

Background: FcRn regulates the long serum half-life of albumin.
Results: The C-terminal DIII of HSA is the principal domain for FcRn binding, whereas two loops in DI at the N terminus modulate the interaction.
Conclusion: DI of albumin contributes to optimal FcRn binding.
Significance: We highlight the importance of DI for pH-dependent binding to FcRn.
Keywords: Albumin, Biodegradation, Bioengineering, Fc Receptor, pH Regulation, Pharmacokinetics
Abstract
Albumin is an abundant blood protein that acts as a transporter of a plethora of small molecules like fatty acids, hormones, toxins, and drugs. In addition, it has an unusual long serum half-life in humans of nearly 3 weeks, which is attributed to its interaction with the neonatal Fc receptor (FcRn). FcRn protects albumin from intracellular degradation via a pH-dependent cellular recycling mechanism. To understand how FcRn impacts the role of albumin as a distributor, it is of importance to unravel the structural mechanism that determines pH-dependent binding. Here, we show that although the C-terminal domain III (DIII) of human serum albumin (HSA) contains the principal binding site, the N-terminal domain I (DI) is important for optimal FcRn binding. Specifically, structural inspection of human FcRn (hFcRn) in complex with HSA revealed that two exposed loops of DI were in proximity with the receptor. To investigate to what extent these contacts affected hFcRn binding, we targeted selected amino acid residues of the loops by mutagenesis. Screening by in vitro interaction assays revealed that several of the engineered HSA variants showed decreased binding to hFcRn, which was also the case for two missense variants with mutations within these loops. In addition, four of the variants showed improved binding. Our findings demonstrate that both DI and DIII are required for optimal binding to FcRn, which has implications for our understanding of the FcRn-albumin relationship and how albumin acts as a distributor. Such knowledge may inspire development of novel HSA-based diagnostics and therapeutics.
Introduction
Albumin is synthesized by hepatocytes in the liver, which secrete an impressive 13–14 g of albumin into the blood every day. In the blood albumin plays a pivotal role as a transporter of a range of small insoluble molecules such as fatty acids, metal ions, hormones, heme, and bilirubin in addition to being a carrier of small chemical drugs (1, 2). Albumin (molecular mass 66.5 kDa) is built up of three homologous domains (DI, DII, and DIII), where each is composed of two subdomains with distinct helical folding patterns connected by flexible loops (see Fig. 1A). Furthermore, albumin has a serum half-life of nearly 3 weeks in humans. This feature is of great interest to the pharmaceutical industry, as the exceptional half-life can be utilized to extend the serum persistence of therapeutics that are chemically or genetically linked to albumin (3–5). Examples are biopharmaceuticals such as cytokines, hormones, and antibody fragments. Also small albumin binding scaffolds, peptides, or fatty acids fused or conjugated directly to a drug of interest, which target or associate with albumin when injected into the bloodstream, are used to improve pharmacokinetics (3–6).
FIGURE 1.
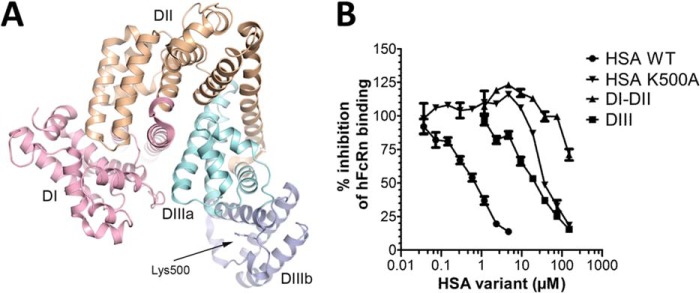
The impact of HSA domains on binding to hFcRn. A, a ribbon structure illustration of full-length WT HSA with its three subdomains DI, DII, and DIII highlighted in pink, orange, and two shades of blue, respectively. The position of amino acid residue Lys-500 is indicated. The figure was made using PyMOL with the crystal structure data of WT HSA (31). B, competitive ELISA showing binding of hFcRn to WT HSA coated in wells in the absence or presence of titrated amounts of WT HSA, HSA-K500A, DI-DII, or DIII at pH 6.0. The numbers represent the mean of triplicates.
The long serum half-life is a feature that albumin shares with the IgG class of antibodies (150 kDa), whereas other serum proteins have a half-life of hours or a few days. This is solely due to their molecular size above the renal clearance threshold and binding to a cellular receptor named the neonatal Fc receptor (FcRn)8 (7–9). Mice deficient in FcRn expression were shown to have 4–5- and 2–3-fold lower serum levels than normal mice of IgG and albumin, respectively (7, 8). A genetic link is also found in humans, as the rare disease, familial hypercatabolic hypoproteinemia, is characterized by abnormally low levels of IgG and albumin, which correlate with a lack of FcRn expression (10). Thus, FcRn is a regulator of the serum levels and circulatory half-lives of these structurally and functionally unrelated proteins. The finding is of particular importance for our understanding of how albumin and its cargo is distributed throughout the body and how albumin can be engineered and utilized for diagnostic or therapeutic applications.
FcRn belongs to a family of major histocompatibility complex class I-related molecules and consists of a unique transmembrane heavy chain that is non-covalently associated with the common soluble β2-microglobulin (11, 12). The two ligands bind simultaneously and in a non-cooperative manner to structurally separate binding sites on FcRn (13–15). A hallmark of both interactions is the strict pH dependence, binding at acidic pH and no binding or release at physiological pH (7, 14, 16–18). This is mediated by histidine residues that become positively charged at acidic pH and are located to the elbow region of the IgG Fc, to DIII of albumin, and to the receptor itself (14, 17–19).
Long serum half-life is maintained via a cellular recycling mechanism that relies on the pH-dependent binding. FcRn resides predominantly within intracellular endosomes, into which IgG and albumin enter after pinocytosis and where they bind FcRn as a consequence of the low pH. The ternary complex is recycled to the cell surface where exposure to the physiological pH of the blood triggers release. Thus, FcRn rescues both its ligands from intracellular degradation, whereas proteins that do not bind the receptor are directed to lysosomes. This recycling pathway has been shown to take place in hematopoietic cells and endothelial cells lining the vascular space (8, 20, 21).
Engineering of the interactions between FcRn and its ligands has resulted in altered binding properties that favor efficient recycling and thereby extended half-life. A number of human IgG molecules with increased binding affinity at acidic pH, but not physiological pH, have been identified that show extended half-life and improved therapeutic efficacy (22–27). Regarding albumin, recent insights into the mechanism of the FcRn-albumin interaction have resulted in publication of the first examples of engineered HSA mutant variants with altered pH-dependent binding to hFcRn, which translates into decreased or increased serum half-life in both mice and rhesus monkeys (18, 28).
Furthermore, in regard to conjugation or genetic fusion of biopharmaceuticals to HSA, it is important to control how FcRn binding is affected. Importantly, we recently showed that fusion of a short peptide or an antibody-derived single-chain variable fragment to either the N- or C-terminal end of HSA or both ends has only minor influence on binding to hFcRn (29).
To gain insight into how FcRn binds albumin pH dependently, we and others have previously reported on mapping of the core FcRn-albumin interaction interface using site-directed mutagenesis (14, 16, 18). These studies revealed that the C-terminal DIII contains the principal binding site for FcRn. Three fully conserved histidine residues on DIII are players in both direct binding to FcRn and in internal interactions of the protein (16, 18). Another interesting finding was that a recombinant form of DIII bound hFcRn but with reduced affinity compared with full-length HSA (16, 30). Furthermore, based on a large set of mutagenesis studies, we constructed a docking model of the FcRn-HSA complex using the available crystallographic data of hFcRn and wild type (WT) HSA (16). The best model suggested that the N-terminal DI may also make direct contacts with hFcRn, which was also confirmed by two recently published co-crystal structures of hFcRn in complex with a WT HSA molecule or a HSA mutant containing four amino acid substitutions (HSA13) that binds the receptor but with reduced pH discrimination (18, 31). Notably, the co-crystal structures show a fit that differs somewhat from the docking model due to changes in conformation and orientation of the three HSA subdomains upon binding to FcRn, including alterations within DIII whereby DIIIb rotates relative to DIIIa (18, 31).
In the present study we have investigated how DI of HSA impacts binding to hFcRn. Stretches of amino acid residues in two exposed loops within DI were targeted by mutagenesis, and the mutant HSA variants were expressed and compared with WT HSA with respect to binding to hFcRn using in vitro binding assays. A total of 12 amino acid residues were replaced with alanine residues. Four mutations negatively affected binding to hFcRn, whereas four improved binding. Two of the mutants shown to bind hFcRn less well are naturally existing missense HSA variants. Thus, we demonstrate that not only DIII but also DI of albumin is important for optimal pH-dependent binding to FcRn, and we pinpoint amino acid residues within two exposed loops of DI that are important for receptor binding. Our data have implications for our understanding on how FcRn regulates the long serum half-life of albumin and transports its cargo throughout the body, and such knowledge will translate into design of novel albumin-based therapeutics with improved properties.
EXPERIMENTAL PROCEDURES
Construction and Production of Recombinant FcRn Molecules
The construction of a eukaryotic pcDNA3 vector encoding a recombinant truncated form of WT hFcRn, which contains the cDNA encoding the three extracellular domains (α1-α3) C-terminally fused to a cDNA encoding glutathione S-transferase (GST) from Schistosoma japonicum, has been described (32, 33). The vector also contains a cDNA encoding human β2-microglobulin and the Epstein-Barr virus origin of replication sequence. GST-tagged hFcRn was produced by transient transfection of human embryonic kidney 293E (HEK293E) cells using polyethyleneimine Max (Polysciences), and the receptor was purified from harvested supernatant using a GSTrap FF column as described (32, 33).
Monomeric His-tagged hFcRn was produced using a Baculovirus expression vector system (34). A viral stock encoding His-tagged hFcRn was a kind gift from Dr. Sally Ward (University of Texas, Southwestern Medical Center, Dallas, TX). Briefly, hFcRn was purified using a HisTrap HP column supplied with Ni2+ ions (GE Healthcare). The column was preequilibrated with 1× PBS with 0.05% sodium azide, and the pH of the supernatant was adjusted with 1× PBS, 0.05% sodium azide (pH 10.9) to pH 7.2 before being applied to the HisTrap HP column with a flow rate of 5 ml/min. The column was washed using 200 ml of 1× PBS followed by 50 ml of 25 mm imidazole, 1× PBS (pH 7.3), and hFcRn was eluted with 250 mm imidazole, 1× PBS (pH 7.4). The collected protein was buffer-exchanged to 1× PBS using Amicron Ultra-10 filter units (Millipore) followed by isolation of the monomeric fraction. A HiLoad 26/600 Superdex 200 prep grade column (GE Healthcare) was used to isolate the monomeric fraction before the protein was concentrated using Amicon Ultra columns (Millipore) and stored at 4 °C.
Construction and Production of HSA Variants
Recombinant WT HSA, HSA-K500A, and the fragments DI-DII, DIII, and DIII-K500A were constructed, produced in yeast, and purified essentially as previously described (16).
GST-tagged HSA variants were constructed by subcloning of cDNA fragments (GenScript) into a pcDNA3 vector that contains a cDNA encoding WT HSA in-frame of a cDNA segment encoding GST (30). cDNA fragments encoding DI substitutions (R81A, E82A, E82K, T83A, Y84A, D108A, N109A, P110A, N111A, L112A, P113A, and R114G) were ordered from GenScript and subcloned into the same vector. Vectors were transiently transfected into human embryonic kidney 293E cells using polyethyleneimine Max (Polysciences), and the HSA variants were purified from harvested supernatant using a GSTrap FF column as described (30). Samples of 2 μg of each GST-tagged HSA variant were applied on a 12% SDS-PAGE (Bio-Rad). Protein concentrations were determined using a NanoDrop N-1000 spectrophotometer (NanoDrop Technologies).
ELISA
Microtiter wells (Nunc) were coated with 100 μl of non-fused WT HSA (100 μg/ml) and incubated overnight at 4 °C. Then the wells were washed 3 times with PBS, 0.005% Tween 20 (PBS/T) (pH 6.0) before blocking with 4% skimmed milk (Acumedia) for 1 h at room temperature. After washing as above, GST-tagged hFcRn (0.5 μg/ml) were diluted in PBS/T (pH 6.0) with 4% skimmed milk alone or in the presence of titrated amounts of WT HSA, HSA-K500A (16), DIII, or DI-DII (16). After incubation for 1 h at room temperature the wells were washed as above. Bound receptor was detected using a horseradish peroxidase-conjugated goat anti-GST antibody (GE Healthcare) that was added for 1 h at room temperature followed by washing with PBS/T (pH 6.0). Visualization was done using tetramethylbenzidine substrate (Calbiochem), and the absorbance was measured at 450 nm after adding 100 μl of 1 m HCl using the Sunrise spectrophotometer (TECAN).
Screening of GST-fused HSA variants were carried out by coating a human IgG1 mutant variant (M252Y/S254T/T256E/H433K/N434F) with specificity for 4-hydroxy-3-iodo-5-nitrophenylacetic acid (10 μg/ml) in microtiter wells (Nunc). The plates were incubated overnight at 4 °C before the wells were blocked with PBS, 4% skimmed milk for 1 h at room temperature followed by washing 4 times in PBS/T (pH 6.0). A constant amount of His-tagged hFcRn (20 μg/ml) was diluted in PBS/T, 4% skimmed milk (pH 6.0), added to the wells, and incubated for 2 h at room temperature before the wells were washed as above. Subsequently, 5 μg/ml GST-tagged WT HSA and the mutants were diluted in PBS/T, 4% skimmed milk (pH 6.0) and added to the wells for 2 h at room temperature. After washing as above, a horseradish peroxidase-conjugated anti-GST antibody (GE Healthcare) diluted (1:3000) in PBS/T, 4% skimmed milk (pH 6.0), was then added and incubated for 1 h. After washing, bound HSA variants were detected using tetramethylbenzidine substrate (Calbiochem). The absorbance was measured at 620 nm using the Sunrise spectrophotometer (TECAN).
SPR
SPR analyses were performed on a BIAcore 3000 instrument (GE Healthcare), and CM5 chips (GE Healthcare) were immobilized with GST-tagged hFcRn or HSA variants. The proteins (2 μg/ml) were injected in 10 mm sodium acetate at pH 5.0 (GE Healthcare), and unreacted moieties on the CM5 surface were blocked with 1 m ethanolamine following the manufacturer's procedure.
For all experiments, phosphate buffer (67 mm phosphate buffer, 0.15 m NaCl, 0.005% Tween 20) at pH 6.0 or 7.4 was used as running buffer or dilution buffer. Kinetic measurements were performed by injecting serial dilutions of WT HSA (1.0 μm to 0.03 μm) or a recombinant form of DIII HSA (200.0 μm to 0.3 μm) (16) over immobilized hFcRn (1000 RU). Alternatively, soluble monomeric hFcRn (1.0 μm to 0.03 μm) was injected over immobilized GST-fused HSA variants (500 relative units) at pH 6.0. For all experiments, buffer pH 6.0 was used with a flow rate of 50 μl/min at 25 °C. The flow cells were regenerated using PBS/T (pH 7.4). Nonspecific binding and bulk buffer effects were corrected by subtracting responses obtained from the control CM5 surfaces and blank injections. Kinetic rate constants were estimated using a simple Langmuir 1:1 ligand binding model or a steady state affinity model provided by the BIAevaluation 4.1 software. The closeness of the fit, described by the statistical value χ2, which represents the mean square, was lower than 4.0 in all affinity estimations.
Stability Analysis of HSA Variants in Human Serum
Before use in experiments, human serum was depleted for IgG and albumin. Briefly, IgG was removed using a HiTrapTM HP column supplied with Protein G-Sepharose (GE Healthcare). The column was pre-equilibrated with 20 mm sodium phosphate (pH 7.0), and serum was diluted in 1× PBS with 0.05% sodium azide (1:10) before being applied to the column at a flow rate of 1 ml/min. The flow through was collected, and subsequently, HSA was removed using a CaptureSelect™ HSA Affinity Matrix (Invitrogen). This column was pre-equilibrated with 1× PBS containing 0.05% sodium azide before application of the serum at a flow rate of 1 ml/min. The flow through was collected and stored at −80 °C. 5 μg of GST-tagged WT and HSA mutant variants were diluted in serum in a total volume of 50 μl. Four samples of each variant were prepared and incubated at 37 °C for 0, 12, 24, and 48 h. After serum incubation the samples were stored at −20 °C before analysis in ELISA as described above.
CD Spectroscopy
Circular dichroism (CD) spectra were recorded using a Jasco J-810 spectropolarimeter (Jasco International) calibrated with ammonium d-camphor-10-sulfonate (Icatayama Chemicals). Measurements were performed with HSA-GST variants (0.15 mg/ml) in 10 mm PBS (pH 6.0) without NaCl added at 25 °C, 50 °C, and 70 °C using a quartz cuvette (Starna) with a path length of 0.1 cm. Each sample was scanned 3 times at 50 nm/min (bandwidth of 1 nm, response time of 4 s) with the wavelength range set to 190–260 nm. The data were averaged, and the spectrum of a sample-free control was subtracted. Secondary structural elements were calculated using the neural network program CDNN Version 2.1 and the supplied neural network based on the 33-member basis set (35).
Structural Analysis
Coordinates of the crystal structures of hFcRn in complex with WT HSA (PDB ID 4N0F) (31) or a HSA variant (HSA13) containing four mutations (PDB ID 4K71) (18) were retrieved from the Protein Data Bank. The structures were inspected using PyMOL (Schrodinger Inc.).
RESULTS
DIII Competes More Weakly than Full-length HSA for Binding to hFcRn
Albumin has a heart-shaped structure that is made up of three subdomains; DI, DII, and DIII (Fig. 1A). We have previously demonstrated that the C-terminal DIII is the principal domain that is engaged in pH-dependent binding to hFcRn (16, 30). Here we used a competitive ELISA to investigate how recombinant forms of HSA DIII and DI-DII competed for binding to hFcRn compared with full-length HSA. The results showed that nearly 20-fold more DIII than complete HSA was needed to give 50% reduction in hFcRn binding. However, DIII competed more efficiently than HSA K500A, a full-length variant with a mutation within DIII that causes a 30-fold reduction in hFcRn affinity (16). The DI-DII construct inhibited binding only at concentrations above 100 μm (Fig. 1B).
Next, we used SPR and injected equimolar amounts of WT HSA, DIII, and DIII containing the K500A substitution over immobilized hFcRn at pH 6.0. The resulting sensorgrams showed that DIII has a much faster kinetic profile than full-length WT HSA, whereas the introduced single point mutation in DIII eliminated binding to the receptor (Fig. 2A). To determine the binding affinity of DIII, we injected serial dilutions of DIII over immobilized hFcRn (Fig. 2B), and the equilibrium binding responses were fitted to a steady state affinity binding model (Fig. 2C), resulting in a calculated affinity constant (KD) of 12 μm, which is >10-fold higher than previously reported for the WT HSA-hFcRn pair (29). Hence, the faster binding kinetics of DIII explains its decreased ability to compete for binding to hFcRn in the ELISA (Fig. 1B).
FIGURE 2.

DIII binds weaker than full-length HSA to hFcRn. A, representative SPR sensorgrams show binding of equimolar amounts of WT HSA, DIII, and DIII-K500A injected over immobilized hFcRn at pH 6.0. B, representative SPR sensorgrams show binding of titrated amounts of monomeric HSA DIII injected over immobilized hFcRn at pH 6.0. C, the equilibrium binding responses of HSA DIII to hFcRn plotted versus injected concentration of DIII. The curve was fitted to a steady state affinity binding model supplied with the BIAevaluation software. RU, resonance units.
Structural Inspection Proposes That DI Is Involved in Binding to FcRn
We previously reported on a docking model of the hFcRn-HSA WT complex, which was derived from experimental evidence demonstrating that DIII is the principal binding domain (16). In addition, the model suggests that DI is in proximity to hFcRn via two exposed loops encompassing amino acid residues 80–89 (loop I) and 105–114 (loop II), respectively (highlighted in Fig. 7A).
FIGURE 7.

Structural inspection of HSA-hFcRn complexes. A, overall illustration of the co-crystal structure of the extracellular part of hFcRn in complex with WT HSA. The heavy chain is shown in green, and the β2m subunit is gray. The three domains of hFcRn are indicated; α1, α2, α3. The three α-helical domains of HSA, DI, DII, and DIII are shown in pink, orange, and two shades of blue, respectively. B, a close-up of the structural interface of WT HSA DI and hFcRn shows the interaction hFcRn-Thr-153 and HSA-Asp-89, and the intramolecular hydrogen bond between HSA-Arg-81 and HSA-Asp-89. C, a close-up of the structural area of WT HSA shows the interdomain hydrogen bond between Lys-466 of DIII and Asp-108 of DI. D, a close-up of the structural area of WT HSA DI and hFcRn shows HSA-Asn-111, which makes two favorable contacts with hFcRn-Ser-58 as well as a repulsive contact with hFcRn-Lys-63. E, a close-up of a superposition of the WT HSA-hFcRn (pink) and HSA13-hFcRn (dark red) co-crystal structures shows that loop II has a very different conformation in the two complexes. The figures were made using PyMOL with the crystal structure data of WT HSA-hFcRn and HSA13-hFcRn (18, 31).
Screening of Mutant HSA DI Variants
To address the impact of amino acids in the two loops on FcRn binding, we targeted 10 individual residues by mutating each to alanine (loop I: R81A, E82A, T83A, and Y84A; and loop II: D108A, N109A, P110A, N111A, L112A, and P113A). The resulting 10 HSA variants were expressed as GST-fusions by transfection of HEK293E cells followed by purification on a GSTrap FF column. The fractions were analyzed by SDS-PAGE followed by Coomassie staining, which revealed that all migrated as distinct bands with molecular weights corresponding to their expected sizes (Fig. 3A).
FIGURE 3.

Engineered HSA DI variants and binding to hFcRn. A, GST-tagged WT HSA and DI mutant variants produced in HEK293E cells and purified on a GSTrap FF column were analyzed by 12% (w/v) SDS-PAGE. ELISA measurements show binding of WT HSA and the loop I (R81A, E82A, T83A, and Y84A) (B) and loop II (D108A, N109A, P110A, N111A, L112A, and P113A) (C) DI mutant variants to hFcRn at pH 6.0. The numbers given represent the mean of triplicates.
We then used an ELISA where serial dilutions of the HSA variants were added to hFcRn captured on a human IgG1 variant engineered to bind hFcRn strongly. Binding of WT HSA and the mutant variants was detected using an HRP-conjugated anti-GST antibody, and the results obtained showed that WT HSA and the mutant variants bound hFcRn at pH 6.0 (Fig. 3, B and C), which is in agreement with the fact that both ligands can bind FcRn simultaneously (13, 14, 17). For the loop I variants, R81A, E82A, and Y84A gave rise to reduced binding responses, with the most negative effect detected for R81A, whereas T83A showed slightly enhanced binding compared with the WT (Fig. 3B). Furthermore, screening of the loop II variants showed that mutation of Asp-108 had the greatest negative effect followed by P110A and P113A, whereas N109A, N111A, and L112A showed enhanced binding to hFcRn (Fig. 3C). Repeating the ELISA at pH 7.4 showed that neither the WT nor the mutant variants bound hFcRn at neutral pH (data not shown).
SPR-based Determination of Kinetics
To determine the binding kinetics, we used SPR where titrated amounts of monomeric hFcRn were injected over immobilized HSA variants at pH 6.0. The obtained sensorgrams fitted well to a 1:1 Langmuir binding model where the estimated kinetic constants showed that the loop I mutant variants E82A, T83A, and Y84A had no major impact on the binding kinetics, whereas R81A gave a nearly 2-fold reduced affinity compared with WT HSA (Fig. 4, Table 1). Of the loop II variants, D108A showed the largest negative impact, with a >4-fold reduced binding affinity, whereas P110A did not alter the kinetics (Fig. 4, F and H, Table 1). Interestingly, alanine substitution at positions Asn-109, Asn-111, Leu-112, and Pro-113 improved binding by 1.5–2.4-fold, where L112A gave the most pronounced effect (Fig. 4, G and I–K, Table 1). The kinetic data were in agreement with the trend of binding observed in ELISA with the exception of P113A that showed reduced binding in ELISA compared with the WT (Fig. 3C). In addition, when comparing the binding kinetics of the WT GST-fusion with that of non-fused HSA, a minor negative impact on hFcRn binding was observed that is in agreement with a previous report on C-terminal HSA fusions (29).
FIGURE 4.

SPR binding kinetics of HSA DI variants to hFcRn. Representative SPR sensorgrams show hFcRn binding of WT HSA (A), R81A (B), E82A (C), T83A (D), Y84A (E), D108A (F), N109A (G), P110A (H), N111A (I), L112A (J), and P113A (K) at pH 6.0. Injections were performed at 25 °C, and the flow rate was 50 μl/min. RU, resonance units.
TABLE 1.
Binding kinetics of HSA DI mutants toward hFcRn
| HSA variantsa | ka | kd | KDb | χ2c |
|---|---|---|---|---|
| 104/Ms | 10−3/s | nm | ||
| HSAa | ||||
| WT | 4.3 ± 0.1 | 5.4 ± 0.1 | 125.6 | 1.9 |
| HSA-GSTa | ||||
| WT | 4.6 ± 0.1 | 6.6 ± 0.1 | 143.4 | 2.1 |
| R81A | 3.9 ± 0.1 | 9.3 ± 0.2 | 238.5 | 2.1 |
| E82A | 4.7 ± 0.2 | 6.4 ± 0.2 | 136.2 | 1.9 |
| E82K Vibo Valentia | 3.3 ± 0.1 | 1.1 ± 0.1 | 333.3 | 3.1 |
| T83A | 4.5 ± 0.0 | 6.5 ± 0.1 | 144.4 | 1.8 |
| Y84A | 3.5 ± 0.1 | 4.0 ± 0.1 | 114.3 | 2.1 |
| D108A | 3.3 ± 0.1 | 20.5 ± 0.5 | 621.2 | 1.0 |
| N109A | 5.2 ± 0.2 | 5.0 ± 0.1 | 96.2 | 2.9 |
| P110A | 4.4 ± 0.1 | 6.5 ± 0.2 | 147.0 | 2.1 |
| N111A | 5.9 ± 0.2 | 4.9 ± 0.1 | 83.1 | 2.8 |
| L112A | 5.0 ± 0.2 | 2.5 ± 0.1 | 50.0 | 2.4 |
| P113A | 6.6 ± 0.3 | 0.4 ± 0.0 | 60.1 | 3.5 |
| R114G Yanomama-2 | 5.3 ± 0.1 | 1.4 ± 0.2 | 264.1 | 3.1 |
a The HSA variants were immobilized (∼500 RU) on chips, and serial dilutions of hFcRn were injected.
b The kinetic rate constants were obtained using a simple first-order (1:1) bimolecular interaction model. The kinetic values represent the average of triplicates.
c χ2 values resulting from curve fitting using the first-order (1:1) bimolecular interaction model. χ2 is a measure of the average squared residual (the difference between the experimental data and the fitted curve).
Two Natural Occurring HSA DI Single Mutant Variants
So far >60 genetic variants of HSA have been characterized (37). Two of these are missense mutants in which a single nucleotide change resulted in an amino acid substitution within the Dl loops; one in loop I (Vibo Valentia, E82K), and one in loop II (Yanomama-2, R114G) (38, 39). We produced these two variants as recombinant GST-fusions that migrated with a profile similar to WT HSA (Fig. 5A). Subsequently, binding to hFcRn was assessed by ELISA, which revealed that both bound the receptor slightly weaker than WT HSA at pH 6.0, whereas none bound at pH 7.4 (Fig. 5B; data not shown). Using SPR, their binding kinetics were determined, and both showed an increase in KD; E82K 2-fold (Fig. 5C, Table 1) and R114G 1.8-fold (Fig. 5D, Table 1).
FIGURE 5.

Two naturally existing HSA DI variants show reduced hFcRn binding. A, GST-tagged WT HSA, Vibo Valentia (E82K), and Yanomama-2 (R114G) were produced in HEK293E cells, purified on a GSTrap FF column, and analyzed by 12% (w/v) SDS-PAGE. B, ELISA measurements show binding of WT HSA, E82K, and R114G to hFcRn at pH 6.0. The numbers given represent the mean of triplicates. Representative SPR sensorgrams show hFcRn binding to E82K (C) and R114G (D) at pH 6.0. Injections were performed at 25 °C, and the flow rate was 50 μl/min. RU, resonance units.
Protein Structure and Stability Analysis
To verify whether the introduced point mutations affected secondary structure and stability of the HSA-GST variants, we performed CD analysis of the WT and four of the mutants for which binding kinetics differed the most from the WT; D108A and E82K (decreased FcRn affinity) and L112A and P113A (improved FcRn affinity). CD was performed at 25 °C, 50 °C, and 70 °C, and we found the dominating structural element for all variants to be α-helix, reflected by a peak of absorption at wavelength 190 nm and two absorption minima at 208 nm and 222 nm (Fig. 6, A–C). This is in agreement with published crystal structures showing that the dominating structure element for both HSA and GST is α-helices (40, 41). WT HSA-GST was found to have 82% α-helix and the mutants 74–82% (Table 2). L112A and P113A showed similar content as the WT (82–80%), whereas the two weak binders D108A and E82K had slightly lower α-helical content, 75 and 74%, respectively. The same patterns were observed at 50 °C, where the α-helix content was only slightly reduced for all variants, reflecting retained secondary structure at this temperature. At 70 °C, the spectra correspond to unfolded proteins, which is in agreement with reports showing that both DI and DII are irreversibly denatured at this temperature (42).
FIGURE 6.

CD spectra and serum stability analysis of HSA DI variants. Representative CD spectra of WT and the HSA mutants E82K, D108A, L112A, and P113A at 25 °C (A), 50 °C (B), and 70 °C (C) obtained by CD measurements at pH 6.0. MRE, mean residual ellipticity. D, ELISA measurements showing relative hFcRn binding of WT and the HSA mutants at pH 6.0 after 0, 12, 24, and 48 h incubation in human serum at 37 °C. The numbers given represent the mean of triplicates.
TABLE 2.
The α-helix content of HSA-GST variants
| HSA-GST variants | α-Helix |
||
|---|---|---|---|
| 25 °C | 50 °C | 70 °C | |
| % | |||
| WT | 82.3 | 76.5 | 20.5 |
| E82K | 74.7 | 68.4 | 43.1 |
| D108A | 75.3 | 71.9 | 18.6 |
| L112A | 82.6 | 78.5 | 21.6 |
| P113A | 80.4 | 78.5 | 15.9 |
To further investigate their stability, the HSA-GST variants were incubated in human serum for 0 h, 12 h, 24 h, and 48 h at 37 °C, and subsequently the effect on FcRn binding was examined in ELISA. All variants showed reduced binding to hFcRn as a function of incubation time (Fig. 6D). After 48 h 40% reduction was observed for WT HSA, whereas three of the mutants (E82K, L112A, and P113A) showed only 20–25% reduced binding to hFcRn. The D108A mutant gave rise to as much as 60% loss of activity after 48 h, which may indicate that this mutation has the most pronounced negative impact on protein stability.
Structural Explanations of the Impact of DI Amino Acid Substitutions
After the publishing of our docking model of the hFcRn-HSA complex (16), two studies recently reported on co-crystal structures of hFcRn in complex with HSA (18, 31), one with WT HSA and one with an engineered HSA variant (HSA13). The latter HSA variant binds with reduced pH dependence and with a 300-fold enhanced affinity at acidic pH. The two experimental structures confirm that DI makes contacts with hFcRn via the same two loops that were predicted from the docking model, but no interaction analysis was presented in these studies to investigate the impact of the DI interactions.
To explain our binding data, we inspected the co-crystal structure of the WT HSA-hFcRn pair (Fig. 7, A–D). For the stretch of amino acids corresponding to loop I, we found that Arg-81 forms an intramolecular salt bridge with Asp89 (Fig. 7B). Disruption of the intramolecular interaction by mutation to alanine could possibly affect the positioning of the loop. Additionally Arg-81 forms hydrogen bonds with Thr-153 of hFcRn (Fig. 7B). Thus, the weaker binding detected for R81A is likely due to a combination of these two factors. The side chain of Glu-82 is not involved in any direct interactions, although the backbone carbonyl group forms a weak hydrogen bond with His-161 of hFcRn. Furthermore, mutations affecting the position of loop I have the potential to affect the intermolecular salt bridge between Glu-86 in HSA and Lys-150 in FcRn. For the loop II mutant variants, substitution of Asp-108 with an alanine had the largest negative effect. As for Glu-82, the Asp-108 side chain does not form any direct interaction with FcRn but instead forms intramolecular hydrogen bonds with His-105 and Lys-466 within DI and DIII of HSA, respectively (Fig. 7C). The removal of these interactions may destabilize the intramolecular interaction between the HSA domains and affect the backbone confirmation, disrupting the hydrogen bond between the backbone carbonyl of Asp-108 and the Lys-63 side chain of FcRn.
Mutation of Asn-109 within loop II of DI improved binding to FcRn, but it is difficult to explain the favorable impact of N109A in the context of the WT HSA FcRn complex structure. Asn-111 forms two favorable contacts with both the side chain and backbone of Ser-58 in the α1-domain of hFcRn and one unfavorable repulsive contact with hFcRn Lys-63 (Fig. 7D). Mutation to an alanine may relieve the latter unfavorable repulsion but only at the expense of two lost hydrogen bond interactions between HSA and hFcRn. Because N111A results in improved binding, it suggests that removal of these interactions may allow improved interactions in neighboring locations.
Enhanced binding was also measured for L112A and P113A, but neither of these residues is in direct contact with the receptor. A possible explanation for stronger binding may be that these substitutions, going from larger side chains to the smaller alanine methyl group, give rise to a more flexible and adjustable loop. However, the R114G Yanomama-2 variant, which would be expected to increase flexibility of the loop region, showed reduced binding compared with WT HSA.
Interestingly, when the co-crystal structure of hFcRn in complex with WT HSA was compared with that of HSA13, we found that loop II of WT HSA and HSA13 have different conformations (Fig. 7E). Consequently, the residues Asp-108 and Asn-111 can take on different orientations and positions in the complexes. Notably, the HSA13 structure contains two crystallographically independent copies of the complex, of which one with an alternative orientation for Asn-111 is shown in Fig. 7E. Thus, there is clearly potential for loop II to adopt a number of conformations and, therefore, interactions during complexation.
DISCUSSION
FcRn is a bifunctional receptor that binds two completely unrelated soluble proteins, IgG and albumin. Strikingly, binding of both ligands is strictly pH-dependent, a mechanism that has evolved to secure constitutive receptor-mediated recycling and thus rescue from intracellular degradation. The process is efficient as exemplified by studies revealing that the receptor salvages one and one-half times the amount of IgG produced by plasma cells daily and half the amount of albumin produced by the hepatocytes (43, 44). A complete understanding of the molecular interplay between FcRn and its ligands is necessary so as to unravel how the receptor acts in homeostatic regulation and how the ligands are transported and distributed throughout the body.
Although IgG is the main antibody of the blood that is involved in fighting infections, albumin serves as a molecular taxi that transports a plethora of cargo in the bloodstream for delivery to their destination. These uncoupled functions are maintained by keeping the concentrations in blood stable and high at all times by balancing their production rates with the level of FcRn expression.
In this study we show that two of three HSA domains take direct part in binding to hFcRn. Compared with full-length HSA, we show that DIII on its own has a 12-fold weaker binding affinity toward hFcRn. This prompted us to investigate the contribution of DI.
Inspection of a previously published docking model of the FcRn-HSA complex (16) led us to suggest that two exposed loops within the N-terminal DI may make direct contact with hFcRn. Since then two co-crystal structures of hFcRn in complex with HSA have been published, one with WT HSA and one with HSA13, a mutant HSA molecule containing four substitutions (18, 31). The latter binds hFcRn with >300-fold improved affinity at pH 6.0 but with loss of pH dependence as it also binds with a significant affinity at pH 7.4. Thus, HSA13 has a reduced serum half-life compared with WT HSA in hFcRn transgenic mice (18). Both co-crystal structures show that hFcRn makes contacts with DI via the same two loops as predicted from the docking model. Variation of the orientation of loop II and the associated interaction network between the WT HSA and HSA13 complexes suggests that loop II is a structural element with some flexibility.
In this study we targeted a selection of the amino acids that are positioned within the two DI loops by mutagenesis, which revealed that replacement of several residues with a neutral alanine modulated hFcRn binding either by gaining or loosing binding affinity at acidic pH, whereas none of the mutants bound at pH 7.4. Thus, our findings show that although the C-terminal DIII contains the principal binding site, which is very important for pH-dependent binding to FcRn, residues in DI also play a substantial role in binding.
None of the mutations hampered the secretion of the recombinant HSA-GST variants from HEK293E cells as they were produced in similar amounts, and SDS-PAGE analysis demonstrated that the mutants migrated as distinct bands similar to that of the WT. Furthermore, measurements of their α-helical content by CD spectroscopy showed that the two weakest binders (D108A and E82K) have 7–8% less α-helices than the WT fusion. In addition, serum stability was measured by incubation of the HSA GST variants in serum followed by binding to hFcRn in ELISA, which showed that D108A was the least stable of the mutants.
By inspecting the co-crystal structures, the observed effects of the mutations could be explained for some but not for all variants. Our analysis of mutant HSA variants suggests that substitution to alanine may impact local conformation rather than affect direct interactions, which in some instances can have a positive impact on the hFcRn-HSA interaction. The conformational flexibility of the subdomain connector loops in HSA has previously been noted (45).
Furthermore, we were intrigued by the fact that two rare naturally occurring HSA variants exist with single amino acid substitutions within the DI loops (38, 39). We expressed these as recombinant molecules and demonstrated that they bound with reduced FcRn affinity. If found in a heterozygote individual, the HSA variants may thus have decreased half-lives as they will compete with WT HSA for binding to hFcRn. Interestingly, homozygotes for the Yanomama-2 variant (R114G), found among the Brazilian tribe with the same name, have been shown to bind bilirubin poorly. This raises an important question on how structural alterations of HSA and the binding of ligands affect FcRn binding and subsequent biodistribution, a topic that should be addressed in future studies.
For determination of binding kinetics of full-length HSA variants, GST-fusions were immobilized on CM5 chips by amine coupling, and for the WT HSA-hFcRn pair the derived KD value was shown to be almost 7-fold higher than when the receptor was immobilized and monomeric HSA injected. The difference in binding strength, which depends on the SPR set-up, is in line with previous studies (16, 17, 29). The reason for this phenomenon is unknown, but it may be due to an effect of the amine coupling procedure where free amine groups of exposed amino acids are targeted. Similar but more drastic is the two ways of immobilization for measurements of the IgG-FcRn interaction, where nearly 300-fold differences are detected (13, 36, 46, 47). Thus, caution must be employed when comparing kinetic values across different experimental set-ups.
Most therapeutic molecules have a molecular weight below the renal clearance threshold and thus are rapidly eliminated from the circulation, a feature that limits their therapeutic potential. This bottleneck may be overcome by the use of HSA as a carrier to increase the circulatory half-life and bioavailability of such molecules. Strategies to achieve this may be by association, chemical conjugation, or genetic fusion to HSA (3–5). In this regard our growing understanding of the pivotal role of FcRn in maintaining the circulatory half-life of albumin may guide the development of HSA-based therapeutics with improved properties and pharmacokinetics.
Although we have shown that the C-terminal end of DIII is very important for binding to hFcRn, genetic fusion of a peptide or an antibody-derived single-chain variable fragment to the C-terminal end slightly affects binding negatively, whereas when fused to the N terminus, binding was more or less unaffected (29). Reduced receptor binding upon fusion may be compensated by introducing amino acid substitutions in HSA that improve pH-dependent binding to hFcRn. One such example is a HSA variant with a single point mutation within DIII (K573P) that shows 12-fold improved binding toward hFcRn, which translates into 3.4 days longer half-life in rhesus monkeys (28). Our data presented here showing the requirement of DI for optimal binding to FcRn may pave the way for a next generation of engineered HSA variants that includes DI, such as the DI mutations with increased binding affinity described in this study.
Acknowledgment
We are grateful to Sathiaruby Sivaganesh for excellent technical assistance.
This work was supported in part by the Research Council of Norway through its Centres of Excellence funding scheme (Project 179573). I. S., J. T. A., B. D., K. B., and J. C. are co-inventors of pending patent applications related to the data described in this paper.
- FcRn
- neonatal Fc receptor
- hFcRn
- human FcRn
- HSA
- human serum albumin
- DI
- DII, DIII, domains I, II, and III
- SPR
- surface plasmon resonance
- RU
- resonance units.
REFERENCES
- 1. Ghuman J., Zunszain P. A., Petitpas I., Bhattacharya A. A., Otagiri M., Curry S. (2005) Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 353, 38–52 [DOI] [PubMed] [Google Scholar]
- 2. Peters T., Jr. (1996) All About Albumin: Biochemistry, Genetics and Medical Applications, Academic Press, San Diego, CA [Google Scholar]
- 3. Andersen J. T., Sandlie I. (2009) The versatile MHC class I-related FcRn protects IgG and albumin from degradation: implications for development of new diagnostics and therapeutics. Drug Metab. Pharmacokinet. 24, 318–332 [DOI] [PubMed] [Google Scholar]
- 4. Elsadek B., Kratz F. (2012) Impact of albumin on drug delivery: new applications on the horizon. J. Control. Release 157, 4–28 [DOI] [PubMed] [Google Scholar]
- 5. Sleep D., Cameron J., Evans L. R. (2013) Albumin as a versatile platform for drug half-life extension. Biochim. Biophys. Acta 1830, 5526–5534 [DOI] [PubMed] [Google Scholar]
- 6. Andersen J. T., Pehrson R., Tolmachev V., Daba M. B., Abrahmsén L., Ekblad C. (2011) Extending half-life by indirect targeting of the neonatal Fc receptor (FcRn) using a minimal albumin binding domain. J. Biol. Chem. 286, 5234–5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chaudhury C., Mehnaz S., Robinson J. M., Hayton W. L., Pearl D. K., Roopenian D. C., Anderson C. L. (2003) The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J. Exp. Med. 197, 315–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Montoyo H. P., Vaccaro C., Hafner M., Ober R. J., Mueller W., Ward E. S. (2009) Conditional deletion of the MHC class I-related receptor FcRn reveals the sites of IgG homeostasis in mice. Proc. Natl. Acad. Sci. U.S.A. 106, 2788–2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ward E. S., Ober R. J. (2009) Chapter 4: multitasking by exploitation of intracellular transport functions the many faces of FcRn. Adv. Immunol. 103, 77–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wani M. A., Haynes L. D., Kim J., Bronson C. L., Chaudhury C., Mohanty S., Waldmann T. A., Robinson J. M., Anderson C. L. (2006) Familial hypercatabolic hypoproteinemia caused by deficiency of the neonatal Fc receptor, FcRn, due to a mutant β2-microglobulin gene. Proc. Natl. Acad. Sci. U.S.A. 103, 5084–5089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Simister N. E., Mostov K. E. (1989) Cloning and expression of the neonatal rat intestinal Fc receptor, a major histocompatibility complex class I antigen homolog. Cold Spring Harbor Symp. Quant. Biol. 54, 571–580 [DOI] [PubMed] [Google Scholar]
- 12. Story C. M., Mikulska J. E., Simister N. E. (1994) A major histocompatibility complex class I-like Fc receptor cloned from human placenta: possible role in transfer of immunoglobulin G from mother to fetus. J. Exp. Med. 180, 2377–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Andersen J. T., Daba M. B., Berntzen G., Michaelsen T. E., Sandlie I. (2010) Cross-species binding analyses of mouse and human neonatal Fc receptor show dramatic differences in immunoglobulin G and albumin binding. J. Biol. Chem. 285, 4826–4836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Andersen J. T., Dee Qian J., Sandlie I. (2006) The conserved histidine 166 residue of the human neonatal Fc receptor heavy chain is critical for the pH-dependent binding to albumin. Eur. J. Immunol. 36, 3044–3051 [DOI] [PubMed] [Google Scholar]
- 15. Anderson C. L., Chaudhury C., Kim J., Bronson C. L., Wani M. A., Mohanty S. (2006) Perspective: FcRn transports albumin: relevance to immunology and medicine. Trends Immunol. 27, 343–348 [DOI] [PubMed] [Google Scholar]
- 16. Andersen J. T., Dalhus B., Cameron J., Daba M. B., Plumridge A., Evans L., Brennan S. O., Gunnarsen K. S., Bjørås M., Sleep D., Sandlie I. (2012) Structure-based mutagenesis reveals the albumin-binding site of the neonatal Fc receptor. Nat. Commun. 3, 610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chaudhury C., Brooks C. L., Carter D. C., Robinson J. M., Anderson C. L. (2006) Albumin binding to FcRn: distinct from the FcRn-IgG interaction. Biochemistry 45, 4983–4990 [DOI] [PubMed] [Google Scholar]
- 18. Schmidt M. M., Townson S. A., Andreucci A. J., King B. M., Schirmer E. B., Murillo A. J., Dombrowski C., Tisdale A. W., Lowden P. A., Masci A. L., Kovalchin J. T., Erbe D. V., Wittrup K. D., Furfine E. S., Barnes T. M. (2013) Crystal structure of an HSA/FcRn complex reveals recycling by competitive mimicry of HSA ligands at a pH-dependent hydrophobic interface. Structure 21, 1966–1978 [DOI] [PubMed] [Google Scholar]
- 19. Sand K. M., Dalhus B., Christianson G. J., Bern M., Foss S., Cameron J., Sleep D., Bjørås M., Roopenian D. C., Sandlie I., Andersen J. T. (2014) Dissection of the neonatal Fc receptor (FcRn)-albumin interface using mutagenesis and anti-FcRn albumin-blocking antibodies. J. Biol. Chem. 289, 17228–17239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Akilesh S., Christianson G. J., Roopenian D. C., Shaw A. S. (2007) Neonatal FcR expression in bone marrow-derived cells functions to protect serum IgG from catabolism. J. Immunol. 179, 4580–4588 [DOI] [PubMed] [Google Scholar]
- 21. Kobayashi K., Qiao S. W., Yoshida M., Baker K., Lencer W. I., Blumberg R. S. (2009) An FcRn-dependent role for anti-flagellin immunoglobulin G in pathogenesis of colitis in mice. Gastroenterology 137, 1746–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dall'Acqua W. F., Woods R. M., Ward E. S., Palaszynski S. R., Patel N. K., Brewah Y. A., Wu H., Kiener P. A., Langermann S. (2002) Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J. Immunol. 169, 5171–5180 [DOI] [PubMed] [Google Scholar]
- 23. Ghetie V., Popov S., Borvak J., Radu C., Matesoi D., Medesan C., Ober R. J., Ward E. S. (1997) Increasing the serum persistence of an IgG fragment by random mutagenesis. Nat. Biotechnol. 15, 637–640 [DOI] [PubMed] [Google Scholar]
- 24. Hinton P. R., Johlfs M. G., Xiong J. M., Hanestad K., Ong K. C., Bullock C., Keller S., Tang M. T., Tso J. Y., Vásquez M., Tsurushita N. (2004) Engineered human IgG antibodies with longer serum half-lives in primates. J. Biol. Chem. 279, 6213–6216 [DOI] [PubMed] [Google Scholar]
- 25. Hinton P. R., Xiong J. M., Johlfs M. G., Tang M. T., Keller S., Tsurushita N. (2006) An engineered human IgG1 antibody with longer serum half-life. J. Immunol. 176, 346–356 [DOI] [PubMed] [Google Scholar]
- 26. Petkova S. B., Akilesh S., Sproule T. J., Christianson G. J., Al Khabbaz H., Brown A. C., Presta L. G., Meng Y. G., Roopenian D. C. (2006) Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int. Immunol. 18, 1759–1769 [DOI] [PubMed] [Google Scholar]
- 27. Zalevsky J., Chamberlain A. K., Horton H. M., Karki S., Leung I. W., Sproule T. J., Lazar G. A., Roopenian D. C., Desjarlais J. R. (2010) Enhanced antibody half-life improves in vivo activity. Nat. Biotechnol. 28, 157–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Andersen J. T., Dalhus B., Viuff D., Ravn B. T., Gunnarsen K. S., Plumridge A., Bunting K., Antunes F., Williamson R., Athwal S., Allan E., Evans L., Bjørås M., Kjærulff S., Sleep D., Sandlie I., Cameron J. (2014) Extending serum half-life of albumin by engineering FcRn binding. J. Biol. Chem. 289, 13492–134502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Andersen J. T., Cameron J., Plumridge A., Evans L., Sleep D., Sandlie I. (2013) Single-chain variable fragment albumin fusions bind the neonatal Fc Receptor (FcRn) in a species-dependent manner: implications for in vivo half-life evaluation of albumin fusion therapeutics. J. Biol. Chem. 288, 24277–24285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Andersen J. T., Daba M. B., Sandlie I. (2010) FcRn binding properties of an abnormal truncated analbuminemic albumin variant. Clin. Biochem. 43, 367–372 [DOI] [PubMed] [Google Scholar]
- 31. Oganesyan V., Damschroder M. M., Cook K. E., Li Q., Gao C., Wu H., Dall'Acqua W. F. (2014) Structural insights into neonatal Fc receptor-based recycling mechanisms. J. Biol. Chem. 289, 7812–7824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Andersen J. T., Justesen S., Fleckenstein B., Michaelsen T. E., Berntzen G., Kenanova V. E., Daba M. B., Lauvrak V., Buus S., Sandlie I. (2008) Ligand binding and antigenic properties of a human neonatal Fc receptor with mutation of two unpaired cysteine residues. FEBS J. 275, 4097–4110 [DOI] [PubMed] [Google Scholar]
- 33. Berntzen G., Lunde E., Flobakk M., Andersen J. T., Lauvrak V., Sandlie I. (2005) Prolonged and increased expression of soluble Fc receptors, IgG and a TCR-Ig fusion protein by transiently transfected adherent 293E cells. J. Immunol. Methods 298, 93–104 [DOI] [PubMed] [Google Scholar]
- 34. Kim J. K., Firan M., Radu C. G., Kim C. H., Ghetie V., Ward E. S. (1999) Mapping the site on human IgG for binding of the MHC class I-related receptor, FcRn. Eur. J. Immunol. 29, 2819–2825 [DOI] [PubMed] [Google Scholar]
- 35. Böhm G., Muhr R., Jaenicke R. (1992) Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 5, 191–195 [DOI] [PubMed] [Google Scholar]
- 36. Zhou J., Mateos F., Ober R. J., Ward E. S. (2005) Conferring the binding properties of the mouse MHC class I-related receptor, FcRn, onto the human ortholog by sequential rounds of site-directed mutagenesis. J. Mol. Biol. 345, 1071–1081 [DOI] [PubMed] [Google Scholar]
- 37. Kragh-Hansen U., Minchiotti L., Galliano M., Peters T., Jr. (2013) Human serum albumin isoforms: genetic and molecular aspects and functional consequences. Biochim. Biophys. Acta 1830, 5405–5417 [DOI] [PubMed] [Google Scholar]
- 38. Galliano M., Minchiotti L., Porta F., Rossi A., Ferri G., Madison J., Watkins S., Putnam F. W. (1990) Mutations in genetic variants of human serum albumin found in Italy. Proc. Natl. Acad. Sci. U.S.A. 87, 8721–8725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Takahashi N., Takahashi Y., Isobe T., Putnam F. W., Fujita M., Satoh C., Neel J. V. (1987) Amino acid substitutions in inherited albumin variants from Amerindian and Japanese populations. Proc. Natl. Acad. Sci. U.S.A. 84, 8001–8005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sugio S., Kashima A., Mochizuki S., Noda M., Kobayashi K. (1999) Crystal structure of human serum albumin at 2.5 A resolution. Protein Eng. 12, 439–446 [DOI] [PubMed] [Google Scholar]
- 41. Cardoso R. M., Daniels D. S., Bruns C. M., Tainer J. A. (2003) Characterization of the electrophile binding site and substrate binding mode of the 26-kDa glutathione S-transferase from Schistosoma japonicum. Proteins 51, 137–146 [DOI] [PubMed] [Google Scholar]
- 42. Flora K., Brennan J. D., Baker G. A., Doody M. A., Bright F. V. (1998) Unfolding of acrylodan-labeled human serum albumin probed by steady-state and time-resolved fluorescence methods. Biophys. J. 75, 1084–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim J., Hayton W. L., Robinson J. M., Anderson C. L. (2007) Kinetics of FcRn-mediated recycling of IgG and albumin in human: pathophysiology and therapeutic implications using a simplified mechanism-based model. Clin. Immunol. 122, 146–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim J., Bronson C. L., Hayton W. L., Radmacher M. D., Roopenian D. C., Robinson J. M., Anderson C. L. (2006) Albumin turnover: FcRn-mediated recycling saves as much albumin from degradation as the liver produces. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G352–G360 [DOI] [PubMed] [Google Scholar]
- 45. Curry S., Mandelkow H., Brick P., Franks N. (1998) Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat. Struct. Biol. 5, 827–835 [DOI] [PubMed] [Google Scholar]
- 46. Vaughn D. E., Bjorkman P. J. (1997) High-affinity binding of the neonatal Fc receptor to its IgG ligand requires receptor immobilization. Biochemistry 36, 9374–9380 [DOI] [PubMed] [Google Scholar]
- 47. Firan M., Bawdon R., Radu C., Ober R. J., Eaken D., Antohe F., Ghetie V., Ward E. S. (2001) The MHC class I-related receptor, FcRn, plays an essential role in the maternofetal transfer of γ-globulin in humans. Int. Immunol. 13, 993–1002 [DOI] [PubMed] [Google Scholar]