

Background: How RPM-1 interacts with FSN-1 remains unknown.
Results: Structure-function and transgenic analysis define the biochemical relationship between RPM-1 and FSN-1.
Conclusion: RPM-1 uses a conserved mechanism to bind FSN-1 that is independent of RPM-1 ubiquitin ligase activity.
Significance: Our biochemical and genetic analysis has led to identification of RIP, an in vivo inhibitor of the RPM-1·FSN-1 ubiquitin ligase complex.
Keywords: axon, Caenorhabditis elegans (C. elegans), Cell Signaling, Neurodegeneration, Ubiquitin Ligase, F-box, FSN-1, Neuronal Development
Abstract
The Pam/Highwire/RPM-1 (PHR) proteins include: Caenorhabditis elegans RPM-1 (Regulator of Presynaptic Morphology 1), Drosophila Highwire, and murine Phr1. These important regulators of neuronal development function in synapse formation, axon guidance, and axon termination. In mature neurons the PHR proteins also regulate axon degeneration and regeneration. PHR proteins function, in part, through an ubiquitin ligase complex that includes the F-box protein FSN-1 in C. elegans and Fbxo45 in mammals. At present, the structure-function relationships that govern formation of this complex are poorly understood. We cloned 9 individual domains that compose the entire RPM-1 protein sequence and found a single domain centrally located in RPM-1 that is sufficient for binding to FSN-1. Deletion analysis further refined FSN-1 binding to a conserved 97-amino acid region of RPM-1. Mutagenesis identified several conserved motifs and individual amino acids that mediate this interaction. Transgenic overexpression of this recombinant peptide, which we refer to as the RPM-1·FSN-1 complex inhibitory peptide (RIP), yields similar phenotypes and enhancer effects to loss of function in fsn-1. Defects caused by transgenic RIP were suppressed by loss of function in the dlk-1 MAP3K and were alleviated by point mutations that reduce binding to FSN-1. These findings suggest that RIP specifically inhibits the interaction between RPM-1 and FSN-1 in vivo, thereby blocking formation of a functional ubiquitin ligase complex. Our results are consistent with the FSN-1 binding domain of RPM-1 recruiting FSN-1 and a target protein, such as DLK-1, whereas the RING-H2 domain of RPM-1 ubiquitinates the target.
Introduction
Caenorhabditis elegans Regulator of Presynaptic Morphology 1 (RPM-1)3 along with Drosophila Highwire, murine Phr1, and human Pam (MCYBP2) are part of a conserved family of E3 ubiquitin ligases referred to as Pam/Highwire/RPM-1 (PHR) proteins (1). Studies in worms, flies, fish, and mice have identified roles for the PHR proteins in synapse formation (2–5), axon guidance and extension (6–11), and axon termination (3, 12–14).
The PHR proteins function through multiple signaling mechanisms to control neuronal development, one of which is ubiquitination, and negative regulation of the MAP3K Dlk (DLK-1 in worms and Wallenda in flies) (7, 15–17). In C. elegans, RPM-1 functions as part of an ubiquitin ligase complex that includes F-box Synaptic protein 1 (FSN-1) (18). The functional relationship between RPM-1 and FSN-1 is conserved in flies and mammals (19, 20).
Despite significant and important progress with genetic approaches, our knowledge of the structure-function relationship between PHR proteins and F-box proteins, such as FSN-1, remains limited. Nonetheless, knowledge gained from structure-function analysis is likely to be valuable for developing specific inhibitors of PHR ubiquitin ligase complexes. Studies in worms and flies have shown that inhibiting PHR protein function results in improved axon regeneration (21–23) and reduced axon degeneration after trauma (24, 25). Thus, an inhibitor that specifically blocks the PHR ubiquitin ligase complex might prove valuable for improving axon regeneration and reducing axon degeneration in the context of trauma and disease.
Here, we detail our discovery of a 97-amino acid (aa) region of RPM-1 that is sufficient for binding to FSN-1. The conservation of key residues in RPM-1 that mediate binding to FSN-1 suggests that this could be a conserved mechanism of interaction. The results of transgenic and genetic analysis are consistent with this peptide inhibiting the RPM-1·FSN-1 complex in vivo. Hence, we have termed it the RPM-1·FSN-1 complex inhibitory peptide (RIP). To our knowledge RIP represents the first inhibitor of a PHR ubiquitin ligase complex.
EXPERIMENTAL PROCEDURES
Genetics and Axon Morphology Analysis
Genetic analysis was performed using the N2 isolate of C. elegans and standard procedures (26). The alleles used in this study included: fsn-1(gk429), rpm-1(ju44), glo-4(ok623), and dlk-1(ju476). The transgene muIs32 (Pmec-7GFP) was used for analyzing axon and synaptic branch morphology in anterior lateral microtubule (ALM) and posterior lateral microtubule (PLM) neurons. Live, young adult animals were anesthetized using 1% (v/v) 1-phenoxy-2-propanol in M9 buffer and mounted on glass slides with 2% agar. Animals were visualized using a 40× magnification oil-immersion lens and an epifluorescent microscope (Leica CRF 5000). Images were acquired using a CCD camera (Leica DFC345 FX).
Cloning
The 9 domains (D1–9) and subdomains (D5a, -b, and -c) of RPM-1 were amplified by RT-PCR from C. elegans total RNA using Superscript III reverse transcriptase (Invitrogen). cDNAs were inserted into pCR8 Topo GY and sequenced to ensure they were mutation-free. Clones in pCR8 Topo GY were recombined using LR recombinase with pBG-GY14 to create pBG-GY189 (GFP-D1), pBG-GY190 (GFP-D2), pBG-GY191 (GFP-D3), pBG-GY192 (GFP-D4), pBG-GY193 (GFP-D5), pBG-GY194 (GFP-D6), pBG-GY195 (GFP-D7), pBG-GY196 (GFP-D8), pBG-GY197 (GFP-D9), pBG-GY389 (GFP-D5a), pBG-GY412 (GFP-D5b), and pBG-GY384 (GFP-D5c). For transgenic expression of RIP (D5c) in C. elegans, pBG-GY134 (Prgef-1FLAG GY) was recombined with pBG-GY349 (pCR8 Topo GY RIP (D5c)) to generate pBG-GY440 (Prgef-1FLAG::RIP (D5c)). pCZ161 encoding full-length RPM-1::GFP driven by its native promoter was engineered to contain a point mutation D2214A (pBG-190). For point mutagenesis, an HpaI-SpeI fragment of RPM-1 was amplified by PCR using pCZ161 as a template and inserted into pCR2.1. The HpaI-SpeI fragment was point-mutated using oligonucleotides with the desired changes and a QuikChange II XL site-directed mutagenesis kit (Agilent Technologies). After mutations were confirmed by sequencing the mutated HpaI-SpeI fragment was subcloned back into pCZ161 to create pBG-190. Similar mutagenesis procedures were performed on pBG-GY349 to generate RIP point mutants. These point mutations were also sequenced to confirm the accuracy of mutagenesis.
Transgenics
Transgenic animals were generated by standard microinjection procedures (27). Transgenic animals were constructed by injecting a plasmid of interest or PCR product with plasmid encoding Pttx-3RFP (50 ng/μl). For dominant negative experiments PCR products were amplified by a long PCR kit (Roche) and injected at 0.625–10 ng/μl as specified. The following plasmids were used as templates for long PCR: Prgef-1FLAG::RIP (pBG-GY440) or point mutants and Prgef-1FLAG::mCherry (pBG-GY371). Transgenic animals used for biochemistry were generated by injecting plasmids: Prgef-1FLAG::FSN-1 (pBG-GY422) at 25 ng/μl and Prpm-1RPM-1::GFP (pCZ161) or Prpm-1RPM-1::GFP D2214A (pBG-190) at 25 ng/μl into rpm-1; fsn-1 mutants. For coimmunoprecipitation (coIP) of GFP::D5c with FLAG::FSN-1 transgenic animals were generated by injecting plasmids: Prgef-1FLAG::FSN-1 and Prgef-1GFP::D5c (RIP) at 25 ng/μl into wild-type animals. Notably, expression of FLAG::FSN-1 was higher on a wild-type background than an fsn-1 mutant background.
Biochemistry
For biochemistry in HEK 293 cells or from transgenic C. elegans, FLAG proteins were immunoprecipitated with a mouse monoclonal anti-FLAG antibody (M2, Sigma) and immunoblotted with a rabbit polyclonal anti-FLAG antibody (Cell Signaling). GFP fusion proteins were precipitated with a mouse monoclonal anti-GFP antibody (3E6, MP Biomedicals) and immunoblotted with a mixture of mouse monoclonal anti-GFP antibodies (Roche Applied Science). Precipitates were boiled in SDS Laemmli sample buffer (Bio-Rad) and run on a 3–8% Tris acetate gel (Invitrogen) for RPM-1::GFP coIP or a 4–12% Bis-Tris gel (Invitrogen) for coIP of RPM-1 domains. Gels were transferred to PVDF membranes in Tris acetate transfer buffer (16–20 h at 30 V for full-length RPM-1::GFP) and immunoblotted. Blots were visualized with HRP-conjugated anti-mouse or anti-rabbit secondary antibodies, enhanced chemiluminescent reagent (ECL), and x-ray film. When necessary due to the size of target proteins, light chain reactive secondary antibodies were used (Millipore). Western Lightning Plus ECL was used for HEK 293 experiments, and Supersignal FemtoWest ECL (Pierce) was used for transgenic C. elegans experiments.
For experiments with HEK 293 cells, 6-cm dishes of cells were transfected with a mixture of Lipofectamine 2000 (Invitrogen) and a total of 9–11 μg of DNA that included: plasmid encoding FLAG-FSN-1 (5.2 μg DNA), plasmid encoding a GFP-RPM-1 domain (3–6 μg DNA), and varying amounts of pBluescript (amount required to reach total of 9–11 μg of DNA). A variable amount of DNA was transfected for particular constructs to ensure similar levels of expression. 36–48 h after transfection, cells were lysed with 1.0% Nonidet P-40 buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 10% glycerol, 1 mm DTT, EDTA-free protease inhibitor pellets (Roche Applied Science), pepstatin, microcystin, NaVO4, NaF, sodium molybdate, and β-glycerophosphate). 1000 μg of total protein from transfected 293 cells was used for individual coIP experiments. Lysates were incubated with primary antibody for 30 min and precipitated for 4 h with 10 μl of protein G agarose (Roche Applied Science) at 4 °C.
For biochemistry using transgenic worms, animals containing extrachromosomal arrays were propagated using Escherichia coli (strain HB101) on 10-cm NGM agar plates. Worms were harvested directly off 10-cm plates or moved to liquid culture for 1–2 days if larger amounts of material were needed. Animals were harvested by centrifugation and washed 3× in M9 buffer. Animals were ground using a mortar and pestle and lysed using sonication and 0.1% Nonidet P-40 lysis buffer (50 mm Tris, pH 7.5, 0.1% Nonidet P-40, 150 mm NaCl, 10% glycerol, 1 mm DTT, and EDTA-free protease inhibitor pellets). For coIP of RPM-1::GFP with FLAG::FSN-1, 20–120 mg of total protein from transgenic worm lysates was used. For coIP of FLAG::FSN-1 with GFP::D5c from transgenic worm lysates, 25 mg of total lysate was used. Lysates were incubated with 3 μl of M2 (anti-FLAG) antibody or 3 μl of 3E6 (anti-GFP) antibody for 30 min and precipitated for 4 h at 4 °C with 10 μl of protein G-agarose.
RESULTS
A Single Domain in RPM-1 Is Sufficient for Binding to FSN-1
Previous genetic and biochemical experiments have shown that C. elegans RPM-1 and Drosophila Hiw are part of a complex that includes the F-box protein FSN-1 (18, 19). In mammals, the Myc binding domain of Pam binds to Fbxo45, the ortholog of FSN-1 (20). Despite significant progress in understanding the function of the PHR ubiquitin ligase complex, the biochemical mechanisms underlying the formation of this complex remain minimally explored.
Because the interaction between RPM-1 and FSN-1 is evolutionarily conserved, we hypothesized that FSN-1 would bind to a region in RPM-1 that corresponded with a conserved portion of the Myc binding domain of Pam. ClustalW2 analysis of the PHR proteins showed that the Myc binding domain of Pam (aa 2413–2712) was composed of a C-terminal region that was not present in Hiw or RPM-1 (Fig. 1A, highlighted in green) and an N-terminal region that was well conserved (22.4% identical and 52.3% conservation between Pam and RPM-1) (Fig. 1A, highlighted in orange). This N-terminal region contained several conserved motifs that might mediate binding between RPM-1 and FSN-1 (see Fig. 3A). To test this possibility, we cloned cDNAs encoding nine individual protein domains (D1–9) that represented the entire RPM-1 coding sequence (Fig. 1A). The boundaries of each domain were engineered at locations where conservation was strongly decreased. We transiently transfected HEK 293 cells with plasmids that express a GFP fusion protein for each of the 9 RPM-1 domains and FLAG epitope-tagged FSN-1. CoIP was used to determine if FSN-1 binds to a specific RPM-1 domain. As predicted by our bioinformatic analysis, FSN-1 coprecipitated with RPM-1 domain 5 (D5), which shared homology with the N-terminal portion of the Pam Myc binding domain (Fig. 1B, top panel). We previously showed that D5 is sufficient for binding to RAE-1, so we also refer to this domain as the RAE-1 binding domain (Fig. 1, A and B) (28). We noted that coexpression of D5 with FSN-1 consistently resulted in increased expression of FSN-1 (Figs. 1B and 2B, bottom panels). This result is consistent with prior work which showed that the Myc binding domain of Pam stabilizes Fbxo45 protein levels, presumably by sequestering it from degradation by an endogenous E3 ligase (20).
FIGURE 1.

Identification of a domain in RPM-1 that is sufficient for binding to FSN-1. A, shown is a schematic of human Pam and C. elegans RPM-1. Annotated protein domains include: the RCC-1 like domain (RLD, black), two PHR family specific domains (PHR, gray), the RAE-1 binding domain (RBD, purple), the Myc binding domain (MBD, green and orange), nuclear localization signal (NLS, yellow), and the RING-H2 ubiquitin ligase domain (RING, red). Highlighted in blue is a motif in the RAE-1 binding domain that is essential for binding to RAE-1. The portion of the MBD in Pam that is highlighted in orange is well conserved with RPM-1, and the portion highlighted in green is not conserved. Also shown are nine cDNA constructs encompassing the entire length of RPM-1, each of which corresponds to an individual, conserved domain of RPM-1 (D1–D9). B, coIP was performed from lysates of transfected HEK 293 cells that expressed FLAG tagged FSN-1 (FLAG-FSN-1) or FLAG tagged RAE-1 (FLAG-RAE-1) and fusion proteins of GFP and RPM-1 D1-D9. Note that FLAG-FSN-1 coprecipitated exclusively with domain 5 of RPM-1 (GFP-D5) (top panel). Consistent with a previous study, FLAG-RAE-1 also coprecipitated with GFP-RPM-1 domain 5 (GFP-D5) (top panel, last lane) (28). Shown are representatives of experiments that were independently performed at least three times.
FIGURE 3.

Identification of conserved residues in RPM-1 D5 required for binding to FSN-1. A, schematic of full-length RPM, RPM-1 domain 5 (D5), and a portion of RPM-1 domain 5 that is sufficient for binding to FSN-1 (D5c). Shown below is a sequence alignment of the D5c region of RPM-1 with its orthologs Pam (also called MYCBP-2) and Highwire that was generated using ClustalW2. Underlined are portions of motifs that were mutated and analyzed in B. Highlighted in red are amino acid residues that were individually point mutated and analyzed in C and D. B and C, coIP using lysates from transfected HEK 293 cells expressing FLAG-FSN-1 and a fusion protein of GFP and domain 5 of RPM-1 (GFP-D5). Wild-type GFP-D5 or the indicated point mutants were analyzed for coprecipitation with FLAG-FSN-1. Note in C that the point mutants of GFP-D5 W2239A, D2214A, and F2207A strongly reduced co-precipitation with FLAG-FSN-1 compared with wild-type GFP-D5 (top panel). The interaction between GFP-D5 (I2208A) and FLAG-FSN-1 was more modestly reduced. D, coIP was performed from whole worm lysates of transgenic C. elegans expressing FLAG::FSN-1 and wild-type RPM-1::GFP or RPM-1::GFP point mutated to inhibit binding to FSN-1 (RPM-1 D2214A). Note that wild-type RPM-1::GFP and RPM-1::GFP D2214A coprecipitate with FLAG::FSN-1 equally. Shown are representatives of experiments that were independently performed at least three times.
FIGURE 2.

RPM-1 D5 contains a 97-aa region that is sufficient for binding to FSN-1. A, shown is a schematic of RPM-1 domain 5 (RPM-1 D5) and three smaller fragments of D5 (D5a–D5c). B, coIP performed from lysates of transfected HEK293 cells that expressed FLAG-FSN-1 and fragments of RPM-1 domain 5 fused with GFP (GFP-D5a–GFP-D5c). Note that GFP-D5b and GFP-D5c coprecipitated with FLAG-FSN-1 with similar efficiency to full-length GFP-D5 (top panel). C, coIP performed from lysates of transgenic C. elegans. FLAG::FSN-1 coprecipitated efficiently only when coexpressed with GFP::D5c. Shown are representatives of experiments that were independently performed at least three times.
To further map the interaction between FSN-1 and RPM-1, we generated smaller fragments of RPM-1 D5 (Fig. 2A). As shown in Fig. 2B, the N-terminal half of D5 (D5a) did not bind to FSN-1, whereas the C-terminal half (D5b) was sufficient for binding. We then generated a smaller, 97-aa portion of D5b (D5c) that contained only a sequence conserved with the Myc binding domain of human Pam. GFP-D5c coprecipitated with FLAG-FSN-1 with similar efficiency to GFP-D5 and GFP-D5b (Fig. 2B).
To test whether this interaction occurs in vivo in neurons, we generated transgenic C. elegans that used a pan-neuronal promoter (Prgef-1) to simultaneously express both GFP::D5c and FLAG::FSN-1. As shown in Fig. 2C, when GFP::D5c was precipitated with an anti-GFP antibody, robust coprecipitation of FLAG::FSN-1 was detected. In contrast, when FLAG::FSN-1 was expressed alone in transgenic worms, no coprecipitation occurred (Fig. 2C). These results demonstrate that D5c, a conserved 97-aa region of RPM-1, is sufficient for binding to FSN-1 in a heterologous expression system and in the neurons of C. elegans.
Residues in RPM-1 D5 Required for Binding to FSN-1
We next wanted to identify the motifs and residues in D5c that are required for binding to FSN-1. Sequence alignment using ClustalW2 identified five motifs in RPM-1 D5c that were highly conserved with Drosophila Hiw and human Pam (Fig. 3A, underlined). As an initial mapping strategy, we generated GFP-D5 that was simultaneously point-mutated at multiple residues in one of the five conserved motifs. Point mutants included W2239A/C2240A/L2241A, R2220A/L2221A, D2214A/D2215A, F2207A/I2208A, and G2182A/R2184A/R2186A. Binding of FLAG-FSN-1 to D5 point mutants was analyzed using coIP from lysates of transiently transfected HEK 293. GFP-D5 fusion proteins were precipitated from transfected cell lysates using an anti-GFP antibody. Although FLAG-FSN-1 coprecipitated well with wild-type GFP-D5, binding was strongly reduced for all GFP-D5 point mutants (Fig. 3B, top panel). Thus, all five conserved motifs in RPM-1 D5 that we tested were required for binding to FSN-1.
To further map where FSN-1 binds to RPM-1 and to minimize structural impacts caused by mutation of multiple residues simultaneously, we generated point mutants of RPM-1 D5 in which only a single amino acid was mutated to alanine (Fig. 3A, red boxes). This was done for three of the five motifs in D5 that were required for binding to FSN-1. As shown in Fig. 3C, coprecipitation of FSN-1 was strongly reduced for three individual point mutants of RPM-1 D5: W2239A, D2214A, and F2207A. Coprecipitation of FSN-1 was more mildly reduced for D5 I2208A (Fig. 3C).
Having used a heterologous expression system to identify specific residues in RPM-1 that mediate binding to FSN-1, we wanted to test if the corresponding point mutations in full-length RPM-1 would inhibit binding to FSN-1 in the neurons of C. elegans. To address this we generated transgenic worms that coexpressed a GFP fusion protein with full-length RPM-1 (RPM-1::GFP), and FLAG epitope-tagged FSN-1 (FLAG::FSN-1). RPM-1::GFP was expressed using the native rpm-1 promoter that is expressed exclusively but broadly in neurons (5). FLAG::FSN-1 was expressed using a pan-neuronal promoter (Prgef-1). Coprecipitating RPM-1::GFP was detected when FLAG::FSN-1 was immunoprecipitated from whole worm lysates (Fig. 3D). This result is consistent with a prior study that used anti-FSN-1 antibodies to show that RPM-1 binds to FSN-1 (18). RPM-1::GFP D2214A also coprecipitated with FLAG::FSN-1 and did not show reduced binding compared with wild-type RPM-1::GFP (Fig. 3D). These results suggest that FSN-1 might bind to multiple sites in RPM-1. We did not find another portion of RPM-1 sequence with strong homology to RPM-1 D5c, which suggests that the second FSN-1 binding site in RPM-1 utilizes a different structural mechanism.
Nonetheless, our biochemical results indicate that FSN-1 binds either directly or through an adaptor such as a Skp to a very precise region in RPM-1; that is, the D5c fragment, which we refer to as the FSN-1 binding domain 1 (FBD1) in the context of full-length RPM-1. Moreover, our findings suggest that the D5c peptide might block the interaction between endogenous RPM-1 and FSN-1 in vivo potentially making it a highly specific inhibitor of the RPM-1·FSN-1 ubiquitin ligase complex. Therefore, when D5c is expressed recombinantly, we refer to it as RIP or the RPM-1·FSN-1 complex inhibitory peptide.
Transgenic Expression of RIP Inhibits Axon Termination and Synapse Formation
Previous studies have shown that rpm-1, fsn-1, and glo-4 loss-of-function (lf) mutants have defects in axon termination and synapse formation in the mechanosensory neurons of C. elegans (3, 12). Furthermore, fsn-1 and glo-4 function in parallel genetic pathways to mediate the function of rpm-1 (12). The genetic relationship between fsn-1 and glo-4 and our biochemical results showing that RIP (D5c) binds to FSN-1 provided a basis for determining whether exogenous expression of RIP would inhibit the function of the endogenous RPM-1·FSN-1 ubiquitin ligase complex. To test this hypothesis, we analyzed how transgenic overexpression of RIP affects axon termination in the mechanosensory neurons that sense soft touch in C. elegans.
In C. elegans there are two PLM mechanosensory neurons each of which extends a single axon that terminates extension well before the cell body of the ALM mechanosensory neurons (Fig. 4A). The morphology of the PLM neurons can be rapidly and accurately analyzed using a transgene, muIs32 (Pmec-7GFP), which expresses GFP specifically in the mechanosensory neurons (29). In fsn-1−/− or glo-4−/− single mutants, two axon termination phenotypes were observed in the PLM neurons consistent with prior studies. The primary, most frequent phenotype was a less severe defect in which the PLM axon failed to terminate extension and grew past the ALM cell body, a defect we refer to as overextension (Fig. 4, A and B) (12, 30). A second, more severe phenotype in which the PLM axon overextended and then hooked toward the ventral cord, which we refer to as a hook defect, was also observed but at very low expressivity (Fig. 4B). Similar to prior work, we observed that glo-4−/−; fsn-1−/− double mutants had strongly enhanced expressivity of hook defects, which was the primary phenotype in these animals (Fig. 4, A and B) (12, 30). The frequency of hook defects in glo-4−/−; fsn-1−/− double mutants is similar to rpm-1−/− mutants (3, 12, 30).
FIGURE 4.

Transgenic overexpression of RIP (RPM-1 D5c) inhibits axon termination in the PLM neurons of C. elegans. A, a schematic (adapted from Baker et al. (31)) showing the mechanosensory neurons of C. elegans (anterior left, dorsal top). The blue box highlights the region of the animal shown below, which was visualized using muIs32 (Pmec-7GFP) and epifluorescent microscopy. Note that the AVM cell body is only present on one side of the animal and is not always shown. Images are shown for the most prevalent phenotype observed for each genotype. Shown are two different types of PLM axon termination defects: overextension (arrow), and more severe hook defects (arrowhead). Note that transgenic OE of RIP on a wild-type background primarily resulted in overextension, whereas overexpression of RIP on a glo-4 mutant background primarily resulted in a more severe hook defect. AVM, anterior ventral microtubule; PVM, posterior ventral microtubule. B, quantitation of PLM axon termination defects for the indicated genotypes (hook = black, overextension = gray). C, analysis of PLM axon termination defects in glo-4 mutants carrying transgenic extrachromosomal arrays that were generated by injecting DNA encoding RIP at a range of concentrations as indicated. D, the blue box in the schematic highlights the region shown below that was visualized using muIs32 and epifluorescent microscopy. In wild-type animals, the PLM neuron extends its synaptic branch ventrally. Also shown is an example of a glo-4 mutant that transgenically overexpressed RIP in which the PLM synaptic branch is absent (arrowhead). E, quantitation of synaptic branch defects for the indicated genotypes. F, IP with an anti-FLAG antibody was used to detect FLAG::RIP in whole worm lysates of representative transgenic lines for the indicated genotypes. B, C, and E, shown are the averages of 5 or more independent counts (20–30 neurons/count) for each genotype. For transgenes, averages are shown for data pooled from four or more transgenic lines. Unless noted otherwise, transgenic animals were generated by injecting PCR product at 10 ng/μl. Error bars represent the S.E., and significance was determined using an unpaired Student's t test. ***, p < 0.001; *, p < 0.05; ns, = not significant. Scale bars are 10 μm.
To determine whether RIP inhibits the function of the endogenous RPM-1·FSN-1 complex, we used a transgenic approach in which the rgef-1 promoter (a strong, pan-neuronal promoter) was used to overexpress RIP. If RIP inhibits the RPM-1·FSN-1 complex, we expected transgenic overexpression of RIP to yield phenotypes that were similar to fsn-1 (lf) mutations. Notably, we did not expect RIP overexpression to yield phenotypes that occurred with the same expressivity as rpm-1 (lf) because RPM-1 functions through several FSN-1 independent mechanisms including the GLO Rab pathway, the microtubule-binding protein RAE-1, the phosphatase PPM-2, and the ANC-1/β-catenin pathway (12, 28, 31, 32). We generated transgenic animals that overexpressed FLAG epitope-tagged RIP by injecting PCR product at relatively high concentrations (10 ng/μl). As a control for promoter effects, we also generated transgenes that overexpressed mCherry. The primary phenotype observed when RIP was transgenically overexpressed in wild-type animals was overextension of the PLM axon (Fig. 4A). Quantitation showed that overextension occurred with increased frequency when RIP was overexpressed (45.4 ± 2.2%) but not mCherry (24.7 ± 2.8%; Fig. 4B). Transgenic overexpression of RIP in wild-type animals also resulted in significant, but lower frequency hook defects (Fig. 4B). Notably, defects caused by overexpression of RIP in wild-type animals resulted in a similar frequency of defects as observed in fsn-1 (lf) mutants (Fig. 4B). When we overexpressed RIP or mCherry in fsn-1−/− mutants, we observed no differences between the two transgenes with regard to the hook phenotype and an extremely small increase in the overextension phenotype (Fig. 4, A and B). Next, we tested the effect of RIP overexpression in glo-4−/− animals. glo-4−/− mutants that overexpress mCherry were similar to non-transgenic glo-4−/− mutants and primarily showed overextension defects, with lower expressivity of the more severe hook defects (Fig. 4, A and B). In contrast, transgenic overexpression of RIP in glo-4−/− mutants resulted in enhanced frequency of hook defects, whereas the expressivity of less severe overextension defects was decreased (compare 87.9 ± 2.1% hook for glo-4 + overexpressed (OE) RIP with 12.4 ± 2.4% for glo-4 + OE Cherry; Fig. 4, A and B). To test the potency of transgenic RIP, we engineered glo-4−/− animals with transgenic arrays that were generated by injecting DNA encoding RIP at a range of concentrations. In all cases including when arrays were constructed at relatively low concentrations (0.625 ng/μl), we observed strong enhancer effects in the expressivity of PLM hook defects (Fig. 4C). Notably, the enhancer effects caused by overexpression of RIP in glo-4−/− animals were comparable with levels of enhancement observed in glo-4−/−; fsn-1−/− double mutants (Fig. 4B). Collectively, these results demonstrate that overexpression of RIP behaves genetically like fsn-1 (lf).
In wild-type animals, each PLM neuron also extends a single synaptic branch that innervates interneurons of the ventral nerve cord (Fig. 4D). It was previously shown that rpm-1−/− mutants lack a PLM synaptic branch at high frequency (3, 12). Likewise, glo-4−/−; fsn-1−/− double mutants have enhanced frequency of PLM synaptic branch defects (Fig. 4E) (12, 30). It was previously noted that the absence of the PLM synaptic branch in rpm-1−/− mutants was likely due to a failure to form or stabilize PLM synaptic connections as opposed to defects in synaptic branch extension (3). Thus, this phenotype is likely to reflect a defect in synapse formation.
Transgenic overexpression of RIP in glo-4−/− mutants primarily resulted in the absence of the PLM synaptic branch (Fig. 4D). Quantitation showed an enhanced frequency of PLM synaptic branch defects when RIP was overexpressed compared with when mCherry was overexpressed in glo-4−/− mutants (compare 77.1 ± 2.7% for glo-4 + OE RIP with 4.1 ± 1.7% for glo-4 + OE Cherry; Fig. 4E). When RIP was overexpressed in wild-type animals, the frequency of synaptic branch defects was mildly, but significantly, increased (compare 21.2 ± 4.0% for wild-type + OE RIP with 6.6 ± 1.8% for wild-type + OE Cherry; Fig. 4E). In contrast, RIP overexpression in fsn-1−/− mutants did not significantly change the frequency of synaptic branch defects compared with overexpression of mCherry (Fig. 4E).
The two ALM neurons sense anterior soft touch and terminate extension at a precise location in the head of the animal (Fig. 5A). In wild-type animals, the ALM axon terminates well before the anterior tip of the animal's nose (Fig. 5A). Consistent with previous work, we observed ALM axon termination defects in glo-4−/− or fsn-1−/− single mutants (Fig. 5B) (12). Categorizing ALM axon termination defects into less severe short hooks and more severe big hooks facilitated identification of genetic enhancer effects in glo-4−/−; fsn-1−/− double mutants (Fig. 5B) (30). Previous studies showed that ALM axon termination defects in glo-4−/−; fsn-1−/− double mutants occur at a similar frequency and severity as rpm-1−/− mutants (3, 12, 30).
FIGURE 5.

Transgenic overexpression of RIP (RPM-1 D5c) inhibits axon termination in the ALM neurons of C. elegans. A, a schematic shows the mechanosensory neurons of C. elegans (anterior left, dorsal top) that were analyzed using the transgene muIs32 (Pmec-7GFP). The blue box highlights the region of the animal shown below that was visualized using epifluorescent microscopy. Shown is an example of normal ALM axon termination in a wild-type animal. Also shown is a glo-4 animal that is overexpressing RIP where the ALM axon fails to terminate properly, overextends, and hooks toward the posterior of the animal (big hook, arrow). The scale bar is 10 μm. B, quantitation of ALM axon termination defects for the indicated genotypes (big hook = black, small hook = gray). A and B, shown are the averages of 5 or more independent counts (20–30 neurons/count) for each genotype. For transgenes, averages are shown for data pooled from 4 or more transgenic lines. Transgenic animals were generated by injecting PCR products at 10 ng/μl. Error bars represent the S.E., and significance was determined using an unpaired Student's t test. ***, p < 0.001; *, p < 0.05; ns, not significant.
When RIP was transgenically overexpressed in glo-4−/− mutants, we primarily observed big hooks in ALM neurons (Fig. 5A). The frequency of these defects was enhanced compared with glo-4−/− mutants that overexpressed mCherry (compare 86.3 ± 2.1% big hook for glo-4 + OE RIP with 34.0 ± 3.6% for glo-4 + OE Cherry; Fig. 5B). Overexpression of RIP on a wild-type background gave both short hook and big hook phenotypes that were relatively low in frequency but significant (compare 7.0% ± 1.5% short hook for wild-type + OE RIP with 1.6 ± 0.8% short hook for wild-type + OE Cherry; Fig. 5B). Transgenic overexpression of RIP in fsn-1−/− mutants did not alter the frequency of ALM axon termination defects compared with overexpression of mCherry (Fig. 5B).
Transgenic overexpression of FLAG::RIP in PLM and ALM neurons had strong functional effects, but we wanted to ensure that changes in transgenic RIP expression in different genetic backgrounds did not account for these findings. Therefore, we biochemically assessed expression of FLAG::RIP in a representative transgenic line for each genotype. As shown in Fig. 4F, we detected expression of transgenic FLAG::RIP in whole worm lysates of all RIP transgenic genotypes assessed in our functional analysis. Importantly, we did not observe lower expression of FLAG::RIP in genotypes with lower expressivity of axon termination defects, such as fsn-1−/− mutants that overexpress RIP. Thus, our functional transgenic results are not simply due to variation in expression of FLAG::RIP on different genetic backgrounds. We note that FLAG::RIP migrated at the expected size of 13.5 kDa but was detected as a doublet. This was most likely the result of protein degradation that occurred during preparation of protein extracts from whole animals. Alternatively, the doublet might reflect post-translational modification of RIP that only occurs in the neurons of C. elegans.
In summary, several of our findings are consistent with overexpression of RIP acting similar to fsn-1 (lf). 1) Transgenic overexpression of RIP on a wild-type background resulted in defects that occurred with similar frequency to non-transgenic fsn-1−/− mutants. 2) We observed no enhancer effects when RIP was overexpressed on an fsn-1−/− mutant background. 3) Strong enhancer effects occurred when RIP was overexpressed on a glo-4−/− mutant background, and enhancer effects occurred with similar frequency to glo-4−/−; fsn-1−/− double mutants. Taken together, these results are consistent with RIP binding to FSN-1 and inhibiting formation of a functional RPM-1·FSN-1 ubiquitin ligase complex.
Transgenic RIP Functions through DLK-1
Previous studies have shown that the RPM-1·FSN-1 complex functions through ubiquitination and inhibition of the MAP3K DLK-1 (16, 19, 31). To provide further evidence that RIP functions by inhibiting endogenous FSN-1, we tested if the effects of overexpression of RIP were altered by dlk-1 (lf). As shown in Fig. 6, transgenic overexpression of RIP in glo-4−/− mutants resulted in an enhanced frequency of the more severe ALM and PLM axon termination defects as well as PLM synaptic branch defects. Enhancer effects caused by RIP overexpression were completely suppressed in glo-4−/−; dlk-1−/− double mutants (Fig. 6). This result was not due to suppression of glo-4 by dlk-1, as similar levels of defects were observed in glo-4−/− single mutants and glo-4−/−; dlk-1−/− double mutants (Fig. 6). These results are consistent with RIP impairing endogenous FSN-1, which functions through ubiquitination and inhibition of DLK-1.
FIGURE 6.

Effects of transgenic overexpression of RIP (RPM-1 D5c) on axon termination are mediated by DLK-1. Quantitation of ALM axon termination defects (dark gray), PLM axon termination defects (black), and PLM synaptic branch defects (light gray) for the indicated genotypes. Note that defects caused by transgenic overexpression of RIP in glo-4 mutants are completely suppressed when RIP is overexpressed in glo-4; dlk-1 double mutants. Averages are shown for data pooled from six or more transgenic lines. Transgenic animals were generated by injecting PCR products at 10 ng/μl. Error bars represent the S.E., and significance was determined using an unpaired Student's t test. ***, p < 0.001; ns = not significant.
RIP Function Is Inhibited by Point Mutations That Block Binding to FSN-1
We wanted to test whether point mutations in RIP (D5c), which reduce binding to FSN-1 in HEK 293 cells (Fig. 3C), affected the function of transgenic RIP in worms. To do so we analyzed PLM axon termination in glo-4−/− mutants that carried transgenic extra-chromosomal arrays that overexpressed wild-type RIP or RIP point mutants. To maximize our ability to detect changes in RIP efficacy, transgenic arrays were generated with 2.5 ng/μl PCR product encoding RIP, a lower concentration at which maximal effects from RIP overexpression were still observed (Fig. 4C). As shown in Fig. 7A, overexpression of wild-type RIP resulted in enhanced PLM axon termination defects compared with non-transgenic glo-4−/− animals. In contrast, enhancer effects were significantly lower in glo-4−/− mutants that overexpressed RIP point mutants (Fig. 7A). RIP W2239A had the weakest enhancer effect compared with wild-type RIP, which suggested that this point mutant was the most functionally impaired (Fig. 7A). RIP D2214A was the point mutant with the highest level of enhancement (Fig. 7A). Thus, RIP D2214A was the least functionally impaired point mutant. Although coIP from HEK 293 cells showed that point mutation of Asp-2214, Phe-2207, or Trp-2239 strongly impaired binding to FSN-1 (Fig. 3), analysis of axon termination in PLM neurons highlighted the functional importance of individual residues in RIP. Presumably this is because the neurons of C. elegans are a more physiologically relevant setting and, therefore, more sensitive to changes in the efficiency of RIP binding to FSN-1. Nonetheless, functional analysis of RIP point mutants provided further evidence that is consistent with exogenous RIP binding to FSN-1 and inhibiting formation of a functional RPM-1·FSN-1 complex.
FIGURE 7.
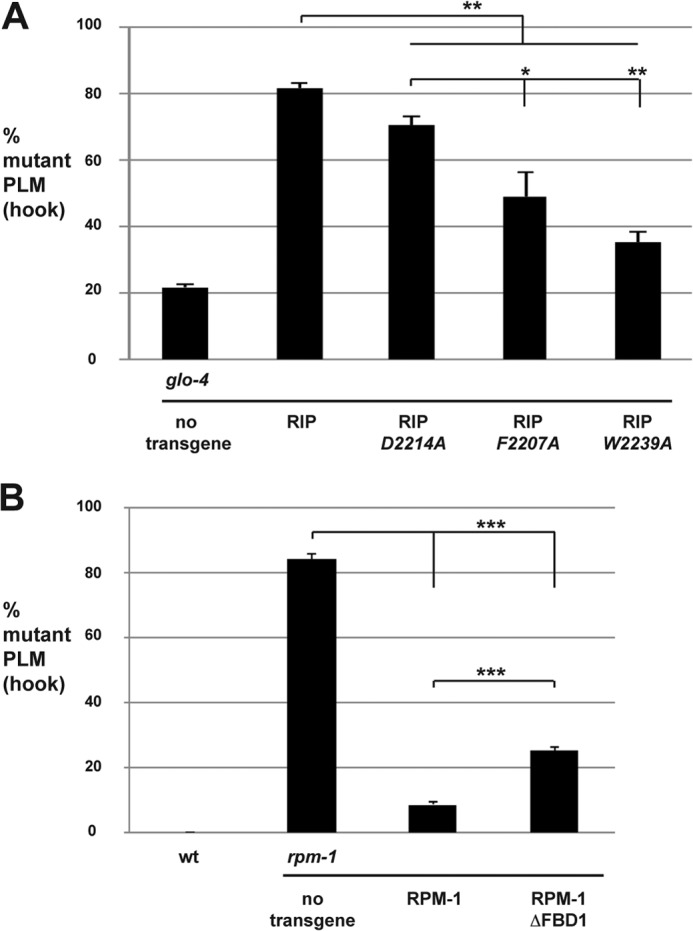
Mutations in RIP that reduce binding to FSN-1 impair the efficacy of transgenic RIP. Quantitation of PLM axon termination defects (hook) for the indicated genotypes. A, mutations in RIP that impair binding to FSN-1 reduce the level of RIP enhancer effects in glo-4 mutants. Transgenic animals were generated by injecting PCR products at 2.5 ng/μl. B, RPM-1 ΔFBD1 rescues defects in PLM axon termination caused by rpm-1 (lf) less efficiently than wild-type RPM-1. Transgenic animals were generated by injecting plasmid at 25 ng/μl. A and B, averages are shown for data pooled from five or more transgenic lines. Significance was determined using an unpaired Student's t test. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
RPM-1 Lacking FBD1 Is Functionally Impaired
RPM-1 functions though several downstream signaling molecules and pathways, one of which is FSN-1 (12, 16, 18, 28, 31, 32). A previous study showed that mutating RPM-1 to reduce binding with a single RPM-1-binding protein, RAE-1, resulted in a partial loss of RPM-1 function (28). This prompted us to test whether RPM-1 that lacked FBD1 would be fully functional. To do so, we generated transgenic rpm-1−/− mutants that carried extrachromosomal arrays and used the native rpm-1 promoter to express wild-type RPM-1 or RPM-1 lacking FBD1 (RPM-1 ΔFBD1). Consistent with prior studies, we observed that transgenic expression of RPM-1 strongly rescued the PLM hook defects in rpm-1−/− mutants (Fig. 7B). In contrast, RPM-1 ΔFBD1 significantly, but partially, rescued the hook defects in rpm-1−/− mutants (Fig. 7B). Thus, RPM-1 ΔFBD1 is only partially functional.
DISCUSSION
RIP, an in Vivo Inhibitor of the RPM-1·FSN-1 Ubiquitin Ligase Complex
Previous studies highlighted the functional relationship between the PHR proteins and FSN-1, but the biochemical mechanism of how PHR proteins bind to FSN-1 has remained poorly understood (18–20). Here, we describe the identification of one mechanism by which FSN-1 binds to RPM-1. We show that this interaction is mediated by several key motifs in the D5c fragment of RPM-1, which we refer to as RIP when expressed recombinantly and as FBD1 within the context of the overall RPM-1 protein (Fig. 8). Importantly, the residues in RPM-1 that mediate binding to FSN-1 are highly conserved, which suggests that this mechanism is likely to be relevant to all PHR proteins.
FIGURE 8.
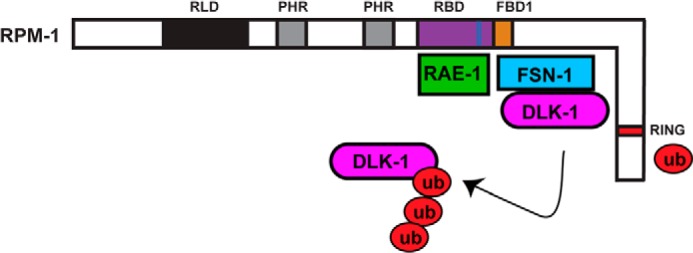
Summary of structure-function analysis of RPM-1 and FSN-1. Schematic shows the annotated and known functional domains of RPM-1: the RAE-1 binding domain (RBD) is highlighted in purple, the RAE-1 binding motif is highlighted in blue, and the FSN-1 binding domain 1 (FBD1) is highlighted in orange. Note that RIP corresponds to the FBD1 region of RPM-1. Our structure-function analysis coupled with previous work (28) demonstrates that RAE-1 and FSN-1 bind at very close but different locations on RPM-1. Our data support a model whereby FSN-1 scaffolds DLK-1 at FBD1 in the center of RPM-1 whereas the E3 ligase domain (RING) of RPM-1 acts independently to ubiquitinate DLK-1.
Several of our results suggest that RIP functions as an inhibitor of the RPM-1·FSN-1 ubiquitin ligase complex in vivo. 1) Transgenic overexpression of RIP caused defects in axon termination that occurred with similar frequency to defects caused by fsn-1 (lf) (Figs. 4 and 5). 2) Transgenic overexpression of RIP in glo-4 (lf) mutants, but not fsn-1 (lf) mutants, caused enhanced defects in axon termination. This is consistent with previous work showing that glo-4 and fsn-1 function in parallel pathways to regulate axon termination (12). 3) Enhanced axon termination defects caused by overexpression of RIP in a glo-4 (lf) mutant background are suppressed by dlk-1 (lf) (Fig. 6). This result is consistent with previous studies that showed FSN-1 regulates axon termination by inhibiting DLK-1 (19, 31). 4) Point mutations that reduce binding of RIP to FSN-1 impair the function of transgenic RIP in vivo (Fig. 7A). 5) Finally, RPM-1 that lacks FBD1 is only partially functional (Fig. 7B). These results provide a body of consistent evidence to support the conclusion that transgenically expressed RIP binds to endogenous FSN-1 and prevents it from forming a functional complex with RPM-1. RIP now represents the first reagent to our knowledge that specifically inhibits the function of a PHR ubiquitin ligase complex.
A version of RIP that targets mammalian PHR proteins might be a useful reagent on several levels. First, viral delivery of RIP or an inducible system for RIP expression might be used to study the post-developmental function of the ubiquitin ligase activity of PHR proteins. Second, previous work has shown that loss of function in rpm-1 or Hiw results in improved axon regeneration (21–23). This suggests that transgenic overexpression of RIP might improve regeneration. Finally, loss of function in PHR proteins in flies and mice dramatically blocks axon degeneration, and this is likely to be mediated by Fbxo45 (24, 25). Thus, specifically inhibiting the Phr1·Fbxo45 complex with RIP could have potential therapeutic implications for blocking or slowing the progression of axon damage following trauma and possibly in the context of neurodegenerative diseases.
Implications for Myc Binding to PHR Proteins
We used HEK 293 cells as a heterologous expression system for biochemistry with C. elegans proteins and identified a 97-aa region of RPM-1, annotated as FBD1, that is sufficient for binding to FSN-1 (Fig. 8). We found that five highly conserved motifs within FBD1 are required for binding to FSN-1. FBD1 is contained within a larger domain we previously showed binds to RAE-1 (28). Our results here show that FSN-1 relies upon a different binding site in RPM-1 than RAE-1 (Figs. 1–3) (28). Thus, although RAE-1 and FSN-1 are likely to be in close physical proximity, FSN-1 is unlikely to act as an adaptor for recruitment of RAE-1 into RPM-1 protein complexes and vice versa. Our biochemical results are consistent with prior genetic and proteomic results, which showed that RAE-1 is not a target of RPM-1 ubiquitin ligase activity (28, 33).
Our findings also have important implications for the relationship between PHR proteins and Myc. Myc was originally found to bind to a region of Pam referred to as the Myc binding domain (34); more recently this domain was shown to also bind the F-box protein Fbxo45 (20). The N-terminal half of the Pam Myc binding domain is conserved with Hiw and RPM-1 and the C-terminal half of this domain is not conserved. Our results show that FBD1, which corresponds with the conserved N-terminal portion of the Myc binding domain of Pam, is sufficient for binding to FSN-1. Furthermore, all the conserved motifs we identified in FBD1 are required for binding to FSN-1. Thus, a likely structural model is that Myc binds to the C-terminal portion of the Myc binding domain of Pam, which is not conserved in Hiw and RPM-1. In this scenario, Myc would only bind to vertebrate PHR proteins, which is consistent with the absence of Myc in proteomic screens for Hiw and RPM-1 binding proteins.4,5
An alternative possibility is that FSN-1 might mediate binding of Myc to PHR proteins, in which case Myc would be ubiquitinated by the PHR proteins. In this scenario we would have expected mutations in orthologs of Myc to be identified in previous suppressor screens with rpm-1 or Hiw (lf), which has not occurred to our knowledge. Furthermore, we would not have expected full-length Pam to bind to Myc in the absence of proteasome inhibitors, which has been observed (34). Thus, it is unlikely that FSN-1/Fbxo45 mediates binding of Myc to mammalian PHR proteins.
The FSN-1 Binding Domain of RPM-1 and Formation of Ubiquitin Ligase Complexes
Based on our analysis it is unclear whether we have mapped a direct interaction site between RPM-1 and FSN-1 or a region of RPM-1 that binds to an adaptor protein to recruit FSN-1. Previous work showed that PHR proteins form complexes that include FSN-1 and Skp proteins, such as SKR-1 in C. elegans (18, 20, 35). A Cullin, CUL-1, has also been implicated in the RPM-1·FSN-1 ubiquitin ligase complex. However, Cullins are absent in the non-canonical Hiw·DFsn and Pam·Fbxo45 ubiquitin ligase complexes. Given the structural nature of Skp·Cullin·F-box complexes (36), we would expect CUL-1 to bind directly to RPM-1 and act as an adaptor for SKR-1, which would then recruit FSN-1. In PHR ubiquitin ligase complexes that lack a Cullin, such as Pam·Fbxo45, we would expect Skp1 to mediate binding of Fbxo45 to Pam. Interestingly, our biochemical results using transgenic C. elegans showed that mutation of a residue, Asp-2214, that is required for FBD1 to bind to FSN-1 does not impair binding of full-length RPM-1 to FSN-1. Thus, although FBD1 is sufficient for binding to FSN-1, it is unlikely to be the only site that mediates binding of FSN-1 to RPM-1. There are two molecular models that explain this observation. First, CUL-1/SKR-1 might mediate binding of FSN-1 to RPM-1 at multiple interaction sites. Alternatively, FSN-1 might bind directly to RPM-1 at one location, and CUL-1/SKR-1 could mediate binding of FSN-1 to RPM-1 at another location(s).
There are several caveats to our experiments in HEK 293 cells that could explain why we only identified a single domain that bound to FSN-1. First, because we generated nine individual domains that compose RPM-1, it is possible that another FSN-1 interaction site might span a junction between two domains. Second, we used a non-native expression system for biochemical mapping. Hence, we might fail to detect a second FSN-1 binding site because post-translational modifications required for the interaction did not occur in 293 cells. Alternatively, adaptor proteins (such as Skp or Cul proteins) required for interaction between FSN-1 and a second site in RPM-1 might not be expressed in these cells or might be unable to bind to C. elegans RPM-1 or FSN-1.
The RING-H2 domain of RPM-1 is the catalytic domain that mediates ubiquitin conjugation to target proteins (16). FSN-1 did not bind to domain 9 of RPM-1, which contains the RING motif. This suggests that FSN-1 binds to FBD1 of RPM-1 to recruit DLK-1, whereas the RING-H2 domain at the C terminus of RPM-1 is available for ubiquitin conjugation to DLK-1 (Fig. 8). This model is consistent with previous work using HEK 293 cells which showed that a C-terminal fragment of RPM-1 (aa 2970–3766) lacking FBD1 binds to DLK-1 in the presence of proteasome inhibitors (16). Although FBD1 and the RING domain are a large distance from one another in the primary protein sequence of RPM-1, it is plausible that these two domains could be in close proximity within the tertiary structure of RPM-1. Our findings on RPM-1 and FSN-1 are consistent with prior work, which showed that other RING-H2 E3 ligases bind F-box proteins and ubiquitination targets at locations that are structurally distinct from the catalytic RING-H2 domain (36, 37).
Our results also have implications for the non-canonical ubiquitin ligase complexes formed by Hiw and Pam that lack Cullins (20, 35). The Rbx·Cul1·Skp1·Skp2 crystal structure shows that Rbx1, a relatively small E3 ligase, is bound by a relatively large Cullin (36). This results in an arch-like structure that brings the F-box protein Skp2 and the ubiquitination target into close proximity with Rbx1. Given the large size of RPM-1 compared with Rbx1 and the extensive amount of protein sequence between the RPM-1 RING domain and FBD1, it is plausible that PHR proteins might not require a Cullin to generate a protein complex structure that is conducive to target recruitment and ubiquitination.
RIP as a Potential Therapeutic Reagent
To date, designing specific inhibitors of PHR ubiquitin ligase activity has been challenging due to the large size and complex biochemistry of these proteins. Although many regions of Hiw have dominant negative effects on synapse formation at the neuromuscular junction (38), a region in Hiw that specifically regulates FSN-1 function has remained elusive. We now show that RIP specifically inhibits the RPM-1·FSN-1 complex in vivo. The conservation of the motifs and residues that mediate the interaction between RIP and FSN-1 suggest that a mammalian version of RIP might be used to block the function of the Pam·Fbxo45 ubiquitin ligase complex in vivo. Previous studies showed that loss of PHR protein function mediated by Fbxo45 prevented axon degeneration after injury, and in some types of neurons improved axon regeneration (21–25). Given the role of PHR proteins in axon degeneration and regeneration, a reagent such as RIP might have potential as a broad-spectrum treatment for neurodegenerative diseases.
Acknowledgments
We thank the C. elegans knock-out consortium for generating deletion alleles and the C. elegans Genetics Center for providing strains.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 NS072129 (to B. G.). This work was also supported by National Science Foundation Grant IOS-1121095 (to B. G.).
B. Grill, unpublished observation.
C. Wu, personal communication.
- RPM-1
- Regulator of Presynaptic Morphology 1
- FSN-1
- F-box synaptic protein 1
- PHR
- Pam/Highwire/RPM-1
- RIP
- RPM-1·FSN-1 complex inhibitory peptide
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- IP
- immunoprecipitate
- FBD1
- FSN-1 binding domain 1
- lf
- loss-of-function
- PLM
- posterior lateral microtubule
- ALM
- anterior lateral microtubule
- OE
- overexpressed
- aa
- amino acid
- Hiw
- Highwire.
REFERENCES
- 1. Po M. D., Hwang C., Zhen M. (2010) PHRs: bridging axon guidance, outgrowth and synapse development. Curr. Opin. Neurobiol. 20, 100–107 [DOI] [PubMed] [Google Scholar]
- 2. Burgess R. W., Peterson K. A., Johnson M. J., Roix J. J., Welsh I. C., O'Brien T. P. (2004) Evidence for a conserved function in synapse formation reveals Phr1 as a candidate gene for respiratory failure in newborn mice. Mol. Cell. Biol. 24, 1096–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schaefer A. M., Hadwiger G. D., Nonet M. L. (2000) rpm-1, a conserved neuronal gene that regulates targeting and synaptogenesis in C. elegans. Neuron 26, 345–356 [DOI] [PubMed] [Google Scholar]
- 4. Wan H. I., DiAntonio A., Fetter R. D., Bergstrom K., Strauss R., Goodman C. S. (2000) Highwire regulates synaptic growth in Drosophila. Neuron 26, 313–329 [DOI] [PubMed] [Google Scholar]
- 5. Zhen M., Huang X., Bamber B., Jin Y. (2000) Regulation of presynaptic terminal organization by C. elegans RPM-1, a putative guanine nucleotide exchanger with a RING-H2 finger domain. Neuron 26, 331–343 [DOI] [PubMed] [Google Scholar]
- 6. Li D., Wang F., Lai M., Chen Y., Zhang J. F. (2005) A protein phosphatase 2cα-Ca2+ channel complex for dephosphorylation of neuronal Ca2+ channels phosphorylated by protein kinase C. J. Neurosci. 25, 1914–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lewcock J. W., Genoud N., Lettieri K., Pfaff S. L. (2007) The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron 56, 604–620 [DOI] [PubMed] [Google Scholar]
- 8. Bloom A. J., Miller B. R., Sanes J. R., DiAntonio A. (2007) The requirement for Phr1 in CNS axon tract formation reveals the corticostriatal boundary as a choice point for cortical axons. Genes Dev. 21, 2593–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. D'Souza J., Hendricks M., Le Guyader S., Subburaju S., Grunewald B., Scholich K., Jesuthasan S. (2005) Formation of the retinotectal projection requires Esrom, an ortholog of PAM (protein associated with Myc). Development 132, 247–256 [DOI] [PubMed] [Google Scholar]
- 10. Culican S. M., Bloom A. J., Weiner J. A., DiAntonio A. (2009) Phr1 regulates retinogeniculate targeting independent of activity and ephrin-A signalling. Mol. Cell. Neurosci. 41, 304–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shin J. E., DiAntonio A. (2011) Highwire regulates guidance of sister axons in the Drosophila mushroom body. J. Neurosci. 31, 17689–17700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grill B., Bienvenut W. V., Brown H. M., Ackley B. D., Quadroni M., Jin Y. (2007) C. elegans RPM-1 regulates axon termination and synaptogenesis through the Rab GEF GLO-4 and the Rab GTPase GLO-1. Neuron 55, 587–601 [DOI] [PubMed] [Google Scholar]
- 13. Kim J. H., Wang X., Coolon R., Ye B. (2013) Dscam expression levels determine presynaptic arbor sizes in Drosophila sensory neurons. Neuron 78, 827–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Refai O., Rohs P., Mains P. E., Gaudet J. (2013) Extension of the Caenorhabditis elegans pharyngeal M1 neuron axon is regulated by multiple mechanisms. G3 3, 2015–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Collins C. A., Wairkar Y. P., Johnson S. L., DiAntonio A. (2006) Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron 51, 57–69 [DOI] [PubMed] [Google Scholar]
- 16. Nakata K., Abrams B., Grill B., Goncharov A., Huang X., Chisholm A. D., Jin Y. (2005) Regulation of a DLK-1 and p38 MAP kinase pathway by the ubiquitin ligase RPM-1 is required for presynaptic development. Cell 120, 407–420 [DOI] [PubMed] [Google Scholar]
- 17. Huntwork-Rodriguez S., Wang B., Watkins T., Ghosh A. S., Pozniak C. D., Bustos D., Newton K., Kirkpatrick D. S., Lewcock J. W. (2013) JNK-mediated phosphorylation of DLK suppresses its ubiquitination to promote neuronal apoptosis. J. Cell Biol. 202, 747–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liao E. H., Hung W., Abrams B., Zhen M. (2004) An SCF-like ubiquitin ligase complex that controls presynaptic differentiation. Nature 430, 345–350 [DOI] [PubMed] [Google Scholar]
- 19. Wu C., Daniels R. W., DiAntonio A. (2007) DFsn collaborates with Highwire to down-regulate the Wallenda/DLK kinase and restrain synaptic terminal growth. Neural Dev. 2, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Saiga T., Fukuda T., Matsumoto M., Tada H., Okano H. J., Okano H., Nakayama K. I. (2009) Fbxo45 forms a novel ubiquitin ligase complex and is required for neuronal development. Mol. Cell. Biol. 29, 3529–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hammarlund M., Nix P., Hauth L., Jorgensen E. M., Bastiani M. (2009) Axon regeneration requires a conserved MAP kinase pathway. Science 323, 802–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xiong X., Wang X., Ewanek R., Bhat P., Diantonio A., Collins C. A. (2010) Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J. Cell Biol. 191, 211–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nix P., Hisamoto N., Matsumoto K., Bastiani M. (2011) Axon regeneration requires coordinate activation of p38 and JNK MAPK pathways. Proc. Natl. Acad. Sci. U.S.A. 108, 10738–10743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xiong X., Hao Y., Sun K., Li J., Li X., Mishra B., Soppina P., Wu C., Hume R. I., Collins C. A. (2012) The Highwire ubiquitin ligase promotes axonal degeneration by tuning levels of Nmnat protein. PLoS Biol. 10, e1001440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Babetto E., Beirowski B., Russler E. V., Milbrandt J., DiAntonio A. (2013) The Phr1 ubiquitin ligase promotes injury-induced axon self-destruction. Cell Rep. 3, 1422–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brenner S. (1974) The genetics of Caenorhabditis elegans. Genetics 77, 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mello C. C., Kramer J. M., Stinchcomb D., Ambros V. (1991) Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10, 3959–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grill B., Chen L., Tulgren E. D., Baker S. T., Bienvenut W., Anderson M., Quadroni M., Jin Y., Garner C. C. (2012) RAE-1, a novel PHR binding protein, is required for axon termination and synapse formation in Caenorhabditis elegans. J. Neurosci. 32, 2628–2636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ch'ng Q., Williams L., Lie Y. S., Sym M., Whangbo J., Kenyon C. (2003) Identification of genes that regulate a left-right asymmetric neuronal migration in Caenorhabditis elegans. Genetics 164, 1355–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tulgren E. D., Baker S. T., Rapp L., Gurney A. M., Grill B. (2011) PPM-1, a PP2Cα/β phosphatase, regulates axon termination and synapse formation in Caenorhabditis elegans. Genetics 189, 1297–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Baker S. T., Opperman K. J., Tulgren E. D., Turgeon S. M., Bienvenut W., Grill B. (2014) RPM-1 uses both ubiquitin ligase and phosphatase-based mechanisms to regulate DLK-1 during neuronal development. PLoS Genet. 10, e1004297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tulgren E. D., Turgeon S. M., Opperman K. J., Grill B. (2014) The nesprin family member ANC-1 regulates synapse formation and axon termination by functioning in a pathway with RPM-1 and β-catenin. PLoS Genet. 10, e1004481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tian X., Li J., Valakh V., DiAntonio A., Wu C. (2011) Drosophila Rae1 controls the abundance of the ubiquitin ligase Highwire in post-mitotic neurons. Nat. Neurosci. 14, 1267–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guo Q., Xie J., Dang C. V., Liu E. T., Bishop J. M. (1998) Identification of a large Myc-binding protein that contains RCC1-like repeats. Proc. Natl. Acad. Sci. U.S.A. 95, 9172–9177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brace E. J., Wu C., Valakh V., DiAntonio A. (2014) SkpA restrains synaptic terminal growth during development and promotes axonal degeneration following injury. J. Neurosci. 34, 8398–8410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zheng N., Schulman B. A., Song L., Miller J. J., Jeffrey P. D., Wang P., Chu C., Koepp D. M., Elledge S. J., Pagano M., Conaway R. C., Conaway J. W., Harper J. W., Pavletich N. P. (2002) Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 416, 703–709 [DOI] [PubMed] [Google Scholar]
- 37. Duda D. M., Scott D. C., Calabrese M. F., Zimmerman E. S., Zheng N., Schulman B. A. (2011) Structural regulation of cullin-RING ubiquitin ligase complexes. Curr. Opin. Struct. Biol. 21, 257–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu C., Wairkar Y. P., Collins C. A., DiAntonio A. (2005) Highwire function at the Drosophila neuromuscular junction: spatial, structural, and temporal requirements. J. Neurosci. 25, 9557–9566 [DOI] [PMC free article] [PubMed] [Google Scholar]