming malonyl-thioester, generally malonyl-acyl carrier protein (-ACP) or methylmalonyl-ACP. PKSs are classified as type I or type II systems depending on their functionality architecture (5, 6). Type I PKSs are composed of multifunctional (multidomain) polypeptides, whereas type II PKSs are a complex of monofunctional enzymes. Type III PKSs are single condensation enzymes that lack ACP and incorporate malonyl-CoA directly.
In type I PKS catalysis, an acyltransferase (AT) domain in the module activates its substrate, a malonyl-thioester derivative, and charges the cognate ACP domain with it. The malonyl-thioester derivatives include malonyl-CoA, methylmalonyl-CoA, ethylmalonyl-CoA, chloroethylmalonyl-CoA, methoxymalonyl-ACP, hydroxymalonyl-ACP, and aminomalonyl-ACP (6). The malonyl-thioester derivative that is attached to the ACP domain is then incorporated into a growing polyketide chain through a decarboxylative Claisen condensation reaction. Type I PKS systems include post-condensation modification domains such as β-ketoacyl-ACP reductase (KR), β-hydroxyacyl-ACP dehydratase (DH), trans-2-enoyl-ACP reductase (ER), and others (6). The terminal thioesterase domain liberates the PKS product. In many cases the thioesterase domain liberates the product in a macrolactone form.
In modular PKSs, which are limited to the type I PKS system, one module is responsible for one condensation plus the following modifications (7). In each elongation step performed by a multimodule PKS, the extent of reductive modification is determined by the particular combination of extra catalytic domains embedded in the cognate module. These combinations include KR, KR/DH, and KR/DH/ER (6). The overall domain organization thereby determines a backbone structure of the product. This Lego-like feature of the modular system provides a way to generate the structural diversity of polyketides in nature.
Iterative bacterial type I PKSs have also been reported for aromatic polyketide synthesis, and the chemical strategy of this non-reducing-type PKS is comparable with that of type II PKS (8). Fungal type I PKSs are iterative, but their products are not limited to aromatic compounds (9). The highly reducing, iterative fungal PKSs synthesize complex polyketide structures by selectively employing post-condensational modification activities in a highly programmed manner (10, 11). In these fungal PKSs, the selective usage of the modification activities in each condensation step generates complex polyketide structures. However, the control mechanism of this selective usage is largely veiled.
The application of the polyketide biosynthetic logic to the biosynthesis of GAL-A and -B led to the hypothesis that the selection of an alternative precursor, occurring during the installation of C-5 and C-6, results in the concurrent production of GAL-A and -B (Fig. 1A). The fourth extension units were thus proposed to be methoxymalonyl-ACP and methylmalonyl-CoA for GAL-A and -B, respectively. The cloning of the methoxymalonyl-ACP biosynthesis locus (galG to K) in S. galbus revealed that this locus was neighbored by a gene set (galA to E) that encodes a single module PKS, a cytochrome P450 protein, and a Rieske N-terminal domain protein (12). GalA contains an AT domain and a thiolation domain, which may act as an ACP (Fig. 1B) (note that GAL and Gal represent the galbonolide compounds and the deduced proteins of the galbonolide biosynthetic genes, respectively, in the present report). A domain architecture analysis with the InterProScan interface predicted that GalB contains ER/KS/KR/DH domains. A truncated KS domain was also found between the KS and KR domains. The C-terminal DH domain is least conserved but displays significant homology to the PF14765 domain in the Pfam protein family database. In fact, GalB (protein sequence identity of E5DHQ1) is listed in the ER/KS/KR/DH domain protein family in the Pfam server. GalC is similar to FabH, but the catalytic Cys residue is replaced with Ser.
FIGURE 1.

The proposed biosynthetic pathway of GAL. A, a schematic representation of the GAL macrolactone backbone assembly line and the proposed biosynthetic pathway. B, the proposed role of GalA-E in GAL biosynthesis; the intermediacy of GAL-C in GAL-B biogenesis is described. For simplicity, the stereochemistry in each chiral center is not shown in the structure but is instead indicated by R or S. GalB harbors a truncated KS domain. GalC is homologous to known KSs but lacks the catalytic Cys residue. Both are indicated as ψKS to denote that they are non-canonical KSs.
It was previously shown that an inactivation of galI, one of the methoxymalonyl-ACP biosynthetic genes, selectively abolished GAL-A biosynthesis, whereas a vector-integrating gene disruption of galB dramatically reduced the level of GAL-A and -B (12). This galB disruption study implied that galA-E are involved in GAL biosynthesis; however, their biochemical roles remained veiled. In this study gene replacement experiments demonstrate that galA-E are GAL biosynthetic genes. Intermediate isolation, [13C]propionate feeding, and S. galbus genome mining experiments led us to propose that GalA-E constitute a novel PKS system. This system iteratively incorporates methylmalonate units and is capable of carrying out highly programmed β-keto group processing using a single set of KR/DH/ER domains.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Culture Media, and Growth Conditions
S. galbus KCCM 41354 was acquired from the Korean Culture Center of Microorganisms (Seoul, Korea), and C. neoformans IFO 40092 was obtained from the Culture Collection of the Research Centre for Pathogenic Fungi and Microbial Toxicoses (Chiba University, Chiba, Japan). S. galbus and C. neoformans were maintained on GYM plates (0.4% glucose, 0.4% yeast extract, 1% malt extract, 0.2% calcium carbonate, and 2% Bacto agar) at 30 °C. For GAL production, the preculture of S. galbus was grown at 28 °C with vigorous shaking in GAL production medium, which contained 0.8% glucose, 0.4% yeast extract, 1% malt extract, and 1 mm l-isoleucine (13). After 3 days, the production culture was initiated in GAL production medium, with volume of inoculum from the preculture, and maintained for 48 h at the same condition. C. neoformans was cultured in Bennett medium (1% d-glucose, 0.2% peptone, 0.1% yeast extract, and 0.1% beef extract) at 28 °C overnight with vigorous shaking.
Escherichia coli DH5α, E. coli W25113/pIJ790, and E. coli ET12567 (dam−, dcm−, hsdS−)/pUZ8002 (14) were used for routine subcloning, PCR-targeting mutagenesis (15), and intergeneric conjugation (16), respectively. These E. coli strains and their derivative strains were routinely grown at 37 °C in Luria-Bertani (LB) medium with vigorous shaking. When necessary, antibiotics were included in the LB culture at the following final concentrations: ampicillin, 100 μg/ml; apramycin, 50 μg/ml; kanamycin, 25 μg/ml; chloramphenicol, 20 μg/ml.
Intergeneric Conjugation and Gene Inactivation
The primer pairs used in this study are listed in Table 1. The PCR-targeting mutagenesis method was used in the preparation of gene inactivation constructs (15). Intergeneric conjugation was used to introduce plasmids into S. galbus (12). Specifically, the conjugation experiment was started with the introduction of a gene inactivation construct into E. coli ET12567/pUZ8002. Five hundred microliters of the overnight culture (the E. coli ET12567/pUZ8002 transformant) was inoculated into 50 ml of LB medium with kanamycin, apramycin, ampicillin, and 10 mm MgCl2. This culture was maintained at 37 °C in a rotary shaker. When the optical density (at 600 nm) reached 0.6, cells were collected by centrifugation at 4000 rpm for 20 min, and the cell pellet was washed twice with LB medium containing 10 mm MgCl2. Then the E. coli cells were resuspended in 5 ml of LB medium with 10 mm MgCl2 and used as the donor cell of the intergeneric conjugation. S. galbus was cultivated in 50 ml of GYM liquid medium for 3 days and collected by centrifugation at 4000 rpm for 30 min. To remove the antibiotics, the mycelium was washed with 50 ml of 10% glycerol followed by 50 ml of 2× YT (1.6% Tryptone, 1% yeast extract, 0.5% NaCl, and pH 7.0). The mycelium was then resuspended in 5 ml of 2× YT and used as the recipient cell. The donor E. coli cell and the recipient S. galbus mycelium, at an equal volume ratio and varying volume ratios (10:1 to 0.1:1), were mixed and laid on MS agar (2% soya flour, 2% mannitol, and 2% Bacto agar) plates containing 10 mm MgCl2. After 12–16 h, 0.5 mg each of nalidixic acid and apramycin was applied to each plate, which contained ∼30 ml of the medium. The exoconjugants were isolated with their apramycin resistance phenotype and tested for a kanamycin-sensitive phenotype when necessary.
TABLE 1.
Primers used in this study
The nucleotide positions in GenBankTM accession number GU300145 are shown as superscript when relevant. The positions of nucleotides inside primers are in parentheses, and the nucleotides are denoted by bold letters. The engineered sequences for the restriction sites are underlined. The positions for aacIV-oriT sequence are italic.
| Primer | Sequence (5′ to 3′) | Usage (reference) |
|---|---|---|
| KgalB-F | 20489TCCGCGTCCGCACCATCCGGGTCCGCACACGTATCTCCCATTCCGGGGATCCGTCGACC (20451) | PCR-targeting mutagenesis of galB |
| KgalB-R | 18051CGGCCGGACCGCGGTGTCCGTCCTGGTCACGACGGCTTCTGTAGGCTGGAGCTGCTTC (18089) | PCR-targeting mutagenesis of galB |
| KgalC-F | 15040CTCAAGGCACTGCGCCACGCGGACGTCACGGGCGCCGACATTCCGGGGATCCGTCGACC (15002) | PCR-targeting mutagenesis of galC |
| KgalC-R | 14314GTTCCCCGGCCCGCAGCGTGCCGGAGCGGAACTCCCGGTTGTAGGCTGGAGCTGCTTC (14352) | PCR-targeting mutagenesis of galC |
| KgalD-F | 14120TTGCCTCTGCTGCGTGATCCGCTGGCGTTCATCACGTCGATTCCGGGGATCCGTCGACC (14082) | PCR-targeting mutagenesis of galD |
| KgalD-R | 12832TCGGCGTCGGCGGACCTCCGCTCGGTGAGCCGCAGGCGCTGTAGGCTGGAGCTGCTTC (12870) | PCR-targeting mutagenesis of galD |
| galB-F | 17951TGAGCTCGGCCTGGTTCCCC17970 | Analytical PCR for galB inactivation |
| galB-R | 20522TCCACGTCATCCGCACCATC20501 | Analytical PCR for galB inactivation |
| galC-F | 14180CACTCGTGTCTGTCATGGAA14199 | Analytical PCR for galC inactivation |
| galC-R | 15220TTGTGGCAGGACATCACGAT15201 | Analytical PCR for galC inactivation |
| galD-F | 12691CGAGCAGATGCTTCACCAGG12710 | Analytical PCR for galD inactivation |
| galD-R | 14219CTCGACGAAGAAGTGAGGTT14200 | Analytical PCR for galD inactivation |
| 417-F | ATTGCGGCCGCCCGGGTGCGGGCAGACCCGC20573 (20554) | Supporting the galB expression cloning |
| 417-R | ATTACTAGTAGATCTCGTCCAGGGCAGGGCGGAGGCTGC21465 (21488) | Supporting the galB expression cloning |
| EgalC-F | ATTTCTAGACGGTGCGTGGATACCGCCGATC15170 (15191) | Expression of galC |
| EgalC-R | ATTCTGCAGCACTCGTGTCTGTCATGGAAAACC14203 (14180) | Expression of galC |
| EgalD-F | ATTTCTAGAGCTGGAGATCACCCGCGATCCCGC14241 (14264) | Expression of galD |
| EgalD-R | ATTCTGCAGAGCAGATGCTTCACCAGGGGCTTC12716 (12693) | Expression of galD |
| 1348-F | ATTAGATCTCCAGGTACTGGACCTCGGGA22805 (22785) | Supporting the galBCD expression cloning |
| 1348-R | ATTACTAGTGTGAACGTATCCCGTCGGTT23084 (23103) | Supporting the galBCD expression cloning |
| sgl-F | 4040GGGAACATCATCACGATCGGCCACTCGATCGGCGGGATGATTCCGGGGATCCGTCGACC (4075) | PCR-targeting deletion of orf-3 to orf-1 |
| sgl-R | 6739GGCCTCGTCAGCGTTCTCCGGTCGCGCGAAGTACTCTCATGTAGGCTGGAGCTGCTTC (6704) | PCR-targeting deletion of orf-3 to orf-1 |
| sgr-F | 24395CGCCGGGTTCTCCGAGAGCTTCGTCGAAGGCTTCTTATGATTCCGGGGATCCGTCGACC (24430) | PCR-targeting deletion of orf1 to orf5 |
| sgr-R | 29586TTGGGGTCGAGAGTGATCCGCGCCTCGGGGAAGCGGTCATGTAGGCTGGAGCTGCTTC (29551) | PCR-targeting deletion of orf1 to orf5 |
| ving-F | GGGTTCTTCGGGATCTCGCCGCGTGAGGCGTTGGCGATGATTCCGGGGATCCGTCGACC | PCR-targeting deletion of vinP2 |
| ving-R | GGTGGGTCAGCGCGGGGCCCGAGGGGGCGGGAGGGGTCATGTAGGCTGGAGCTGCTTC | PCR-targeting deletion of vinP2 |
| sglpo-F | 3801GAATCTGGTGCGAACCCGTC3820 | Analytical PCR for deletion of orf-3 to orf-1 |
| sglpo-R | 7050CAGTCGTGCAGGATCGTCTT7031 | Analytical PCR for deletion of orf-3 to orf-1 |
| sglne-F | 5001TTCTTGAAGGCACGCACGAA5020 | Analytical PCR for deletion of orf-3 to orf-1 |
| sglne-R | 6269TGTCAGTCATGACAGCCAGT6250 | Analytical PCR for deletion of orf-3 to orf-1 |
| sgrpo-F | 24152CACACTGAAGGACCTGAAAC24171 | Analytical PCR for deletion of orf1 to orf5 |
| sgrpo-R | 29870TCGCGTTGCACAACTGTGCT29851 | Analytical PCR for deletion of orf1 to orf5 |
| sgrne-F | 26291TGCATACGCAGACAGCATCT26310 | Analytical PCR for deletion of orf1 to orf5 |
| sgrne-R | 27462TTTGAACTGTCGAGCGAGAG27443 | Analytical PCR for deletion of orf1 to orf5 |
| vingpo-F | CTGGGACATCGACAGTCTCT | Analytical PCR for deletion of vinP2 |
| vingpo-R | CATGAGTAGTTCACCCGTCC | Analytical PCR for deletion of vinP2 |
| vingne-F | TGATCAAGATGGTGATGGCG | Analytical PCR for deletion of vinP2 |
| vingne-R | CTCATACACACCGACGAGAT | Analytical PCR for deletion of vinP2 |
| ACP-KS-F | GCSTCSCGSGACCTSGGCTTCGACTC | Degenerate PCR for type I PKS gene (18) |
| ACP-KS-R | SGASGASGAGCASGCSGTSTCSAC | Degenerate PCR for type I PKS gene (18) |
The cosmid library of S. galbus total DNA in SuperCos1 (Stratagene), the gal cosmid clone of pHJK1011, and its nucleotide sequence (GenBankTM accession number GU300145) were previously described (12). The isolation of the cosmid clones harboring the vicenistatin biosynthetic genes (vin) (17) was achieved by colony hybridization with a type I PKS-KS fragment, which was obtained through degenerate PCR. The PCR amplification of the ACP-KS region from S. galbus total DNA was conducted with the degenerate primer pair of ACP-KS-F/R shown in Table 1 (18). The cloned 700-bp DNA fragment displays a high similarity (86% identity) to a part of the vinP1 sequence (17) in the BLAST-N search. Among several vin cosmid clones isolated, pSYK-363 was used for the targeted deletion of vinP2. End sequencing with the primers for T7 and T3 promoter and the S. galbus draft genome sequence, which is described under “Results,” were used to deduce the nucleotide sequence of pSYK-363.
The cosmid clones (pHJ1011 and pSYK-363) were transformed to E. coli BW25113/pIJ790 by electroporation (15). The EcoRI/HindIII fragment of pIJ773 was used as a template in the PCR amplification of the apramycin resistance gene cassette-conjugal transfer origin (aacIV-oriT) using the primer pairs that were endowed with target-specific sequences (15). The PCR products were introduced by electroporation into the E. coli BW25113/pIJ790 transformants harboring the relevant cosmid. Analytical PCR was performed to confirm the gene inactivation constructs and the cognate S. galbus mutants.
For gene complementation of the knock-out mutants of galB, galC, and galD (ΔgalB::aacIV, ΔgalC::aacIV, and ΔgalD::aacIV, respectively), galB, galC, galD, and galA-E expression constructs were prepared in pWHM3-ermEp* (19). The full lengths of galC and galD were amplified by PCR from pHJK1011 with the primer pairs listed in Table 1. The PCR products were digested with XbaI and PstI, the sites introduced in the PCR primers, and ligated into the same sites of pWHM3-ermEp*. Into the EcoRI site of the resulting plasmids, the XbaI fragment of aacIV-oriT from pIJ773 (15) was subcloned after Klenow treatment. The resulting galC and galD expression plasmids are designated as ermEp-galC and ermEp-galD, respectively. The control plasmid pSK03 was generated by inserting the aacIV-oriT fragment into pWHM3-ermEp*.
For galA-E expression, the 8.8-kb BglII-NcoI fragment (containing the intact galBC region and being truncated in galA and galD) and the 3.2-kb NcoI fragment (the full-length galE and a partial galD) were assembled in the BglII-NcoI site of pLithmus28 by two-step subcloning. The resulting plasmid pSK1348 comprises the intact galBCDE region and an N-terminal truncated galA. To complete the galA cloning in pSK1348, we employed the PCR product obtained with the primer pair of 1348-F and -R in Table 1. This PCR product contains 80 bp of 5′-UTR of galA with the engineered SpeI site and the N-terminal galA sequence up to the internal BglII site. The PCR product, digested with SpeI and BglII, was ligated into the same sites of pSK1348 to generate pSK1349. The insert of pSK1349 was retrieved as the XbaI-SpeI fragment and subcloned into the XbaI site of pWHM3-ermEp* to yield pSK1–349B, into which the aacIV-oriT fragment was subcloned as mentioned for ermEp-galC and ermEp-galD to ultimately produce the galA-E expression construct (ermEp-galABCDE).
The preparation of the galB expression construct began with pSK1349. pSK1349 was digested with KpnI and then self-ligated to yield pSK1–417A. This process eliminated galCDE in the construct. From pSK1–417A, the SpeI-NotI fragment (the intact galA and an 842-bp N-terminal galB; the NotI site is internal to galB) was removed and replaced with a PCR product to constitute the intact galB. The PCR product was obtained with the primer pair of 417-F and 417-R (Table 1), and in this PCR product the engineered SpeI site located 99 bp upstream at the 5′-UTR of galB and the NotI site was internal to galB. The insert of the resulting plasmid was retrieved as the KpnI-SpeI fragment and ligated into the HindIII site of pSK03 after Klenow treatment to generate the galB expression construct (ermEp-galB).
Chemical Analysis Procedure
For the extraction of GAL, the mycelium from the production culture was collected by centrifugation and extracted in a minimal volume of methanol. The culture supernatant was extracted with an equal volume of ethyl acetate two times. The organic extracts were dried under reduced pressure and dissolved in methanol for the antifungal activity assay and chromatographic analysis.
For the antifungal activity assay, the overnight Bennett medium culture of C. neoformans was suspended in GYM soft agar (0.4% of Bacto agar) at a 1% v/v dilution and overlaid on a GYM plate. On this assay plate of C. neoformans, the extracts were applied on a filter disc (ϕ, 1 cm). TLC analysis was conducted in a silica gel 60 F254 TLC-plate (Merck) with a mixture of ethyl acetate and benzene (1:3) as the developing solvent (3). TLC plates were developed two times for better separation. The developed TLC plates were placed on the C. neoformans assay plate upside down. The assay plates were then incubated at 30 °C until the clear inhibition zone was developed, which generally took 48 h.
HPLC and MS analysis of the extracts were performed with the Agilent 1100 series LC system coupled with a Bruker HCT 3000 ion trap mass spectrometer. Mass analysis was performed in positive electrospray ionization mode. The mass scan range was m/z 100–500, the dry temperature was 350 °C, the nebulizer gas was 40 p.s.i., and the dry gas was 9 liter/min. A Gemini C-18 column (150 × 3.0 mm, 5.0 μm; Phenomenex) was used. A mobile phase consisting of acetonitrile (A) and water (B) was run with gradient elution. The elution started at 20% A, from 20% A to 60% A for 20 min, was maintained at 60% A for 20 min, and then from 60% A to 100% A for 20 min. The temperature and the flow rate were maintained at 25 °C, 0.4 ml/min.
Isolation of GALs and Nuclear Magnetic Resonance (NMR) Measurement
Isolation of GAL C and 13C-labeled GAL-A and -B was achieved with the semipreparative HPLC experiment that employed the Varian HPLC ProStar system. The flow rate was run at 3 ml/min within the YMC-Pack ODS-A column (250 × 10 mm, 5.0 μm) and monitored with UV detection at 230 nm. The mobile phase consisting of 25 mm ammonium acetate in water, pH 5.5, (A) and acetonitrile (B) was run with gradient elution. The elution started at 75% B, was maintained for 15 min, from 75% B to 95% B for 15 min, and then was maintained at 95% B for 10 min. The eluate of the HPLC separation was collected with dozens of injections, and the collected eluates were pooled and dried in vacuo. For the isolation of GAL-C from the ΔgalD::aacIV mutant, the methanol extracts of mycelium that were obtained from the 2.4-liter culture were applied to the HPLC preparation. One- and two-dimensional NMR analysis of GAL-C was conducted in CDCl3 with the 600-MHz Bruker AVANCE 600 NMR (Bremen, Germany). The NMR measurement was performed in the National Instrumentation Center for Environmental Management (NICEM), Seoul National University, Seoul, Republic of Korea. Additional 1H NMR measurements were done in CD3OD with the 400 MHz Varian VNMRS-400 NMR in the Instrumentation Center of Industry and Academic Cooperation, Myongji University, Yongin, Republic of Korea. High resolution fast atom bombardment mass spectrum of GAL-C was recorded by using the JEOL JMS-600W spectrometer in the National Center for Inter-University Research Facilities (NCIRF), Seoul National University, Seoul, Republic of Korea.
The preparation of 13C-enriched GAL-A and -B was performed by feeding 80 mg of [U-13C3] sodium propionate (13C3, 99%; Cambridge Isotope Laboratories) to the 1.6-liter culture of the wild-type S. galbus (WT). The methanol cell extract and the ethyl acetate supernatant extract were combined, dried under the reduced pressure, and dissolved in methanol for the HPLC preparation as described for the isolation of GAL-C. One- and two-dimensional NMR analysis of GAL-A and -B was conducted in CD3OD with the 600-MHz Bruker AVANCE 600 NMR, as previously described for GAL-C. Heteronuclear single quantum coherence and heteronuclear multiple bond correlation data were used to identify 13C chemical shifts for each carbon atom in GAL-A and -B. 13C-13C coupling was determined to deduce the intact incorporation of [13C3]propionate in GAL-A and -B.
RESULTS
Gene Inactivation of galB, -C, and -D and the Identification of GAL-C in a galD Mutant
It was previously shown that a vector-integrating knock-out of galB, generated with the temperature-sensitive replicon vector pKC1139, severely impaired GAL production (12). To assure that galA-E encode GAL biosynthetic enzymes, gene-replacement mutants of galB, galC, and galD (ΔgalB::aacIV, ΔgalC::aacIV, and ΔgalD::aacIV, respectively) were prepared (data not shown).
GAL-A activity was abolished in all of the galB, -C, and -D mutants (Fig. 2, A and B), and in trans complementation with ermEp-galABCDE restored GAL-A activity (Fig. 2, C–E, lane 3). It is worthwhile to note that the mycelia extract of S. galbus exhibited antifungal activity apart from GAL-A. This unknown compound did not migrate under the TLC conditions employed (Fig. 2B) (12). When a single gene complementation was conducted, ermEp-galB and ermEp-galC restored GAL-A activity in ΔgalB::aacIV and ΔgalC::aacIV, respectively; however, ermEp-galD failed to do so (Fig. 2, C–E, lane 4). It is possible that the integration of aacIV-oriT into the galD region gave rise to a polar effect, thereby repressing downstream transcription. We also conjecture that GalD may work together with GalE, the N-terminal Rieske domain protein, and that due to the electron shuttling through GalE the co-translation of galDE is necessary for the maintenance of GalD activity.
FIGURE 2.

GAL-A activity assays for the galB-D mutants and gene complementation studies. GAL-A activity was assessed by using C. neoformans as the indicator organism. The culture supernatant was extracted with ethyl acetate (A and C–E), and the mycelial paste was extracted with methanol (B). The assay results of WT, ΔgalB::aacIV (ΔgalB), ΔgalC::aacIV (ΔgalC), and ΔgalD::aacIV (ΔgalD) are shown in A and B, where the amounts of mutant extract applied were four times that of WT. The in trans gene complementation results of ΔgalB::aacIV (C), ΔgalC::aacIV (D), and ΔgalD::aacIV (E) are shown alongside the descriptions of the complementation constructs (F). The lane identities for C–E are: WT (1), each mutant (2), each mutant with ermEp-galABCDE (3), each mutant with the target gene under ermEp control (4), and each mutant with the vector control (5). The position of GAL-A is indicated by arrows and lines.
The absence of GAL-A and -B in the mutants was also verified by a LC-MS experiment (Fig. 3). In this HPLC experiment, the ΔgalD::aacIV mutant accumulated a compound that eluted at minute 52. In tandem mass analysis, this compound (named GAL-C) generated a daughter ion peak at m/z 313 similar to GAL-B (Fig. 4, B and D). This suggests that the GAL-C compound is related to GAL-B. It seems likely that the loss of CO2 in GAL-C (m/z 357) resulted in a fragmentation ion of m/z 313. GAL-C was also found in the extracted ion chromatogram from the WT mycelium extract, but the concentration was too low to be detected by UV (Figs. 3, A-1 and 4C).
FIGURE 3.

HPLC-MS analysis of the galB-D mutants. Total ion chromatograms (TIC), UV detection at 230 nm, and extracted ion chromatograms (EIC; m/z 403, 387, and 357 for GAL-A, GAL-B, and GAL-C, respectively) of the mycelium methanol extract (−1) and the supernatant ethyl acetate extract (−2) from WT (A), ΔgalB::aacIV (B), ΔgalC::aacIV (C), and ΔgalD::aacIV (D) cells are shown.
FIGURE 4.

MS spectra of GAL-A, -B, and -C. Molecular ion mass spectra (−1) and fragmentation ion mass spectra (−2 and −3) of GAL-A (A), GAL-B (B), and GAL-C (C) from WT and GAL-C from ΔgalD::aacIV (D). Mass analysis was performed in positive electrospray ionization mode.
In the ΔgalD::aacIV mutant extract, another compound(s) was found at minute 51 by UV detection. The nature of this compound was unclear because the peak could not be identified in the ion chromatogram. Furthermore, a similar peak was found in the extracted ion chromatogram (m/z 357) of the supernatant extract; however, no reasonable mass spectrum could be obtained from this peak. We also tried to isolate this compound through HPLC preparation during GAL-C isolation, but the eluate failed to give any reasonable signal in the 1H NMR analysis (data not shown).
Two distinctive ion peaks of m/z 335 and m/z 357 in the GAL-C mass spectrum were assigned as [M+H]+ and [M+Na]+, respectively. The molecular mass of GAL-C was predicted to be 334 Da. Through high resolution fast atom bombardment mass analysis of the peak eluate, we detected a m/z 335.2584 (calculated 335.2586) for the [M+H]+ ion. This confirmed the molecular formula of GAL-C as C21H34O3. This deduced molecular formula indicates that GAL-C lacks two oxygen atoms and has two additional hydrogen atoms (a double bond saturation) when compared with GAL-B.
A milligram of GAL-C was isolated from a 2.4-liter culture of ΔgalD::aacIV through HPLC preparation. NMR measurements indicate that GAL-C has one carbonyl carbon (C-4) found at δC (ppm) 207.5 and one ester group (C-2, δC 161.9). Heteronuclear multiple bond correlation measurements indicated a correlation of C-4 to 17-H3 and 18-H3. The positional identity of C-18 was confirmed with a COSY correlation of 18-H3 to 5-H. Two doublet allylic hydrogen signals are evident at 5.00 (8-H) and 4.81 ppm (12-H), and each allylic signal correlates to a methyl group (19-H3 and 21-H3, respectively) through long-range coupling. NMR measurements in CDCl3 were unable to resolve the three methyl proton signals upfield. Thus, additional 1H NMR measurements were done in CD3OD. Overall, the NMR analysis of purified GAL-C confirmed its structure, which is shown in Fig. 1B (Table 2). We, therefore, suggest that GalD (or GalDE) mediates the conversion of GAL-C into GAL-B through an oxidative isomerization on C-11 and C-12 and hydroxylations on C-5 and C-18. These gene inactivation experiments substantiate the hypothesis that GalA-E are the biosynthetic genes for GAL.
TABLE 2.
1H (600 MHz) and 13C (125 MHz) spectral data (CDCl3) for GAL-C
The structure of GAL-C is provided with carbon position numbering and 1H-1H COSY coupling data highlighted in bold lines (arrows indicate long-range coupling).

To examine whether any gene flanking galA-E and galG-J has a role in GAL biosynthesis, the flanking regions (orf-1 to orf-3 and orf6 to orf10 in GenBankTM accession number GU300145) were eliminated and replaced with the aacIV marker. The resulting mutants retained GAL-A activity (data not shown). We thus concluded that the flanking loci are not involved in GAL biosynthesis.
[U-13C]Propionate Feeding Experiment and 13C Incorporation in GAL-A and -B
To determine whether GalA-E are involved in the biosynthesis of a special, yet unidentified, precursor unit [U-13C]propionate was fed to WT cultures, and the propionate incorporation in GAL-A and GAL-B was examined by NMR analysis (Fig. 5 and Table 3). Propionate can be converted to propionyl-CoA and subsequently to (2S)-methylmalonyl-CoA by endogenous propionate-CoA synthetase and propionyl-CoA carboxylase (20). Thus, intact incorporation of propionate into the polyketide backbone is indicative of a methylmalonyl-CoA origin. In proton-decoupled 13C NMR measurements, 13C-13C coupling is represented by doublet splitting surrounding the cognate natural abundance 13C signal. Intact incorporation of [U-13C]propionate was evident in GAL-A and -B as judged by 13C-13C coupling, especially at the position originating from C-1 and C-3 of [U-13C]propionate. The positions originating from C-2 of [U-13C]propionate formed multiple splits due to tandem coupling. It appeared that all of the methyl carbons (C-16, -17, -18, -19, -20, -21, and -22), which were predicted to originate from C-3 of [U-13C]propionate, showed distinctive 13C-13C coupling, with the exception of C-19 in GAL-A (Fig. 5). In addition, all of the positions originating from C-1 of [U-13C]propionate (C-2, -5, -7, -9, -11, -13, and -15), except for C-7 in GAL-A, produced doublet signals (Fig. 5). Note that C-6, C-7, and C-19 in GAL-A originate from methoxymalonyl-ACP, which is generated from 1,3-bis(phosphoglycerate) (6). This result excludes the possibility that GalA-E mediate the biogenesis of an unknown, rare malonate derivative and suggests that GalA-C are directly involved in polyketide chain assembly.
FIGURE 5.

Extracted spectrum of the proton-decoupled 13C NMR measurements of GAL-A (A) and -B (B), which were isolated after administration of U-13C-labeled sodium propionate. Enriched doublet signals surrounding the natural abundance signals were observed at the carbon positions corresponding to the C-1 and C-3 positions of propionate. The carbon numbers are shown above with the calculated coupling constants (J in Hz), and the chemical shifts are shown below. A, the singlet signals of C-6, C-7, and C-19, which originated from methoxymalonyl-ACP, are also shown. B, splitting patterns of the carbonyl carbons (C-2 and C-4) of GAL-B were not resolved due to their low intensities.
TABLE 3.
1H (600 MHz) and 13C (125 MHz) spectral data (CD3OD) for GAL-A and -B
ND, not determined.
| C no. | GAL-A |
GAL-B |
||
|---|---|---|---|---|
| δ 1H(multi., JHH in Hz) | δ 13C | δ 1H(multi., JHH in Hz) | δ 13C | |
| ppm | ppm | ppm | ppm | |
| 2 | 170.4 | ND | ||
| 3 | 3.97 (q, 7) | 51.2a | 4.00 (q, 7) | 50.7a |
| 4 | 209.5 | ND | ||
| 5 | 85.1 | 85.8 | ||
| 6 | 2.57 (d, 15), 2.50 (d, 15) | 34.2 | 2.71 (br d, 14), 2.05 (d, 15) | 42.7 |
| 7 | 150.2 | 129.7 | ||
| 8 | 4.51 (d, 10) | 123.5 | 4.99a | 137.7 |
| 9 | 2.74 (m) | 31.0 | 2.53 (m) | 33.4 |
| 10 | 2.25 (br d, 12), 2.11 (d, 13) | 47.0 | 2.24 (d, 13), 2.15 (d, 13) | 46.6 |
| 11 | 145.3 | ND | ||
| 12 | 5.67 (br s) | 128.5 | 5.67 (br s) | 128.3 |
| 13 | 136.3 | 136.3 | ||
| 14 | 4.89a | 82.2 | 4.89a | 82.1 |
| 15 | 1.68 (m) | 27.5 | 1.59 (m) | 27.6 |
| 16 | 0.95 (t, 7) | 10.4 | 0.96 (t, 7) | 10.4 |
| 17 | 1.43 (d, 7) | 15.3 | 1.45 (d, 7) | 15.8 |
| 18 | 3.98 (d, 12), 3.65 (d, 12) | 68.4 | 3.95 (d, 12), 3.60 (d, 13) | 69.1 |
| 19 | 3.48 (s) | 57.5 | 1.67 (s) | 19.3 |
| 20 | 0.79 (d, 7.0) | 20.5 | 0.78 (d, 7) | 19.7 |
| 21 | 4.81/5.03a | 117.3 | 4.79/5.03a | 117.2 |
| 22 | 1.81 (s) | 16.3 | 1.81 (s) | 16.5 |
a Overlapped with solvent peak and deduced by HSQC or HMBC.
Screening for a Modular Type I PKS Gene Cluster in S. galbus
In the pursuit of identifying the GAL PKS gene cluster, we searched for modular type I PKS genes in the S. galbus genome by screening a cosmid library (described under “Experimental Procedures”). The end sequencing analysis of the resulting 10 positive cosmid clones indicated the presence of vicenistatin biosynthetic genes in all of the clones (data not shown). The vicenistatin biosynthetic gene cluster was previously isolated and characterized in Streptomyces halstedii (17), and its biosynthetic program is clearly distinct from that proposed for GAL. We thus concluded that S. galbus harbors a vicenistatin biosynthetic gene cluster and that our DNA hybridization approach was incapable of detecting a type I modular PKS gene other than the vicenistatin gene cluster in S. galbus.
To investigate all of the modular type I PKS genes in S. galbus, we obtained a draft genome sequence of S. galbus using the 454 GS-FLX (Roche Applied Science) system. This generated 809,880 reads containing 351,239,903 bases (peak depth, 33X). De novo sequence assembly was performed with the Newbler 2.3 software, and 799,571 reads were used to generate 107 contigs covering 10,055,701 bases. In the 90 large contigs covering 10,051,861 bases, N50 is 243,319 bases long. The version described in this paper is version JRHJ01000000 (DBLINK BioProject: PRJNA259693). The previously reported galA-E and galG-K cluster (GenBankTM accession number GU300145) is located inside contig00111, which is 346 kb long. There is no hint of a modular type I PKS gene in this contig. All of the contigs were then analyzed with antiSMASH 2.0 (21), and the output can be found at AntiSMASH. This result predicts putative type I PKS genes in four contigs: 00038, 00039, 00064, and 00106. The PKS sequences in contigs 00038, 00039, and 00106 correspond to the vicenistatin gene cluster, the presence of which in S. galbus was aforementioned. Contig 00064 contains a single module type I PKS gene encoding ACP/KS/AT/ACP/thioesterase, which bears high homology (81% identity and 85% similarity) to SCAB78961 of Streptomyces scabies 87.22 origin. The AT domain of SCAB78961 and its S. galbus homolog are predicted to be malonate-specific. SCAB78961 is neighbored by genes for esterase, AMP-dependent ligase, and acetyl-CoA carboxylase. This genetic organization is conserved in several Streptomyces genomes, including S. galbus. This domain organization and the stand-alone feature of SCAB78961 are reminiscent of mycolic acid PKS (PKS13) in Mycobacterium tuberculosis (22) and suggest a potential role in special fatty acid biogenesis for the S. galbus homolog. These overall features of SCAB78961 led us to exclude its role in GAL biosynthesis.
We further analyzed the draft S. galbus genome sequences with a BLAST-P search. This analysis reassured us that the S. galbus genome encodes a single type I modular PKS, which is involved in vicenistatin biosynthesis (data not shown). Notably, a search of the KS domain identified all of the eight KS domains in the vicenistatin PKS. No other modular PKS KS genes were identified. This draft genome analysis supports the concept that S. galbus harbors a single type I modular PKS gene cluster that encodes for vicenistatin biosynthesis.
To exclude any possibility that the vicenistatin gene cluster was involved in GAL biosynthesis, vinP2 was eliminated in S. galbus in a gene replacement experiment. We chose vinP2 because the module organization of VinP2 corresponds to the predicted catalytic activities for the third and fifth extension steps of GAL biosynthesis (Fig. 1A). The resulting mutant was capable of producing GAL-A activity (Fig. 6), leading us to conclude that the vicenistatin PKS genes have no role in GAL biosynthesis.
FIGURE 6.
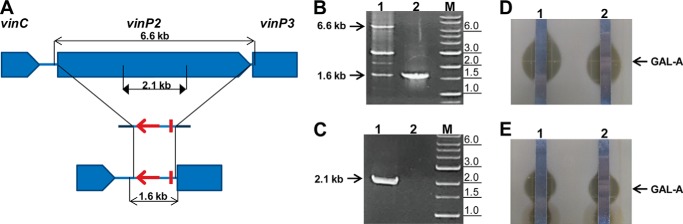
Targeted deletion scheme (A), PCR confirmation (B and C), and TLC-coupled antifungal assay (D and E) of the vinP2 mutant. A, the relevant genetic organization of the WT gene (top) and the vinP2 deletion mutant (bottom) are shown with a simple representation of the cognate gene inactivation construct (middle). For simplicity, only the target gene is shown for each gene inactivation construct. aacIV and oriT in the PCR targeting cassette are denote by a bold arrow and bar, respectively. PCR primer positions are shown along with the estimated sizes of the PCR products. B and C, agarose gel electrophoretic analysis of the PCR products from total DNA samples. The primer pairs are complementary to the region outside (B) or inside (C) the deletion: lane 1, WT; lane 2, the vinP2::aacIV mutant; lane M, DNA size marker. B, the 1.6-kb band in WT resulted from nonspecific PCR amplification; the PCR experiment was performed in a low stringency condition for the amplification of the 6.6-kb fragment in WT. A, the PCR strategy is shown, with triangular arrows indicating the target fragment inside the deletion. Thus, no PCR amplifications should be observed for the deletion mutant (C). The primer pairs used in this analytical PCR are vingpo-F/R (B) and vingne-F/R (C) (Table 1). The DNA size marker contains DNA fragments of 0.1, 0.2, 0.3, 0.4, 0.5, 0.65, 0.8, 1.0, 1.5, 2.0, 3.0, 4.0, 5.0, 6.0, 8.0, and 10.0 kb. The estimated sizes of the PCR products are indicated on the left by arrows, and the relevant DNA size marker fragments are indicated on the right in kb. D and E, the TLC-coupled antifungal assay against C. neoformans was performed with ethyl acetate extract of the culture supernatant (D) and methanol extract of the mycelium (E). The position of GAL-A is indicated by arrows.
DISCUSSION
Chain assembly during the biosynthesis of GAL was predicted to be modular in nature. The polyketide biosynthesis paradigm dictates that all three degrees of β-keto group modifications are necessary for GAL biosynthesis (Fig. 1A). This type of divergence in β-keto group modification is not known for bacterial iterative PKS systems and has some precedents only in fungal type I PKS systems (10, 11). An iterative PKS system that incorporates methylmalonyl-CoA has not been reported, whereas the ability of FAS to incorporate methylmalonyl-CoA has been observed for mycocerosic acid synthase (23). Furthermore, GAL biosynthesis was thought to involve relaxed precursor specificity at a specific round of the chain extension step, resulting in the generation of GAL-A and GAL-B. This implied the participation of a special AT domain at this extension step (12). Overall, it was hard to envision that GalA-E would make up the main body of the PKS involved in GAL biosynthesis. It was thus hypothesized that GAL biosynthesis was mediated by a modular type I PKS system and that GalA-E participated in a specific step(s) of PKS function. Our gene inactivation experiments solidified the idea that GalA-E are directly involved in GAL biosynthesis (Figs. 2 and 3). We had envisioned the possibility that GalA-E generate a novel derivative of malonyl thioester, such as hydroxymethylmalonyl-ACP or 2,3-dihydroxymethylmalonyl-ACP for the C-4, -5, and -18 positions. This hypothesis was motivated by the report that allylmalonyl-ACP biosynthesis for FK506 is mediated by a single module PKS system that is similar to GalA and B (24). The identification of GAL-C (Fig. 3 and Table 2), however, provided the first evidence disproving this scenario (Fig. 1, A and B). GAL-C would not accumulate in ΔgalD::aacIV if GalA-E are involved in the generation of a precursor for the C-4, -5, and -18 positions. We could not exclude the possibility that GAL-C was generated through the incorporation of methylmalonyl-CoA instead of the presumed precursor that is generated by GalA-E; however, the probability of this occurring is not high because GAL-C is thought to accumulate in all of the galB, -C, and -D mutants in this scenario. The [13C]propionate incorporation experiment clarified this issue, as our results indicated that methylmalonate is the precursor of choice for the C-4, -5, and -18 positions (Fig. 5). Our genome sequence analysis and the subsequent gene inactivation study (Fig. 6) demonstrated the presence of only one modular type I PKS gene cluster in S. galbus that is not related to GAL biosynthesis. We thus envisioned that GalA-E constitute the PKS system that generates GAL in concert with GalG-K (methoxymalonyl-ACP biosynthesis).
The genome BLAST-P analysis of galA-E indicated that these five genes constitute synteny, similar to galG-K. The galA-E synteny exists in several Streptomyces genomes including Streptomyces violaceusniger Tu 4113, Streptomyces rapamycinicus NRRL 5491, and Streptomyces niveus NCIMB 11891 (Fig. 7). Notably, this synteny is clustered with the methoxymalonyl-ACP biosynthesis locus in S. niveus as seen in S. galbus. In the other two strains, the homolog of galJ (the ACP gene in methoxymalonyl-ACP biosynthesis) is co-localized with the synteny. This suggests that galA-E and galG-K are cooperating syntenies and that GAL is a cryptic metabolite of these Streptomyces spp.
FIGURE 7.
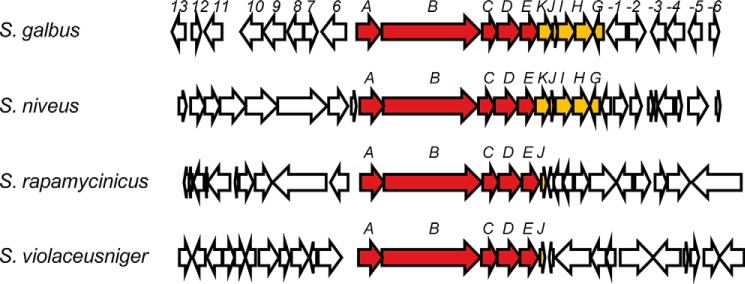
Genetic organization of the regions surrounding the galA-E synteny in S. galbus, S. violaceusniger, S. rapamycinicus, and S. niveus. galA-E and galG-K are indicated by filled arrows, and other genes are indicated by unfilled arrows. The S. galbus gal cluster region (GenBankTM accession number GU300145) is compared with the regions bearing galA-E synteny in the genome sequences (locus tag, GenBankTM accession number) of S. violaceusniger Tü 4113 (Strvi_5006 to Stri_4980, CP002994.1), S. rapamycinicus NRRL 5491 (M271_41420 to M271_41195, CP006557.1), and S. niveus NCIMB 11891 (M877_35370 to M877_35500, CM002280.1). There is no significant homology in the regions flanking galA-E and galG-K among the four Streptomyces genomes.
As noted previously, galAB homologues can be found in several Burkholderia species (12), including the Burkholderia pseudomallei group that causes melioidosis and its close relatives, Burkholderia thailandensis and Burkholderia oklahomensis. A member of the Burkholderia cepacia complex, Burkholderia multivorans also harbors galAB homologues. These galAB homologues accompany the genes for a cytochrome P450/KR domain protein and a flavin monooxygenase (12). The oxidation function of these accompanying genes appears analogous to the proposed role of GalDE. It is thus tempting to suggest that these Burkholderia spp. also produce a GAL-like polyketide.
In summary, the isolation of the methoxymalonyl-ACP biosynthesis locus in the pursuit of a GAL biosynthetic gene cluster led us to identify galA-E synteny. This synteny is conserved in some Streptomyces genomes and has analogs in some pathogenic Burkholderia species. GalB bears all of the three β-keto group modification activities in its KR, DH, and ER domains, whereas the AT domain and a thiolation site are evident in GalA. Our experiments substantiate that no catalytic function can be assigned for GalA-C other than a canonical PKS function. It is thus proposed that GAL is not synthesized by a modular type I PKS system and that GalA-C constitutes a novel bacterial iterative type I PKS that incorporates methylmalonate and exerts highly programmed β-keto group modifications in a manner similar to the highly reducing type I fungal PKSs. It will be interesting to elucidate the GAL analog in the Burkholderia species and to determine its biological activity.
Acknowledgments
The S. galbus genome sequencing was performed using Roche Applied Science 454 FLX sequencer at the Korea Institute of Science and Technology Gangneung Institute.
This work was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) supported by the Ministry of Education (2011-0021169; 2013R1A1A2059458) and by a grant from the Next-Generation BioGreen 21 Program (PJ009643), Rural Development Administration, Republic of Korea.
The nucleotide sequence(s) reported in this paper has been submitted to the DDBJ/GenBankTM/EBI Data Bank with accession number(s) JRHJ01000000.
- GAL
- galbonolide
- ACP
- acyl carrier protein
- gal
- galbonolide biosynthetic gene
- vin
- vicenistatin biosynthetic gene
- PKS
- polyketide synthase
- KS
- β-ketoacyl-thioester synthase
- KR
- β-ketoacyl-ACP reductase
- DH
- β-hydroxyacyl-ACP dehydratase
- ER
- trans-2-enoyl-ACP reductase
- AT
- acyltransferase
- contig
- group of overlapping clones.
REFERENCES
- 1. Achenbach H., Mühlenfeld A., Fauth U., Zähner H. (1988) The galbonolides. Novel, powerful antifungal macrolides from Streptomyces galbus ssp. eurythermus. Ann. N. Y. Acad. Sci. 544, 128–140 [DOI] [PubMed] [Google Scholar]
- 2. Takatsu T., Nakayama H., Shimazu A., Furihata K., Ikeda K., Furihata K., Seto H., Otake N. (1985) Rustmicin, a new macrolide antibiotic active against wheat stem rust fungus. J. Antibiot. 38, 1806–1809 [DOI] [PubMed] [Google Scholar]
- 3. Abe Y., Nakayama H., Shimazu A., Furihata K., Ikeda K., Furihata K., Seto H., Otake N. (1985) Neorustmicin A, a new macrolide antibiotic active against wheat stem rust fungus. J. Antibiot. 38, 1810–1812 [DOI] [PubMed] [Google Scholar]
- 4. Mandala S. M., Thornton R. A., Milligan J., Rosenbach M., Garcia-Calvo M., Bull H. G., Harris G., Abruzzo G. K., Flattery A. M., Gill C. J., Bartizal K., Dreikorn S., Kurtz M. B. (1998) Rustmicin, a potent antifungal agent, inhibits sphingolipid synthesis at inositol phosphoceramide synthase. J. Biol. Chem. 273, 14942–14949 [DOI] [PubMed] [Google Scholar]
- 5. Shen B., Kwon H. J. (2002) Macrotetrolide biosynthesis: a novel type II polyketide synthase. Chem. Rec. 2, 389–396 [DOI] [PubMed] [Google Scholar]
- 6. Hertweck C. (2009) The biosynthetic logic of polyketide diversity. Angew. Chem. Int. Ed. Engl. 48, 4688–4716 [DOI] [PubMed] [Google Scholar]
- 7. Cane D. E. (2010) Programming of erythromycin biosynthesis by a modular polyketide synthase. J. Biol. Chem. 285, 27517–27523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhao Q., He Q., Ding W., Tang M., Kang Q., Yu Y., Deng W., Zhang Q., Fang J., Tang G., Liu W. (2008) Characterization of the azinomycin B biosynthetic gene cluster revealing a different iterative type I polyketide synthase for naphthoate biosynthesis. Chem. Biol. 15, 693–705 [DOI] [PubMed] [Google Scholar]
- 9. Cox R. J. (2007) Polyketides, proteins and genes in fungi: programmed nano-machines begin to reveal their secrets. Org. Biomol. Chem. 5, 2010–2026 [DOI] [PubMed] [Google Scholar]
- 10. Ma S. M., Li J. W., Choi J. W., Zhou H., Lee K. K., Moorthie V. A., Xie X., Kealey J. T., Da Silva N. A., Vederas J. C., Tang Y. (2009) Complete reconstitution of a highly reducing iterative polyketide synthase. Science 326, 589–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gao Z., Wang J., Norquay A. K., Qiao K., Tang Y., Vederas J. C. (2013) Investigation of fungal iterative polyketide synthase functions using partially assembled intermediates. J. Am. Chem. Soc. 135, 1735–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karki S., Kwon S. Y., Yoo H. G., Suh J. W., Park S. H., Kwon H. J. (2010) The methoxymalonyl-acyl carrier protein biosynthesis locus and the nearby gene with the β-ketoacyl synthase domain are involved in the biosynthesis of galbonolides in Streptomyces galbus, but these loci are separate from the modular polyketide synthase gene cluster. FEMS Microbiol. Lett. 310, 69–75 [DOI] [PubMed] [Google Scholar]
- 13. Fauth U., Zähner H., Mühlenfeld A., Achenbach H. (1986) Galbonolide A and B: two non-glycosidic antifungal macrolides. J. Antibiot. 39, 1760–1764 [DOI] [PubMed] [Google Scholar]
- 14. Flett F., Mersinias V., Smith C. P. (1997) High efficiency intergeneric conjugal transfer of plasmid DNA from Escherichia coli to methyl DNA-restricting streptomycetes. FEMS Microbiol. Lett. 155, 223–229 [DOI] [PubMed] [Google Scholar]
- 15. Gust B., Challis G. L., Fowler K., Kieser T., Chater K. F. (2003) PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. U.S.A. 100, 1541–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bierman M., Logan R., O'Brien K., Seno E. T., Rao R. N., Schoner B. E. (1992) Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116, 43–69 [DOI] [PubMed] [Google Scholar]
- 17. Ogasawara Y., Katayama K., Minami A., Otsuka M., Eguchi T., Kakinuma K. (2004) Cloning, sequencing, and functional analysis of the biosynthetic gene cluster of macrolactam antibiotic vicenistatin in Streptomyces halstedii. Chem. Biol. 11, 79–86 [DOI] [PubMed] [Google Scholar]
- 18. Hyun C. G., Suh J. W. (1999) Development of PCR-based screening methods for macrolide type polyketides in actinomycetes. Agric. Chem. Biotechnol. 42, 119–124 [Google Scholar]
- 19. Rajkarnikar A., Kwon H. J., Suh J. W. (2007) Role of adenosine kinase in the control of Streptomyces differentiations: loss of adenosine kinase suppresses sporulation and actinorhodin biosynthesis while inducing hyperproduction of undecylprodigiosin in Streptomyces lividans. Biochem. Biophys. Res. Commun. 363, 322–328 [DOI] [PubMed] [Google Scholar]
- 20. Murli S., Kennedy J., Dayem L. C., Carney J. R., Kealey J. T. (2003) Metabolic engineering of Escherichia coli for improved 6-deoxyerythronolide B production. J. Ind. Microbiol. Biotechnol. 30, 500–509 [DOI] [PubMed] [Google Scholar]
- 21. Blin K., Medema M. H., Kazempour D., Fischbach M. A., Breitling R., Takano E., Weber T. (2013) antiSMASH 2.0: a versatile platform for genome mining of secondary metabolite producers. Nucl. Acids Res. 41, W204–W212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gokhale R. S., Saxena P., Chopra T., Mohanty D. (2007) Versatile polyketide enzymatic machinery for the biosynthesis of complex mycobacterial lipids. Nat. Prod. Rep. 24, 267–277 [DOI] [PubMed] [Google Scholar]
- 23. Fernandes N. D., Kolattukudy P. E. (1998) A newly identified methyl-branched chain fatty acid synthesizing enzyme from Mycobacterium tuberculosis var. bovis BCG. J. Biol. Chem. 273, 2823–2828 [DOI] [PubMed] [Google Scholar]
- 24. Mo S., Kim D. H., Lee J. H., Park J. W., Basnet D. B., Ban Y. H., Yoo Y. J., Chen S. W., Park S. R., Choi E. A., Kim E., Jin Y. Y., Lee S. K., Park J. Y., Liu Y., Lee M. O., Lee K. S., Kim S. J., Kim D., Park B. C., Lee S. G., Kwon H. J., Suh J. W., Moore B. S., Lim S. K., Yoon Y. J. (2011) Biosynthesis of the allylmalonyl-CoA extender unit for the FK506 polyketide synthase proceeds through a dedicated polyketide synthase and facilitates the mutasynthesis of analogues. J. Am. Chem. Soc. 133, 976–985 [DOI] [PMC free article] [PubMed] [Google Scholar]