

Background: There is a need for new small molecule pre-mRNA splicing inhibitors as biotools.
Results: High throughput screening resulted in the identification of small molecule splicing inhibitors that are active in vitro and in cells.
Conclusion: New small molecules for studying pre-mRNA splicing in vitro and in cells are identified.
Significance: Small drug-like molecules are identified that modulate splicing in vitro and in cells.
Keywords: High Throughput Screening, RNA, RNA Splicing, Small Molecule, Spliceosome, DDD00107587, Madrasin
Abstract
Eukaryotic pre-mRNA splicing is an essential step in gene expression for all genes that contain introns. In contrast to transcription and translation, few well characterized chemical inhibitors are available with which to dissect the splicing process, particularly in cells. Therefore, the identification of specific small molecules that either inhibit or modify pre-mRNA splicing would be valuable for research and potentially also for therapeutic applications. We have screened a highly curated library of 71,504 drug-like small molecules using a high throughput in vitro splicing assay. This identified 10 new compounds that both inhibit pre-mRNA splicing in vitro and modify splicing of endogenous pre-mRNA in cells. One of these splicing modulators, DDD00107587 (termed “madrasin,” i.e. 2-((7methoxy-4-methylquinazolin-2-yl)amino)-5,6-dimethylpyrimidin-4(3H)-one RNAsplicing inhibitor), was studied in more detail. Madrasin interferes with the early stages of spliceosome assembly and stalls spliceosome assembly at the A complex. Madrasin is cytotoxic at higher concentrations, although at lower concentrations it induces cell cycle arrest, promotes a specific reorganization of subnuclear protein localization, and modulates splicing of multiple pre-mRNAs in both HeLa and HEK293 cells.
Introduction
The removal of introns from pre-mRNA is an essential step in gene expression in eukaryotes. Deep sequencing data have shown that >95% of all human genes are spliced, often in a developmental, tissue-specific, and/or signal transduction-dependent manner (1). Pre-mRNA splicing is mediated by the spliceosome, a large RNA-protein complex that in humans consists of five snRNAs (U1, U2, U4, U5, and U6) and numerous proteins (2). The spliceosome coordinates the two separate transesterification reactions required for intron removal and the completion of the splicing cycle. The splicing of pre-mRNA takes place in the cell nucleus and involves the stepwise assembly of a separate spliceosome complex on each intron in a pre-mRNA transcript. In most, if not all cases, spliceosomes assemble on nascent transcripts while transcription of the gene is still in progress.
The maintenance of high fidelity pre-mRNA splicing is important to allow accurate protein expression. Point mutations affecting pre-mRNA sequences involved in splice site recognition and splicing are thought to be responsible for ∼15% of all inherited human diseases (3, 4). As well as its essential role in removing introns to permit protein expression, different patterns of alternative splicing allow the creation of multiple mRNAs encoding different proteins from a single gene. Alternative splicing is also often disrupted in many forms of disease, including cancer. For example, this can be due to either altered expression or mis-localization of RNA-binding proteins, resulting in changes in the pattern of alternatively spliced mRNAs (5, 6).
Small molecules can be powerful tools for dissecting complex biological processes, as is well documented for transcription and translation. This is typified by the antibiotics used to study translation in prokaryotes, e.g. puromycin and viomycin, which inhibit the prokaryotic ribosome (7). In contrast, there are no comparable classes of natural antibiotics that inhibit the pre-mRNA splicing process. This is likely because natural antibiotics are mostly produced by fungi to kill bacteria, which lack introns in protein-coding genes and thus do not require splicing for protein expression. In recent years, however, several natural compounds, and their synthetic derivatives, have been reported to inhibit splicing, either in vitro, or in cells, or both. These include FR901464, spliceostatin A (SSA)2 (8), E7107, pladienolide B, pladienolide D (9), GEX1A (10), sudemycin (11), and isoginkgetin (12). With the exception of isoginkgetin, whose target is currently unknown, all the other compounds target the SF3b subunit of the U2 snRNP (13).
Several high throughput screening approaches have also been undertaken to identify small molecules that act as specific splicing inhibitors, albeit with limited success. Most of the molecules that have been identified in these assays either modify splicing in cells but are not shown to do so in vitro, e.g. clotrimazol, flunarizine, and chlorhexidine (14), or else they inhibit splicing in vitro but have not been shown to modulate splicing also in cells, e.g. naphthoquinone, tetrocarcin, and ANSC659999 (15).
Two main types of high throughput assay have been established for the identification of new pre-mRNA splicing modifiers as follows: (a) cell-based splicing assays, using reporter constructs as a readout (12, 16) and (b) high throughput in vitro splicing assays, using either ELISA or quantitative PCR techniques for signal detection (15, 17, 18).
The high throughput in vitro splicing assay developed by Samatov et al. (17) allows the identification of small molecule splicing inhibitors that affect either spliceosome assembly, activation, and/or step 1 catalysis. This is achieved by using the PM5 pre-mRNA, which lacks both the 3′-splice site and 3′ exon of a typical pre-mRNA and therefore stops the splicing reaction after C complex formation. Addition of the PM5 pre-mRNA to a splicing reaction containing FLAG-tagged DDX41, a component of the spliceosomal C complex, allows detection of the C complex using an anti-FLAG antibody. The high throughput screening in vitro splicing assay established by Samatov et al. (17) thus allows the detection of small molecules that interfere with the splicing process at any stage up to C complex formation. We therefore used this assay to screen a highly selected library of 71,504 small molecules developed by the University of Dundee Drug Discovery Unit to identify new splicing inhibitors (19).
Here, we describe the discovery of 13 new small chemical compounds that inhibit splicing in vitro. 10 out of these 13 compounds also inhibit splicing in cultured human cell lines. By way of example, we characterize the effect of one of these compounds, DDD00107587 (madrasin), both on splicing in vitro and in cells.
EXPERIMENTAL PROCEDURES
High Throughput Screening
The high throughput screen was performed using an adapted version of the high throughput in vitro splicing assay published by Samatov et al. (17) and the in-house small molecule compound library of the Drug Discovery Unit, University of Dundee. In short, using a Wellmate bulk reagent dispenser (Thermo Fisher, Waltham, MA), 384-well polystyrene high protein-binding microplates (Greiner) were coated with rabbit polyclonal antibodies to maltose-binding protein (Abcam, Cambridge, UK) before PM5 pre-mRNA bound by MS2-MBP was added to the microplates and incubated for ∼1 h. 0.1 μl of test compounds were added to the pre-mRNA-labeled microplates by using a Hummingbird (Digilab Genomic Solutions Ltd., Marlborough, MA) to produce a final concentration of 50 μm. The splicing reactions containing HEK293 whole cell extract from cells stably expressing FLAG-tagged DDX41 protein and the monoclonal anti-FLAG® M2-peroxidase (Sigma) were prepared at room temperature, and 20 μl/well were added to the plates using the Biomek FX automated workstation (Beckman Coulter, High Wycombe, UK). The plates were incubated at 26 °C for 90 min before they were washed four times with wash buffer. 20 μm SuperSignal® ELISA Femto Maximum Sensitivity Substrate (Pierce, Thermo Fisher, Rockford, IL) was then added, and the luminescence was monitored after 5 min of incubation at room temperature using the TopCount microplate luminometer (PerkinElmer Life Sciences).
Data Analysis
For all primary and potency screen data points, the relative luminescence units per well were measured. From cross-talk-corrected data, a percentage inhibition value for each test compound was determined as follows: % inhibition = 100 −((RLUTEST − RLULOW)/(RLUHIGH − RLULOW)) × 100. RLUHIGH = median of all “high” signal (“uninhibited” control) wells per plate. RLULOW = median of all “low” signal (noMS2-MBP-pre-mRNA) wells per plate. Robust Z′ was calculated for each plate based on the median and scaled median absolute deviation (1.4826 × median absolute deviation) for RLUHIGH and RLULOW from all wells per plate. All data processing was conducted within the activity base using normalized data as defined above. Database querying and report creation was undertaken using SARgen version 7.3.0.2 and SARview version 7.1.1 from IDBS (London, UK). All IC50 curve fitting was undertaken within the ActivityBase XE utilizing the underlying “Math IQ” engine of XLfit version 5.1.0.0 from IDBS. A four-parameter logistic dose-response curve of the following form was utilized for compound potency determination as shown in Equation 1,
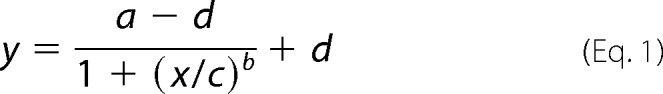 |
where a is the minimum; b is the Hill slope; c is the EC50 value; and d is the maximum. Data are presented as the mean pEC50 with the standard error of the mean of n experiments.
Cell Culture, RNA Isolation, and RT-PCR
HeLa and HEK293 cells were purchased from ATCC and cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 2 mm glutamine (Invitrogen), and 100 μg/ml streptomycin (100× stock, Invitrogen). Total RNA was extracted from cells using the NucleoSpin RNA II kit (Macherey-Nagel, Düren, Germany) according to the manufacturer's instructions. 200 ng of total RNA was reverse-transcribed and amplified using the One-step RT-PCR kit (Qiagen, Venlo, Netherlands) according to the manufacturer's instructions. Primer sequences are as follows: HPV18 E6, forward 5′-GCGACCCTACAAGCTACCTA-3′ and reverse 5′-GCACTGGCCTCTATAGTGCC-3′ (20); E1A, forward 5′-CCGAAGAAATGGCCGCCAGTC-3′ and reverse 5′-GGACGCCGGGTAGGTCTTGC-3′ (21); RIOK3, forward 5′-GCTGAAGGACCATTTATTACTGGAG-3′ and reverse 5′-TTCTTGCTGTGTTCTTTCTCCCACA-3′; Hsp40, 5′-GAACCAAAATCACTTTCCCCAAGGAAGG-3′ and reverse 5′-AATGAGGTCCCCACGTTTCTCGGGTGT-3′ (22); BRD2, 5′-CAAAATTATAAAACAGCCTATGGACATG-3′ and reverse 5′-TTTTCCAGCGTTTGTGCCATTAGGA-3′ (22); MCL1, forward 5′-GAGGAGGAGGAGGACGAGTT-3′ and reverse 5′-ACCAGCTCCTACTCCAGCAA; CCNA2, forward 5′-TCCTCGTGACTGGTTAGTT-3′ and reverse CCCGTGACTGTGTAGAGTGC-3′; AURKA, forward 5′-CTTGGATCAGCTGGAGAGCTT-3′ and reverse 5′-AGCTGATTCTTTGTTTTGGCAAT-3′; p27 forward 5′-CAGCTTGCCCGAGTTCTACT-3′ and reverse 5′-GTCCATTCCATGAAGTCAGCG-3′.
In Vitro Transcription and Nonradioactive in Vitro Splicing Reaction
The adenovirus pre-mRNA Ad1 was transcribed in vitro using the RNAMaxx High Yield Transcription kit (Agilent, Santa Clara, CA) according to the manufacturer's instruction, followed by a DNase I digestion and RNA clean up using the RNeasy kit (Qiagen).
Standard splicing reactions were carried out in 30% HeLa nuclear extract (Computer Cell Culture Centre, Seneffe, Belgium) in the presence of either DMSO or compound and incubated at 30 °C for 90 min. The splicing reaction was followed by a heat inactivation step of 5 min at 95 °C before the samples were subjected to proteinase K (Qiagen) digestion for 30 min at 55 °C and another heat inactivation step at 95 °C for 5 min. The spliced and nonspliced RNA was amplified by using the one-step RT-PCR kit (Qiagen) according to the manufacturer's instructions. PCR products were separated by electrophoresis using 1% agarose gels containing SYBR safe DNA gel stain (Invitrogen).
Radioactive in Vitro Splicing Reaction and Native Gels
Radioactive in vitro splicing reactions were performed as described previously (1) using either 32P-labeled pBSAd1 pre-mRNA substrate or 32P-labeled MINX pre-mRNA substrate (23). Splicing complexes were analyzed on a low melting point agarose gel (1.5%, w/v) as described previously (24) and visualized by phosphorimaging (Typhoon 8600, GE Healthcare).
Cell Fixation and Immunofluorescence Analysis
HeLa cells, treated either with DMSO or compound, were grown on coverslips in DMEM for 24 h at 37 °C before fixing with 4% paraformaldehyde in PHEM buffer for 10 min at room temperature. After rinsing the cells with PBS, the cells were permeabilized with 0.5% Triton X-100 in PBS prior to incubation with the primary antibodies, i.e. anti-SC35 (Abcam, Cambridge, UK), anti-coilin (ProteinTech, Chicago, IL), anti-Y12 (Dundee Cell Products, Dundee, UK), anti-TMG (Calbiochem), and anti-SMN (BD Biosciences). After incubation with the primary antibody for 1 h at room temperature, the coverslips were washed twice with 0.5% of Tween 20 in PBS for 5 min before they were incubated with the dye-conjugated secondary antibody for 30 min. Cells were then stained with DAPI (Sigma), and the coverslips were mounted in Vectashield medium (Vector Laboratories, Peterborough, UK). The samples were visualized using a fluorescence microscope (Zeiss, Jena, Germany; Axiovert-DeltaVision Image Restoration; and Applied Precision, LLC).
Flow Cytometry Analysis
HeLa cells were seeded in 6-well plates, and after 24 h cells were treated with compound. The cells were harvested after 4, 8, and 24 h and then washed twice with PBS, resuspended in ice-cold 70% ethanol, and fixed at room temperature for 30 min. Fixed cells were then washed twice with PBS, and resuspended in propidium iodide solution (50 μg/ml propidium iodide and 100 μg/ml ribonuclease A in PBS). Cells were incubated in propidium iodide solution for 30 min and then analyzed by flow cytometry on a FACSCalibur (BD Biosciences). The flow cytometry data were analyzed using FlowJo (Treestar Inc., Ashland, OR).
Pulse Labeling of HeLa Cells with EdU and EU
Newly synthesized DNA or RNA was detected by using either the Click-iT EdU imaging kit (Invitrogen) or the Click-iT RNA imaging kit (Invitrogen). In brief, HeLa cells were grown for 24 h in the presence of either DMSO or compound and pulse-labeled using either EdU or EU for 20 min. The cells were then fixed, and the Click-iT detection was performed according to the manufacturer's manual.
RESULTS
High Throughput Screening
To identify small molecules that inhibit the human spliceosome, we performed a high throughput in vitro screen using a splicing assay (17), optimized for robotic screening as outlined in Fig. 1. Thus, the PM5 pre-mRNA was immobilized on anti-MBP antibody-coated 384-well microplates via the MS2-MBP fusion protein. This was incubated in a splicing reaction containing HEK293 whole cell splicing extract, including a FLAG-tagged version of the DEAD box ATPase DDX41. As the PM5 pre-mRNA lacked the 3′ exon and the 3′-splice site, the second step of the splicing reaction could not be performed, and as a consequence, the splicing reaction was stalled after C complex formation. The presence or absence of the C complex on the PM5 pre-mRNA was detected using a horseradish peroxidase-labeled anti-FLAG antibody, and the luminescence was monitored using the TopCount microplate luminometer (PerkinElmer Life Sciences).
FIGURE 1.
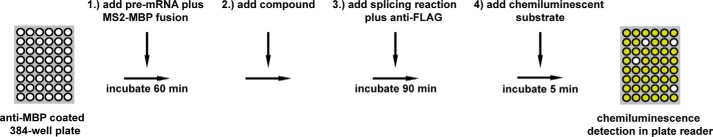
Schematic representation of in vitro splicing high throughput screening. 384-well plates coated with anti-MBP antibodies were incubated with a mix of pre-mRNA and MS2-MBP fusion protein for 1 h. After addition of 50 μm compound, the splicing reaction containing anti-FLAG antibodies was added, and the splicing reaction was incubated for 90 min at 26 °C. The addition of chemiluminescent substrate for 5 min is followed by signal detection with the TopCount microplate luminometer.
A single point screen was performed with a selected library of 71,504 small molecules, each at a final concentration of 50 μm. The test library used a combination of a diverse set (63,874 compounds) and several target-type focused sets, i.e. nuclear hormone receptor (2,407), epigenetic modifier (2,664), and kinase (2,559). We chose to follow 352 compounds from the primary screen. This resulted in all molecules giving >52% splicing inhibition being investigated further (Tables 1 and 2).
TABLE 1.
Summary of screen statistics (average ± S.D.)
| Criterion | Result | |
|---|---|---|
| Valid plates/total plates | 217/252 | |
| Signal/background | >10 | 62 ± 37 |
| Robust high control CV (%) | <15 | 7.2 ± 2.5 |
| Robust Z′ | >0.5 | 0.75 ± 0.08 |
TABLE 2.
Summary of screen outcome
| Output | Result | % total |
|---|---|---|
| No. of compounds screened | 71,504 | |
| Hit identification threshold | 52% | |
| Putative hits >52% | 352 | 0.49 |
| Putative hits >75% | 112 | 0.16 |
| Putative hits >90% | 54 | 0.07 |
To reduce false-positive results e.g. caused by compounds that restrict the accessibility of the DDX41 antibody to its target, the 352 hit compounds were rescreened using a separate in vitro splicing assay format with a nonradioactive RT-PCR-based in vitro splicing reaction (Fig. 2). For the secondary screen, a different pre-mRNA, i.e. the adenovirus-related Ad1 transcript made from plasmid pBSAd1 (25), was used as a template pre-mRNA, and separate assays were carried out in the presence of (a) HeLa nuclear extract and (b) HEK293 total cell lysate. A final concentration of 500 μm compound was chosen for this screen to maximize detection of active compounds. The splicing reactions, which were carried out for 90 min, were followed by a proteinase K digestion and a RT-PCR to measure the ratio of spliced/unspliced RNA (see “Experimental Procedures”). This secondary screen reduced the original number of 352 potential hits to 29 molecules that consistently showed inhibition of splicing in each assay format (supplemental Table S1).
FIGURE 2.

Secondary screen. A, schematic representation of the RT-PCR in vitro splicing assay. 20-μl splicing reaction was incubated in the presence of 500 μm compound at 30 °C for 90 min before the splicing reaction was stop by heat inactivation. After proteinase K digestion, the spliced and unspliced RNA is amplified by RT-PCR. B shows examples of compounds that either inhibit pre-mRNA splicing or do not interfere with the splicing reaction. The star indicates splicing reactions that were inhibited. Lane M, marker (hyperladder, 1 kb).
DMSO stocks of all 29 compounds were tested by liquid chromatography-mass spectrometry (LC-MS) to confirm the identity, and the composition of each was as expected. 20 of the 29 compounds passed the quality assurance test (supplemental Table S1). For six compounds, the LC-MS test identified a large percentage of impurities, and another three compounds failed due to incorrect molecular weight. The 20 remaining hit compounds were grouped by chemical similarity into six chemical series. Compounds were triaged to remove molecules predicted to be either chemically reactive or unstable. In addition, molecules that have demonstrated frequent activity across a broad range of assays, possibly as a result of being fluorescent, redox active, or aggregating, were removed. One series stood out, however, with three of the most active hit compounds all belonging to a series of aminoquinazolines. These compounds have no obvious undesirable structural features with respect to potential chemical development and have promising physical properties, as represented by DDD00107597, which has a ClogP of 3.3 and a Mr of 281.
Validation of Hit Compounds
Next, we sourced 33 analogues of DDD00107597, both from commercial sources and through in-house synthesis of new compounds. The 33 analogues and DDD00107597 were designed to investigate which structural features are required for splicing modulation. A third round of in vitro screening was then performed to confirm the ability of these related compounds to inhibit pre-mRNA splicing. This time the compounds were all tested in two different in vitro splicing assays as follows: (a) an in vitro splicing assay, incubated with HeLa nuclear extract and the radiolabeled adenovirus-derived MINX pre-mRNA (26), and (b) a nonradioactive RT-PCR based in vitro splicing assay with the Ad1 pre-mRNA, as described above for the secondary screen. Out of this group of 34 compounds, 13 inhibited splicing in at least one assay, with four of these compounds consistently inhibiting pre-mRNA splicing in both assay systems. Of the nine compounds that showed assay-specific splicing inhibition, seven inhibited splicing only in the nonradioactive PCR-based in vitro splicing assay using the Ad1 pre-mRNA, and two compounds only inhibited splicing of the MINX pre-mRNA (supplemental Table S2). The reason for these assay-specific inhibitory effects was not clear, and these compounds were not investigated further. We focused instead on the compounds that consistently inhibited splicing in both in vitro assay formats.
DDD00107587 Inhibits Splicing in Vitro
Compound DDD00107587 (Fig. 3A), one of the four compounds that inhibited splicing of both the MINX and the Ad1 pre-mRNAs (supplemental Table S2), was studied in more detail (Fig. 3). In vitro splicing reactions with either radiolabeled MINX or Ad1, pre-mRNAs, were carried out for 2 h in the presence of increasing DDD00107587 concentrations (Fig. 3B). Splicing of the Ad1 pre-mRNA was completely inhibited at a final concentration of 62.5 μm, whereas splicing of the MINX pre-mRNA was completely inhibited at 150 μm.
FIGURE 3.
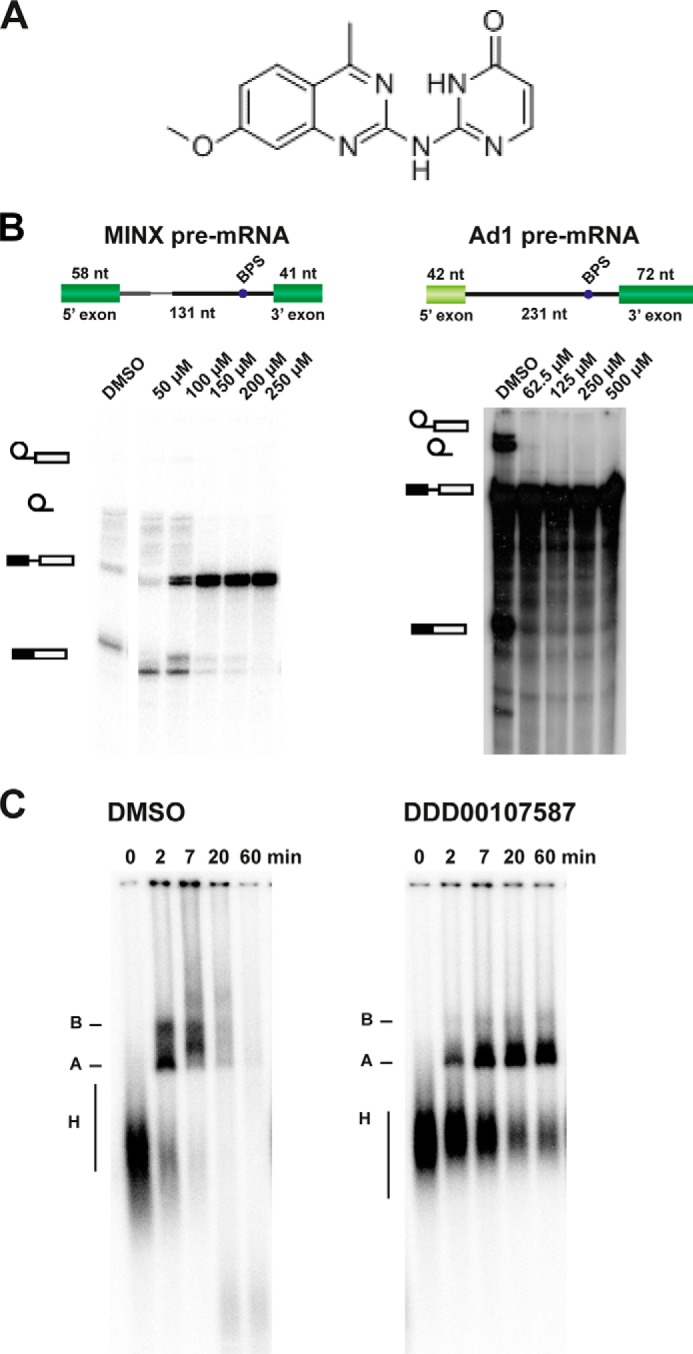
Effect of DDD00107587 on in vitro splicing. A, chemical structure of DDD00107587. B, denaturing gel analysis of 32P-labeled MINX or Ad1 RNA isolated from splicing reactions incubated with dimethyl sulfoxide (DMSO) or different concentrations of DDD00107587. The positions of the lariat intron, lariat, pre-mRNA, and fully spliced mRNA are shown top to bottom on the left. C, formation of splicing complex on the MINX pre-mRNA in the presence of DMSO or 250 μm DDD00107587. The reactions were stopped at the time points indicated, and the products were analyzed on a native agarose gel. The positions of the splicing complexes B, A, and H are indicated on the left.
DDD00107587 Stalls Spliceosome Assembly at the A Complex Formation
The spliceosome assembles onto pre-mRNA in a stepwise pathway that involves the formation of assembly intermediate sub-complexes. Native agarose gels were used to separate spliceosomes and assemble intermediate complexes to investigate the effect of DDD00107587 on the assembly of splicing complexes on the MINX pre-mRNA. When the splicing assays were carried out in the presence of a DMSO negative control, the native gel shows separation of the H, A, and B complexes. At early times after the start of the splicing reaction (2 and 7 min), mainly A and B complexes are visible. In the presence of 250 μm DDD00107587, which blocks the formation of spliced mRNA, the kinetics of spliceosome assembly differ dramatically. After 2 min, H complexes and small amounts of A complex can be observed. At later times, A complexes accumulate; however, no progression to either the B complex or mature spliceosome was detected (Fig. 3C).
These data suggest that DDD00107587 inhibits the formation of spliceosome complexes subsequent to complex A formation. A block of assembly after A complex formation has also been reported for isoginkgetin (12) and spliceostatin A (27). In the case of SSA, however, it was shown that the A complex that forms lacks critical RNA-protein interactions and is therefore relatively instable (28).
DDD00107587 Alters Pre-mRNA Splicing in Vivo
Next, we investigated the effect of DDD00107587 on the splicing of endogenous pre-mRNAs in vivo. HeLa and HEK293 cells were each treated with either DMSO, as a negative control, or with DDD00107587 at a final concentration of 10, 20, or 30 μm, for either 4, 8, or 24 h. After incubation, the cells were harvested, and total RNA was extracted. 200 ng of total RNA was used in RT-PCR assays to detect both intron inclusion in transcripts from the RIOK3, BRD2, and Hsp40 genes and changes in the alternative splicing of the MCL1, CCNA2, AURKA, and p27 pre-mRNAs. In addition, primers were chosen to detect the splicing of the HPV18 E6 (human papillomavirus type 18) RNA in HeLa cells and the adenovirus E1A RNA in HEK293 cells.
Fig. 4A shows the effect on pre-mRNA splicing changes induced by DDD00107587 in HeLa cells. Although no splicing inhibition of the Hsp40 pre-mRNA could be detected at the concentrations and time points tested, inhibition of splicing of each of the RIOK3, HPV18, and BRD2 pre-mRNAs was observed after 4 h of treatment at 30 μm. Only a slight increase in inhibition of pre-mRNA splicing over time was seen for most HPV18 E6 and BRD2 pre-mRNAs. DDD00107587 potently inhibited splicing of the RIOK3 pre-mRNA, with >95% splicing inhibition at 30 μm for 24 h and ∼80% splicing inhibition at 20 μm for 24 h.
FIGURE 4.
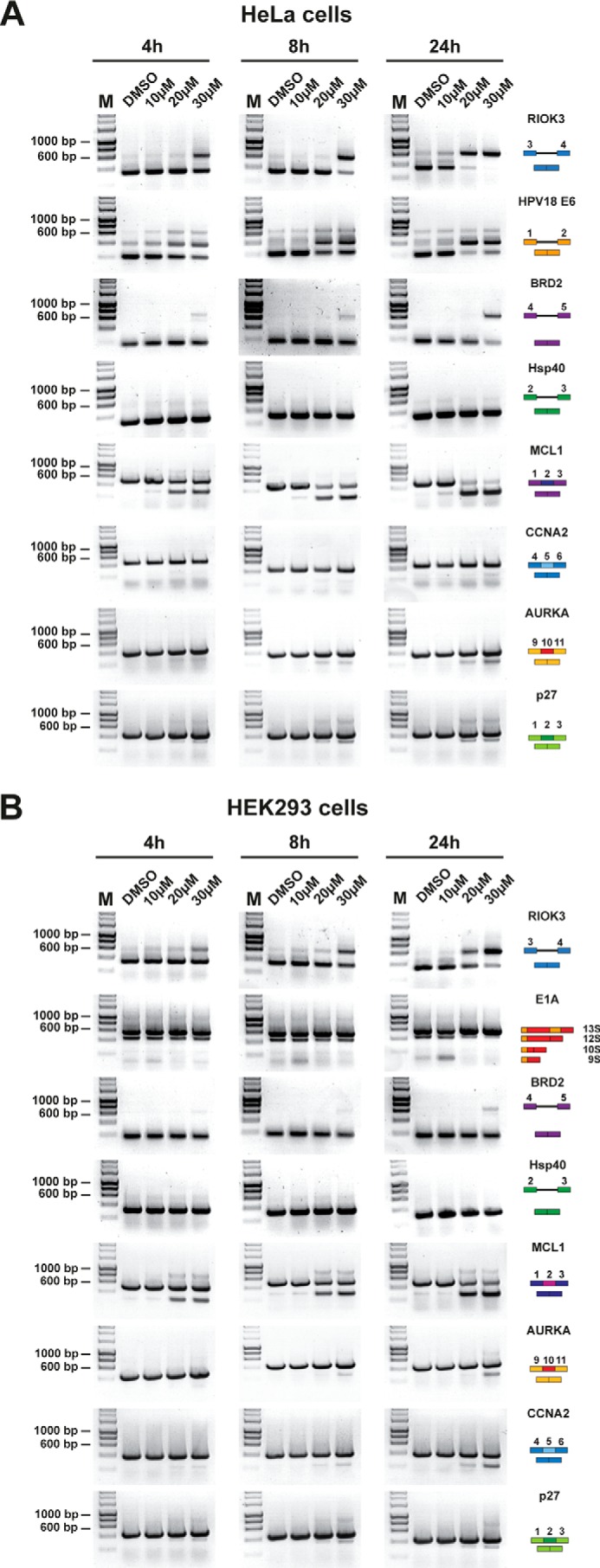
Splicing inhibition in cells treated with DDD00107587. Semiquantitative RT-PCR analysis of cells treated with increasing concentrations of DDD00107587 and DMSO for 4, 8, and 24 h. HeLa cells were investigated for intron inclusion of RIOK3, HPV18 E6, BRD2, and Hsp40 as well as the exon skipping of MCL1, AURKA, CCNA2, and p27 (A). HEK293 cells were examined for intron inclusion of RIOK3, BRD2, and Hsp40, the alternative splicing the adenovirus E1A gene and the exon skipping of MCL1, AURKA, CCNA2, and p27 (B). The positions of different cDNA products are pictured on the right of each gel image, and molecular weight markers are shown on the left. Lane M, marker (hyperladder, 1 kb).
Exon skipping after DDD00107587 treatment was observed for all four genes (MCL1, CCNA2, AURKA, and p27) examined. The strongest effect was observed on exon 2 skipping of the MCL1 pre-mRNA. 4 h after addition of DDD00107587 to HeLa cells, a significant shift from the long splice variant of MCL1 (MCL1L) to the short splice variant (MCL1S) was observed at concentrations of 20 and 30 μm. Over time this shift increased until almost exclusively the short isoform was expressed at these concentrations. At 10 μm, low levels of MCL1S were detected, which did not increase over time. Weak exon skipping was observed for AURKA and p27 after 4 h at concentrations of 20 and 30 μm DDD00107587. Changes in alternative splicing of AURKA and p27 increased slightly over time. DDD00107587 treatment resulted only in low amounts of alternatively spliced CCNA2 transcripts after 24 h at 30 μm.
The results for HEK293 cells are very similar to those in HeLa cells (Fig. 4B), with little or no pre-mRNA splicing inhibition detected in the case of the Hsp40 pre-mRNA, but clear inhibition of splicing of the RIOK3 and BRD2 pre-mRNAs was detected 24 h after treatment at a final concentration of 30 μm. The alternative splicing of the E1A gene was also changed in the presence of DDD00107587 in a dose-dependent manner. Whereas treatment of HEK293 cells with 10 μm DDD00107587 led to a shift toward formation of the 9 S splice variant, 20 and 30 μm DDD00107587 resulted in the accumulation of 13 S splice products. In HEK293 cells, exon skipping for the four transcripts tested was only detected for the two highest concentrations of DDD00107587, 20 and 30 μm. As in HeLa cells, DDD00107587 had the greatest effect on the alternative splicing of MCL1 with significantly more MCL1S expressed than MCL1L after 24 h of treatment. Exon skipping of AURKA, CCNA2, and p27 was mostly observed at 30 μm.
DDD00107587 Causes Cell Cycle Arrest
We next tested the effect of DDD00107587 on cell cycle progression in exponentially dividing cells. HeLa cells were treated either with a DMSO negative control or with a final concentration of 10, 20, or 30 μm DDD00107587 for 4, 8, and 24 h before labeling with propidium iodide and analysis by flow cytometry (Fig. 5A). No cell cycle changes were observed after 4 h of treatment at any concentration tested. In the presence of 10 μm DDD00107587 8 h after treatment, the proportion of cells in G2, M, and S phases increased, with a concomitant decrease in the number of G1 phase cells. This effect increased over time, with >40% of cells in G2 and M phase and >50% in S phase 24 h after treatment with DDD00107587. This contrasted with 14% in G2 and M and 23% in S phase in the DMSO control (Fig. 5B). Cells treated with higher concentrations showed a delayed cell cycle arrest. 8 h of treatment with either 20 or 30 μm DDD00107587 exhibited the same cell cycle profile as the DMSO control. At 24 h, however, cells treated with 20 or 30 μm DDD00107587 also showed G2, M, and S phase arrest, with ∼40% of cells in S and ∼30% in G2 and M phase in the presence of 20 μm and 30% of cells in S and 24% in G2 and M phase when treated with 30 μm DDD00107587.
FIGURE 5.

DDD00107587 induces S/G2 and M phase arrest. Cell cycle analysis was performed on HeLa cells treated with DMSO and 10, 20, and 30 μm DDD00107587 for 4, 8, and 24 h. Cellular DNA content was measured by propidium iodide staining followed by flow cytometry analysis. A, histograms of DNA content. B, cell cycle distributions quantified using the Watson model (51).
To test whether cells arrested by DDD000107587 showed any inhibition of DNA synthesis, HeLa cells were pulse-labeled with EdU either 4, 8, or 24 h after treatment with different concentrations of DDD000107587 (Fig. 6). DDD00107587 showed a time- and dose-dependent effect on reducing DNA synthesis, with the greatest reduction in DNA synthesis at 30 μm. After 4 h of treatment with 30 μm DDD00107587, a modest reduction in DNA synthesis was detected (∼20%), which increased over time until after 24 h no incorporation of EdU could be detected at this concentration. Reduction in DNA synthesis levels in HeLa cells was first observed after 8 h of treatment with 20 μm, with essentially complete inhibition after 24 h. At the lowest concentration tested, 10 μm, a decrease in DNA synthesis could only be detected after 24 h.
FIGURE 6.

DDD00107587 reduces DNA synthesis. HeLa cells were treated with DMSO and 10, 20, and 30 μm DDD00107587 for 4, 8, and 24 h. Areas of active DNA replication were marked by pulse labeling with EdU 20 min before the cells were fixed. Scale bars, 40 μm.
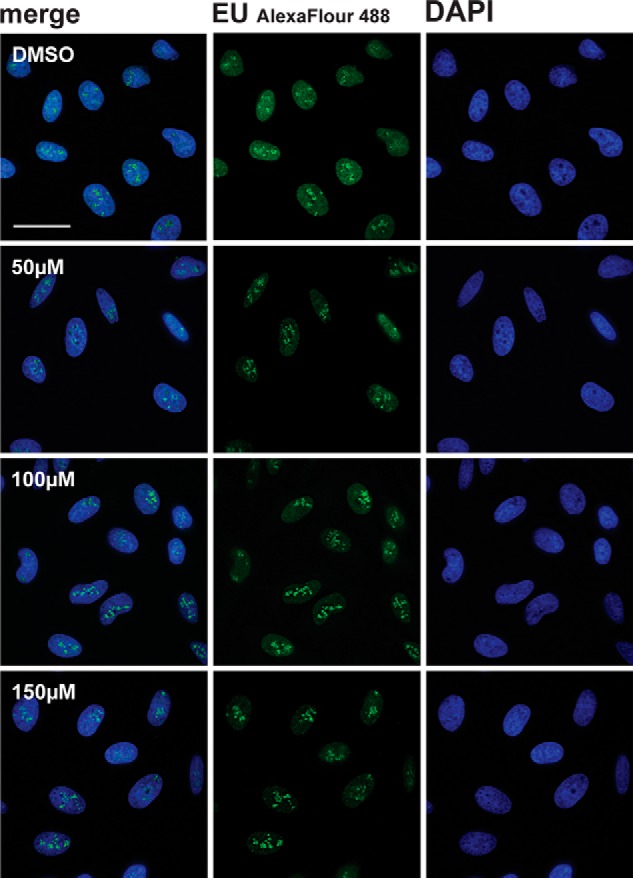
Next, we tested whether DDD00107587 had a similar inhibitory effect on levels of newly transcribed RNA in HeLa cells by pulse labeling with EU (see “Experimental Procedures”). In contrast with the reduced levels of DNA synthesis, total RNA synthesis levels appeared less affected by DDD00107587 (Fig. 7). In particular, nucleoplasmic transcription levels showed minimal reduction, consistent with the RT-PCR data (Fig. 4) showing an accumulation of unspliced pre-mRNA. Interestingly, the main inhibitory effect on nuclear RNA caused by DDD00107587 was a decrease in nucleolar RNA levels, likely representing decreased rRNA synthesis (Fig. 7).
FIGURE 7.

DDD00107587 does not inhibit transcription of RNA polymerase II. RNA synthesis in HeLa cells treated with DMSO or 10, 20, and 30 μm for either 4 or 24 h was analyzed by EU pulse labeling. Scale bars, 30 μm.
Loss of Cajal Bodies after Treatment with DDD00107587
Next, we investigated the effect of DDD00107587 on subnuclear structures and specifically on the organization of pre-mRNA splicing factors within the nucleus. To analyze the effect of DDD00107587 on splicing speckles, HeLa cells were treated for 4 h with either a DMSO negative control or with 10, 20, or 30 μm DDD00107587 before cells were fixed and stained with an antibody to label the splicing factor SC35 (Fig. 8). Interestingly, no enlargement and rounding up of the speckles was detected (Fig. 8A), in contrast to what is typically observed when transcription is inhibited (Fig. 8B). However, DDD00107587 induced a dose-dependent disruption of CBs. Thus, antibodies to each of four CB markers (coilin, SMN, TMG cap, and Y12) label bright foci, corresponding to CBs (Fig. 9, arrowheads), in cells treated with the DMSO control. Intact CBs were still visible when HeLa cells were treated for 4 h with 10 μm DDD00107587. However, after 4 h of treatment with either 20 or 30 μm DDD00107587, the bright CB foci were no longer observed, and coilin instead forms numerous microdots throughout the nucleoplasm (Fig. 9A). This change in coilin localization was consistently observed in essentially all cells, whereas a subset of these cells also showed SMN forming a multitude of tiny dots in the nucleoplasm (Fig. 9D). Other cells treated for 4 h with either 20 or 30 μm DDD00107587 displayed instead large SMN aggregates in the cytoplasm (Fig. 9D). The coilin microfoci mostly did not colocalize with SMN and never colocalized in cells showing SMN aggregates in the cytoplasm (Fig. 10A). In contrast with coilin and SMN, Y12 and TMG are not only located in CBs but also in nuclear speckles. After treatment for 4 h with either 20 or 30 μm DDD00107587, both the Y12 and TMG antibodies no longer stained CBs, although both still labeled a similar pattern of speckles compared with the DMSO control. Furthermore, the morphology of the splicing speckles showed little or no change (Fig. 9, B and C). In the case of Y12, some cells also showed cytoplasmic aggregates in the presence of 30 μm DDD00107587 for 4 h. These Y12 aggregates colocalize with the cytoplasmic SMN aggregates (Fig. 10B, arrows).
FIGURE 8.
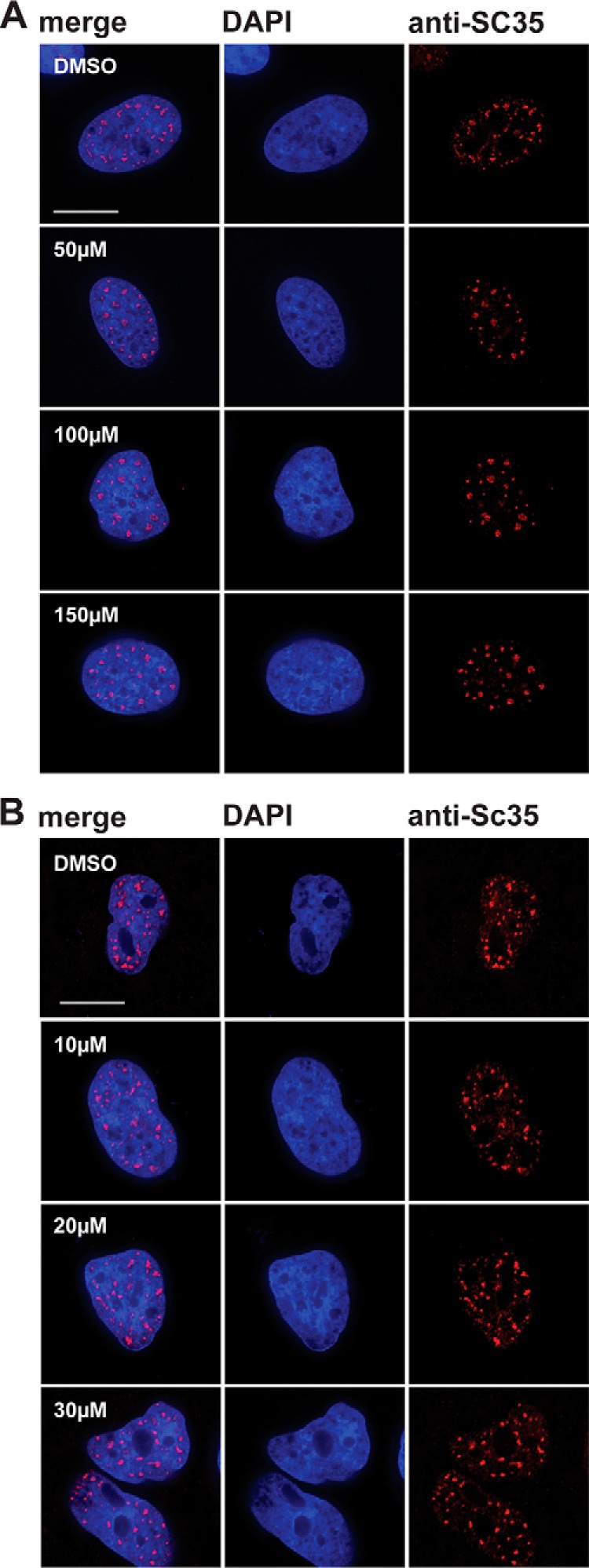
Splicing speckles morphology is not changed by treatment with DDD00107587. SC35 immunofluorescence of HeLa cells treated with DMSO or 10, 20, and 30 μm DDD00107587 (B) and 50, 100, and 150 μm DRB (A) for 4 h. Scale bars, 15 μm.
FIGURE 9.

Cajal body disruption after treatment with DDD00107587. HeLa cells treated with DMSO or 10, 20, and 30 μm for 4 h were stained with anti-coilin (A), anti-Y12 (B), anti-TMG (C), and anti-SMN (D) antibodies, respectively. Arrowheads denote intact CBs. Scale bars, 15 μm.
FIGURE 10.
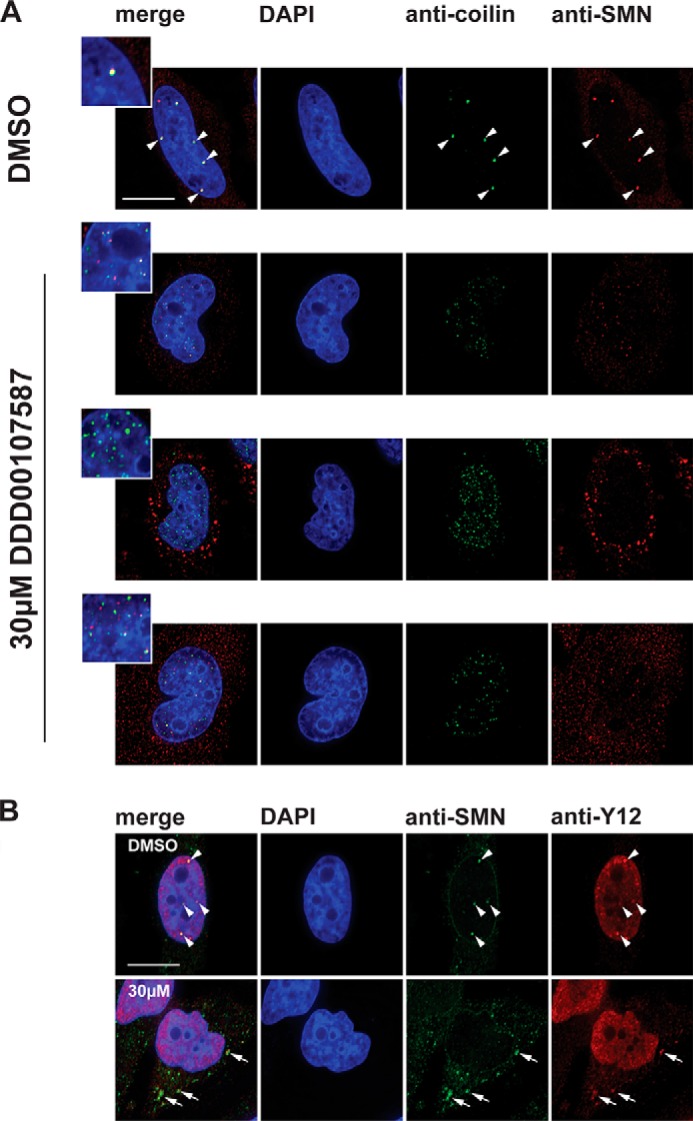
Subcellular localization of SMN, coilin, and Y12 after treatment with DDD00107587. HeLa cells were treated for 4 h with DMSO or 30 μm DDD00107587. The localization of SMN and coilin (A) as well as SMN and Y12 (B) was visualized by immunostaining. Arrows mark the colocalization of cytoplasmic SMN and Y12 aggregates in the presence of DDD00107587. Arrowheads denote intact CBs. Scale bars, 15 μm.
As it is known that RNA polymerase II activity is required for CB integrity, we next compared the effect of DDD00107587 on the redistribution of CB components with that induced by treating cells with transcription inhibitors. HeLa cells were treated with either 50, 100, or 150 μm DRB for 4 h to block RNA polymerase II (Fig. 11). This showed that the CB disruption in response to DRB treatment is different from that induced by DDD00107587. Thus, the staining with both the Y12 and anti-TMG antibodies show that in the presence of either 100 or 150 μm DRB for 4 h, the nuclear speckles become more rounded and increase in size (Fig. 12, C and D). Coilin and SMN were still visible in foci after 4 h of incubation with 50 μm DRB, but they no longer colocalized with snRNP antigens Y12 and TMG. After the total disruption of CBs caused by 4 h of treatment with either 100 or 150 μm DRB, coilin relocated into nucleolar caps (Fig. 12A), as observed previously (29), whereas SMN could mainly be detected in a diffuse cytoplasmic pool (Fig. 12B).
FIGURE 11.
DRB inhibits transcription of RNA polymerase II. HeLa cells were incubated for 4 h with DRB before the cells were incubated with EU that was incorporated into newly synthesized RNA for 20 min. Cells were then fixed, and labeled RNA was detected with the help of a fluorescent microscope. Scale bar, 40 μm.
FIGURE 12.

Cajal body disruption after treatment with DRB. HeLa cells were incubated with DMSO or 50, 100, and 150 μm DRB for 4 h and stained with anti-coilin (A), anti-SMN (B), anti-Y12 (C), and anti-TMG (D) antibodies respectively. Arrowheads denote intact CBs. Scale bar, 15 μm.
DISCUSSION
Using a recently described ELISA-based high throughput screening in vitro splicing assay (17), we screened a diverse small molecule library of 71,504 compounds. This primary screen was followed with independent, secondary screens using a combination of different in vitro splicing assay formats. We have thereby identified new small molecule splicing inhibitors, which are active in vitro and in cells. These small molecule splicing inhibitors are structurally distinct from previously described natural compound splicing inhibitors. One of these new splicing inhibitors, DDD00107587 (2-((7methoxy-4-methylquinazolin-2-yl)amino)-5,6-dimethylpyrimidin-4(3H)-one, was characterized in more detail with respect to its effect on spliceosome assembly and nuclear structure. For practical reasons we have renamed DDD00107587 as madrasin (2-((7methoxy-4-methylquinazolin-2-yl)amino)-5,6-dimethylpyrimidin-4(3H)-oneRNAsplicing inhibitor).
Several different classes of compounds have been reported to inhibit splicing in vitro and/or to modify alternative splicing of specific pre-mRNA transcripts in cells. These compounds include acetylation/deacetylation inhibitors (23), kinase inhibitors (23, 30), phosphatase inhibitors (31), topoisomerase I inhibitors (32), and cardiotonic steroids (16, 33). However, very few natural compounds and their synthetic derivatives have been reported to inhibit splicing both in vitro and in vivo e.g. FR901464 (8), GEX1A (10), pladienolide B, pladienolide D (9), and isogengketin (12).
Madrasin is not related in structure to these previously described inhibitors. It prevents formation of both splicing intermediates and products in vitro and interferes with one or more early steps in the pathway of spliceosome assembly, allowing the formation of the A complex but blocking the formation of larger spliceosome complexes. Other previously described splicing inhibitors have also been shown to target early events in the spliceosome assembly pathway, resulting from their having the same target, i.e. SF3b. This is a multiprotein subunit of the U2 snRNP that is present in the A complex. For example, SSA and pladienolide (27) both stall spliceosome assembly after A complex formation. However, despite having the same target SF3b, meayamycin has been reported to stop the splicing process after E complex formation (34). To date, the molecular target has only been identified for the splicing modulators isolated from the broth of bacteria, e.g. FR901464, pladienolide, and GX1A and their derivatives, which all bind the SF3b subcomplex of U2 snRNP.
Splicing inhibition has long been associated with cell cycle arrest. Not only has the down-regulation of splicing factors such as SFRS1, SFRS2, and U2AF35 (35–37) by siRNA depletion been shown to arrest cells in G2 and M, but treatment with splicing inhibitors such as SSA (HeLa cells), GEX1A (WI-38 cells), and E7107, as well as pladienolide (WiDr cells), all arrest cells in G1, G2, and M phase (8, 38–40). The cell cycle arrest caused by the inhibitors shown to target SF3b has been mainly attributed to the inhibition of splicing of the p27 pre-mRNA. p27 is a cyclin-dependent kinase inhibitor important for the regulation of cell cycle progression. Altered p27 splicing results in the expression of a C-terminally truncated version of the p27 protein (8). Recently, the effect of SSA on splicing was studied in more detail using a splicing-sensitive microarray, which showed that in addition to inhibition of p27 splicing, the splicing of other genes important for cell cycle progression, including CCNA2 and AURKA, was also affected (28).
We demonstrate here that madrasin shows a dose- and time-dependent inhibitory effect on cell cycle progression in HeLa cells. Low doses (10 μm) of madrasin show a clear accumulation of cells in S, G2, and M phases after 8 and 24 h of treatment. Higher doses of madrasin also result in the accumulation of cells arrested in S, G2, and M. However, the cell cycle arrest at 20 and 30 μm madrasin could only be detected after 24 h, which suggests a block in the transition of all cell cycle stages. Our data indicate that the cell cycle arrest induced by 10 μm madrasin might be different from that observed for the two higher concentrations. Like the SF3b targeting compounds, madrasin also modulates the alternative splicing of MCL1, CCNA2, AURKA, and p27. However, exon skipping of these genes was almost exclusively detected at 20 and 30 μm and therefore is unlikely to account for the cell cycle arrest induced by 10 μm madrasin. The cell cycle arrest seen with the higher concentrations coincides with a reduction in DNA synthesis levels as shown by the reduced incorporation of EdU in cells treated with 20 and 30 μm madrasin for 8 h. In contrast, a reduction in DNA synthesis, as judged by reduced EdU incorporation, was only detected after 24 h of treatment, whereas cell cycle arrest was observed after 8 h in the presence of 10 μm madrasin. Even though inhibition of DNA synthesis caused either by DNA damage or by the depletion of deoxyribonucleotide precursors is a well known cause of cell cycle arrest, it is likely that the cell cycle arrest caused by madrasin results from altered splicing of one or more transcripts that encode proteins needed for normal cell cycle progression. Therefore, the observed inhibition of DNA synthesis is more likely an indirect effect.
It has been previously reported that either treatment of cells with splicing inhibitors, e.g. SSA, or the down-regulation of gene expression caused by transcription inhibitors, e.g. DRB, changes the morphology of splicing speckles, which become round and enlarged (41). Splicing speckles are granules located in interchromatin regions, which are enriched in splicing factors (41). Treatment with madrasin caused little or no reduction in RNA polymerase II transcription, as judged by nucleoplasmic EU incorporation in HeLa cells. In agreement with these findings, madrasin treatment also did not result in morphological changes of nuclear speckles. However, treatment with madrasin did result in the disruption of Cajal bodies in HeLa cells.
In contrast to polymerase II transcription, a noticeable reduction in newly synthesized rRNA was observed after 24 h of incubation with madrasin. A possibility for this decrease in rRNA synthesis might be indirect effects caused by splicing inhibition. Besides the down-regulation of factors important for rRNA transcription, splicing inhibition might also cause a decrease in the production of small nucleolar RNAs. Small nucleolar RNAs are essential for the maturation of rRNA in the nucleolus and most small nucleolar RNAs in mammalian cells are located in introns and processed from spliced introns (42).
CBs play an important role not only in the maturation of small nucleolar RNAs, small Cajal body-specific RNAs, and telomerase RNAs but also in the maturation of the spliceosomal snRNPs (43). It is known that the integrity of CBs depends on ongoing RNA synthesis, and inhibition of transcription leads to the disruption of CBs (41). As RNA polymerase II transcription is not prevented by madrasin treatment, another mechanism must be responsible for causing the loss of CBs. Redistribution of coilin throughout the nucleus independently of transcription inhibition has been described after up-regulation of PA28γ (29). Another difference between the disruption of CBs caused by transcription inhibition and by madrasin treatment is the effect on coilin localization. Coilin relocates to nucleolar caps after transcription inhibition, but not, as shown here, after madrasin treatment, when coilin relocates to form numerous microfoci throughout the nucleoplasm.
During the cell cycle CBs disassemble in M phase and reappear as multiple small foci in early G1 phase. It is unlikely that the changes seen in the localization of coilin after madrasin treatment are indirectly due to changes in the cell cycle, however, as no cell cycle arrest was observed after 4 h treatment with madrasin by which time CB disruption and changes in coilin localization were present in all cells. In addition, the nuclear localization of other CB components analyzed also behave differently after treatment of cells with madrasin, as compared with transcription inhibition. For example, after transcription inhibition, the snRNPs detected with the Y12 antibody are located in enlarged, rounded splicing speckles, whereas Y12 instead labels cytoplasmic aggregates, as well as morphologically normal splicing speckles, after a 4 h treatment of HeLa cells with 30 μm madrasin (Fig. 9). To date, none of the previously described molecules that alter splicing have been shown to cause a similar disruption of CBs. Therefore, madrasin provides a unique chemical tool with which to study the relationship between splicing and CBs in greater detail.
Interestingly, madrasin does not inhibit splicing of all transcripts we tested in vivo. For example, pre-mRNA splicing of the Hsp40 pre-mRNA was relatively unaffected by the same concentration and time of treatment with madrasin that strongly altered the splicing of RIOK3. It was previously assumed that general splicing inhibitors, like SSA, which target the U2 snRNP splicing factor SF3b 155, would likely cause inhibition of all pre-mRNA splicing, because U2 snRNP is thought to be needed for splicing of all GT-AG introns. However Corrionero et al. (28) showed recently using splicing arrays to examine global splicing events that, instead of affecting all transcripts, SSA alters splicing of only a subset of genes, 3 h after treatment. To analyze the full extent of the splicing changes induced by madrasin and compare this with the effects of other splicing inhibitors, it will be important in the future to assess also changes in global transcription levels and alternative splicing patterns.
Our results highlight the potential of madrasin as a tool that can be used to dissect the splicing mechanism in vitro and in cells. However, as madrasin is not a natural product, rather it was identified from a library of drug-like small molecules, its further development may also prove useful in understanding the therapeutic potential of splicing inhibition. Increasingly, pre-mRNA splicing is becoming an attractive target for drug discovery (44–47). Splicing inhibitors, including meamyacin (34), isoginkgetin (48), and pladienolides (49), have been shown to have anticancer properties. In the case of pladienolides, it has been demonstrated that the targeting of SF3b is the direct mechanism of their antitumor activity (40). Most recently, while this manuscript was under review, Naryshkin et al. (50) have reported the identification of small molecules that alter the splicing of transcripts from the SMN2 gene, which is linked with genetic disease causing neuromuscular degeneration and mortality. Consistent with the known effects of other splicing inhibitors, madrasin exhibits cytostatic activity. In addition, madrasin also alters the splicing of at least two viral oncogenes, i.e. E1A (adenovirus) and the HPV18 E6 (human papillomavirus type 18), which may be interesting with regard to the effects of madrasin-related compounds on viral reproduction and their potential to transform cells.
In summary, we have identified a new class of small molecule splicing modulators that inhibit splicing of multiple introns both in vitro and in cells. Further characterization of this compound, and its related derivatives, may lead to the development of more potent and transcript-selective compounds with uses in studying the splicing mechanism and potential applications as antiviral and/or anti-cancer agents.
Supplementary Material
Acknowledgments
We thank Dr. Tony Ly for discussion and advice on flow cytometry analysis. We thank Rosie Clarke (Centre for Advanced Scientific Technologies, University of Dundee) for advice on flow cytometry analysis.
This work was supported by EUFP6 EURASNET Grant LSHG-CT-2005-518238 and by Wellcome Trust Programme Grant 073980/Z/03/Z, GRE Strategic Award 097945/B/11/Z, and Biological Chemistry and Drug Discovery Strategic Award 100476/Z/12/Z.

This article contains supplemental Tables S1 and S2.
- SSA
- spliceostatin A
- snRNP
- small nuclear ribonucleoprotein
- CB
- Cajal Body
- EU
- 5-ethynyl uridine
- EdU
- 5-ethynyl-2′-deoxyuridine
- DRB
- 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole
- MBP
- maltose-binding protein
- RLU
- relative luminescence unit
- SMN
- survival of motor neuron 1, telomeric.
REFERENCES
- 1. Pan Q., Shai O., Lee L. J., Frey B. J., Blencowe B. J. (2008) Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 40, 1413–1415 [DOI] [PubMed] [Google Scholar]
- 2. Wahl M. C., Will C. L., Lührmann R. (2009) The spliceosome: design principles of a dynamic RNP machine. Cell 136, 701–718 [DOI] [PubMed] [Google Scholar]
- 3. Krawczak M., Thomas N. S., Hundrieser B., Mort M., Wittig M., Hampe J., Cooper D. N. (2007) Single base-pair substitutions in exon-intron junctions of human genes: nature, distribution, and consequences for mRNA splicing. Hum. Mutat. 28, 150–158 [DOI] [PubMed] [Google Scholar]
- 4. Lim K. H., Ferraris L., Filloux M. E., Raphael B. J., Fairbrother W. G. (2011) Using positional distribution to identify splicing elements and predict pre-mRNA processing defects in human genes. Proc. Natl. Acad. Sci. U.S.A. 108, 11093–11098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ahn E. Y., Yan M., Malakhova O. A., Lo M. C., Boyapati A., Ommen H. B., Hines R., Hokland P., Zhang D. E. (2008) Disruption of the NHR4 domain structure in AML1-ETO abrogates SON binding and promotes leukemogenesis. Proc. Natl. Acad. Sci. U.S.A. 105, 17103–17108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. David C. J., Manley J. L. (2010) Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev. 24, 2343–2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jurica M. S. (2008) Searching for a wrench to throw into the splicing machine. Nat. Chem. Biol. 4, 3–6 [DOI] [PubMed] [Google Scholar]
- 8. Kaida D., Motoyoshi H., Tashiro E., Nojima T., Hagiwara M., Ishigami K., Watanabe H., Kitahara T., Yoshida T., Nakajima H., Tani T., Horinouchi S., Yoshida M. (2007) Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat. Chem. Biol. 3, 576–583 [DOI] [PubMed] [Google Scholar]
- 9. Kotake Y., Sagane K., Owa T., Mimori-Kiyosue Y., Shimizu H., Uesugi M., Ishihama Y., Iwata M., Mizui Y. (2007) Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 3, 570–575 [DOI] [PubMed] [Google Scholar]
- 10. Hasegawa M., Miura T., Kuzuya K., Inoue A., Won Ki S., Horinouchi S., Yoshida T., Kunoh T., Koseki K., Mino K., Sasaki R., Yoshida M., Mizukami T. (2011) Identification of SAP155 as the target of GEX1A (Herboxidiene), an antitumor natural product. ACS Chem. Biol. 6, 229–233 [DOI] [PubMed] [Google Scholar]
- 11. Fan L., Lagisetti C., Edwards C. C., Webb T. R., Potter P. M. (2011) Sudemycins, novel small molecule analogues of FR901464, induce alternative gene splicing. ACS Chem. Biol. 6, 582–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. O'Brien K., Matlin A. J., Lowell A. M., Moore M. J. (2008) The biflavonoid isoginkgetin is a general inhibitor of pre-mRNA splicing. J. Biol. Chem. 283, 33147–33154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dehm S. M. (2013) Test-firing ammunition for spliceosome inhibition in cancer. Clin. Cancer Res. 19, 6064–6066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Younis I., Berg M., Kaida D., Dittmar K., Wang C., Dreyfuss G. (2010) Rapid-response splicing reporter screens identify differential regulators of constitutive and alternative splicing. Mol. Cell. Biol. 30, 1718–1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Effenberger K. A., Perriman R. J., Bray W. M., Lokey R. S., Ares M., Jr., Jurica M. S. (2013) A high-throughput splicing assay identifies new classes of inhibitors of human and yeast spliceosomes. J. Biomol. Screen. 18, 1110–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stoilov P., Lin C. H., Damoiseaux R., Nikolic J., Black D. L. (2008) A high-throughput screening strategy identifies cardiotonic steroids as alternative splicing modulators. Proc. Natl. Acad. Sci. U.S.A. 105, 11218–11223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Samatov T. R., Wolf A., Odenwälder P., Bessonov S., Deraeve C., Bon R. S., Waldmann H., Lührmann R. (2012) Psoromic acid derivatives: a new family of small-molecule pre-mRNA splicing inhibitors discovered by a stage-specific high-throughput in vitro splicing assay. Chembiochem 13, 640–644 [DOI] [PubMed] [Google Scholar]
- 18. Berg M. G., Wan L., Younis I., Diem M. D., Soo M., Wang C., Dreyfuss G. (2012) A quantitative high-throughput in vitro splicing assay identifies inhibitors of spliceosome catalysis. Mol. Cell. Biol. 32, 1271–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brenk R., Schipani A., James D., Krasowski A., Gilbert I. H., Frearson J., Wyatt P. G. (2008) Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem 3, 435–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Butz K., Ristriani T., Hengstermann A., Denk C., Scheffner M., Hoppe-Seyler F. (2003) siRNA targeting of the viral E6 oncogene efficiently kills human papillomavirus-positive cancer cells. Oncogene 22, 5938–5945 [DOI] [PubMed] [Google Scholar]
- 21. Gendra E., Colgan D. F., Meany B., Konarska M. M. (2007) A sequence motif in the simian virus 40 (SV40) early core promoter affects alternative splicing of transcribed mRNA. J. Biol. Chem. 282, 11648–11657 [DOI] [PubMed] [Google Scholar]
- 22. Gencheva M., Lin T. Y., Wu X., Yang L., Richard C., Jones M., Lin S. B., Lin R. J. (2010) Nuclear retention of unspliced pre-mRNAs by mutant DHX16/hPRP2, a spliceosomal DEAH-box protein. J. Biol. Chem. 285, 35624–35632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kuhn A. N., van Santen M. A., Schwienhorst A., Urlaub H., Lührmann R. (2009) Stalling of spliceosome assembly at distinct stages by small-molecule inhibitors of protein acetylation and deacetylation. RNA 15, 153–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bessonov S., Anokhina M., Will C. L., Urlaub H., Lührmann R. (2008) Isolation of an active step I spliceosome and composition of its RNP core. Nature 452, 846–850 [DOI] [PubMed] [Google Scholar]
- 25. Konarska M. M., Sharp P. A. (1987) Interactions between small nuclear ribonucleoprotein particles in formation of spliceosomes. Cell 49, 763–774 [DOI] [PubMed] [Google Scholar]
- 26. Zillmann M., Zapp M. L., Berget S. M. (1988) Gel electrophoretic isolation of splicing complexes containing U1 small nuclear ribonucleoprotein particles. Mol. Cell. Biol. 8, 814–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roybal G. A., Jurica M. S. (2010) Spliceostatin A inhibits spliceosome assembly subsequent to prespliceosome formation. Nucleic Acids Res. 38, 6664–6672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Corrionero A., Miñana B., Valcárcel J. (2011) Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev. 25, 445–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cioce M., Boulon S., Matera A. G., Lamond A. I. (2006) UV-induced fragmentation of Cajal bodies. J. Cell Biol. 175, 401–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Muraki M., Ohkawara B., Hosoya T., Onogi H., Koizumi J., Koizumi T., Sumi K., Yomoda J., Murray M. V., Kimura H., Furuichi K., Shibuya H., Krainer A. R., Suzuki M., Hagiwara M. (2004) Manipulation of alternative splicing by a newly developed inhibitor of Clks. J. Biol. Chem. 279, 24246–24254 [DOI] [PubMed] [Google Scholar]
- 31. Mermoud J. E., Cohen P., Lamond A. I. (1992) Ser/Thr-specific protein phosphatases are required for both catalytic steps of pre-mRNA splicing. Nucleic Acids Res. 20, 5263–5269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pilch B., Allemand E., Facompré M., Bailly C., Riou J. F., Soret J., Tazi J. (2001) Specific inhibition of serine- and arginine-rich splicing factors phosphorylation, spliceosome assembly, and splicing by the antitumor drug NB-506. Cancer Res. 61, 6876–6884 [PubMed] [Google Scholar]
- 33. Anderson E. S., Lin C. H., Xiao X., Stoilov P., Burge C. B., Black D. L. (2012) The cardiotonic steroid digitoxin regulates alternative splicing through depletion of the splicing factors SRSF3 and TRA2B. RNA 18, 1041–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Albert B. J., McPherson P. A., O'Brien K., Czaicki N. L., Destefino V., Osman S., Li M., Day B. W., Grabowski P. J., Moore M. J., Vogt A., Koide K. (2009) Meayamycin inhibits pre-messenger RNA splicing and exhibits picomolar activity against multidrug-resistant cells. Mol. Cancer Ther. 8, 2308–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li X., Wang J., Manley J. L. (2005) Loss of splicing factor ASF/SF2 induces G2 cell cycle arrest and apoptosis, but inhibits internucleosomal DNA fragmentation. Genes Dev. 19, 2705–2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xiao R., Sun Y., Ding J. H., Lin S., Rose D. W., Rosenfeld M. G., Fu X. D., Li X. (2007) Splicing regulator SC35 is essential for genomic stability and cell proliferation during mammalian organogenesis. Mol. Cell. Biol. 27, 5393–5402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pacheco T. R., Moita L. F., Gomes A. Q., Hacohen N., Carmo-Fonseca M. (2006) RNA interference knockdown of hU2AF35 impairs cell cycle progression and modulates alternative splicing of Cdc25 transcripts. Mol. Biol. Cell 17, 4187–4199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mizui Y., Sakai T., Iwata M., Uenaka T., Okamoto K., Shimizu H., Yamori T., Yoshimatsu K., Asada M. (2004) Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. III. In vitro and in vivo antitumor activities. J. Antibiot. 57, 188–196 [DOI] [PubMed] [Google Scholar]
- 39. Ghosh A. K., Li J. (2011) A stereoselective synthesis of (+)-herboxidiene/GEX1A. Org. Lett. 13, 66–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yokoi A., Kotake Y., Takahashi K., Kadowaki T., Matsumoto Y., Minoshima Y., Sugi N. H., Sagane K., Hamaguchi M., Iwata M., Mizui Y. (2011) Biological validation that SF3b is a target of the antitumor macrolide pladienolide. FEBS J. 278, 4870–4880 [DOI] [PubMed] [Google Scholar]
- 41. Spector D. L., Lamond A. I. (2011) Nuclear speckles. Cold Spring Harb. Perspect. Biol. 3, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hirose T., Shu M. D., Steitz J. A. (2003) Splicing-dependent and -independent modes of assembly for intron-encoded box C/D snoRNPs in mammalian cells. Mol. Cell 12, 113–123 [DOI] [PubMed] [Google Scholar]
- 43. Machyna M., Heyn P., Neugebauer K. M. (2013) Cajal bodies: where form meets function. Wiley Interdisciplinary Reviews. RNA 4, 17–34 [DOI] [PubMed] [Google Scholar]
- 44. Bonnal S., Vigevani L., Valcárcel J. (2012) The spliceosome as a target of novel antitumour drugs. Nat. Rev. Drug Discov. 11, 847–859 [DOI] [PubMed] [Google Scholar]
- 45. Hernandez-Lopez H. R., Graham S. V. (2012) Alternative splicing in human tumour viruses: a therapeutic target? Biochem. J. 445, 145–156 [DOI] [PubMed] [Google Scholar]
- 46. Hagiwara M. (2005) Alternative splicing: a new drug target of the post-genome era. Biochim. Biophys. Acta 1754, 324–331 [DOI] [PubMed] [Google Scholar]
- 47. Tazi J., Durand S., Jeanteur P. (2005) The spliceosome: a novel multi-faceted target for therapy. Trends Biochem. Sci. 30, 469–478 [DOI] [PubMed] [Google Scholar]
- 48. Yoon S. O., Shin S., Lee H. J., Chun H. K., Chung A. S. (2006) Isoginkgetin inhibits tumor cell invasion by regulating phosphatidylinositol 3-kinase/Akt-dependent matrix metalloproteinase-9 expression. Mol. Cancer Ther. 5, 2666–2675 [DOI] [PubMed] [Google Scholar]
- 49. Sakai T., Asai N., Okuda A., Kawamura N., Mizui Y. (2004) Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. II. Physico-chemical properties and structure elucidation. J. Antibiot. 57, 180–187 [DOI] [PubMed] [Google Scholar]
- 50. Naryshkin N. A., Weetall M., Dakka A., Narasimhan J., Zhao X., Feng Z., Ling K. K., Karp G. M., Qi H., Woll M. G., Chen G., Zhang N., Gabbeta V., Vazirani P., Bhattacharyya A., Furia B., Risher N., Sheedy J., Kong R., Ma J., Turpoff A., Lee C. S., Zhang X., Moon Y. C., Trifillis P., Welch E. M., Colacino J. M., Babiak J., Almstead N. G., Peltz S. W., Eng L. A., Chen K. S., Mull J. L., Lynes M. S., Rubin L. L., Fontoura P., Santarelli L., Haehnke D., McCarthy K. D., Schmucki R., Ebeling M., Sivaramakrishnan M., Ko C. P., Paushkin S. V., Ratni H., Gerlach I., Ghosh A., Metzger F. (2014) Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 345, 688–693 [DOI] [PubMed] [Google Scholar]
- 51. Watson J. V., Chambers S. H., Smith P. J. (1987) A pragmatic approach to the analysis of DNA histograms with a definable G1 peak. Cytometry 8, 1–8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.