

Background: Dysregulation of neuronal acetylation homeostasis promotes neurodegeneration.
Results: Histone hyperacetylation up-regulates PKCδ in dopaminergic neurons and augments susceptibility to oxidative damage.
Conclusion: Epigenetic regulation of PKCδ plays a proapoptotic role in neuronal cell death.
Significance: The up-regulation of PKCδ expression by hyperacetylation provides an epigenetic molecular basis of neurodegenerative disease.
Keywords: Epigenetics, Histone Deacetylase Inhibitor (HDAC Inhibitor), Neurodegeneration, Oxidative Stress, Parkinson Disease, PKCδ, Histone Acetylation
Abstract
The oxidative stress-sensitive protein kinase Cδ (PKCδ) has been implicated in dopaminergic neuronal cell death. However, little is known about the epigenetic mechanisms regulating PKCδ expression in neurons. Here, we report a novel mechanism by which the PKCδ gene can be regulated by histone acetylation. Treatment with histone deacetylase (HDAC) inhibitor sodium butyrate (NaBu) induced PKCδ expression in cultured neurons, brain slices, and animal models. Several other HDAC inhibitors also mimicked NaBu. The chromatin immunoprecipitation analysis revealed that hyperacetylation of histone H4 by NaBu is associated with the PKCδ promoter. Deletion analysis of the PKCδ promoter mapped the NaBu-responsive element to an 81-bp minimal promoter region. Detailed mutagenesis studies within this region revealed that four GC boxes conferred hyperacetylation-induced PKCδ promoter activation. Cotransfection experiments and Sp inhibitor studies demonstrated that Sp1, Sp3, and Sp4 regulated NaBu-induced PKCδ up-regulation. However, NaBu did not alter the DNA binding activities of Sp proteins or their expression. Interestingly, a one-hybrid analysis revealed that NaBu enhanced transcriptional activity of Sp1/Sp3. Overexpression of the p300/cAMP-response element-binding protein-binding protein (CBP) potentiated the NaBu-mediated transactivation potential of Sp1/Sp3, but expressing several HDACs attenuated this effect, suggesting that p300/CBP and HDACs act as coactivators or corepressors in histone acetylation-induced PKCδ up-regulation. Finally, using genetic and pharmacological approaches, we showed that NaBu up-regulation of PKCδ sensitizes neurons to cell death in a human dopaminergic cell model and brain slice cultures. Together, these results indicate that histone acetylation regulates PKCδ expression to augment nigrostriatal dopaminergic cell death, which could contribute to the progressive neuropathogenesis of Parkinson disease.
Introduction
Protein kinase C (PKC) isozymes, a family of at least 12 serine/threonine kinases, are key regulators of a broad spectrum of cellular functions, including proliferation, differentiation, cell cycle progression, gene transcription and translation, altered cell shape and migration, and cell death (1). According to their structures and sensitivities to calcium and diacylglycerol (DAG),2 these kinases have been categorized into three subfamilies, namely conventional PKCs (α, βI, βII, and γ), novel PKCs (δ, ϵ, η, and θ), and atypical PKCs (ζ and ι/λ). Conventional PKCs require calcium and DAG for complete activation; novel PKCs are fully activated by DAG only, and atypical PKCs are calcium- and DAG-insensitive and regulated in a different manner. Among the various PKC isoforms, PKCδ is known to play a critical role as a mediator of apoptotic responses in various cell types (2). The role of PKCδ signaling in the neurodegenerative processes of Parkinson disease (PD) is now better understood. We previously showed that this kinase is preferentially expressed in dopaminergic neurons in the substantia nigra (3). We and others also have established that caspase-3-dependent proteolytic cleavage and the ensuing activation of PKCδ mediate the dopaminergic cell death triggered by a variety of dopaminergic neurotoxic insults in neuronal culture models as well as in animal models (4–12). PKCδ inhibition by genetic manipulations (e.g. siRNA knockdown and overexpression of dominant-negative, caspase-3 cleavage-resistant, or catalytically dead form of PKCδ) or by using a peptide inhibitor directed against PKCδ proteolytic cleavage effectively attenuated neurotoxin-induced dopaminergic apoptosis (13–15). Furthermore, primary dopaminergic neurons isolated from PKCδ-deficient mice were resistant to TNFα toxicity (9). Consistent with these studies, we demonstrated in an in vivo study that administering rottlerin, a PKCδ-specific inhibitor, protected against MPTP-induced loss of tyrosine hydroxylase-positive neurons in the nigrostriatum and the depletion of striatal dopamine and its metabolites, as well as MPTP-induced locomotor deficits (16). In a methamphetamine-intoxicated animal model, PKCδ deficiency and rottlerin effectively attenuated methamphetamine-induced dopaminergic damage and behavioral deficits, further supporting that PKCδ could represent a valid pharmacological target for the treatment of dopaminergic neuronal degeneration (17, 18). Interestingly, we also showed that PKCδ negatively modulates dopamine synthesis by inhibiting the rate-limiting enzyme, tyrosine hydroxylase (3). In addition to PD, deregulation of PKCδ activity has been linked to various diseases, including cancer, stroke, diabetic complications, autoimmune diseases, atherosclerosis, and myocardial infarction (19–25). Although regulation of PKCδ activity is generally through post-translational modifications (e.g. phosphorylation and proteolysis), there are also reports of changes in the expression of PKCδ in several pathophysiological conditions (26–33). Thus, it is of both physiological and pathological interest to study the molecular basis of PKCδ induction and expression.
PKCδ is ubiquitously expressed in most tissues as well as in many cell types. The PKCδ gene, which is located on human chromosome 3, includes 18 exons and 17 introns and spans ∼32 kb. A gap of larger than 17 kb between the transcription start and translation start sites of the human PKCδ gene, coupled with an unusually long 5′-untranslated region, underscores the complicated structure of this gene (34). The 5′-flanking region of the PKCδ gene lacks a typical TATA box but has a GC-rich region in the proximal region. Recently, we characterized the regulatory mechanisms underlying basal PKCδ expression in neurons (35). Serial deletion analysis of the ∼2-kb mouse PKCδ promoter revealed the presence of multiple positive and negative regulatory elements contributing to PKCδ transcriptional activity. Notably, basal transcription of the PKCδ gene is directed from a core promoter located between nucleotides −147 and +289, relative to transcription initiation. We also revealed that four GC boxes residing in this core promoter specifically bind the Sp transcription factors (e.g. Sp1, Sp3, and Sp4) and play a pivotal role in mediating constitutive PKCδ gene expression in neuronal cells. Additionally, it has been reported that PKCδ can be regulated by the transcription factors NFκB, c-Jun/ATF2, and p53 family proteins (34, 36–39). To the best of our knowledge, the involvement of epigenetic mechanisms in controlling PKCδ expression has thus far not been explored.
Post-translational modification of histone proteins, such as acetylation, methylation, and phosphorylation, has been recognized as an integral mechanism in the epigenetic regulation of gene transcription and other important cellular functions. Acetylation of histone tails, catalyzed by histone acetyltransferases (HATs), promotes a more relaxed chromatin structure, which facilitates the recruitment of transcription factors and increases gene transcription. Conversely, turnover of acetylated histones by histone deacetylase (HDACs) leads to chromatin condensation and correlates with transcriptional repression. An optimal balance between HAT and HDAC activity is required for cell survival, and disruption of this balance has been implicated in neurodegenerative diseases (40). We previously reported that neurotoxic pesticide exposure induced hyperacetylation of histones H3 and H4, which contributed to cell death in cell culture models of PD (41–43). Similarly, overexpression of a HAT, Tip60, was found to induce neuronal apoptosis in Drosophila (44). In line with these findings, experiments in a variety of cellular and rodent models of neurodegeneration have described a beneficial role for many individual HDACs (45–49). Paradoxically, other groups have reported that either loss of HAT activity or increased HDAC activity is associated with several neurodegenerative conditions (50–52). Hence, the role that specific HATs or HDACs play in neurodegenerative diseases remains equivocal. A similar debate exists about the functional response of pharmacological manipulation with HDAC inhibitors, whereas the effects of the HDAC inhibitor appear to be mainly neuroprotective. A growing body of evidence has also suggested that increased acetylation levels by the HDAC inhibitor can be detrimental for neurons (53–56).
This study was designed to determine whether the proapoptotic PKCδ gene is regulated by altered histone acetylation homeostasis and, if so, to decipher the molecular mechanisms responsible for this epigenetic regulation. Our results indicate that HDAC inhibition markedly induced the PKCδ gene expression in the striatum and substantia nigra of animals, in mouse corticostriatal slices, in primary nigral and striatal neuron cultures, and in human dopaminergic LUHMES and mouse NIE115 and MN9D cells. In vitro experiments reveal that butyrate induced hyperacetylation of histone H4 in association with the PKCδ promoter. The minimal region of the PKCδ promoter mediating butyrate induction was mapped to an 81-bp GC-rich region, and four functioning GC boxes within this region regulated the butyrate effect. Furthermore, we present evidence indicating that butyrate increased the transactivating capacity of Sp proteins to activate the PKCδ promoter without changes in their DNA binding activities or protein expression levels. Most notably, we found that increased levels of PKCδ by HDAC inhibition increased the sensitivity to oxidative stress in human dopaminergic neuronal cells and mouse slice cultures. These data represent a novel molecular basis of the proapoptotic PKCδ gene up-regulation in neurodegenerative processes through acetylation-mediated epigenetic dysregulation during neurotoxic stress.
EXPERIMENTAL PROCEDURES
Reagents
6-Hydroxydopamine (6-OHDA), 1-methyl-4-phenylpyridinium (MPP+), propidium iodide (PI), MISSIONTM lentiviral packaging mix, mithramycin A, NaBu, trichostatin A (TSA), dibutyryl cAMP, tetracycline, poly-l-ornithine, fibronectin, mouse β-actin antibody, and tolfenamic acid were purchased from Sigma. Valproic acid (VPA), scriptaid, and apicidin were obtained from Alexis Biochemicals (Plymouth Meeting, PA). The Bradford protein assay kit was purchased from Bio-Rad. Lipofectamine 2000 reagent, Alexa 680-conjugated anti-mouse secondary antibody, penicillin, streptomycin, fetal bovine serum, l-glutamine, Hanks' balanced salt solution, Neurobasal medium, B27 supplement, N-2 supplement, DMEM, minimal essential medium, and Advanced DMEM/F-12 were obtained from Invitrogen. Antibodies against PKC isoforms (δ, ϵ, η, α, and ζ), Sp1, Sp3, Sp4, c-Myc, and HA tag were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The pan-acetyl histone H4 antibody was obtained from Active Motif (Carlsbad, CA), and the Fluoro-Jade stain, rabbit polyclonal antibody for acetyl-lysine, mouse p300, and histone H3 antibodies were obtained from Millipore (Billerica, MA). IRDye800-conjugated anti-rabbit secondary antibody was obtained from Rockland Labs (Gilbertsville, PA). Recombinant human glial cell line-derived neurotrophic factor and basic FGF were purchased from R&D Systems (Minneapolis, MN). Cell Titer 96 Aqueous One Solution proliferation assay kit was obtained from Promega (Madison, WI). [3H]Dopamine was obtained from PerkinElmer Life Sciences. The HDAC antibody sampler kit, which was used to evaluate the levels of HDACs, was obtained from Cell Signaling Technology (Danvers, MA).
Plasmids
The mouse PKCδ promoter reporter constructs used in this study have been extensively described (35). To construct Sp1-luc consisting of three consensus Sp1-binding sites from the SV40 promoter and its mutant plasmid mSp1-luc, complementary oligonucleotides (Sp1-luc: sense, 5′-ATATATCTCGAGCGCGTGGGCGGAACTGGGCGGAGTTAGGGGCGGGAAAGCTTATATAT-3′, and antisense, 5′-ATATATAAGCTTTCCCGCCCCTAACTCCGCCCAGTTCCGCCCACGCGCTCGAGATATAT-3′; mSp1-luc: sense, 5′-ATATATCTCGAGCGCGTGTTTTGAACTGTTTTGAGTTAGGTTTTGGAAAGCTTATATAT-3′, and antisense, 5′-ATATATAAGCTTTCCAAAACCTAACTCAAAACAGTTCAAAACACGCGCTCGAGATATAT-3′) were synthesized, annealed, and cloned into the XhoI and HindIII sites of pGL3-Basic. The constructs for mammalian expression pN3-Sp1, pN3-Sp4, and pN3-Sp3, encoding both long and short isoforms of Sp3 (57), as well as the “empty” control vectors pN3, were generously provided by Dr. G. Suske (Philipps-Universität Marburg, Germany). To generate the expression vectors for dominant-negative forms Sp1 (amino acids 603–785) and Sp3 (amino acids 540–781), pN3-DN-Sp1 and pN3-DN-Sp3, the appropriated cDNA fragments were PCR-generated from pN3-Sp1 and pN3-Sp3 with the following primer pairs, respectively: pN3-DN-Sp1, forward, 5′-ATATATCTCGAGACCATGGCATGCACCTGCCCCTACT-3′, and reverse, 5′-ATATATAAGCTTTCAATGGTGATGGTGATGATGGAAGCCATTGCCACTGAT-3′; pN3-DN-Sp3, forward, 5′-ATATATCTCGAGACCATGGAGAATGCTGACAGTCCTG-3′, and reverse, 5′-ATATATAAGCTTTCAATGGTGATGGTGATGATGCTCCATTGTCTCATTTCC-3′. The PCR products were then subcloned into the pN3 vector. The MISSIONTM shRNA plasmid set (SHCLNG-NM_006524) that consists of five PKCδ-targeting shRNA lentiviral plasmids and the scrambled nontarget shRNA control plasmid (pLKO.1-puro, SHC002V) were purchased from Sigma. Target sequences of PKCδ shRNA clones include the following: PKCδ #1, 5′-CCGGCAGAGCCTGTTGGGATATATCCTCGAGGATATATCCCAACAGGCTCTGTTTTTTG-3′; PKCδ #2, 5′-CCGGCAACAGCCGGGACACTATATTCTCGAGAATATAGTGTCCCGGCTGTTGTTTTTG-3′; PKCδ #3, 5′-CCGGCAGAGCCTGTTGGGATATATCCTCGAGGATATATCCCAACAGGCTCTGTTTTTG-3′; PKCδ #4, 5′-CCGGCTTCGGAGGGAAATTGTAAATCTCGAGATTTACAATTTCCCTCCGAAGTTTTTG-3′; and PKCδ #5, 5′-CCGGGGCCGCTTTGAACTCTACCGTCTCGAGACGGTAGAGTTCAAAGCGGCCTTTTT-3′. The p300 wild-type expression plasmid pCl-p300 and its HAT deletion mutant, pCl-p300ΔHAT, were kindly provided by Dr. Joan Boyes (Institute of Cancer Research, London, UK) and generated as described previously (58), and the empty vector pCIneo was a gift from Dr. Christian Seiser (University of Vienna, Austria). The expression plasmid pcDNA3-CBP was a gift from Dr. Xiang-Jiao Yang (59). The expression vectors for HDAC1 (pcDNA3-Myc-His-HDAC1), HDAC4 (pcDNA3-Myc-His-HDAC4), and the empty vector pcDNA3-Myc-His were generously provided by Dr. Tony Kouzarides (60). Dr. Saadi Khochbin kindly provided the expression vectors for HDAC5 (pcDNA3-HA-HDAC5) and HDAC7 (pcDNA3-HA-HDAC7) (61). The Gal4 fusion constructs pM-Sp1 and pM-Sp3, as well as the Gal4-dependent reporter construct pG5-luc containing five Gal4 DNA-binding sites, were gifts from Dr. Toshiyuki Sakai (62), and the empty vector pM was kindly provided by Dr. Bruce Paterson (NCI, National Institutes of Health). To construct the Gal4 DNA-binding domain-fused Sp1- or Sp3-truncated mutants, Gal4-Sp1N(83–785), Gal4-Sp1AB(83–494), Gal4-Sp1ABSer/Thr(83–351), Gal4-Sp1A(83–251), Gal4-Sp1ASer/Thr(83–145), Gal4-Sp1AGln(146–251), Gal4-Sp1B(252–494), Gal4-Sp1BGln(352–494), Gal4-Sp1DBD(603–785), Gal4-Sp3AB(1–499), Gal4-Sp3ABSer/Thr(1–371), Gal4-Sp3A(1–251), Gal4-Sp3AGln(81–251), Gal4-Sp3(1–80), Gal4-Sp3B(252–499), Gal4-Sp3BGln(372–499), and Gal4-Sp3-DBD(540–781), the appropriate cDNA fragments were PCR-generated from pN3-Sp1 and pN3-Sp3 and cloned into the pM vector. All construction sequences were confirmed by DNA sequencing.
Animal Experiments
Six- to 8-week-old C57BL/6 male mice were housed in a temperature-controlled, 12-h light/dark room, and were allowed free access to food and water. NaBu was dissolved in sterile saline and administered to C57BL/6 mice by i.p. injection at a dose of 1.2 g/kg for 6–24 h. An equal volume of saline was given to control animals. Mice were then sacrificed, and the brain areas of interest were immediately and carefully dissected out and stored at −80 °C. PKCδ−/− C57BL/6 mice were originally obtained from Dr. Keiichi Nakayama at the Medical Institute of Bioregulation, Fukuoka, Japan (22, 63). Animal care procedures strictly followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Iowa State University IACUC.
Mouse Striatal and Nigral Neurons in Primary Cultures and Treatments
Plates (6-well for striatal neurons and 12-well for nigral neurons) were coated overnight with 0.1 mg/ml poly-d-lysine. Striatal or substantia nigral tissue was dissected from gestational 14-day-old mice embryos and kept in ice-cold Ca2+-free Hanks' balanced salt solution. Cells were then dissociated in Hanks' balanced salt solution containing trypsin, 0.25% EDTA for 30 min at 37 °C. After enzyme inhibition with 10% heat-inactivated fetal bovine serum in Dulbecco's modified Eagle's medium, the cells were suspended in Neurobasal medium supplemented with 2% B27 supplement, 500 μm l-glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin, plated at 2 × 106 cells in 2 ml/well, and incubated in a humidified CO2 incubator (5% CO2 and 37 °C). Half of the culture medium was replaced every 2 days, and experiments were conducted between culture days 5 and 6. After exposure to various doses of HDAC inhibitors (NaBu, VPA, Scriptaid, TSA, or apicidin) for 24–48 h, the primary cultures were collected for Western blot or real time RT-PCR analysis.
Cell Lines, siRNAs, Transient Transfections, and Reporter Gene Assays
All cells were grown at 37 °C in a humidified 95% air, 5% CO2 atmosphere. The mouse dopaminergic MN9D cell line was a kind gift from Dr. Syed Ali (National Center for Toxicological Research/Food and Drug Administration, Jefferson, AR), and the mouse neuroblastoma NIE115 cell line was a generous gift from Dr. Debomoy Lahiri (Indiana University School of Medicine, Indianapolis, IN). MN9D and NIE115 cells were cultured in DMEM supplemented with 10% fetal bovine serum, 2 mm l-glutamine, 50 IU penicillin, and 50 IU streptomycin, as described previously (35). The Lund human mesencephalic (LUHMES) cell line, derived from female human embryonic ventral mesencephalic cells by conditional immortalization (tetracycline-controlled v-Myc overexpression) and subsequent clonal selection, was obtained from the American Type Culture Collection. This cell line can be differentiated into post-mitotic neurons with a clear dopaminergic phenotype that was described previously in detail (64–66). Undifferentiated LUHMES cells were propagated in Advanced DMEM/F-12 supplemented with 1× N-2 supplement, 2 mm l-glutamine, and 40 ng/ml recombinant basic FGF on plastic flasks or multiwell plates precoated with 50 μg/ml poly-l-ornithine and 1 μg/ml fibronectin. Differentiation of LUHMES cells was initiated by the addition of differentiation medium containing advanced DMEM/F-12, 1× N-2 supplement, 2 mm l-glutamine, 1 mm dibutyryl cAMP, 1 μg/ml tetracycline, and 2 ng/ml recombinant human glial cell line-derived neurotrophic factor. After 2 days, cells were trypsinized and seeded onto multiwell plates at a cell density of 1.5 × 105 cells/cm2. LUHMES cells differentiate into a dopaminergic phenotype after an additional 3 days culture in differentiation medium. For 6-OHDA treatment studies, at day 5 of differentiation, differentiated LUHMES cells were pretreated in the presence or absence of NaBu (1 mm) or rottlerin (0.3 μm) for 1 h and then coincubated with 6-OHDA (30 μm) for another 24 h before experiments.
The dicer-substrate RNA (DsiRNA) and scrambled negative control siRNA (NC1) were purchased from Integrated DNA Technologies (Coralville, IA). The sense strands of the individual siRNA sequences were as follows: HDAC1, 5′-AGAGGAAAGUCUGUUACUACUACGA-3′; and HDAC2, 5′-CGAUCAAUAAGACCAGAUAAUAUGT-3′. We carried out siRNA transfections in MN9D cells with Lipofectamine 2000 reagent according to the manufacturer's protocol. In brief, cells were plated in 6-well plates at 7 × 104 cells/well 1 day before transfection. For each well, 100 pmol of siRNA duplex mixed with 5 μl of Lipofectamine 2000 were added to the cells. We also performed a second identical transfection 24 h later. Cell lysates collected 96 h after the initial transfection were analyzed using Western blotting to confirm the extent of HDAC1, HDAC2, and PKCδ expression.
For luciferase reporter experiments, transient transfections of NIE115 and MN9D cells were performed using Lipofectamine 2000 reagent according to the manufacturers' instructions. Cells were plated at 0.3 × 106 cells/well in 6-well plates 1 day before transfection. Each transfection was performed with 4 μg of reporter constructs. Cells were harvested at 24 h post-transfection, lysed in 200 μl of Reporter Lysis Buffer (Promega), and assayed for luciferase activity. For cotransfection assays, various amounts of expression plasmids as indicated in the figures were added to the reporter plasmids. The total amount of DNA was adjusted by adding empty vector. In HDAC inhibitor treatment experiments, indicated doses of HDAC inhibitors were added 24 h after DNA transfection, and cells were collected at designated time points and assayed for luciferase activity.
Luciferase activity was measured on a Synergy 2 Multi-Mode Microplate Reader (BioTek, Winooski, VT) using the luciferase assay system (Promega). The ratio of luciferase activity to total amount of proteins was used as a measure of normalized luciferase activity.
Lentivirus-based shRNA Transduction
For the production of shRNA lentiviruses, the 293FT cell line (Invitrogen) was transfected with individual clones from the PKCδ-targeting shRNA plasmid set or the scrambled control plasmid along with MISSION lentiviral packaging mix (Sigma) using Lipofectamine 2000 reagent according to the manufacturers' recommendations. The lentiviral particles in the medium were collected by centrifuging at 48 h after transfection. Lentiviral transduction of LUHMES cells was performed as described previously (67) with slight modifications. Briefly, LUHMES cells were pre-differentiated for 2 days and infected with lentiviral particles at a multiplicity of infection of 2.5 in the presence of Polybrene (8 μg/ml). Two or 4 days post-infection, cells were subjected to quantitative real time RT-PCR, Western blotting, or 6-OHDA treatment studies.
Organotypic Brain Slice Cultures and Treatments
Organotypic brain slices were prepared as described previously (68, 69) with slight modifications. Briefly, adolescent C57BL/6 wild-type or PKCδ−/− mice were anesthetized and decapitated. For Fluoro-Jade staining experiments, we used 9–12-day-old C57BL/6 wild-type and PKCδ−/− pups. The brains were quickly removed and embedded in 2% low melting point agarose (Invitrogen, prepared freshly in GBSS) in a specimen syringe. The CompresstomeTM Vf-300 (Precisionary Instruments Inc., Greenville, NC) was used to obtain 250-μm-thick coronal sections of corticostriatal brain regions in ice-cold Gey's balanced salt solution (137 mm NaCl, 5 mm KCl, 0.845 mm Na2HPO4, 1.5 mm CaCl2·2H2O, 0.66 mm KH2PO4, 0.28 mm MgSO4·7H2O, 1.0 mm MgCl2·6H2O, 2.7 mm NaHCO3, and pH adjusted to 7.4) supplemented with 1 mm kynurenic acid. After trimming off the extra agarose, the slices were transferred to Millicell-CM Biopore PTFE membrane inserts (Millipore) in 6-well culture plates. Each culture well contained 1 ml of slice culture medium (50% minimal essential medium, 25% basal Eagle's medium, 25% horse serum, 0.65% glucose, supplemented with penicillin/streptomycin and GlutaMAX). Organotypic slice cultures were maintained in a humidified 37 °C incubator with 5% CO2 and 95% air. Half of the media was exchanged every day, and experiments were conducted after 7–14 days in culture. For MPP+ treatment studies, corticostriatal slices were pretreated in the presence or absence of NaBu (1 mm) or rottlerin (5 μm) for 3 h and then cotreated with 300 μm MPP+ for another 24-h period.
Propidium Iodide Uptake and Fluoro-Jade Staining Assays
The propidium iodide uptake and Fluoro-Jade staining methods were used to determine the extent of neuronal damage after MPP+ treatment in brain slice cultures as described previously (69, 70). PI is a fluorescent molecule that is excluded from cells with intact membranes, but it labels nucleic acids in cells that have damaged cell membranes to produce red fluorescence (71, 72). Fluoro-Jade is an anionic fluorochrome, which selectively and specifically stains degenerating neurons in brain slices (69, 70, 72). In brief, after treatment, the slice cultures were incubated with 5 μg/ml PI in culture medium for 20 min at 37 °C. Fluoro-Jade staining was performed according to the method of Noraberg et al. (69). PI and Fluoro-Jade fluorescent images were viewed using a Nikon TE2000 microscope (Tokyo, Japan) with ×2 or ×20 magnification and captured with a SPOT color digital camera (Diagnostic Instruments, Sterling Heights, MI). For quantitative analysis of PI and Fluoro-Jade fluorescence, we measured average pixel intensities from randomly selected regions using ImageJ software (National Institutes of Health).
Immunostaining and Microscopy
For immunohistochemistry, the corticostriatal organotypic slices were washed with PBS and fixed in 4% paraformaldehyde for 1–2 h. After washing, the membrane inserts containing slices were blocked with the blocking agent (2% goat serum and 0.1% Triton X-100 in PBS) for 1 h. Membrane inserts were then incubated with the antibodies against PKCδ (1:1000, Santa Cruz Biotechnology) and β-III tubulin (1:1000, Millipore) for 2–5 days at 4 °C. Fluorescently conjugated secondary antibodies (Alexa Fluor 555-conjugated anti-mouse antibody, 1:2000, and Alexa Fluor 488-conjugated anti-rabbit antibody, 1:2000) were used to visualize the proteins. Hoechst 33342 (10 μg/ml) was used as a nuclear stain. The membranes were then removed from the inserts and mounted directly on microscope slides. Finally, images were viewed using a Nikon TE2000 microscope at ×20 magnification. The images were captured with a SPOT color digital camera and processed using ImageJ software.
For immunostaining of PKCδ and β-III tubulin in differentiated LUHMES cells, cells were fixed with 4% paraformaldehyde for 30 min at room temperature. After washing, the cells were permeabilized with 0.2% Triton X-100 in PBS, washed, and blocked with blocking agent (2% bovine serum albumin, 0.5% Triton X-100, and 0.05% Tween 20 in PBS) for 1 h. Cells were then incubated with the antibody against PKCδ (1:1000, Santa Cruz Biotechnology) and β-III tubulin (1:1000, Millipore) overnight at 4 °C, followed by incubation with fluorescently conjugated secondary antibodies (Alexa Fluor 555-conjugated anti-mouse antibody, 1:2000, and Alexa Fluor 488-conjugated anti-rabbit antibody, 1:2000) for 1 h at room temperature. After this, Hoechst 33342 (10 μg/ml) was added for 3 min at room temperature to stain the nucleus. Images were viewed using a Nikon TE2000 microscope, captured with a SPOT color digital camera, and processed using the ImageJ software.
Quantitative Real Time RT-PCR
Total RNA was isolated from fresh cell pellets using the Absolutely RNA Miniprep kit (Stratagene, La Jolla, CA). First strand cDNA synthesis was synthesized using an AffinityScript quantitative PCR cDNA synthesis kit (Stratagene). Real time PCR was performed in an Mx3000P quantitative PCR system (Stratagene) using the Brilliant SYBR Green quantitative PCR Master Mix kit (Stratagene), with cDNAs corresponding to 150 ng of total RNA, 12.5 μl of 2× master mix, 0.375 μl of reference dye, and 0.2 μm of each primer in a 25-μl final reaction volume. All reactions were performed in triplicate. Mouse β-actin or human 18 S rRNA was used as an internal standard for normalization. The sequences for mouse PKCδ primers are as follows: forward, 5′-TCTGGGAGTGACATCCTAGACAACAACGGG-3′, and reverse, 5′-CAGATGATCTCAGCTGCATAAAACGTAGCC-3′. Validated QuantiTect primer sets for human PKCδ, mouse β-actin, and human 18 S rRNA (Qiagen) were also used. The PCR cycling conditions contained an initial denaturation at 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 30 s, annealing at 60 °C for 30 s, and extension at 72 °C for 30 s. Fluorescence was detected during the annealing step of each cycle. Dissociation curves were run to verify the singularity of the PCR product. The data were analyzed using the comparative threshold cycle (Ct) method (73).
Acid Extraction of Histones
Acid extraction of histones was performed as described previously (74) with modifications. Briefly, fresh cell pellets were suspended with 5 volumes of lysis buffer (10 mm HEPES, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, 1× halt protease inhibitor mixtures) and hydrochloric acid at a final concentration of 0.2 m and subsequently lysed on ice for 30 min. After centrifugation at 11,000 × g for 10 min at 4 °C, the histone mixtures were collected from the supernatant.
Immunoblotting
Organotypic corticostriatal slices were lysed using the tissue extraction reagent (Invitrogen) containing protease and phosphatase inhibitor mixture (Thermo Scientific). Cell lysates and brain homogenates were prepared as described previously (38). Immunoblotting was performed as described previously (15). Briefly, the samples containing equal amounts of protein were fractionated through a 7.5–15% SDS-PAGE and transferred onto a nitrocellulose membrane (Bio-Rad). Membranes were blotted with the appropriate primary antibody and developed with either IRDye 800 anti-rabbit or Alexa Fluor 680 anti-mouse secondary antibodies. The immunoblot imaging was performed with an Odyssey Infrared Imaging system (LI-COR, Lincoln, NE).
Chromatin Immunoprecipitation (ChIP)
ChIP assays were conducted with chromatin isolated from NIE115 cells using the ChIP-IT Express enzymatic kit from Active Motif according to the manufacturer's instructions with slight modifications. Briefly, after cross-linking, the nuclei were prepared and subjected to enzymatic digestion to generate chromatin fragments between 200 and 1500 bp. The sheared chromatin was collected by centrifuge, and a 10-μl aliquot was removed to serve as a positive input sample. Aliquots of 70 μl of sheared chromatin were immunoprecipitated with protein-G magnetic beads and 3 μg of pan-acetyl histone H4 antibody (Active Motif) or 3 μg of normal mouse IgG. Equal aliquots of each chromatin sample were saved for no-antibody controls. The immunoprecipitated DNA was analyzed by PCR to amplify a region (+2 to +289) within the PKCδ promoter. Primers for amplification are as follows: forward, 5′-ATATATCTCGAGTCCTGGGCTCCATTGTGTGTG-3′, and reverse, 5′-GTCTATAAGCTTACCTCACCCAGGTGCCGG-3′. Conditions of linear amplification were determined empirically for these primers. PCR conditions are as follows: 94 °C for 3 min; 94 °C for 30 s, 59 °C for 30 s, and 68 °C for 30 s for 35 cycles. PCR products were resolved by electrophoresis in a 1.2% agarose gel and visualized after ethidium bromide staining.
DNA Affinity Precipitation Assay
Nuclear and cytoplasmic proteins were prepared using the NE-PER nuclear and cytoplasmic extraction kit (Thermo Scientific, Waltham, MA). 5′-Biotinylated oligonucleotides corresponding to the sequence between +204 and +238 of the PKCδ promoter were synthesized by Integrated DNA Technologies (Coralville, IA) and annealed. Twenty pmol of oligonucleotides were incubated with 100 μg of Dynabeads M-280 (Dynal Biotech, Oslo, Norway) in B&W buffer at room temperature for 10 min. Unconjugated DNA was washed off with a magnetic particle concentrator (Dynal Biotech). After blocking with 0.5% BSA in TGED buffer (20 mm HEPES, pH 7.9, 1 mm EDTA, 10% glycerol, 0.01% Triton X-100) at 4 °C for 2 h, the DNA-conjugated beads were incubated with 350 μg of nuclear extracts from NIE115 cells treated with or without 1 mm NaBu for 4 h at 4 °C. After an extensive wash by TGED buffer, the beads were eluted with 50 μl of 2× SDS loading buffer. Complexing proteins were resolved on a 7.5% SDS-polyacrylamide gel and examined by immunoblotting with polyclonal anti-Sp3 and -Sp4 antibodies.
Determination of Cell Viability
Cell viability assay was performed using the Cell Titer 96 Aqueous One Solution proliferation assay kit from Promega, according to the manufacturer's instructions. This colorimetric assay is based on the cellular conversion of an MTS tetrazolium compound into a colored formazan product that is soluble in culture medium. Briefly, differentiated LUHMES cells were seeded onto 96-well plates at a cell density of 3.6 × 104 per well. Treatment of differentiated LUHMES cells was performed at day 5 of differentiation. After treatment, Cell Titer 96 Aqueous One Solution Reagent was added to each well, and the plates were incubated in a humidified incubator (5% CO2 and 37 °C) for 2–3 h. Measurements were made at 490 nm with a 96-well plate reader (SpectraMax 190 spectrophotometer, Molecular Devices).
High Affinity [3H]Dopamine Uptake Assays
Dopamine uptake measurements were measured as described previously (75) with some modifications. Briefly, differentiated LUHMES cells grown in 6-well plates were washed twice with Krebs-Ringer buffer (16 mm NaH2PO4, 16 mm Na2HPO4, 120 mm NaCl, 4.7 mm KCl, 1.8 mm CaCl2, 1.2 mm MgSO4, 1.3 mm EDTA, and 5.6 mm glucose, pH 7.4), followed by incubation with 10 nm [3H]dopamine in Krebs-Ringer buffer for 30 min at 37 °C. Nonspecific uptake was determined by adding 10 μm dopamine uptake blocker mazindol. Afterward, the cultures were washed three times with fresh ice-cold Krebs-Ringer buffer and then lysed with 1 n NaOH. The radioactivity was measured with a liquid scintillation counter (Tri-Crab 4000, Packard Instrument Co.) after the addition of a 5-ml scintillation mixture to each vial. The specific dopamine uptake was expressed as mean values of counts, subtracted from nonspecific counts measured in the presence of mazindol.
Statistical Analysis
Unless otherwise stated, all data were determined from three independent experiments, each done in triplicate, and expressed as means ± S.E. All statistical analyses were performed using the GraphPad Prism 4.0 software (GraphPad Software, San Diego). One-way analysis of variance test followed by Tukey's multiple comparison test were used for statistical comparisons, and differences were considered significant if p < 0.05.
RESULTS
PKCδ mRNA and Protein Levels Are Stimulated by Exposure to HDAC Inhibitors in Vivo and in Vitro
Previously, we reported that activation of PKCδ mediates dopaminergic neurodegeneration in multiple models of PD (4–9, 12). Given the importance of histone acetylation in modulating signaling events (40), we asked whether histone acetylation could influence PKCδ signaling in neurons. In our first set of experiments, we assessed the effect of increased histone acetylation through HDAC inhibition on the PKCδ protein levels in a variety of primary and cultured neuronal cells. NaBu, a short-chain fatty acid, potently inhibits HDAC activity at millimolar concentrations (76). Treatment with 1 mm NaBu markedly increased the levels of total PKCδ protein in primary mouse nigral (Fig. 1A, left panel) and striatal (Fig. 1A, right panel) neurons following 24–48 h of drug exposure. Because butyrate has multiple pharmacological actions that may not be due to inhibition of HDAC (77), we further analyzed whether other HDAC inhibitors had similar effects on PKCδ protein expression. For this, we exposed striatal neurons to increasing concentrations of multiple HDAC inhibitors including TSA, valproic acid, scriptaid, and apicidin for 48 h, and PKCδ protein levels were determined by Western blot analysis. As seen with NaBu, Fig. 1B shows a dose-dependent induction of PKCδ protein in cells treated with VPA, another short-chain fatty acid. Induction of native PKCδ by VPA was accompanied by a dose-dependent increase in PKCδ proteolytic cleavage. Scriptaid, which is structurally unrelated to NaBu, also enhanced total PKCδ levels in the dose range tested (Fig. 1C), whereas induction of PKCδ proteolytic cleavage was only observed at the lower dose (1.23 μm). PKCδ levels were also dramatically up-regulated after treatment of striatal neurons with nanomolar levels of TSA or apicidin, two other structurally unrelated HDAC inhibitors (Fig. 1, D and E). Next, analysis of mouse neuroblastoma NIE115 cells demonstrates that 48 h of NaBu (1 mm) treatment elevated PKCδ protein levels up to ∼2-fold compared with untreated cells (Fig. 1F). Additional analysis of various PKC isoforms in mouse dopaminergic MN9D cells shows that NaBu increased PKCδ protein levels, whereas PKCα, -ϵ, and -η were not affected (Fig. 1G). Interestingly, PKCζ, a typical PKC isoform, was elevated after 24 h NaBu treatment, suggesting that the effect of HDAC inhibitors on PKCδ gene expression is somewhat isoform-specific.
FIGURE 1.

Exposure to HDAC inhibitors increases PKCδ protein expression in primary neurons and cell lines. A, primary mouse nigral (left panel) and striatal (right panel) neurons were exposed to 1 mm NaBu for 24 or 48 h, after which whole protein lysates were prepared and subjected to Western blot analysis of PKCδ and actin expression. Representative immunoblots are shown. B–E, primary mouse striatal neurons were exposed to the designated concentrations of HDAC inhibitors VPA (B), Scriptaid (C), TSA (D), or apicidin (E) for 48 h, after which protein lysates were prepared and analyzed for PKCδ and actin expression by immunoblot. Representative immunoblots are shown. F, left panel, mouse neuroblastoma NIE115 cells were treated with 1 mm NaBu for 24 or 48 h, lysed, and analyzed by immunoblot for levels of PKCδ and actin. Right panel, densitometric analysis. PKCδ bands were quantified and normalized to that of β-actin. Values are shown as means ± S.E. of two independent experiments. G, mouse dopaminergic MN9D cells were treated with 1 mm NaBu for 24 or 48 h and analyzed for levels of PKC isoforms (δ, α, η, ϵ, and ζ) and actin. Representative immunoblots are shown. *, p < 0.05; control (con) versus butyrate-treated samples.
Following the characterization of PKCδ protein up-regulation during HDAC inhibition in multiple neuronal models, we then determined whether the effect of HDAC inhibitors on PKCδ up-regulation occurs at the level of gene transcription. PKCδ mRNA expression was measured using real time RT-PCR at various doses and time points. As shown in Fig. 2, exposure of primary nigral (Fig. 2A, left panel) and striatal (Fig. 2A, right panel) cultures to 1 mm NaBu for 24 or 48 h significantly increased PKCδ mRNA expression. The magnitude of the inductions varied from 4- to 6-fold relative to untreated neurons. Furthermore, when nigral and striatal cells were administered increasing concentrations of NaBu (0.2–5 mm) for 24 h, we found a dose-dependent increase in PKCδ mRNA, peaking at 1 mm (Fig. 2B). In addition, similar inductions of endogenous PKCδ mRNA by NaBu were observed in both NIE115 and MN9D neuronal cells (Fig. 2C). These increases peaked at ∼3-fold after the 48-h sodium butyrate treatment.
FIGURE 2.

HDAC inhibition increases PKCδ mRNA expression. A–B, primary mouse nigral (left panel) and striatal (right panel) neurons were exposed to 1 mm NaBu for 24 or 48 h (A) or to different concentrations of NaBu for 48 h (B). Real time RT-PCR analysis of PKCδ mRNA level was performed. β-Actin mRNA level served as internal control. C, NIE115 (left panel) and MN9D (right panel) cells were exposed to 1 mm NaBu for 24 or 48 h, and PKCδ mRNA expression was evaluated by real time RT-PCR analysis. β-Actin mRNA level served as internal control. All values are expressed as a percentage of the activity of controls and represented as means ± S.E. of three independent experiments performed in triplicate. *, p < 0.05; **, p < 0.01; ***, p < 0.001; control (con) versus butyrate-treated samples.
To further address whether the effect of HDAC inhibition on PKCδ expression observed above reflects the regulation of PKCδ expression in vivo, we administered NaBu (1.2 g/kg, a single i.p. injection) to male C57BL/6 mice and quantified the brain levels of PKCδ at various time points (6–24 h) after injection. Interestingly, we found that PKCδ had increased 3-fold in the mouse substantia nigra by 24 h post-injection, whereas tyrosine hydroxylase and actin remained unchanged under these conditions (Fig. 3A). Moreover, the striatal regions exhibited a similar NaBu-induced up-regulation of PKCδ (Fig. 3B). Notably, PKCδ showed little change in cerebrum regions (Fig. 3C), indicating a brain region-specific mechanism for HDAC inhibition-induced PKCδ up-regulation. Collectively, these data clearly demonstrate that HDAC inhibition induces PKCδ gene expression in nigrostriatal dopaminergic neuronal cells in both cell culture and animal models.
FIGURE 3.

Effects of in vivo sodium butyrate injection on PKCδ protein level. A and B, C57 black mice were administered 1.2 g/kg NaBu or an equivalent volume of saline via intraperitoneal injection for 6–24 h. Substantia nigra (A), striatum (B), and cerebrum (C) tissues from each mouse were harvested and prepared and analyzed for PKCδ, tyrosine hydroxylase (TH), and actin expression by immunoblot. Top panel, representative immunoblots are shown. Bottom panel, quantitation data. The results are normalized to β-actin and expressed as a percentage of the untreated mice. All data are represented as means ± S.E. from four mice per group. **, p < 0.01; saline-treated versus butyrate-treated.
Butyrate Induces Hyperacetylation of PKCδ Promoter Histones
The next set of experiments was designed to ascertain the molecular mechanisms whereby histone acetylation stimulates PKCδ gene expression. For mechanistic studies, we used NIE115 or MN9D neuronal cells because these neuronal cells were highly amenable to transfection of gene constructs. Because butyrate inhibits the activity of many HDAC isoforms (class I and IIa), we first confirmed the influence of HDAC inhibition on histone acetylation in NIE115 cells. As expected, the systemic acetylation of histone H3 and H4 in NIE115 cells was up-regulated following exposure to 1 mm NaBu, whereas total histone H3 levels were not changed (Fig. 4A). The acetylation of histone proteins promotes an open chromatin structure and thereby leads to transcriptional activation. We then sought to determine whether change in PKCδ expression occurs through a chromatin-specific regulation. To this end, we performed ChIP assays using chromatin isolated from NIE115 neuronal cells and an antibody specific for histone H4 acetylation. As depicted in Fig. 4B, exposure of NIE115 cells to 1 mm NaBu resulted in a dramatic enrichment of histone H4 acetylation at the PKCδ promoter (Fig. 4B). Together, these data indicate that chromatin remodeling is at least in part responsible for the transcription activation of PKCδ gene brought about by NaBu treatment.
FIGURE 4.

Sodium butyrate increases levels of total histone acetylation and histone acetylation of PKCδ promoter-associated chromatin. A, NIE115 cells were exposed to 1 mm NaBu for 24 h. Total histones were prepared for blotting with specific anti-acetyl-lysine and anti-H3 antibodies. A representative immunoblot is shown. B, ChIP analysis of hyperacetylated histone H4 on PKCδ promoter. NIE115 cells were treated with 1 mm NaBu for 24 h, after which chromatin was prepared and sheared by enzymatic digestion. The sheared DNA was then immunoprecipitated with antibody against pan-acetylated histone H4, normal mouse IgG, or without antibody (No Ab). After reversal of cross-linking, immunoprecipitated DNA fragments were analyzed by PCR amplification with primers specific for the PKCδ promoter region generating a 312-bp fragment. A representative gel electrophoresis is shown. con, control.
HDAC Inhibition Activates PKCδ Promoter Transcription, Delineation of the Histone Acetylation-responsive Elements on the PKCδ Promoter
We further explored whether HDAC inhibition stimulated PKCδ transcription by directly activating the PKCδ promoter. To address this, the effect of HDAC inhibitors on PKCδ promoter activity was assessed in a luciferase reporter construct-based transient transfection assay. Our previously cloned mouse PKCδ promoter/luciferase reporter construct pGL3−1694/+289 (35), which contains 1694 bp of the 5′-flanking sequences and 289 bp of noncoding exon 1 (access number GU182370), or pGL3-Basic empty vector was transfected into NIE115 and MN9D cells. Transfected cells were incubated with increasing concentrations of NaBu (0.2 to 1 mm) for 24 h. The incubation of NaBu enhanced transcription of the luciferase reporter pGL3−1694/+289 in a dose-dependent manner up to ∼5-fold in both MN9D and NIE115 neuronal cells (Fig. 5A). No changes in luciferase activity were noted in cells transfected with pGL3-Basic control, implying the stimulatory effect of NaBu on PKCδ promoter is specific. Interestingly, exposure of MN9D cells to varying doses of VPA, TSA, scriptaid, or apicidin for 24 h revealed a more robust activation of PKCδ promoter activity than that achieved with NaBu (Fig. 5, B–E). The extent of maximum activation for those HDAC inhibitors varied from 8- to 14-fold compared with untreated cells. Taken together, these results indicate that histone acetylation-induced PKCδ gene up-regulation involves transcriptional regulation of PKCδ promoter.
FIGURE 5.

Regulation of PKCδ promoter activity by sodium butyrate treatment and ectopic expression of HDACs. A, PKCδ promoter activity is activated after treatment with NaBu. The PKCδ promoter reporter construct pGL3−1694/+289 or empty vector pGL3-Basic was transfected into MN9D (left panel) and NIE115 (right panel) cells. After 24 h of transfection, the cells were incubated with or without NaBu at concentrations ranging from 0.2 to 1 mm for 24 h. Cells were then harvested, and luciferase activities were determined and normalized by total cellular protein. Values are expressed as a percentage of the activity of the pGL3−1694/+289-transfected control and represented as means ± S.E. of three independent experiments performed in triplicate. **, p < 0.01; ***, p < 0.001; control versus NaBu-treated samples. B–E, other HDAC inhibitors stimulate PKCδ promoter activity in MN9D cells. The PKCδ promoter reporter construct pGL3−1694/+289 was transfected into MN9D cells. After 24 h of transfection, the cells were incubated with VPA (B), TSA (C), apicidin (D), or scriptaid (E) at the designated concentrations for 24 h. Cells were then harvested, and luciferase activities were determined and normalized by total cellular protein. Values are expressed as a percentage of the activity of untreated control (con) and represent the mean ± S.E. of three independent experiments performed in triplicate. ***, p < 0.001; control versus treated samples. F, PKCδ promoter activity is repressed by forced expression of HDAC proteins in NIE115 (black bar) and MN9D (blue bar) cells. Cells were cotransfected with pGL3−1694/+289 and 8 μg of HDAC1, HDAC4, HDAC5, or HDAC7 expression vector or the empty vector (EV) control. Luciferase activity was measured after 24 h of transfection and normalized by total cellular protein. Values are expressed as a percentage of the luciferase activity obtained from cells transfected with 8 μg of empty vector (EV) and represented as means ± S.E. of three independent experiments performed in triplicate. **, p < 0.01; ***, p < 0.001; EV- versus HDAC-transfected samples. G, effects of ectopic expression of HDAC proteins on butyrate-induced PKCδ promoter activation. MN9D (left panel) and NIE115 (right panel) cells were cotransfected with pGL3−1694/+289 and increasing concentrations of HDAC1, HDAC4, HDAC5, or HDAC7 expression vector (from 2 to 8 μg) or the empty vector (EV) control. After 24 h of transfection, the cells were incubated with or without NaBu (1 mm) for 24 h. Cells were then harvested, and luciferase activities were determined and normalized by total cellular protein. Values are expressed as a percentage of the luciferase activity obtained from NaBu-treated cells transfected with 8 μg of empty vector (EV) and are represented as means ± S.E. of three independent experiments performed in triplicate. Variations in the amount of total DNA were compensated with the corresponding empty vector. H, silencing of class I HDAC (HDAC1 and -2) up-regulated PKCδ protein expression in MN9D cells. MN9D cells were transiently transfected with siRNA-HDAC1, siRNA-HDAC2, and scrambled siRNA. Cells were collected 96 h after the initial transfection and then subjected to Western blot analysis. I, densitometric analysis. HDAC1, HDAC2, and PKCδ bands were quantified and normalized to that of β-actin. Values are shown as means ± S.E. of two independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001. J, multiple class I and IIa HDACs are expressed in MN9D and NIE115 cells. MN9D and NIE115 cell lysates were prepared and subjected to immunoblot for various HDACs and β-actin. Representative immunoblots are shown.
The regulation of PKCδ promoter activity by HDAC inhibition was further confirmed by cotransfection with the pGL3−1694/+289 promoter construct and expression vectors for several class I and IIa HDAC isoforms (HDAC1, -4, -5, and -7) under either basal or butyrate-stimulated conditions. Efficient overexpression of these HDACs was verified by Western blot (data not shown). Consistent with the HDAC inhibitor data, exogenous expression of all four HDAC proteins led to a significant inhibition of basal PKCδ promoter activity in both NIE115 and MN9D cells, with HDAC4 and HDAC5 being the most potent repressors (∼60 and 80% repression for HDAC4 and HDAC5, respectively) (Fig. 5F). Furthermore, butyrate-induced activation of PKCδ promoter was reduced by expressing various amounts of HDAC1, HDAC4, or HDAC5 in MN9D cells (Fig. 5G, left panel) and NIE115 cells (Fig. 5G, right panel), although only marginal changes were observed for HDAC7. We also performed siRNA silencing experiments to examine the role that class I HDACs (HDAC1 and -2) play in regulating PKCδ protein expression. As shown in Fig. 5, H and I, silencing of HDAC1 and HDAC2 induced a significant up-regulation of PKCδ protein levels in MN9D cells. These results collectively indicate that PKCδ up-regulation is mediated by multiple HDACs. In support of this view, we found that multiple class I and IIa HDACs, including HDAC1–4, express at various levels in NIE115 and MN9D cells (Fig. 5J). By comparison, the expression of two class IIa isoforms, HDAC5 and HDAC7, was too low to be detected in both cells, demonstrating a distinct HDACs expression profile in these cells.
We were next interested in understanding the mechanism underlying histone acetylation-driven PKCδ promoter activation. First, we delineated the regions at the PKCδ promoter that respond to butyrate-induced acetylation. A series of truncated promoter constructs in the −1694/+289 region were analyzed by transient transfection for their response to NaBu in MN9D and NIE115 cells. As shown in Fig. 6, A and B, in MN9D cells, NaBu strongly increased luciferase activities from the promoter reporter construct pGL3−147/+289 as well as the pGL3+2/+289 plasmid up to 3.9- and 4.2-fold, respectively, which is comparable with that obtained from the full size promoter (pGL3−1694/+289, 4.9-fold). However, the lack of the sequence from +2 to +289 led to a significant loss of butyrate responsiveness. Furthermore, similar results were obtained using NIE115 cells (Fig. 6C). Thus, our mapping suggests that the major histone acetylation-responsive elements are located within the +2/+289 region. We therefore focused our characterization of the PKCδ promoter on this region.
FIGURE 6.
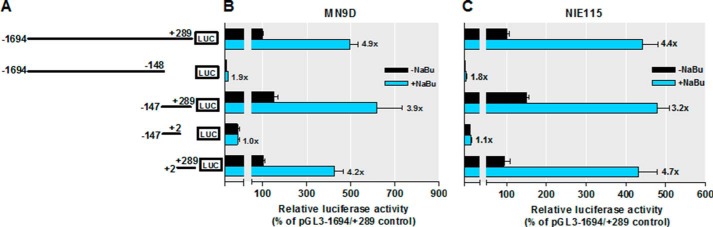
Mapping of sodium butyrate-responsive elements on the PKCδ promoter. A, schematic representation of PKCδ promoter deletion/luciferase reporter constructs. A series of PKCδ promoter deletion derivatives was generated by PCR methods and inserted into the pGL3-Basic luciferase vector. The 5′ and 3′ positions of the constructs with respect to the transcription start site are depicted. B and C, each construct as shown in A was transiently transfected into MN9D (B) and NIE115 (C) cells. After 24 h of transfection, the cells were incubated with (blue bar) or without (black bar) 1 mm NaBu for 24 h and then analyzed for luciferase activities. Values are expressed as a percentage of the activity of pGL3−1694/+289-transfected control and represented as means ± S.E. of three independent experiments performed in triplicate. Above each blue bar is the fold change in activation following NaBu exposure in cells transfected with individual promoter construct.
Sodium Butyrate Stimulates PKCδ Promoter Activity through Four GC Box Elements
In our previous study (35), we found that the PKCδ promoter region between +2 and +289 is GC-rich and contains multiple Sp-binding sites, including four consecutive GC boxes designated GC(1) to GC(4) within ∼250 bp downstream of the transcription start site, as well as a CACCC box located at position +35 bp downstream of the transcription start site (Fig. 7A). We also reported that those Sp sites act as crucial cis-elements regulating constitutive PKCδ transcription in neuronal cells (35). To address whether these Sp sites have any functional role in the acetylation-induced activation of PKCδ, we performed site-directed mutagenesis of pGL3−147/+209 and pGL3+165/+289 constructs. The former possesses the proximal CACCC site, whereas in the latter, only the four GC boxes are present (Fig. 7A). Those mutated and wild-type reporter plasmids were used and assayed for luciferase activity following NaBu treatment. Exposure to NaBu did not activate luciferase activity of the wild-type pGL3−147/+209, and it even reduced its activity in MN9D cells, suggesting that the CACCC site is not involved in the activation by butyrate (Fig. 7B). Indeed, mutation of the CACCC site (mCACCC) did not diminish the NaBu responsiveness (Fig. 7B). Moreover, NaBu significantly activated the luciferase activity of wild-type pGL3+165/+289 up to 3.4- and 4.7-fold in MN9D and NIE115 cells, respectively (Fig. 7, C and D). These findings also indicate a minimal 81-bp NaBu-responsive promoter region from +209 to +289. Alteration of the most distal GC(4) or GC(3) site reduced the NaBu responsiveness by 15 and 24%, respectively, compared with that of wild-type pGL3+165/+289 in MN9D cells (Fig. 7C). In contrast, mutation of either the proximal GC(2) box or GC(1) box caused major decrements in response to NaBu, resulting in about 35 and 33% elimination, respectively, compared with that of wild type. Furthermore, triple mutated reporter constructs, mGC123, mGC124, mGC134, or mGC134, in which only site GC(4), GC(3), GC(2), or GC(1) remains active, respectively, all resulted in a complete loss of NaBu-induced promoter activity in both cell types (Fig. 7D), suggesting that cooperative interactions among the different GC sites are required for NaBu-mediated transactivation of the PKCδ promoter. Taken together, these data suggest that the GC(1) and GC(2) sites, and less significantly the GC(3) and GC(4) sites rather than the CACCC site, are the main NaBu-induced histone acetylation-responsive elements and that these GC boxes cooperate in an additive manner in the transmission of the NaBu response.
FIGURE 7.

Sodium butyrate activates PKCδ promoter through the GC box elements. MN9D and NIE115 cells were transfected with either the wild-type or mutated PKCδ promoter and Sp1 site-driven promoter constructs for 24 h. Cells were then incubated with or without NaBu (1 mm) for 24 h, and the luciferase activities were measured and normalized by total cellular protein. Luciferase activity following transfection of the wild-type construct (pGL3−147/+209, pGL3+165/+289, or Sp1-luc) was arbitrarily set to 100, and all other data are expressed as a percentage thereof. The results are represented as means ± S.E. of three independent experiments performed in triplicate. Above each blue bar is the fold change in activation following NaBu exposure in cells transfected with the individual promoter construct. A, schematic representation of the wild-type PKCδ promoter reporter constructs pGL3−147/+209 and pGL3+165/+289. The multiple Sp sites are depicted by either a circle or square. B, MN9D (left panel) and NIE115 (right panel) cells were transfected with 4 μg of either wild-type (pGL3−147/+209) or mCACCC-mutated luciferase reporter constructs. C, MN9D cells were transfected with either the wild-type (pGL3+165/+289) or single mutated luciferase reporter constructs. D, wild-type (pGL3+165/+289) or triple mutated luciferase reporter constructs, as indicated, were transfected into MN9D (left panel) and NIE115 (right panel) cells. E, Sp1 consensus site-driven luciferase reporter plasmid (Sp1-luc) or its mutant construct (mSp1-luc) was individually transfected into MN9D (left panel) and NIE115 (right panel) cells.
To confirm further that Sp sites indeed mediate transcriptional activation by NaBu, we generated an Sp1 reporter construct (Sp1-luc), composed of three SV40 promoter-derived consensus Sp1-binding elements inserted into the promoter-less luciferase reporter vector (pGL3-Basic). The effects of NaBu on its transcriptional activity were subsequently examined in transient transfection studies performed in MN9D and NIE115 cells. The luciferase activities of Sp1-luc were significantly elevated following NaBu exposure (up to ∼4.0- and 5.0-fold activation in MN9D and NIE115 cells, respectively), whereas mutations of all Sp1 consensus binding sites (mSp1-luc) completely abolished the NaBu-induced transcriptional activation (Fig. 7E). Consistently, expression of HDAC isoforms inhibited both basal and NaBu-induced promoter activity of Sp1-luc (data not shown).
Sp Family Proteins Are Required for Mediating Histone Acetylation Induction of PKCδ Expression
Our recent data implicated that Sp families of transcription factors (Sp1, Sp3, and Sp4) play a crucial role in transcriptional regulation of PKCδ through specific interaction with those multiple GC sites, with Sp3 being the most robust activator (35). These observations led to a hypothesis that the NaBu-induced transcriptional activation of PKCδ might be mediated by Sp transcriptional factors. To test this possibility, we analyzed the functional impact of ectopically expressed Sp proteins on NaBu-induced transcriptional activation in transient transfections. The PKCδ promoter reporter construct pGL3−147/+289, as illustrated in Figs. 6 and 7A, was cotransfected into NIE115 cells along with 4 μg of expression vectors for Sp family proteins (pN3-Sp1, pN3-Sp3, and pN3-Sp4), or alternatively a control empty vector (pN3), in the presence or absence of 1 mm NaBu for 24 h. All of these Sp expression plasmids have been shown to express stable proteins in both NIE115 and MN9D cells (35). In accordance with butyrate induction of PKCδ promoter activity as shown in Figs. 5 and 6, exposing the empty vector-transfected cells to NaBu induced an ∼4.5-fold activation of the pGL3−147/+289 reporter, whereas in the absence of NaBu, overexpression of Sp3 alone led to ∼2.5-fold activation (Fig. 8A). Importantly, elevated synergistic activation of promoter activity up to ∼11.5-fold was seen when cells overexpressing Sp3 protein were treated with NaBu. The synergism was also evident in Sp1- and Sp4-transfected cells after 24 h of incubation with NaBu (Fig. 8A). These findings clearly indicate that activation of the PKCδ promoter by NaBu is mediated by the Sp family of transcription factors. In addition, parallel transfection studies of NIE115 cells with two different amounts of expression vector for wild-type or dominant-negative mutant Sp1/Sp3 were done to confirm the effects of Sp proteins on NaBu transactivation (Fig. 8B). In these experiments, expression of wild-type Sp1 or Sp3 caused a dose-dependent increase in the NaBu-induced enhancement of PKCδ promoter activity. In contrast, expression of a dominant-negative construct pN3-DN-Sp1 or pN3-DN-Sp3, which has an intact DNA binding domain but lacks the complete transactivation domains of Sp1/3, had no effect on the NaBu-mediated induction of PKCδ promoter activity. Interestingly, even the highest dose of these mutant constructs did not affect the basal PKCδ promoter activity (Fig. 8B).
FIGURE 8.

Sp family transcriptional factors mediate responsiveness to sodium butyrate. A, overexpression of Sp1, Sp3, and Sp4 synergistically activated the NaBu induction of PKCδ promoter activity in NIE115 cells. NIE115 cells were cotransfected with pGL3−147/+289 and 8 μg of pN3-Sp1, pN3-Sp3, pN3-Sp4, or empty vector (EV) pN3. After 24 h of transfection, the cells were incubated with or without 1 mm NaBu for 24 h. Luciferase activities were then assayed and normalized by total cellular protein. Luciferase activity following transfection of empty vector without NaBu treatment was assigned the value 1, and all other data are expressed as a fold induction thereof. The results are represented as means ± S.E. of three independent experiments performed in triplicate. B, overexpression of the dominant-negative mutant Sp1 or Sp3 protein (left panel, pN3-DN-Sp1; right panel, pN3-DN-Sp3) lacking the transactivation domains did not enhance the NaBu induction of PKCδ promoter activity in NIE115 cells. NIE115 cells were cotransfected with pGL3−147/+289 and varying concentrations (4–8 μg) of pN3-Sp1, pN3-DN-Sp1, pN3-Sp3, or pN3-DN-Sp3 for 24 h. Cells were then exposed to 1 mm NaBu for 24 h, and luciferase activities were determined and normalized. The results are represented as means ± S.E. of three independent experiments performed in triplicate. Variations in total DNA were compensated with the corresponding empty vector pN3. C and D, mithramycin A (C) and tolfenamic acid (D) inhibited the response to NaBu. NIE115 cells were transfected with the PKCδ promoter reporter construct pGL3−147/+289 for 24 h. After pretreatment with different doses of mithramycin A and tolfenamic acid for 1 h, the cells were incubated with or without NaBu (1 mm) for 24 h. Cells were then harvested, and luciferase activities were determined and normalized by total cellular protein. Values are expressed as a percentage of the activity obtained from control samples without NaBu and mithramycin A or tolfenamic acid treatment and are represented as means ± S.E. of three independent experiments performed in triplicate. **, p < 0.01; ***, p < 0.001; mithramycin A or tolfenamic acid-treated versus untreated samples.
To further corroborate our observation that forced expression of Sp family proteins affects NaBu transactivation of the PKCδ promoter, we employed different types of known Sp-specific inhibitors to test whether they block NaBu-induced PKCδ promoter activity. Pretreatment with mithramycin A, an aureolic antibiotic that is known to bind to the GC-rich motif and selectively inhibit Sp transcription factor binding (35, 78, 79), significantly compromised NaBu-induced transactivation of the PKCδ promoter in a concentration-dependent manner (Fig. 8C). Furthermore, tolfenamic acid, which has been shown to induce Sp protein degradation (80), also inhibited NaBu transactivation (Fig. 8D).
Sodium Butyrate Enhances the Transactivational Activity of Sp Proteins
To further investigate the mechanisms underlying the stimulation of PKCδ promoter activity by NaBu, we determined whether NaBu affects the protein levels of Sp effectors. Previously, we showed that Sp3 and Sp4 are endogenously expressed at appreciable levels in both MN9D and NIE115 cells, but the expression of endogenous Sp1 was not detected in these cells (35); therefore, in this study, the effect of NaBu on the expression of Sp3 and Sp4 was examined. Western blot analyses indicate that NaBu did not change the protein levels of Sp3 or Sp4 (Fig. 9A). We next examined the possibility that NaBu might stimulate PKCδ transcription by elevating the recruitment of Sp proteins to the PKCδ promoter. DNA affinity protein binding assays were performed using a biotin-labeled oligonucleotide spanning the GC(1) and GC(2) elements, between positions +204 and +238 on the PKCδ promoter, and nuclear extracts from NIE115 cells. Surprisingly, the association of Sp3 (Fig. 9B) or Sp4 (data not shown) with this oligonucleotide was unaltered after incubation with NaBu. These findings indicate that stimulation by NaBu resulted from a mechanism other than alteration of Sp protein levels or DNA binding. We then asked whether NaBu could directly increase the transactivating potential of Sp proteins. To address this issue, we utilized a Gal4-based one-hybrid system, in which Sp1 or Sp3 is fused to the DNA-binding domain of the yeast transcription factor Gal4, and the effects of NaBu on the activity of these chimeric proteins were assayed in MN9D and NIE115 cells using a luciferase reporter plasmid pG5-luc containing five Gal4 DNA-binding sites. As shown in Fig. 9C, NaBu had a negligible effect on either the pG5-luc reporter alone or pG5-luc cotransfected with the empty control vector Gal4. In contrast, a vast stimulation of transactivation of Gal4-Sp1 or Gal4-Sp3 upon NaBu treatment was observed (12- and 19-fold in MN9D cells; 32- and 31-fold in NIE115 cells for Gal4-Sp3 and Gal4-Sp1, respectively). However, the transactivation by NaBu was almost abolished when the chimeric proteins Gal4-Sp1DBD or Gal4-Sp3DBD lacking the Sp transactivation domains were used, suggesting the specificity of NaBu on Sp1/3 transactivational ability. It should be noted that under the basal condition, however, Gal4-Sp1 is a stronger activator than Gal4-Sp3. In addition, overexpressing HDAC4 or HDAC5 resulted in a significant reduction in butyrate-induced transactivation of Gal4-Sp1 or Gal4-Sp3, whereas we observed only minimal effects of HDAC1 or HDAC7 overexpression (Fig. 9D). We conclude that NaBu specifically increases the transactivational capacity of Sp1/3 proteins and that multiple HDACs might be involved in regulating Sp transcriptional activity by NaBu.
FIGURE 9.

NaBu increases Sp1/3 transcriptional activity. A, Sp3 and Sp4 expression was unaffected by NaBu treatment. NIE115 cells were incubated with or without 1 mm NaBu for 24 h. Whole cell lysates were prepared and immunoblotted for Sp3, Sp4, or β-actin (loading control). Both short Sp3 (sSp3) and long Sp3 (lSp3) isoforms are shown. B, NaBu treatment did not lead to increased Sp3 DNA binding. NIE115 cells were treated with or without 1 mm NaBu for 24 h, and nuclear extracts from harvested cells were incubated with biotinylated PKCδ promoter probe spanning the GC(1) and GC(2) sites. The presence of Sp3 was detected by immunoblotting analysis. A representative immunoblot is shown. C, stimulation by NaBu of the Sp1/3 transactivational potential. The reporter plasmid pG5-luc, which contains five Gal4-binding sites upstream of a minimal TATA box, and the effector plasmids for Gal4 (pM), Gal4-Sp3 (pM-Sp3), Gal4-Sp3DBD (pM-Sp3DBD), Gal4-Sp1 (pM-Sp1), Gal4-Sp1DBD (pM-Sp1DBD) were cotransfected into NIE115 (left panel) and MN9D (right panel) cells and incubated with or without 1 mm NaBu for 24 h. Luciferase activities were then determined and normalized by cellular protein. Values are expressed as the fold induction of luciferase activity following transfection of the pG5-luc alone without NaBu treatment and are represented as means ± S.E. of three independent experiments performed in triplicate. Above each blue bar is the fold change in activity in the presence of NaBu over that observed in the absence of NaBu. D, effects of overexpression of HDAC isoforms on the NaBu-induced Gal4-Sp1 (left panel) and Gal4-Sp3 (right panel) transactivation. NIE115 cells were cotransfected with reporter plasmid pG5-luc and 8 μg of Gal4-Sp1 or Gal4-Sp3 in combination with 4 μg of HDAC1, HDAC4, HDAC5, or HDAC7 expression plasmids or empty vector control (pcDNA3.1). The cells were then treated with or without NaBu (1 mm) for 24 h, and luciferase activities were determined. Values are expressed as fold induction over the activity obtained following transfection of the Gal4 without NaBu treatment and are represented as means ± S.E. of three independent experiments performed in triplicate. ***, p < 0.001; pCDNA3.1-transfected versus HDACs-transfected samples.
Characterizing Domains of Sp1 and Sp3 for Mediating Responsiveness of PKCδ to Sodium Butyrate
Sp transcription factors (Sp1, Sp3, and Sp4) contain several conserved regions constituting two transactivation domains (A and B boxes) close to the C terminus with regions rich in serine/threonine and glutamine residues, an extreme N-terminal transactivation domain (D box), an N-terminal DNA binding domain (zinc finger), and a domain of highly charged amino acids (C box) immediately adjacent to the zinc finger on its N-terminal side. Additionally, Sp1 and Sp3 each possess an inhibitory domain located in the extreme N terminus of Sp1 and near the C terminus of Sp3, respectively (81, 82). To characterize the regions of Sp1/3 required for NaBu responsiveness, a series of truncated Gal4-Sp1 or Gal4-Sp3 expression constructs was generated and is depicted schematically in Fig. 10, A and C. Similar to the above experiments, the ability of these chimeric proteins to transactivate pG5-luc activity in both the presence and absence of NaBu was assayed in NIE115 cells. As shown in Fig. 10, B and D, the chimeras that retain the entire N-terminal part (A+B+C boxes) or A+B boxes of Sp1 (Gal4-Sp1N and Gal4-Sp1AB) or Sp3 (Gal4-Sp3AB) displayed comparable capacities to activate transcription in response to NaBu in comparison with Gal4-Sp1 or Gal4-Sp3 full-length fusions. Interestingly, the Gal4-Sp3AB chimera lacking the inhibitory domain located adjacent to zinc fingers even confers a higher NaBu responsiveness to the G5-luc reporter construct than that obtained following overexpression of the Gal4-Sp3 full-length protein, suggesting that the inhibitory domain of Sp3 may have a negative regulatory action in mediating NaBu induction of PKCδ promoter activity. Further analysis of the N-terminal region (A+B boxes) revealed that sequences within the three subdomains AGln, BSer/Thr, and BGln, corresponding to amino acids Sp1(146–494) and Sp3(81–499), are essential to the transactivation actions of NaBu, as removal of any one of the three subdomains showed a significant decrease in their capacity to mediate the butyrate-induced transactivation. Furthermore, each of these subdomains was unable to render the G5-luc reporter construct NaBu responsiveness. Interestingly, the ASer/Thr subdomain of Sp1(83–145) had no influence on the ability of NaBu to enhance the transcriptional activity.
FIGURE 10.

Localization of the domains of Sp1 and Sp3 that are activated in response to NaBu stimulation. A and C, schematic diagram of the expression constructs carrying Gal4-Sp1 (A) and Gal4-Sp3 (C) fusion proteins with each of the indicated portions of Sp1 or Sp3. Amino acid positions demarcating each domain are indicated. AS/T, serine/threonine-rich subdomain within the A box; AQ, glutamine-rich subdomain within the A box; BS/T, serine/threonine-rich subdomain within the B box; BQ, glutamine-rich subdomain within the B box; C, C box, ID, inhibitory domain; D, D box. B and D, expression plasmids as shown in A and C were cotransfected into NIE115 cells with the pG5-luc reporter plasmid. Gal4 (pM) is the empty vector control plasmid. At 24 h post-transfection, cells were treated with or without NaBu (1 mm) for 24 h. Luciferase activities were then determined and normalized by cellular protein. Values are expressed as fold induction by NaBu for each transfected sample and are represented as means ± S.E. of three independent experiments performed in triplicate.
Ectopic Expression of p300/CBP Stimulates Sodium Butyrate-mediated Transactivation of Sp1 and Sp3
Given that the HATs p300 and CBP can function as coactivators for Sp transcription factors, we investigated whether there is a cooperative function of p300 or CBP and Sp1/3 in the NaBu-stimulated PKCδ promoter activation. We addressed the question by performing cotransfection assays with expression vectors for p300 or CBP and measuring the transcriptional activity of Sp1/3 with the aforementioned Gal4 luciferase assay one-hybrid system. The coexpression of CBP or p300 significantly enhanced the NaBu-induced transactivation of Gal4-Sp1 and Gal4-Sp3 in a dose-dependent manner (Fig. 11, A and B). Interestingly, we found that coexpression of a p300 mutant lacking HAT activity did not affect the NaBu-stimulated transcriptional activity of Gal4-Sp1 and Gal4-Sp3. These findings indicate that p300/CBP synergistically participates in the Sp-dependent transcription by NaBu.
FIGURE 11.

Expression of CBP/p300 stimulates NaBu-induced transactivation of Sp1 and Sp3. A and B, NIE115 cells were cotransfected with Gal4-Sp1 or Gal4-Sp3 expression constructs, the luciferase reporter plasmid pG5-luc, and the indicated amounts of CMV-driven expression vectors for p300, p300dHAT (A), or CBP (B). The cells were then treated with or without NaBu (1 mm) for 24 h. Luciferase activities were determined. Values are expressed as the fold induction over the activity obtained following transfection of the Gal4 without NaBu treatment and are represented as means ± S.E. of three independent experiments performed in triplicate.
Functional Studies in Human Dopaminergic Neuronal Cells and Organotypic Brain Slice Culture Models Show Acetylation-induced PKCδ Up-regulation Enhances Sensitivity to Oxidative Stress
Our biochemical experiments described above suggest that HDAC inhibition promotes the expression of PKCδ protein and mRNA through Sp-dependent transcription activation in neuronal cells. The ultimate step in our study was to explore the functional relevance of PKCδ up-regulation due to hyperacetylation. Evidence to date has implicated PKCδ as a key regulator of oxidative stress-induced neuronal cell death (10, 12). We therefore reasoned that increased level of PKCδ seen after hyperacetylation enhances the sensitivity property of dopaminergic neurons in response to oxidative stress. To test this hypothesis, we utilized the human dopaminergic cell cultures, namely LUHMES, and analyzed the role of NaBu-induced PKCδ up-regulation on oxidative stress-induced neuronal cell death. Differentiated LUHMES cells display unique properties of human dopaminergic neurons and offer a suitable model to replace primary neuron cultures (64, 65). Consistent with this, our own analysis revealed that LUHMES cells express the dopamine transporter, vesicular monoamine transporter 2 (VMAT-2), tyrosine hydroxylase, and the neuronal marker β-III-tubulin upon 5–6 days of differentiation (data not shown). The dopaminergic specific toxin 6-OHDA, which is widely used as an oxidative stress inducer (83, 84), was chosen as a test compound. First, we confirmed that treatment with 1 mm butyrate for 24 h increased PKCδ protein levels in differentiated LUHMES cells by Western blot (Fig. 12A) and immunocytochemical analysis (Fig. 12B). We then proceeded to analyze the 6-OHDA-induced oxidative toxicity by MTS viability and dopamine reuptake assays. To determine the effects of PKCδ, we applied the PKCδ inhibitor rottlerin, which has been shown to protect against MPTP and methamphetamine dopaminergic neurotoxicity and alleviate LPS-induced neuroinflammation both in vitro and in vivo (16, 18, 85). Our preliminary analyses indicate 0.3 μm rottlerin for a 24-h treatment would be optimal for differentiated LUHMES cells (data not shown). As shown in Fig. 12, C and D, 30 μm 6-OHDA treatment led to significant neurotoxicity in the LUHMES dopaminergic neuronal model, as evidenced from reduced dopamine reuptake and cell viability. In contrast, pretreatment and cotreatment with 1 mm NaBu substantially potentiated 6-OHDA-induced cellular toxicity in these assays. However, exposure to 1 mm NaBu or 0.3 μm rottlerin had only marginal effects in unstressed LUHMES cells when added alone. Rottlerin effectively inhibited the LUHMES cell death brought about by 6-OHDA treatment in the presence of NaBu. Concerning the specificity of rottlerin (86), we further evaluated the effects of PKCδ depletion by lentiviral transduction of short hairpin RNA (shRNA) in LUHMES cells. We used an shRNA set with five individual clones and tested their efficacy to knock down PKCδ mRNA. As shown in Fig. 12E, four of the five tested shRNA clones knocked down PKCδ mRNA by >50%, with sh-PKCδ #1 being the most effective (∼70% silencing). By MTS viability analysis, we found that PKCδ depletion remarkably blocked the 6-OHDA-induced cell death in the absence or in the presence of NaBu. More importantly, we showed that knockdown of PKCδ could also suppress the NaBu-potentiated 6-OHDA cytotoxicity. As depicted in Fig. 12F (left panel), in the presence of NaBu, a significantly increased (p < 0.05) 6-OHDA-induced cell death was again observed in scrambled shRNA-infected cells. However, NaBu had no significant effect (p > 0.05) on cell death induced by 6-OHDA when cells were transduced with sh-PKCδ #1. The effect of sh-PKCδ #1 on PKCδ protein expression was also confirmed by Western blotting. As shown in Fig. 12F (right panel), infection with sh-PKCδ #1 lentivirus for 48 and 96 h almost completely diminished PKCδ. Taken together, these results demonstrate that PKCδ up-regulation resulting from histone hyperacetylation sensitizes dopaminergic neurons to oxidative damage.
FIGURE 12.

Increased PKCδ potentiates 6-OHDA-induced oxidative damage in differentiated human dopaminergic LUHMES cells. A and B, differentiated LUHMES cells were treated with 1 mm NaBu, and PKCδ expression was analyzed by immunoblot (A) and immunocytochemistry (B). For immunoblot, densitometric quantitation of the ratio of band intensity of PKCδ and β-actin from three independent experiments (means ± S.E.; *, p < 0.05) is shown on the bottom panel. For immunocytochemistry, cells were immunostained for PKCδ (green) and neuron-specific marker β-III tubulin (TuJ1, red), and the nuclei were counterstained by Hoechst 33342 (blue). Images were taken using a Nikon TE2000 fluorescence microscope. Magnification, ×40. Scale bar, 20 μm. Representative immunofluorescence images are shown. C and D, differentiated LUHMES cells were pretreated with or without 1 mm NaBu or 0.3 μm rottlerin for 1 h and then cotreated with 30 μm 6-OHDA for 24 h. After treatment, 6-OHDA-induced oxidative toxicity was measured using the dopamine assay (C) and MTS assay (D). E, LUHMES cells were pre-differentiated for 2 days followed by 2 days of transduction with shRNA lentivirus targeting against human PKCδ (sh-PKCδ) or scrambled lentivirus (sh-Scr). Real time RT-PCR analysis of PKCδ mRNA level was performed. 18 S rRNA level served as internal control. F, left panel, LUHMES cells infected with sh-PKCδ #1 or scrambled lentivirus were pretreated with or without 1 mm NaBu for 1 h and then cotreated with 30 μm 6-OHDA for 24 h. Cell viability was analyzed by MTS assay. All data are expressed as the mean ± S.E. of three independent experiments and are shown as a percentage of control or scrambled shRNA-infected control cultures. Right panel, LUHMES cells infected with sh-PKCδ #1 or scrambled lentivirus for 48 or 96 h were analyzed for PKCδ expression. Representative immunoblots are shown. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Finally, we extended our findings from a human dopaminergic cell culture model to an ex vivo organotypic brain slice culture model. Brain slices preserve the tissue architecture of the brain regions and can replicate many aspects of the in vivo context. The organotypic slice cultures of corticostriatal tissue have been used extensively to study the mechanistic basis of neurodegeneration (72). We prepared organotypic slice cultures from adult C57BL6 mice and assayed neuronal cell damage after dopaminergic neurotoxicant MPP+ treatment in the presence and absence of butyrate and/or rottlerin. Cell damage was determined by PI uptake assay. As illustrated in Fig. 13, A and B, control slices showed minimal PI uptake. Likewise, neither NaBu (1 mm) nor rottlerin (5 μm) on its own caused a noticeable increase in PI-labeled cells relative to control slices. However, slices exposed to 300 μm MPP+ displayed great PI labeling neurodegeneration. Consistent with data obtained in LUHMES cells, in the presence of 1 mm NaBu, there was a significant increase in MPP+-induced neurodegeneration. Most notably, rottlerin at 5 μm greatly suppressed the PI-labeling degenerating neurons induced by MPP+ treatment in the presence and absence of NaBu, confirming a critical role for NaBu-mediated PKCδ up-regulation in promoting oxidative stress-induced toxicity. To further establish the role of increased PKCδ levels during oxidative stress, we examined the organotypic striatal slice cultures prepared from PKCδ knock-out (PKCδ−/−) mice. Similarly, PKCδ−/− and wild-type organotypic brain slice cultures were treated with 300 μm MPP+ for 24 h in the presence and absence of 1 mm NaBu and analyzed cell death by PI uptake analysis. In wild-type slice cultures, NaBu treatment consistently augmented MPP+-induced neuronal cell death. In contrast, NaBu-induced enhancement of MPP+ neurotoxicity was significantly attenuated in PKCδ−/− slice cultures (Fig. 13, C and D). To further substantiate the PI staining results, we used Fluoro-Jade, which is capable of selectively staining degenerating neurons in brain tissues and organotypic slices (69, 70, 72, 87). The organotypic brain slice cultures from wild-type and PKCδ knock-out pups were subjected to NaBu and MPP+ as described above, and neurotoxicity was measured by Fluoro-Jade staining. Again, PKCδ deficiency significantly diminished the NaBu-induced increase in MPP+ neurotoxicity (Fig. 13, E and F). The effect of NaBu treatment (1 mm for 24 h) on PKCδ protein expression in wild-type organotypic striatal slice cultures was also confirmed by Western blot and immunohistochemistry analyses (Fig. 13, G and H). These data are consistent with the analyses of LUHMES cells above and establish a functional correlation between PKCδ up-regulation induced by hyperacetylation and altered sensitivity to oxidative stress in neuronal cells.
FIGURE 13.

Increased PKCδ sensitizes organotypic corticostriatal slice cultures to MPP+-induced neurodegeneration. A, organotypic corticostriatal slice cultures from wild-type adult C57 black mice were preincubated in the presence or absence of 1 mm NaBu and/or 5 μm rottlerin for 3 h followed by a 24-h incubation with 300 μm MPP+. Cell death was measured using PI uptake assay. Representative PI uptake fluorescent images are shown. Images were obtained using a Nikon TE2000 fluorescence microscope. Magnification, ×2. Scale bar, 500 μm. B, quantification of PI fluorescence intensity. PI fluorescence was measured in each group using ImageJ software. C, organotypic corticostriatal slice cultures from wild-type and PKCδ knock-out (PKCδ−/−) C57 black mice were pretreated with or without 1 mm NaBu for 3 h and then cotreated with 300 μm MPP+ for 24 h, and cell death was measured using PI uptake assay. Representative PI uptake fluorescent images are shown. Magnification, ×2. Scale bar, 500 μm. D, quantification of PI fluorescence intensity. E, organotypic corticostriatal slice cultures from 9- to 12-day-old wild-type and PKCδ−/− pups were pretreated with or without 1 mm NaBu for 3 h and then cotreated with 300 μm MPP+ for 24 h, and cell death was measured using Fluoro-Jade staining assay. Representative Fluoro-Jade fluorescent images are shown. Magnification, ×20. Scale bar, 100 μm. F, quantification of Fluoro-Jade fluorescence intensity. G, organotypic corticostriatal slice cultures from adult C57 black mice were treated with 1 mm NaBu for 24 h, and levels of PKCδ were analyzed by immunoblot. H, exposure of organotypic corticostriatal slices to 1 mm NaBu increased PKCδ immunoreactivity. After treatment, cultures were immunostained for PKCδ (green) and neuron-specific marker β-III tubulin (red), and the nuclei were counterstained by Hoechst 33342 (blue). Images were taken using a Nikon TE2000 fluorescence microscope. Magnification, ×20. Scale bar, 50 μm. Representative immunofluorescence images are shown. Quantification of normalized PKCδ (PKCδ/TuJ1) immunoreactivity is shown in the right panel. Fluorescence immunoreactivity of PKCδ and TuJ1 was measured in each group using ImageJ software. All data expressed as percent of wild-type control (con) group are mean ± S.E. and representative for results obtained from three separate experiments. ns, not significant. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
DISCUSSION
In this study, we present evidence for a novel model of mouse PKCδ transcriptional regulation by an epigenetic control mechanism involving HDAC inhibition and histone hyperacetylation in vitro and in vivo. These findings are particularly interesting because PKCδ is a major kinase critically involved in apoptotic signaling in various cell types. Indeed, there is considerable evidence supporting the notion that activation of PKCδ via caspase-dependent proteolysis plays an essential role in oxidative stress-induced dopaminergic cell death in PD (11, 12, 14, 15). Lines of evidence have also demonstrated that the PKCδ modulators exhibit a neuroprotective effect in the MPTP mouse model (16). The prominent role PKCδ plays in regulating multiple biological events would suggest that its expression is tightly regulated. Although a number of studies have documented changes in PKCδ levels in response to multiple stimuli, knowledge of the regulation of PKCδ gene expression at the transcriptional level is rather limited. The PKCδ promoter lacks a TATA or TATA-like box and contains GC-rich sequences in the proximal promoter region (34, 88). We have recently characterized the PKCδ promoter in neuronal cells (35, 38). We showed that a proximal 400-bp genomic fragment surrounding the transcription start site functions as a basal PKCδ promoter to sustain the basal expression of PKCδ in neurons, and we identified multiple functional transcription factor-binding sites that contribute to basal PKCδ expression, including two for NFκB and five for Sp family transcription factors. Nevertheless, the epigenetic regulation of PKCδ expression mediated by histone acetylation in neuronal cells has never been reported. Thus, we carried out a comprehensive study to determine whether altered histone acetylation homeostasis has a regulatory role in PKCδ expression in neurons.
We initiated our study by evaluating the possible alterations in PKCδ levels after exposing cultured nigrostriatal neurons to the HDAC inhibitor NaBu, and our result showed that PKCδ protein levels are dramatically increased in NaBu-exposed cells (Fig. 1A). Importantly, this induction of PKCδ also occurred in human dopaminergic LUHMES neurons (Fig. 12, A and B), in mouse organotypic slice cultures (Fig. 13, G and H), and in a mouse model following acute NaBu treatment (Fig. 3, A and B), suggesting that histone acetylation-mediated PKCδ expression occurs both in vitro and in vivo. Interestingly, our in vivo findings suggest a brain region-specific regulation of PKCδ protein expression following NaBu treatment. The precise mechanism underlying regional specific up-regulation of PKCδ gene expression and its relationship to histone acetylation remain unclear. NaBu has been shown to induce hyperacetylation of nuclear histones in other brain regions, such as the cortex and hippocampus (89–91), suggesting that global histone acetylation alone would not explain the region-specific PKCδ gene up-regulation. One possibility is that NaBu induces a region-specific histone acetylation status on the PKCδ gene promoter. Another likely explanation would be that NaBu alters the acetylation of key regulators governing PKCδ expression, such as NFκB and Sp proteins, in a region-specific manner. We also demonstrate that the up-regulation of PKCδ protein levels by NaBu correlates with increased PKCδ mRNA levels. Other structurally unrelated HDAC inhibitors, including apicidin, scriptaid, and TSA, also induced PKCδ mRNA, implying that the NaBu induction of robust PKCδ expression appears not to be due to nonspecific action of NaBu. Interestingly, these findings, combined with previous evidence, indicate a cell type-specific regulation of PKCδ gene expression in response to HDAC inhibition. For instance, in nasopharyngeal carcinoma and AA/C1 adenoma cells, HDAC inhibitors, such as TSA and NaBu, did not alter the PKCδ expression levels but rather led to a distinct cellular sub-localization of the PKCδ protein (92, 93). By contrast, in human hepatoma Hep3B cells, TSA was found to specifically down-regulate PKCδ levels (94). Of note, however, this study did not address the specific mechanisms responsible for TSA-induced PKCδ repression or its potential functional relevance.
We next investigated the molecular mechanism underlying NaBu-mediated PKCδ gene activation. Analysis of systemic histone acetylation levels indicated that NaBu significantly increased cellular histone acetylation levels. Importantly, an increase in PKCδ promoter histone acetylation was observed after NaBu treatment (Fig. 4B). Thus, these results suggest that the HDAC inhibitor mediated chromatin remodeling by enhancing histone acetylation, which constitutes a basic mechanism underlying the NaBu induction of PKCδ. To clarify whether the up-regulation of PKCδ mRNA is accompanied by activation of the PKCδ promoter, we analyzed PKCδ promoter activity using a promoter region (−1494/+289) that we cloned recently (35). Our results indicate that NaBu and other HDAC inhibitors significantly increased the luciferase activity of this reporter (Fig. 5, A–E). Furthermore, luciferase reporter assays using serial deletion PKCδ reporter constructs revealed that the major histone acetylation response elements resided in the 289-bp noncoding exon 1 region (Fig. 6). The proximal region of the PKCδ promoter conferring NaBu responsiveness is GC-rich and contains multiple Sp-binding sites, including one proximal CACCC box and four distal consecutive GC boxes. Previously, we have shown that the CACCC box and the GC boxes act differentially in mediating promoter activation by ectopic expression of Sp transcription factors (35). Therefore, we performed experiments to determine the possible involvement of these Sp sites in the NaBu-mediated induction (Fig. 7). Unexpectedly, a smaller construct, namely pGL3−147/+209, which possesses the upstream CACCC box but lacks the downstream GC boxes, was not activated by NaBu treatment. Moreover, NaBu strongly reduced promoter activity in this promoter context (−147 to +209) in MN9D cells, suggesting that the CACCC box is not required for NaBu induction. Indeed, mutation of the CACCC box had no effect on NaBu-mediated activation of the PKCδ promoter. However, using another small construct, pGL3+165/+289, we found that all GC boxes are required for the full response to NaBu. Moreover, cooperative actions of different GC boxes are required for mediating the NaBu response, because triple mutation of any three GC boxes completely diminished it. Analysis using a luciferase reporter (Sp1-luc) containing three Sp1 consensus sequences further implicates the cluster of four GC boxes in NaBu-induced transcriptional control of the PKCδ promoter.
The Sp family of transcription factors, Sp1, Sp3, and Sp4, are all structurally similar and bind to GC and GT/CACCC boxes found in a variety of promoter and enhancers through three characteristic zinc fingers located at the C terminus of the proteins. Sp1 and Sp3 are ubiquitously expressed, whereas the expression of Sp4 is limited to the brain (95, 96). Growing studies have implicated GC-rich Sp1-binding sites in the regulation of a number of HDAC inhibitor-regulated genes, including the IN4K gene (97), WAF1/Cip gene (62, 98), HMG-CoA synthase gene (99), HSP70 gene (100), EGFR gene (101), and MMP11 gene (102). Dissecting the mechanism of NaBu induction of the PKCδ gene revealed a dependence on Sp proteins. First, histone acetylation-induced PKCδ promoter activity was dramatically enhanced by overexpressing the Sp1, Sp3, or Sp4 protein (Fig. 8A). The most potent effect was provided by exogenous Sp3, whereas Sp1 and Sp4 displayed weaker activation, consistent with our observation that Sp3 is the strongest transactivator of basal PKCδ promoter activity (35). Next, we cloned a dominant-negative isoform of the Sp1 or Sp3 protein, which has an intact DNA binding domain but lacks the full transactivation domains. Its ectopic expression was effective in abolishing the synergistic activation of PKCδ promoter activity by wild-type Sp1/3 and NaBu (Fig. 8B). Finally, we used pharmacological inhibitors to block the Sp signaling pathways and assessed their effects on NaBu-stimulated PKCδ promoter activity. Fig. 8C shows that mithramycin A, an inhibitor of Sp-mediated transcriptional activation (35, 78, 79), directly blocks histone hyperacetylation-induced PKCδ promoter activity. In addition, tolfenamic acid, another Sp inhibitor known to induce degradation of Sp proteins (80), also significantly diminishes the NaBu response (Fig. 8D). We therefore conclude that NaBu activates PKCδ transcription via Sp3, Sp1, and Sp4.
Although neither Sp3 and Sp4 levels nor direct association of Sp3 and Sp4 with the PKCδ promoter was affected by NaBu, treatment with NaBu significantly stimulated Sp1- and Sp3-medated luciferase activity of the Gal4-luc reporter construct in one-hybrid assays (Fig. 9C). Sp1 and Sp3 contain multiple domains, including a zinc finger DNA-binding domain and a bipartite transactivation domain composed of glutamine-rich and serine/threonine-rich regions. Using a serial Gal4-Sp1 or Gal4-Sp3 fusion chimeric, we were able to show that the increased transactivational potency of Sp1 and Sp3 induced by NaBu is specific to the transactivation domains of Sp1 and Sp3. The three subdomains AGln, BSer/Thr, and BGln (amino acids from 146 to 494 in Sp1; amino acids from 81 to 499 for Sp3) are all required for histone acetylation-induced transcription from the PKCδ promoter. It remains unclear how the transcriptional capacities of Sp1 and Sp3 are up-regulated by histone acetylation. Regulation of Sp1 and Sp3 activity could be achieved by protein-protein interactions. Previous reports demonstrated that both Sp1 and Sp3 functionally interact with HDAC1, HDAC2, and HDAC4 (103–106). HDACs act as transcriptional repressors, repressing gene expression by forming complexes with several corepressors, including mSin3A, SMRT, and N-CoR (107). In our experimental conditions, the consistent expression pattern of the HDAC isoforms 1–4, but not 5 and 7 (Fig. 5H), was intriguing and suggests a role for HDACs 1–4 in mediating histone acetylation-induced PKCδ up-regulation. In line with this, we found that PKCδ protein levels were significantly up-regulated after the individual knockdown of the class I HDACs (HDAC1 and -2) (Fig. 5, H and I), as seen following NaBu treatment. At present, however, it is unclear whether HDAC3/4 deacetylase isoforms contribute to NaBu's regulation of the PKCδ promoter. However, we found that overexpression of HDAC4 dramatically reduced NaBu's enhancement of the transcriptional activity of Gal4-Sp1 and Gal4-Sp3 (Fig. 9D). It is also noteworthy that HDAC4 alone is enzymatically inactive, and it acts as a scaffold protein that recruits catalytically active HDACs such as HDAC1 and HDAC3 into the transcriptional corepressor complexes (106, 108). Thus, further investigations are needed to fully elucidate whether HDAC3/4 participates in the PKCδ up-regulation upon HDAC inhibition and, if so, which catalytically active HDAC isoform is required.
Additionally, Sp1 and Sp3 also bind directly to coactivators p300 and its homolog CBP (109, 110). Our results indicate ectopic expression of p300/CBP-stimulated Gal4-Sp1- and Gal4-Sp3-dependent transcription in the presence of NaBu. Interestingly, the p300 stimulation is independent of HAT activity. These data suggest that the cooperative, possibly physical, interactions between Sp proteins and p300/CBP may represent a secondary mechanism responsible for the NaBu-stimulated transactivating activity of Sp1/3. The recruitment of p300/CBP into the transcription complex is also supported by our previous observation that transcription from the PKCδ promoter is significantly activated by overexpression of p300/CBP (35). Taken together, it seems likely that NaBu-induced acetylation alters the transcriptional activities of Sp1 and Sp3 by inhibiting HDAC activity, disrupting the repressor complex containing HDACs, and allowing the recruitment of the coactivators p300/CBP to the transcription complex bound to the GC boxes on the PKCδ promoter.
In addition to protein-protein interactions, regulating the activities of Sp proteins also includes post-translational modifications. For example, acetylation of Sp1/Sp3 stimulates their activity (111, 112), whereas sumoylation of Sp1/Sp3 inactivates them (113). Moreover, phosphorylation of Sp1 also mediates the activation of Sp-dependent transcription (114). Our preliminary in vitro results suggest that NaBu does not cause gross change in the amount of acetylation of Sp1/3, which is supported by our observation that transcriptional activation by NaBu is HAT-independent. However, it remains to be examined whether it is true in the in vivo situation. It is also unclear whether HDAC inhibition modulates specific intracellular signaling pathways to affect the amount of phosphorylation or other modifications of Sp or Sp-interacting proteins.
Thus far, the specific role of HDAC activity in neuronal survival and cell death has been a matter of debate. Experimental evidence has suggested that activation of HDACs or their pharmacological inhibition can have both protective and toxic effects, depending on cell type and proliferation state (46, 56, 115, 116). Different mechanisms have been postulated to explain how HDAC activity promotes or protects against neurodegeneration. One postulated mechanism is through an altered balance between the expression of proapoptotic and anti-apoptotic genes. Accordingly, a key goal for studies on HDAC enzymes and their pharmacological inhibitors is to identify their in vivo downstream effectors. In this regard, we provide evidence here that the proapoptotic gene PKCδ can be up-regulated by HDAC inhibition, and more importantly, we show that increased levels of PKCδ kinase exacerbate neuronal injury and degeneration, thereby providing a new molecular insight into the enhanced vulnerability of dopaminergic neurons during histone acetylation dysregulation. Also, we believe that our study will promote others in the field to take a closer look at the role deacetylase inhibition plays in neurons, which may facilitate optimization of the clinical applicability of deacetylase inhibitors. Notably, although HDAC inhibitors invariably induce hyperacetylated histones and facilitate gene expression, only a small number of genes (2–17%) have been found to alter their expression upon HDAC inhibition (117). Our studies herein add the PKCδ gene to this small list.
Besides transcriptional regulation of PKCδ production, PKCδ activity can be regulated at multiple integrated levels, including caspase-mediated proteolysis and coordinated phosphorylation. Our published (5, 35) and preliminary results indicate that both the neurotoxin MPP+ and the oxidative stress-inducer hydrogen peroxide do not alter the transcriptional machinery to increase PKCδ activity but may activate the kinase by activation loop phosphorylation or caspase-mediated proteolysis. It is likely that the nature of neurotoxic agents, as well as their severity and duration of exposure, may dictate PKCδ gene up-regulation. In this regard, recent evidence has linked exposure to environmental chemicals and toxicants, such as pesticides and metals, with altered acetylation homeostasis in cells (43, 118–120). Particularly, we reported that dieldrin and paraquat, two neurotoxic pesticides implicated in the etiopathogenesis of PD, increase cellular histone acetylation by modulating HDAC and HAT activity (41, 42). Future studies should clarify whether chronic exposure to environmental neurotoxic pesticides increases PKCδ expression in nigral dopaminergic neurons due to hyperacetylation. Nevertheless, our present results show that imbalanced histone acetylation/deacetylation can result in abnormal expression of the apoptotic PKCδ, providing mechanistic insights into hyperacetylation-mediated epigenetic changes and the subsequent degenerative fate of neurons.
In summary, we demonstrate here for the first time that modulation of the HAT/HDAC balance by inhibiting HDAC activity induces proapoptotic PKCδ transcription in neurons. Moreover, PKCδ induction is triggered by the acetylation of histone proteins associated with the PKCδ promoter and the subsequent enhancement of the transcriptional capacities of Sp transcription factors. Our results further demonstrate that up-regulated PKCδ levels produce a proapoptotic state, thereby increasing susceptibility to neurotoxic insults. We propose that induction of proapoptotic PKCδ by HDAC inhibition in dopaminergic neurons may represent a novel epigenetic molecular basis for the neurodegenerative processes in etiology of Parkinson disease.
Acknowledgment
We thank Gary Zenitsky for assistance in the preparation of this manuscript.
This work was supported, in whole or in part, by National Institutes of Health RO1 Grants ES10586 and NS074443 (to A. G. K.), NS65167 (to A. K.), and GM055835 (to A. R.). This work was also supported by the W. Eugene and Linda Lloyd Endowed Chair Professorship (to A. G. K.) and the Dean Professorship (to A. K.).
- DAG
- diacylglycerol
- 6-OHDA
- 6-hydroxydopamine
- HAT
- histone acetyltransferases
- HDAC
- histone deacetylase
- MPP+
- 1-methyl-4-phenylpyridinium
- NaBu
- sodium butyrate
- TSA
- trichostatin A
- PI
- propidium iodide
- PD
- Parkinson disease
- VPA
- valproic acid
- CBP
- cAMP-response element-binding protein-binding protein
- LUHMES
- Lund human mesencephalic
- MPTP
- 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)2H-tetrazolium.
REFERENCES
- 1. Dempsey E. C., Newton A. C., Mochly-Rosen D., Fields A. P., Reyland M. E., Insel P. A., Messing R. O. (2000) Protein kinase C isozymes and the regulation of diverse cell responses. Am. J. Physiol. Lung Cell Mol. Physiol 279, L429–L438 [DOI] [PubMed] [Google Scholar]
- 2. Brodie C., Blumberg P. M. (2003) Regulation of cell apoptosis by protein kinase Cδ. Apoptosis 8, 19–27 [DOI] [PubMed] [Google Scholar]
- 3. Zhang D., Kanthasamy A., Yang Y., Anantharam V., Kanthasamy A. (2007) Protein kinase Cδ negatively regulates tyrosine hydroxylase activity and dopamine synthesis by enhancing protein phosphatase-2A activity in dopaminergic neurons. J. Neurosci. 27, 5349–5362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Anantharam V., Kitazawa M., Wagner J., Kaul S., Kanthasamy A. G. (2002) Caspase-3-dependent proteolytic cleavage of protein kinase Cδ is essential for oxidative stress-mediated dopaminergic cell death after exposure to methylcyclopentadienyl manganese tricarbonyl. J. Neurosci. 22, 1738–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kaul S., Kanthasamy A., Kitazawa M., Anantharam V., Kanthasamy A. G. (2003) Caspase-3-dependent proteolytic activation of protein kinase Cδ mediates and regulates 1-methyl-4-phenylpyridinium (MPP+)-induced apoptotic cell death in dopaminergic cells: relevance to oxidative stress in dopaminergic degeneration. Eur. J. Neurosci. 18, 1387–1401 [DOI] [PubMed] [Google Scholar]
- 6. Kanthasamy A. G., Kitazawa M., Kanthasamy A., Anantharam V. (2005) Dieldrin-induced neurotoxicity: relevance to Parkinson's disease pathogenesis. Neurotoxicology 26, 701–719 [DOI] [PubMed] [Google Scholar]
- 7. Latchoumycandane C., Anantharam V., Jin H., Kanthasamy A. (2011) Dopaminergic neurotoxicant 6-OHDA induces oxidative damage through proteolytic activation of PKCδ in cell culture and animal models of Parkinson's disease. Toxicol. Appl. Pharmacol. 256, 314–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lin M., Chandramani-Shivalingappa P., Jin H., Ghosh A., Anantharam V., Ali S., Kanthasamy A. G., Kanthasamy A. (2012) Methamphetamine-induced neurotoxicity linked to ubiquitin-proteasome system dysfunction and autophagy-related changes that can be modulated by protein kinase Cδ in dopaminergic neuronal cells. Neuroscience 210, 308–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gordon R., Anantharam V., Kanthasamy A. G., Kanthasamy A. (2012) Proteolytic activation of proapoptotic kinase protein kinase Cδ by tumor necrosis factor α death receptor signaling in dopaminergic neurons during neuroinflammation. J. Neuroinflammation 9, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hanrott K., Gudmunsen L., O'Neill M. J., Wonnacott S. (2006) 6-Hydroxydopamine-induced apoptosis is mediated via extracellular auto-oxidation and caspase 3-dependent activation of protein kinase Cδ. J. Biol. Chem. 281, 5373–5382 [DOI] [PubMed] [Google Scholar]
- 11. Hanrott K., Murray T. K., Orfali Z., Ward M., Finlay C., O'Neill M. J., Wonnacott S. (2008) Differential activation of PKCδ in the substantia nigra of rats following striatal or nigral 6-hydroxydopamine lesions. Eur. J. Neurosci. 27, 1086–1096 [DOI] [PubMed] [Google Scholar]
- 12. Kanthasamy A., Jin H., Mehrotra S., Mishra R., Kanthasamy A., Rana A. (2010) Novel cell death signaling pathways in neurotoxicity models of dopaminergic degeneration: relevance to oxidative stress and neuroinflammation in Parkinson's disease. Neurotoxicology 31, 555–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Anantharam V., Kitazawa M., Latchoumycandane C., Kanthasamy A., Kanthasamy A. G. (2004) Blockade of PKCdelta proteolytic activation by loss of function mutants rescues mesencephalic dopaminergic neurons from methylcyclopentadienyl manganese tricarbonyl (MMT)-induced apoptotic cell death. Ann. N.Y. Acad. Sci. 1035, 271–289 [DOI] [PubMed] [Google Scholar]
- 14. Yang Y., Kaul S., Zhang D., Anantharam V., Kanthasamy A. G. (2004) Suppression of caspase-3-dependent proteolytic activation of protein kinase Cδ by small interfering RNA prevents MPP+-induced dopaminergic degeneration. Mol. Cell. Neurosci. 25, 406–421 [DOI] [PubMed] [Google Scholar]
- 15. Kanthasamy A. G., Anantharam V., Zhang D., Latchoumycandane C., Jin H., Kaul S., Kanthasamy A. (2006) A novel peptide inhibitor targeted to caspase-3 cleavage site of a proapoptotic kinase protein kinase Cδ (PKCδ) protects against dopaminergic neuronal degeneration in Parkinson's disease models. Free Radic. Biol. Med. 41, 1578–1589 [DOI] [PubMed] [Google Scholar]
- 16. Zhang D., Anantharam V., Kanthasamy A., Kanthasamy A. G. (2007) Neuroprotective effect of protein kinase Cδ inhibitor rottlerin in cell culture and animal models of Parkinson's disease. J. Pharmacol. Exp. Ther. 322, 913–922 [DOI] [PubMed] [Google Scholar]
- 17. Shin E. J., Duong C. X., Nguyen T. X., Bing G., Bach J. H., Park D. H., Nakayama K., Ali S. F., Kanthasamy A. G., Cadet J. L., Nabeshima T., Kim H. C. (2011) PKCδ inhibition enhances tyrosine hydroxylase phosphorylation in mice after methamphetamine treatment. Neurochem. Int. 59, 39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shin E. J., Duong C. X., Nguyen X. K., Li Z., Bing G., Bach J. H., Park D. H., Nakayama K., Ali S. F., Kanthasamy A. G., Cadet J. L., Nabeshima T., Kim H. C. (2012) Role of oxidative stress in methamphetamine-induced dopaminergic toxicity mediated by protein kinase Cδ. Behav. Brain Res. 232, 98–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kuehn H. S., Niemela J. E., Rangel-Santos A., Zhang M., Pittaluga S., Stoddard J. L., Hussey A. A., Evbuomwan M. O., Priel D. A., Kuhns D. B., Park C. L., Fleisher T. A., Uzel G., Oliveira J. B. (2013) Loss-of-function of the protein kinase Cδ (PKCδ) causes a B-cell lymphoproliferative syndrome in humans. Blood 121, 3117–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Palaniyandi S. S., Sun L., Ferreira J. C., Mochly-Rosen D. (2009) Protein kinase C in heart failure: a therapeutic target? Cardiovasc. Res. 82, 229–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leitges M., Mayr M., Braun U., Mayr U., Li C., Pfister G., Ghaffari-Tabrizi N., Baier G., Hu Y., Xu Q. (2001) Exacerbated vein graft arteriosclerosis in protein kinase Cδ-null mice. J. Clin. Invest. 108, 1505–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miyamoto A., Nakayama K., Imaki H., Hirose S., Jiang Y., Abe M., Tsukiyama T., Nagahama H., Ohno S., Hatakeyama S., Nakayama K. I. (2002) Increased proliferation of B cells and auto-immunity in mice lacking protein kinase Cδ. Nature 416, 865–869 [DOI] [PubMed] [Google Scholar]
- 23. Griner E. M., Kazanietz M. G. (2007) Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer 7, 281–294 [DOI] [PubMed] [Google Scholar]
- 24. Chou W. H., Messing R. O. (2005) Protein kinase C isozymes in stroke. Trends Cardiovasc. Med. 15, 47–51 [DOI] [PubMed] [Google Scholar]
- 25. Kim J. H., Kim J. H., Jun H. O., Yu Y. S., Kim K. W. (2010) Inhibition of protein kinase Cδ attenuates blood-retinal barrier breakdown in diabetic retinopathy. Am. J. Pathol. 176, 1517–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Varga A., Czifra G., Tállai B., Németh T., Kovács I., Kovács L., Bíró T. (2004) Tumor grade-dependent alterations in the protein kinase C isoform pattern in urinary bladder carcinomas. Eur. Urol. 46, 462–465 [DOI] [PubMed] [Google Scholar]
- 27. Pongracz J., Clark P., Neoptolemos J. P., Lord J. M. (1995) Expression of protein kinase C isoenzymes in colorectal cancer tissue and their differential activation by different bile acids. Int. J. Cancer 61, 35–39 [DOI] [PubMed] [Google Scholar]
- 28. D'Costa A. M., Robinson J. K., Maududi T., Chaturvedi V., Nickoloff B. J., Denning M. F. (2006) The proapoptotic tumor suppressor protein kinase C-δ is lost in human squamous cell carcinomas. Oncogene 25, 378–386 [DOI] [PubMed] [Google Scholar]
- 29. Reno E. M., Haughian J. M., Dimitrova I. K., Jackson T. A., Shroyer K. R., Bradford A. P. (2008) Analysis of protein kinase Cδ (PKCδ) expression in endometrial tumors. Hum. Pathol. 39, 21–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koponen S., Goldsteins G., Keinänen R., Koistinaho J. (2000) Induction of protein kinase Cδ subspecies in neurons and microglia after transient global brain ischemia. J. Cereb. Blood Flow Metab. 20, 93–102 [DOI] [PubMed] [Google Scholar]
- 31. Miettinen S., Roivainen R., Keinänen R., Hökfelt T., Koistinaho J. (1996) Specific induction of protein kinase Cδ subspecies after transient middle cerebral artery occlusion in the rat brain: inhibition by MK-801. J. Neurosci. 16, 6236–6245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goldberg M., Steinberg S. F. (1996) Tissue-specific developmental regulation of protein kinase C isoforms. Biochem. Pharmacol. 51, 1089–1093 [DOI] [PubMed] [Google Scholar]
- 33. Azadi S., Paquet-Durand F., Medstrand P., van Veen T., Ekström P. A. (2006) Up-regulation and increased phosphorylation of protein kinase C (PKC) δ, μ, and θ in the degenerating rd1 mouse retina. Mol. Cell. Neurosci. 31, 759–773 [DOI] [PubMed] [Google Scholar]
- 34. Suh K. S., Tatunchak T. T., Crutchley J. M., Edwards L. E., Marin K. G., Yuspa S. H. (2003) Genomic structure and promoter analysis of PKC-δ. Genomics 82, 57–67 [DOI] [PubMed] [Google Scholar]
- 35. Jin H., Kanthasamy A., Anantharam V., Rana A., Kanthasamy A. G. (2011) Transcriptional regulation of pro-apoptotic protein kinase Cδ: implications for oxidative stress-induced neuronal cell death. J. Biol. Chem. 286, 19840–19859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu J., Yang D., Minemoto Y., Leitges M., Rosner M. R., Lin A. (2006) NF-κB is required for UV-induced JNK activation via induction of PKCδ. Mol. Cell 21, 467–480 [DOI] [PubMed] [Google Scholar]
- 37. Ponassi R., Terrinoni A., Chikh A., Rufini A., Lena A. M., Sayan B. S., Melino G., Candi E. (2006) p63 and p73, members of the p53 gene family, transactivate PKCδ. Biochem. Pharmacol. 72, 1417–1422 [DOI] [PubMed] [Google Scholar]
- 38. Jin H., Kanthasamy A., Ghosh A., Yang Y., Anantharam V., Kanthasamy A. G. (2011) α-Synuclein negatively regulates protein kinase Cδ expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. J. Neurosci. 31, 2035–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Min B. W., Kim C. G., Ko J., Lim Y., Lee Y. H., Shin S. Y. (2008) Transcription of the protein kinase C-δ gene is activated by JNK through c-Jun and ATF2 in response to the anticancer agent doxorubicin. Exp. Mol. Med. 40, 699–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Saha R. N., Pahan K. (2006) HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 13, 539–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Song C., Kanthasamy A., Jin H., Anantharam V., Kanthasamy A. G. (2011) Paraquat induces epigenetic changes by promoting histone acetylation in cell culture models of dopaminergic degeneration. Neurotoxicology 32, 586–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Song C., Kanthasamy A., Anantharam V., Sun F., Kanthasamy A. G. (2010) Environmental neurotoxic pesticide increases histone acetylation to promote apoptosis in dopaminergic neuronal cells: relevance to epigenetic mechanisms of neurodegeneration. Mol. Pharmacol. 77, 621–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kanthasamy A., Jin H., Anantharam V., Sondarva G., Rangasamy V., Rana A., Kanthasamy A. (2012) Emerging neurotoxic mechanisms in environmental factors-induced neurodegeneration. Neurotoxicology 33, 833–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pirooznia S. K., Sarthi J., Johnson A. A., Toth M. S., Chiu K., Koduri S., Elefant F. (2012) Tip60 HAT activity mediates APP induced lethality and apoptotic cell death in the CNS of a Drosophila Alzheimer's disease model. PLoS One 7, e41776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim D., Frank C. L., Dobbin M. M., Tsunemoto R. K., Tu W., Peng P. L., Guan J. S., Lee B. H., Moy L. Y., Giusti P., Broodie N., Mazitschek R., Delalle I., Haggarty S. J., Neve R. L., Lu Y., Tsai L. H. (2008) Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron 60, 803–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bardai F. H., Price V., Zaayman M., Wang L., D'Mello S. R. (2012) Histone deacetylase-1 (HDAC1) is a molecular switch between neuronal survival and death. J. Biol. Chem. 287, 35444–35453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Morrison B. E., Majdzadeh N., Zhang X., Lyles A., Bassel-Duby R., Olson E. N., D'Mello S. R. (2006) Neuroprotection by histone deacetylase-related protein. Mol. Cell. Biol. 26, 3550–3564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pandey U. B., Nie Z., Batlevi Y., McCray B. A., Ritson G. P., Nedelsky N. B., Schwartz S. L., DiProspero N. A., Knight M. A., Schuldiner O., Padmanabhan R., Hild M., Berry D. L., Garza D., Hubbert C. C., Yao T. P., Baehrecke E. H., Taylor J. P. (2007) HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859–863 [DOI] [PubMed] [Google Scholar]
- 49. D'Mello S. R. (2009) Histone deacetylases as targets for the treatment of human neurodegenerative diseases. Drug News Perspect. 22, 513–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bardai F. H., D'Mello S. R. (2011) Selective toxicity by HDAC3 in neurons: regulation by Akt and GSK3β. J. Neurosci. 31, 1746–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pirooznia S. K., Elefant F. (2013) Targeting specific HATs for neurodegenerative disease treatment: translating basic biology to therapeutic possibilities. Front. Cell. Neurosci. 7, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Valor L. M., Viosca J., Lopez-Atalaya J. P., Barco A. (2013) Lysine acetyltransferases CBP and p300 as therapeutic targets in cognitive and neurodegenerative disorders. Curr. Pharm. Des. 19, 5051–5064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Boutillier A. L., Trinh E., Loeffler J. P. (2003) Selective E2F-dependent gene transcription is controlled by histone deacetylase activity during neuronal apoptosis. J. Neurochem. 84, 814–828 [DOI] [PubMed] [Google Scholar]
- 54. Salminen A., Tapiola T., Korhonen P., Suuronen T. (1998) Neuronal apoptosis induced by histone deacetylase inhibitors. Brain Res. Mol. Brain Res. 61, 203–206 [DOI] [PubMed] [Google Scholar]
- 55. Wang Y., Wang X., Liu L., Wang X. (2009) HDAC inhibitor trichostatin A-inhibited survival of dopaminergic neuronal cells. Neurosci. Lett. 467, 212–216 [DOI] [PubMed] [Google Scholar]
- 56. Dietz K. C., Casaccia P. (2010) HDAC inhibitors and neurodegeneration: at the edge between protection and damage. Pharmacol. Res. 62, 11–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sapetschnig A., Koch F., Rischitor G., Mennenga T., Suske G. (2004) Complexity of translationally controlled transcription factor Sp3 isoform expression. J. Biol. Chem. 279, 42095–42105 [DOI] [PubMed] [Google Scholar]
- 58. Boyes J., Byfield P., Nakatani Y., Ogryzko V. (1998) Regulation of activity of the transcription factor GATA-1 by acetylation. Nature 396, 594–598 [DOI] [PubMed] [Google Scholar]
- 59. Yang X. J., Ogryzko V. V., Nishikawa J., Howard B. H., Nakatani Y. (1996) A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature 382, 319–324 [DOI] [PubMed] [Google Scholar]
- 60. Miska E. A., Karlsson C., Langley E., Nielsen S. J., Pines J., Kouzarides T. (1999) HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J. 18, 5099–5107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lemercier C., Brocard M. P., Puvion-Dutilleul F., Kao H. Y., Albagli O., Khochbin S. (2002) Class II histone deacetylases are directly recruited by BCL6 transcriptional repressor. J. Biol. Chem. 277, 22045–22052 [DOI] [PubMed] [Google Scholar]
- 62. Sowa Y., Orita T., Minamikawa-Hiranabe S., Mizuno T., Nomura H., Sakai T. (1999) Sp3, but not Sp1, mediates the transcriptional activation of the p21/WAF1/Cip1 gene promoter by histone deacetylase inhibitor. Cancer Res. 59, 4266–4270 [PubMed] [Google Scholar]
- 63. Harischandra D. S., Kondru N., Martin D. P., Kanthasamy A., Jin H., Anantharam V., Kanthasamy A. G. (2014) Role of proteolytic activation of protein kinase Cδ in the pathogenesis of prion disease. Prion 8, 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Scholz D., Pöltl D., Genewsky A., Weng M., Waldmann T., Schildknecht S., Leist M. (2011) Rapid, complete and large-scale generation of post-mitotic neurons from the human LUHMES cell line. J. Neurochem. 119, 957–971 [DOI] [PubMed] [Google Scholar]
- 65. Schildknecht S., Pöltl D., Nagel D. M., Matt F., Scholz D., Lotharius J., Schmieg N., Salvo-Vargas A., Leist M. (2009) Requirement of a dopaminergic neuronal phenotype for toxicity of low concentrations of 1-methyl-4-phenylpyridinium to human cells. Toxicol. Appl. Pharmacol. 241, 23–35 [DOI] [PubMed] [Google Scholar]
- 66. Winner B., Jappelli R., Maji S. K., Desplats P. A., Boyer L., Aigner S., Hetzer C., Loher T., Vilar M., Campioni S., Tzitzilonis C., Soragni A., Jessberger S., Mira H., Consiglio A., Pham E., Masliah E., Gage F. H., Riek R. (2011) In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. U.S.A. 108, 4194–4199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Prots I., Veber V., Brey S., Campioni S., Buder K., Riek R., Böhm K. J., Winner B. (2013) α-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J. Biol. Chem. 288, 21742–21754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Stoppini L., Buchs P. A., Muller D. (1991) A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 37, 173–182 [DOI] [PubMed] [Google Scholar]
- 69. Noraberg J., Kristensen B. W., Zimmer J. (1999) Markers for neuronal degeneration in organotypic slice cultures. Brain Res. Brain Res. Protoc. 3, 278–290 [DOI] [PubMed] [Google Scholar]
- 70. Schmued L. C., Albertson C., Slikker W., Jr. (1997) Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 751, 37–46 [DOI] [PubMed] [Google Scholar]
- 71. Macklis J. D., Madison R. D. (1990) Progressive incorporation of propidium iodide in cultured mouse neurons correlates with declining electrophysiological status: a fluorescence scale of membrane integrity. J. Neurosci. Methods 31, 43–46 [DOI] [PubMed] [Google Scholar]
- 72. Cho S., Wood A., Bowlby M. R. (2007) Brain slices as models for neurodegenerative disease and screening platforms to identify novel therapeutics. Curr. Neuropharmacol. 5, 19–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 74. Zhu W. G., Lakshmanan R. R., Beal M. D., Otterson G. A. (2001) DNA methyltransferase inhibition enhances apoptosis induced by histone deacetylase inhibitors. Cancer Res. 61, 1327–1333 [PubMed] [Google Scholar]
- 75. Ghosh A., Saminathan H., Kanthasamy A., Anantharam V., Jin H., Sondarva G., Harischandra D. S., Qian Z., Rana A., Kanthasamy A. G. (2013) The peptidyl-prolyl isomerase Pin1 up-regulation and proapoptotic function in dopaminergic neurons: relevance to the pathogenesis of Parkinson disease. J. Biol. Chem. 288, 21955–21971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Davie J. R. (2003) Inhibition of histone deacetylase activity by butyrate. J. Nutr. 133, 2485S–2493S [DOI] [PubMed] [Google Scholar]
- 77. Marks P. A., Miller T., Richon V. M. (2003) Histone deacetylases. Curr. Opin. Pharmacol. 3, 344–351 [DOI] [PubMed] [Google Scholar]
- 78. Ray R., Snyder R. C., Thomas S., Koller C. A., Miller D. M. (1989) Mithramycin blocks protein binding and function of the SV40 early promoter. J. Clin. Invest. 83, 2003–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Blume S. W., Snyder R. C., Ray R., Thomas S., Koller C. A., Miller D. M. (1991) Mithramycin inhibits SP1 binding and selectively inhibits transcriptional activity of the dihydrofolate reductase gene in vitro and in vivo. J. Clin. Invest. 88, 1613–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Konduri S., Colon J., Baker C. H., Safe S., Abbruzzese J. L., Abudayyeh A., Basha M. R., Abdelrahim M. (2009) Tolfenamic acid enhances pancreatic cancer cell and tumor response to radiation therapy by inhibiting survivin protein expression. Mol. Cancer Ther. 8, 533–542 [DOI] [PubMed] [Google Scholar]
- 81. Dennig J., Beato M., Suske G. (1996) An inhibitor domain in Sp3 regulates its glutamine-rich activation domains. EMBO J. 15, 5659–5667 [PMC free article] [PubMed] [Google Scholar]
- 82. Murata Y., Kim H. G., Rogers K. T., Udvadia A. J., Horowitz J. M. (1994) Negative regulation of Sp1 trans-activation is correlated with the binding of cellular proteins to the amino terminus of the Sp1 trans-activation domain. J. Biol. Chem. 269, 20674–20681 [PubMed] [Google Scholar]
- 83. Dauer W., Przedborski S. (2003) Parkinson's disease: mechanisms and models. Neuron 39, 889–909 [DOI] [PubMed] [Google Scholar]
- 84. Asaithambi A., Ay M., Jin H., Gosh A., Anantharam V., Kanthasamy A., Kanthasamy A. G. (2014) Protein kinase D1 (PKD1) phosphorylation promotes dopaminergic neuronal survival during 6-OHDA-induced oxidative stress. PLoS One 9, e96947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Burguillos M. A., Deierborg T., Kavanagh E., Persson A., Hajji N., Garcia-Quintanilla A., Cano J., Brundin P., Englund E., Venero J. L., Joseph B. (2011) Caspase signalling controls microglia activation and neurotoxicity. Nature 472, 319–324 [DOI] [PubMed] [Google Scholar]
- 86. Soltoff S. P. (2007) Rottlerin: an inappropriate and ineffective inhibitor of PKCδ. Trends Pharmacol. Sci. 28, 453–458 [DOI] [PubMed] [Google Scholar]
- 87. Ghosh A., Kanthasamy A., Joseph J., Anantharam V., Srivastava P., Dranka B. P., Kalyanaraman B., Kanthasamy A. G. (2012) Anti-inflammatory and neuroprotective effects of an orally active apocynin derivative in pre-clinical models of Parkinson's disease. J. Neuroinflammation 9, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kurkinen K. M., Keinänen R. A., Karhu R., Koistinaho J. (2000) Genomic structure and chromosomal localization of the rat protein kinase Cδ-gene. Gene 242, 115–123 [DOI] [PubMed] [Google Scholar]
- 89. Schroeder F. A., Lin C. L., Crusio W. E., Akbarian S. (2007) Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol. Psychiatry 62, 55–64 [DOI] [PubMed] [Google Scholar]
- 90. Minamiyama M., Katsuno M., Adachi H., Waza M., Sang C., Kobayashi Y., Tanaka F., Doyu M., Inukai A., Sobue G. (2004) Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 13, 1183–1192 [DOI] [PubMed] [Google Scholar]
- 91. Ferrante R. J., Kubilus J. K., Lee J., Ryu H., Beesen A., Zucker B., Smith K., Kowall N. W., Ratan R. R., Luthi-Carter R., Hersch S. M. (2003) Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J. Neurosci. 23, 9418–9427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lee H. H., Chang S. S., Lin S. J., Chua H. H., Tsai T. J., Tsai K., Lo Y. C., Chen H. C., Tsai C. H. (2008) Essential role of PKCδ in histone deacetylase inhibitor-induced Epstein-Barr virus reactivation in nasopharyngeal carcinoma cells. J. Gen. Virol. 89, 878–883 [DOI] [PubMed] [Google Scholar]
- 93. McMillan L., Butcher S. K., Pongracz J., Lord J. M. (2003) Opposing effects of butyrate and bile acids on apoptosis of human colon adenoma cells: differential activation of PKC and MAP kinases. Br. J. Cancer 88, 748–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lin K. T., Wang Y. W., Chen C. T., Ho C. M., Su W. H., Jou Y. S. (2012) HDAC inhibitors augmented cell migration and metastasis through induction of PKCs leading to identification of low toxicity modalities for combination cancer therapy. Clin. Cancer Res. 18, 4691–4701 [DOI] [PubMed] [Google Scholar]
- 95. Suske G. (1999) The Sp-family of transcription factors. Gene 238, 291–300 [DOI] [PubMed] [Google Scholar]
- 96. Suske G., Bruford E., Philipsen S. (2005) Mammalian SP/KLF transcription factors: bring in the family. Genomics 85, 551–556 [DOI] [PubMed] [Google Scholar]
- 97. Yokota T., Matsuzaki Y., Miyazawa K., Zindy F., Roussel M. F., Sakai T. (2004) Histone deacetylase inhibitors activate INK4d gene through Sp1 site in its promoter. Oncogene 23, 5340–5349 [DOI] [PubMed] [Google Scholar]
- 98. Han J. W., Ahn S. H., Kim Y. K., Bae G. U., Yoon J. W., Hong S., Lee H. Y., Lee Y. W., Lee H. W. (2001) Activation of p21(WAF1/Cip1) transcription through Sp1 sites by histone deacetylase inhibitor apicidin: involvement of protein kinase C. J. Biol. Chem. 276, 42084–42090 [DOI] [PubMed] [Google Scholar]
- 99. Camarero N., Nadal A., Barrero M. J., Haro D., Marrero P. F. (2003) Histone deacetylase inhibitors stimulate mitochondrial HMG-CoA synthase gene expression via a promoter proximal Sp1 site. Nucleic Acids Res. 31, 1693–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Marinova Z., Ren M., Wendland J. R., Leng Y., Liang M. H., Yasuda S., Leeds P., Chuang D. M. (2009) Valproic acid induces functional heat-shock protein 70 via Class I histone deacetylase inhibition in cortical neurons: a potential role of Sp1 acetylation. J. Neurochem. 111, 976–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Chou C. W., Wu M. S., Huang W. C., Chen C. C. (2011) HDAC inhibition decreases the expression of EGFR in colorectal cancer cells. PLoS One 6, e18087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Barrasa J. I., Olmo N., Santiago-Gómez A., Lecona E., Anglard P., Turnay J., Lizarbe M. A. (2012) Histone deacetylase inhibitors upregulate MMP11 gene expression through Sp1/Smad complexes in human colon adenocarcinoma cells. Biochim. Biophys. Acta 1823, 570–581 [DOI] [PubMed] [Google Scholar]
- 103. Doetzlhofer A., Rotheneder H., Lagger G., Koranda M., Kurtev V., Brosch G., Wintersberger E., Seiser C. (1999) Histone deacetylase 1 can repress transcription by binding to Sp1. Mol. Cell. Biol. 19, 5504–5511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Sun J. M., Chen H. Y., Moniwa M., Litchfield D. W., Seto E., Davie J. R. (2002) The transcriptional repressor Sp3 is associated with CK2-phosphorylated histone deacetylase 2. J. Biol. Chem. 277, 35783–35786 [DOI] [PubMed] [Google Scholar]
- 105. Won J., Yim J., Kim T. K. (2002) Sp1 and Sp3 recruit histone deacetylase to repress transcription of human telomerase reverse transcriptase (hTERT) promoter in normal human somatic cells. J. Biol. Chem. 277, 38230–38238 [DOI] [PubMed] [Google Scholar]
- 106. Mottet D., Pirotte S., Lamour V., Hagedorn M., Javerzat S., Bikfalvi A., Bellahcène A., Verdin E., Castronovo V. (2009) HDAC4 represses p21(WAF1/Cip1) expression in human cancer cells through a Sp1-dependent, p53-independent mechanism. Oncogene 28, 243–256 [DOI] [PubMed] [Google Scholar]
- 107. Yang X. J., Seto E. (2007) HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26, 5310–5318 [DOI] [PubMed] [Google Scholar]
- 108. Fischle W., Dequiedt F., Hendzel M. J., Guenther M. G., Lazar M. A., Voelter W., Verdin E. (2002) Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell 9, 45–57 [DOI] [PubMed] [Google Scholar]
- 109. Suzuki T., Kimura A., Nagai R., Horikoshi M. (2000) Regulation of interaction of the acetyltransferase region of p300 and the DNA-binding domain of Sp1 on and through DNA binding. Genes Cells 5, 29–41 [DOI] [PubMed] [Google Scholar]
- 110. Walker G. E., Wilson E. M., Powell D., Oh Y. (2001) Butyrate, a histone deacetylase inhibitor, activates the human IGF binding protein-3 promoter in breast cancer cells: molecular mechanism involves an Sp1/Sp3 multiprotein complex. Endocrinology 142, 3817–3827 [DOI] [PubMed] [Google Scholar]
- 111. Ammanamanchi S., Freeman J. W., Brattain M. G. (2003) Acetylated sp3 is a transcriptional activator. J. Biol. Chem. 278, 35775–35780 [DOI] [PubMed] [Google Scholar]
- 112. Hung J. J., Wang Y. T., Chang W. C. (2006) Sp1 deacetylation induced by phorbol ester recruits p300 to activate 12(S)-lipoxygenase gene transcription. Mol. Cell. Biol. 26, 1770–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Spengler M. L., Brattain M. G. (2006) Sumoylation inhibits cleavage of Sp1 N-terminal negative regulatory domain and inhibits Sp1-dependent transcription. J. Biol. Chem. 281, 5567–5574 [DOI] [PubMed] [Google Scholar]
- 114. Chu S. (2012) Transcriptional regulation by post-transcriptional modification–role of phosphorylation in Sp1 transcriptional activity. Gene 508, 1–8 [DOI] [PubMed] [Google Scholar]
- 115. Haberland M., Montgomery R. L., Olson E. N. (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10, 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Chen B., Cepko C. L. (2009) HDAC4 regulates neuronal survival in normal and diseased retinas. Science 323, 256–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Markova N. G., Karaman-Jurukovska N., Pinkas-Sarafova A., Marekov L. N., Simon M. (2007) Inhibition of histone deacetylation promotes abnormal epidermal differentiation and specifically suppresses the expression of the late differentiation marker profilaggrin. J. Invest. Dermatol. 127, 1126–1139 [DOI] [PubMed] [Google Scholar]
- 118. Hou L., Zhang X., Wang D., Baccarelli A. (2012) Environmental chemical exposures and human epigenetics. Int. J. Epidemiol. 41, 79–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ramirez T., Brocher J., Stopper H., Hock R. (2008) Sodium arsenite modulates histone acetylation, histone deacetylase activity and HMGN protein dynamics in human cells. Chromosoma 117, 147–157 [DOI] [PubMed] [Google Scholar]
- 120. Li J., Gorospe M., Barnes J., Liu Y. (2003) Tumor promoter arsenite stimulates histone H3 phosphoacetylation of proto-oncogenes c-fos and c-jun chromatin in human diploid fibroblasts. J. Biol. Chem. 278, 13183–13191 [DOI] [PubMed] [Google Scholar]