

Background: It remains unaddressed whether varying neutralizing potency of mAbs is somehow correlated with differences in their global shapes.
Results: Non-neutralizing mAbs have an open shape, whereas Fab-Fab and Fab-Fc interactions induce a closed shape in HIV-1-neutralizing mAbs.
Conclusion: An unopen shape appears to be a hallmark of neutralizing potency, at least for HIV-1.
Significance: This work provides new insight into the shape-function relationship of mAbs.
Keywords: HIV-1 Protease, Molecular Modeling, Protein Folding, Protein Structure, X-ray Scattering, Global Shape, Limited Proteolysis, Neutralizing Antibodies, Small Angle X-ray Scattering, Solution Scattering
Abstract
Asymmetric disposition of Fab arms in the structures solved for the broadly neutralizing monoclonal antibody (nmAb) IgG1 b12 raised the question of whether the unusual shape observed for b12 is common for all IgG1 mAbs or if there is a difference in the overall shape of nmAbs versus non-nmAbs. We compared small angle x-ray scattering (SAXS) data-based models and limited proteolysis profiles of some IgG1 mAbs known to be having and lacking HIV-1 neutralizing potency. In non-nmAbs, the Fab arms were found to be symmetrically disposed in space relative to central Fc, but in most nmAbs, the Fab arms were asymmetrically disposed, as seen for IgG1 b12. The only exceptions were 2G12 and 4E10, where both Fab arms were closed above Fc, suggesting some Fab-Fc and/or Fab-Fab interaction in the nmAbs that constrained extension of the Fab-Fc linker. Interestingly, these observations were correlated with differential proteolysis profiles of the mAbs by papain. Under conditions when papain could cut both Fab arms of non-nmAbs, only one Fab arm could be removed from neutralizing ones (except for 2G12 and 4E10). Chromatography and small angle x-ray scattering results of papain-digested products revealed that 1) the Fab-Fc or Fab-Fab interactions in unliganded mAbs are retained in digested products, and 2) whereas anti-gp120 non-nmAbs could bind two gp120 molecules, nmAbs could bind only one gp120. Additional experiments showed that except for 2G12 and 4E10, unopen shapes of nmAbs remain uninfluenced by ionic strength but can be reversibly opened by low pH of buffer accompanied by loss of ligand binding ability.
Introduction
Researchers committed to designing antibody-mediated intervention of infectious diseases like HIV are intrigued by the fact that although a handful of IgG1 mAbs can react with antigens displayed on the viral surface (gp120 or gp41) and neutralize broad spectrum of viral strains, other mAbs of same isotype recognizing the same ligands with comparable affinities cannot achieve the same result (1). To date, IgG1 b12 is the only broadly neutralizing mAb (nmAb)3 whose full-length structure by x-ray diffraction and solution SAXS data-based modeling is known (2, 3). A unique characteristic of the structure was the asymmetric disposition of its two Fab arms. Whereas one remained “nestled” close to the VH2 domain of the Fc, the other Fab preferred a spatial disposition extended away from the Fc. Although this unique structure was reasoned to be a snapshot of the range of conformations accessible to IgG1 antibodies (4), solution scattering data analysis ruled out conformational polydispersity among the b12 molecules in solution and confirmed that the predominant solution shape of IgG1 b12 is very similar to the structure refined from crystallography (3). This observation raised questions like whether all IgG1 mAbs possess a b12-like asymmetric shape or if the predominant shape of IgG1 mAbs can vary and, if the answer to the latter is “yes,” then whether the difference in shape can be somehow correlated to the neutralizing efficacy of that particular mAb. Answers to these shape-function-related queries might reveal features necessary to be present in mAbs for neutralizing potential.
Besides the structure of IgG1 b12, crystal structures of the isolated Fab arms of different nmAbs and non-nmAbs are available, which unfortunately cannot be utilized for understanding or commenting on the three-dimensional shape of the full-length mAbs. Importantly, the shapes of some of these mAbs reactive to envelope components of HIV-1 have been interpreted from electron microscopy (EM)-tomography data in their unliganded and gp120-bound form (5, 6). Based on negative stain electron micrographs, the shapes of unliganded IgG1 b12 and 2G12 were interpreted as symmetric shapes with two Fab arms disposed on two sides of Fc and intertwined above Fc, respectively (7). Interestingly, the conclusions drawn for b12 from EM images clearly contrasted with the asymmetric shape seen from crystallography and SAXS. On the other hand, the two Fab arms of 2G12 were seen to be intertwined on the top of the molecule, suggesting some kind of interaction between the two Fab arms of 2G12. The latter observation was supported by the dimeric status of the Fab arm of 2G12 in the crystalline state (PDB codes 1OM3, 1OP3, and 1OP5). Another EM-based study put forth the shapes of gp140-bound IgG1 nmAbs 2F5, b12, and 2G12 (8) and concluded that in the gp140-bound state, 2F5 and b12 adopted a shape where the two gp140 bound Fab arms were symmetrically disposed relative to central Fc, similar to the classic “Y” shape of antibodies. In contrast, gp140-bound 2G12 appeared to adopt a shape where the two Fab arms were locked above the Fc. The closed shape of 2G12 was supported in another EM image analysis-based study, where the mAb was studied in unliganded form (9). Further support for the Fab-Fab interactions leading to conjoined status of the Fab arms was seen from papain digestion of this mAb, where the Fab arms “cleaved off” as dimer. In summary, the structural studies on 2G12 in the unliganded and gp140-bound state concluded that the shape of the mAbs does not undergo a substantial change upon ligand binding. If that is true, then the asymmetric shape seen for b12 from crystallography and SAXS and the symmetric shape of two gp140-bound b12s from the EM-based data raise the question of whether the closed Fab arm of b12 undergoes dramatic opening post-gp120-gp140 binding. To address the above questions and gain insight into the global shape versus known neutralizing potency of the anti-HIV antibodies, we analyzed SAXS data from 17 different mAbs having and lacking HIV-1 neutralizing potency. Data analysis and modeling results showed that Fab domains in nmAbs have differential orientation in space, either due to interaction with central Fc (as seen for b12) or due to Fab-Fab interaction (as seen for 2G12). Additionally, the papain digestion profiles and biochemical-structural analysis of the digested products were also analyzed. Overall, our results reveal that whereas non-nmAbs prefer an open shape with Fab arms extended away in space, Fab-Fc or Fab-Fab interactions lead to unextended spatial disposition of Fab arm(s) and a twisted orientation of the Fc in nmAbs.
EXPERIMENTAL PROCEDURES
SAXS Sample Preparation and Characterization
All of the mAbs and gp120 were purified to homogeneity in PBS buffer using a SHODEX PROTEIN KW-800 GFC column (Showa-Denkko-K.K., Japan) attached to a Waters HPLC system. Purified proteins were reconcentrated to about 1 mg/ml using AMICON membrane concentrators (Millipore, County Cork, Ireland) with a molecular mass cut-off of 30 kDa. The purity and stability of purified proteins in PBS, pH 7.4, were confirmed by single bands at the expected migration positions in SDS-PAGE. After incubation of unliganded proteins for about 2 h, complexes of gp120 and mAbs were purified using a SHODEX column. The stability of purified mAbs and gp120 stored at 4 °C was confirmed by reinjecting purified complexes on FPLC. Later, mixtures of mAbs and gp120 were characterized by gel filtration to assess the extent and stoichiometry of mAb-gp120 binding. Peaks were collected and reinjected to confirm their stability. Similarly, papain-digested products after different time intervals of incubation with bead-bound enzyme were also analyzed by gel filtration. The relative elution volumes of the mAbs, complexes of gp120-mAbs, and enzymatic degradation products of mAbs were compared with those of the molecular mass standards to estimate their apparent mass. The relative elution volumes were calculated as follows,
where Ve is the elution volume of mAb, complex, or fragment; V0 is the void volume determined by the elution volume of blue dextran 2000 (mass ∼2000 kDa); and Vg is the geometric column volume as determined by the elution of free tryptophan (mass ∼0.15 kDa). A standard curve was plotted of KAV versus log10(mass) (Fig. 1A). The linear relationship was utilized to estimate the molecular mass of the eluted entity.
FIGURE 1.
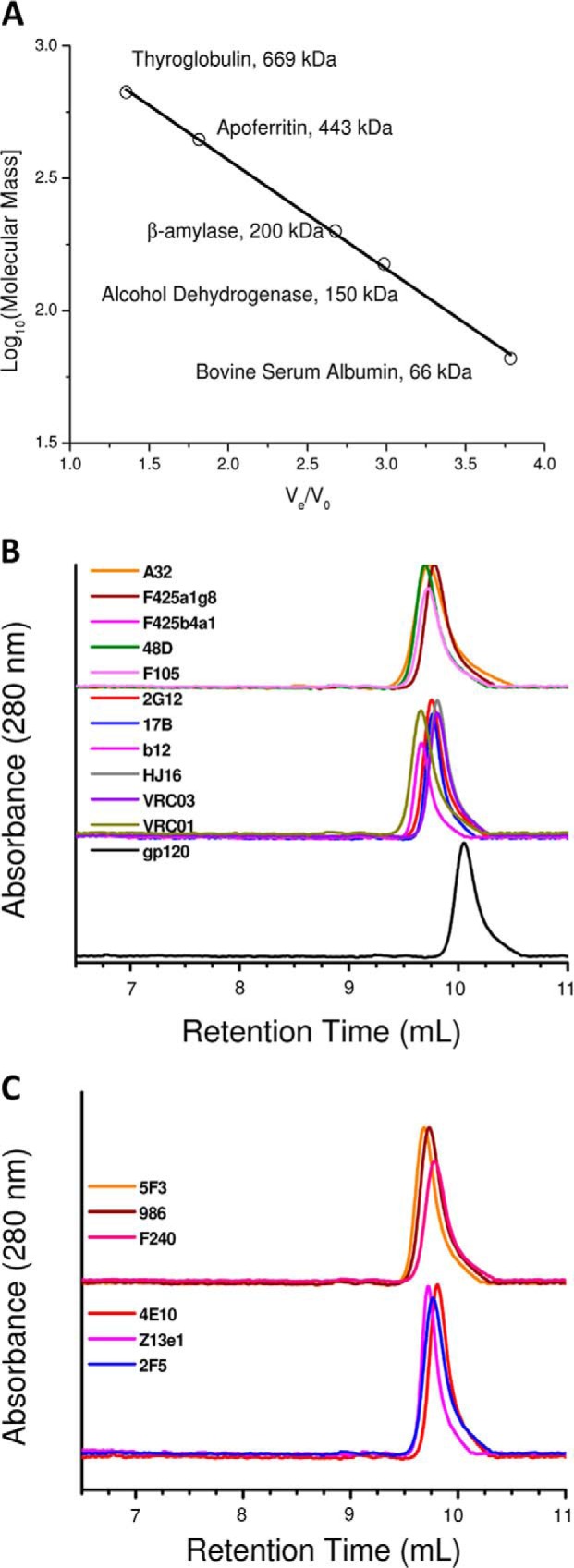
Gel filtration profile of unliganded mAbs. A, relative retention volumes versus molecular masses of standard proteins in a gel filtration chromatography set-up are plotted. The linear fit was utilized to estimate elution profile-based masses of the mAbs, their complexes, and fragments. B, elution profiles of the gp120-reactive mAbs (non-neutralizing (top), neutralizing (middle), and SF162 gp120 (bottom)). C, elution profiles of the gp41-reactive non-neutralizing (top) and neutralizing (bottom) mAbs are shown.
Synchrotron SAXS Data
All SAXS experiments were carried out at the X9 beam line (National Synchrotron Light Source, Brookhaven National Laboratory, Upton, NY) using purified unliganded mAbs and their complexes with gp120 and their matched buffers (eluted fraction of buffer from the same column). For each data set, ∼70 μl of sample and their matched buffer were exposed for 60 s in a quartz flow cell with a flow rate of ∼55 μl/min. Images were recorded on attached Pilatus detectors and processed to obtain SAXS intensity profiles (I(Q)) from protein solutions as a function of Q using Python script-based programs available at the X9 beam line at the National Synchrotron Light Source. Momentum transfer vector, Q, was defined as Q = ((4πsin θ)/λ), where λ was the beam wavelength and θ was the scattering angle. All of the data collection was carried out in triplicate and averaged. After data collection, SDS-PAGE loading buffer was immediately added to the samples recovered after scattering experiments. Similarity in their migration pattern to that seen for the mAbs and complexes, which were never exposed to x-rays and remained in the laboratory, confirmed that no degradations occurred during travel and data collection. Alongside, to estimate beam intensity value at zero angles (I0) and calculate the concentration of unliganded proteins, we acquired SAXS data from a set of FPLC-purified proteins (the tetrameric form of GAPDH in PBS, pH 7.4 (147 kDa; Sigma-Aldrich) and recombinant human plasma gelsolin in EGTA Tris buffer, pH 8.0 (82 kDa)) with previously determined concentration using an absorbance value at 280 nm and their calculated extinction coefficient values.
Additional SAXS Experiments with In-house Instrument
To analyze the effect of varying buffer pH and ionic strength on global shapes of mAbs, SAXS data were collected using the SAXSpace instrument (Anton Paar GmbH, Graz, Austria). Different salt (NaCl) concentrations (100, 137, 300, 600, and 1000 mm) were considered in PBS, and buffers with varying pH but the same amount of NaCl (137 mm) were prepared as described below in different buffers with final pH 4, 5, 5.5, 6, 7, and 8. The source of the SAXS instrument was a sealed tube, and experiments were done using line collimation, and data were collected at the one-dimensional CMOS Mythen detector (Dectris, Baden, Switzerland). In each case, ∼60 μl of samples and their matched buffer were exposed for 1 h at 20 °C in a thermostated quartz capillary with 1-mm diameter. The data were reduced in SAXStreat software to calibrate the position of the primary beam. The data were further processed using SAXSquant software to subtract buffer contribution. The Porod constant (C) was calculated and subtracted from I(Q) to obtain an intensity profile where the scattering of the particle decays as a function of Q−3. The final I(Q) data sets were further processed using the GIFT (Generalized Indirect Fourier Transformation) software package. The same program was used to desmear the data using the primary beam profile and to estimate the pair distribution profile from the SAXS I(Q) of the protein samples.
SAXS Data Analysis and ab Initio Shape Restoration
Kratky plots (I(Q) × Q2 versus Q) of each data set were prepared to examine the globular protein-like scattering nature of the mAbs and their complexes in solution. All of the Guinier approximations were performed using the PRIMUS software package (10). Using GNOM45 software (11), indirect Fourier transformation of the scattering data over the measured Q range computed a pairwise distribution function of interatomic vectors, P(r). During the transformation, the probability of finding a pairwise vector equal to 0 Å and the maximum linear dimension, Dmax, was considered to be zero. This analysis also provided Rg and I0 from the second moment and the start of P(r), respectively. To visualize the predominant solution shape of the mAbs and complexes of gp120-reactive mAbs, for each data set, 10 independent uniform density and chain-ensemble models were generated using DAMMIN and GASBOR software (12, 13). These models were compared with each other and averaged using DAMCLUST programs (14). We also used the SASTBX server-based toolbox to construct envelope shapes of the unliganded mAbs. This method employed the PISA database of shapes to compute solutions that have their SAXS profiles closest to the experimental data. The results from the SHAPEUP module were composed of several candidate shapes that are aligned and clustered using a hierarchical clustering algorithm serving as an internal model consistency check, indicating the quality of the solution and the experimental data. The models with best correlation between experimental data and selected profiles were considered for analysis.
Concurrently, using the SASREF program and coordinates of the Fab arms and Fc portion (abstracted from the crystal structure of IgG1 b12; PDB code 1HZH) and experimentally measured scattering data as a reference, rigid body docking was done to generate models of mAbs (15). Additionally, to compute the putative positions of the component proteins within the experimentally determined shape of mAb-gp120 complexes, the MONSA program, a multiphase version of DAMMIN, was employed to reconstruct models of different phases from an assembly of beads inside a defined search volume (12). The program was used to simultaneously fit three scattering curves of mAbs, gp120, and the complexes of gp120-mAbs to find the best distribution of beads that corresponds to interconnected phases representing the protein-protein complex. For each case, 10 independent runs of the MONSA program provided models with an average normalized spatial discrepancy below 0.65, which were averaged using the DAMAVERM suite of programs to obtain the most predominant model of gp120-mAb complexes. For figure generation, the averaged models generated by averaging different models were converted to the volume/density maps using the MDFF plug-in in VMD software (17), and the density maps were generated using UCSF Chimera visualization software (18).
SEC-MALLS
Absolute molecular masses were determined with static light scattering using a Wyatt Dawn Heleos 8+ multiangle laser light scattering detector (Wyatt Technology) coupled to a Waters HPLC protein purification system with a Shodex HPLC-size exclusion column. Unliganded antibody at concentrations of 0.5 mg/ml and its 1:1 and 1:2 molar mixtures with gp120 were injected to the size exclusion column equilibrated with PBS, pH 7.4, at a flow rate of 1 ml/min. A BSA sample (5 mg/ml) was used as a reference to calibrate the system. The molecular masses of individual peaks in the size exclusion chromatogram were determined using the Astra software package version 6.0.3. A standard value of refractive index (dn/dc = 0.185 ml/g) was used for calculations.
Papain Digestion
For proteolysis experiments of the Fab-Fc linker, papain was obtained from Sigma and immobilized on agarose CL-4B beads (Sigma) using the reductive amination method published previously (19). Briefly, about 200 μg of papain was immobilized per 1 ml of settled beads. Alongside, purified IgG1 mAbs at a concentration of ∼1 mg/ml were dialyzed against digestion buffer (20 mm sodium phosphate, 10 mm EDTA, pH 7.0, containing 20 mm cysteine·HCl). 100 μl of the dialyzed IgG1 samples were added to 10 μl of the immobilized papain agarose and incubated for 30, 60, 90, and 120 min in a shaker water bath at 16 °C. The reaction was stopped by adding crystalline iodoacetamide, and each proteolysis experiment was repeated three times. To rule out differences in results due to immobilization of papain, the exact same experiments were repeated by mixing 100 μg of IgG1 mAbs in digestion buffer and 2 μg of papain in solution. Importantly, proteolysis results were comparable by both protocols, but immobilized papain provided only products from mAbs. The proteolysis products of mAbs from papain digestion were analyzed by Western blots and gel filtration profiles. In some cases, digested fractions were eluted from gel filtration and concentrated using membrane concentrators for SAXS data acquisition.
Purification of Fc Fragment from Digested IgG1 mAbs
High affinity Protein A-Sepharose beads (Bio Vision) with a binding capacity of ≥16 mg of human or rabbit IgG/ml were used for purification of the Fc portion from papain-digested products. Beads packed in columns were equilibrated with binding buffer (PBS, TBS, 0.15 m NaCl, and 50 mm sodium borate, pH 8). Papain-digested products of mAbs were loaded onto the column after dialysis with binding buffer, followed by washing of column with a 5× bed volume of binding buffer now containing 0.5 m NaCl. Bound fragments were eluted with elution buffer (0.1 m citric acid, pH 2.75). Immediately after elution, 100 μl of 1 m Tris, pH 9.0, was added per ml of eluted volume to keep the eluted protein stable in solution. The protein concentration was calculated based on measured absorbance at 280 nm, and the purity was confirmed by SDS-PAGE and Western blot analysis. For SAXS-based structural analysis, the Fc fractions were concentrated using membrane concentrators (30 kDa cut-off; Millipore).
Western Blot Analysis
The presence of the Fc portion in the fragments of IgG1 mAbs after different time intervals of papain proteolysis was affirmed by Western blot analysis. For each case, 10 μl of the digested sample was applied to a 10% SDS-polyacrylamide gel in non-reducing conditions and transferred onto Immobilon-P membrane (Millipore). After transfer, the membrane was blocked with 5% (w/v) nonfat dry skimmed milk powder and incubated at 4 °C for 12 h with polyclonal rabbit anti-human Fc antibody (Sigma). Then the membrane was washed with PBS saline with 0.1% Tween (PBS-T) and incubated for 1 h with a peroxidase-conjugated goat anti-rabbit IgG antibody (Sigma). Chemiluminescence of peroxidase was developed with Luminata-Forte substrate (Millipore). The exposed and developed photo films were scanned using an image scanner (Fujifilm FLA-900). Pixel intensities in the acquired images were analyzed using the software provided with the instrument.
Synthesis of MPER Peptide
To check binding with anti-gp41 mAbs and its digested Fab arms to its ligand, one peptide (NEQELLELDKWASLWNWFNITNWLWYIK) was synthesized (20) by a solid phase peptide synthesis strategy (21) employing Fmoc chemistry at 0.04-mmol scale (using a PS-3 peptide synthesizer, Protein Technologies Inc.). First, C-terminal amino acid was coupled manually to the Rink amide-MBHA resin using TBTU (O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate) and DIPEA (N,N-diisopropylethylamine). The chain elongation of the peptide on the synthesizer was done by using four equivalents of the protected Fmoc-amino acid with 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate as a coupling reagent (22) and n-hydroxybenzotriazole (23) or 1-cyano-2-ethoxy-2-oxoethylidenamino-oxydimethyl-amino-morpholino-carbenium hexafluorophosphate to suppress racemization. After final Fmoc group removal, the peptide was cleaved from the resin by using a mixture of trifluoroacetic acid/water/tri-isopropylsilane (95:2.5:2.5, v/v/v) for 6 h. Crude peptide was made soluble in a water/acetonitrile (70:30) mixture by adding few drops of HFIP (1,1,1,3,3,3-hexafluoro-2-propanol) and then purified using reverse phase HPLC (C-18 (4.6 × 250 mm) column; Waters or Dionex Ultimate 3000 HPLC systems) by using a water/acetonitrile gradient containing 0.1% trifluoroacetic acid. Finally, the organic solvent was evaporated from the eluted fraction using a rotary evaporator and lyophilized to complete dryness to obtain 12 mg of the purified peptide. An observed mass of 3654.2 Da versus the expected mass of 3654.1 Da supported the identity of the purified synthetic peptide (MALDI-TOF; Voyager, AB-Sciex).
N-terminal Sequencing of the Digested Fc Portion
Purified Fc fragments (as described above) were transferred on a PVDF membrane using a semidry blotter (100 mA; 35 min). After the transfer, the membrane was rinsed 2–3 times with double-distilled water. To confirm transfer efficiency, the membrane was stained with 0.1% Ponceau S, destained with a 1:1 methanol/water solution, and air-dried. Finally, membrane sections were then loaded onto a BlottTM cartridge (reaction chamber) of an Applied Biosystems PROCISE 491 cLC protein sequencer, which employs an automated version of the Edman reaction to provide the N-terminal sequence of papain-digested products.
Binding Measurements Using ELISA
Binding of gp120-gp41-reactive mAbs to gp120-MPER Peptide as a Function of pH and Ionic Strength
Antigen (gp120 and MPER region peptide) at a concentration of 10 nm was coated onto 96-well plates (BD Biosciences) overnight at 4 °C in PBS (pH 7.4) following a previously published protocol with slight modifications (24). The wells were washed twice with PBS containing 0.1% Tween 20 (PBS-T) and blocked with 3% BSA for 4 h at 37 °C, followed by washing with PBS-T twice. gp120 binding mAbs (2G12, 17b, b12, VRC01, VRCO3, and HJ16) and gp41-reactive mAbs (4E10, 2F5, and Z13e1) were dialyzed against buffers with different pH values (acetate buffer, pH 4, citrate buffer, pH 5, Tris buffer, pH 6, PBS, pH 7, and HEPES, pH 8) and added to the wells coated with gp120-MPER, respectively, at a concentration of 100 nm/well and were incubated at 37 °C for 2 h. Additionally, to examine the effect of ionic strength on binding of mAbs to their ligands, mAbs were added to each well coated with gp120 and MPER peptide, respectively, in PBS with increasing NaCl concentrations (100 mm, 137 mm, 300 mm, 600 mm, and 1 m) and incubated at 37 °C for 2 h. The binding of antibody was detected with horseradish peroxidase-conjugated goat anti-human IgG against the Fc region of human IgG1 mAbs. The plates were developed by adding 100 μl of substrate (3,3′,5,5′-tetramethylbenzidine) solution (Pierce). The reaction was stopped as the color developed by adding 100 μl of 2 m H2SO4 and measuring the optical density at 450 nm in a microplate reader (Biotek).
Binding of C1q to Intact gp120- and gp41-reactive mAbs and Their Fc Portion
To analyze the binding affinity of IgG1 mAbs to C1q, ELISA was done following a previously published protocol with slight modification (25). The IgG1 mAbs were diluted to a concentration of 10 nm in PBS, and each mAb was coated in an individual well of a 96-well Maxisorp Immunoplate (Nunc, Rochester, NY) incubated at 4 °C overnight. Alongside, the Fc fragments purified from papain digestion of each mAb were also coated onto ELISA plates at a concentration of 10 nm in PBS. The wells were blocked with 5% BSA (Sigma) in PBS for 4 h at 37 °C. The plates were then washed with 1× PBS, 0.1% Tween 20 solution. Then the plates were incubated with a 100 nm solution of human C1q protein (Sigma) for 1 h at 37 °C, washed, and reincubated with goat anti-C1q antibody (Pierce) for 1 h at 37 °C. Secondary antibody (HRP-conjugated) against the Fc region of human IgG1 mAbs at a dilution of 1:5000 in PBS containing 1% BSA was added, and the plates were kept for 1 h at room temperature. The plates were then washed three times with PBS-T and twice with PBS and developed by adding 100 μl of substrate (3,3′,5,5′-tetramethylbenzidine) solution (Pierce). Incubation was done at room temperature, the reaction was stopped as the color developed by adding 100 μl of 2 m H2SO4, and absorbance at 450 nm was measured on a microplate reader (Biotek) as described above.
Models of IgG1 Symmetric mAbs for Accessible Surface Area Calculation
To compare accessible surface area of symmetric and asymmetric IgG1 mAbs, residue resolution models of the mAbs were generated by using the crystal structure of IgG1 b12 (PDB code 1HZH) and using its open chain to replace the closed chain. The final model of symmetric and open IgG1 had both Fab arms positioned at equal distance from the central Fc, and the resultant model matched well with the SAXS-based shape profile of the symmetric non-nmAb A32. To compute solvent-accessible surface areas of proteins, we used the online server POPS, which computed solvent-accessible surface areas at the atomic and residue level (26). Probe radii from 0.4 to 10 nm were used to estimate the accessible surface area.
RESULTS AND DISCUSSION
SAXS Data Analysis of Unliganded Neutralizing and Non-neutralizing IgG1 mAbs
Relative to the elution profile of the standard proteins, the apparent masses of the gp120 and mAbs were calculated to be about 108 kDa and in the range of 135–154 kDa, respectively (Fig. 1). SAXS data collected from three dilutions of each unliganded purified mAb confirmed no aggregation and no role of concentration-dependent shape effects, particularly in the dilute solutions being studied (Fig. 2 and Tables 1–3). Scattering intensity profiles were acquired from the following: 11 gp120-recognizing mAbs (six neutralizing: 17b, b12, VRC01, VRC03, HJ16 and 2G12; five non-neutralizing: A32, F425_a1g8, F425_b4a1, F105, and 48D) and six gp41-recognizing mAbs (three neutralizing: 4E10, 2F5, and Z13e1; three non-neutralizing: 5F3, 98-6, and F-240) (Fig. 2). Among the broadly neutralizing ones, 2F5, Z13e1, and 4E10 are reactive to the transmembrane protein gp41 (27, 28), and 2G12, VRC01, VRC03, 17b, HJ16, and b12 bind to the envelope glycoprotein gp120. Differing from the rest of the gp120-reactive mAbs, 2G12 recognizes hybrid mannose moieties in the heavily glycosylated portion of the HIV-1 gp120 (29). On the other hand, b12, HJ16, VRC01, and VRC03 bind to an epitope that, apart from slight differences, overlaps with the segment recognized by the CD4 receptor on T-cells (30–33), and 17b recognizes the bridging sheet region of CD4-bound gp120 (34). Linearity of the data in the ln(I(Q)) versus Q2 plot (Guinier analysis) with a constraint that Rg*Q is ≤1.3 confirms monodispersity of the globular sample in solution (35). Insets in Fig. 2A show the linear region of the Guinier approximation, presuming globular scattering shapes of the mAbs, and the Rg values are reported in Tables 1–3. Additionally, presuming a rodlike scattering shape of the mAbs in solution, Guinier approximation for rodlike shapes provided the RC values. Using the relationship that the long dimension (L) of the scattering shape is (12(Rg2 − RC2))½, the L values of the unliganded mAbs were estimated in solution (Tables 1–3). For gp120-binding mAbs, the nmAbs clearly appeared shorter in dimensions than the non-neutralizing ones. A similar trend was seen in the gp41-binding mAbs, but the difference was less pronounced for 5F3.
FIGURE 2.

SAXS data analysis and shape restoration of unliganded mAbs. A, SAXS data measured from samples of purified gp120- and gp41-reactive mAbs. Shown are Kratky plots of the SAXS data sets (B) and their P(r) curves (C). The same color is used to present data in each panel.
TABLE 1.
SAXS data analysis-based structural parameters deduced for the gp120-reactive IgG1 mAbs
The concentrations of the mAbs were estimated considering the mass of the individual mAb to be ∼150 kDa. NSD values are of the 10 models computed for the gp120-reactive mAbs.
| Unliganded mAbs | Sample | Guinier analysis |
P(r) analysis |
I0/(c*m) | NSD ± variation | ||||
|---|---|---|---|---|---|---|---|---|---|
| Rg | RC | L | Rg | Dmax | I0 | ||||
| Å | Å | Å | mg/ml | ||||||
| Standard proteins | |||||||||
| EGTA gelsolina | 30.0 | 17.1 | 86 | 30.3 | 105 | 97.2 | 0.62 | ||
| Tetrameric GAPDHa | 29.8 | 15.7 | 88 | 30.2 | 98 | 64.6 | 0.61 | ||
| gp120-reactive neutralizing IgG1 mAbs | |||||||||
| 17b | Sample 1 | 54.8 | 21.1 | 175 | 54.9 | 180 | 75.6 | 0.8 | 0.8 ± 0.022 |
| Sample 2 | 54.8 | 21.1 | 175 | 54.9 | 180 | 38.2 | 0.4 | ||
| Sample 3 | 54.8 | 21.2 | 175 | 54.8 | 180 | 18.7 | 0.2 | ||
| b12 | Sample 1 | 53.7 | 22.1 | 170 | 53.4 | 180 | 54.5 | 0.6 | 0.8 ± 0.039 |
| Sample 2 | 53.7 | 22.1 | 170 | 53.1 | 180 | 27.1 | 0.3 | ||
| Sample 3 | 53.7 | 22.1 | 170 | 53.1 | 180 | 13.5 | 0.1 | ||
| 2G12 | Sample 1 | 47.6 | 20.5 | 150 | 47.8 | 155 | 67.7 | 0.7 | 0.66 ± 0.020 |
| Sample 2 | 47.6 | 20.5 | 150 | 47.7 | 155 | 33.5 | 0.3 | ||
| Sample 3 | 47.4 | 20.5 | 150 | 47.7 | 155 | 16.5 | 0.2 | ||
| VRC01 | Sample 1 | 56.1 | 23.4 | 176 | 56.3 | 180 | 78.0 | 0.8 | 0.76 ± 0.037 |
| Sample 2 | 56.1 | 23.4 | 176 | 56.2 | 180 | 39.3 | 0.4 | ||
| Sample 3 | 56.1 | 23.4 | 176 | 56.3 | 180 | 19.2 | 0.2 | ||
| VRC03 | Sample 1 | 55.2 | 22.4 | 175 | 55.1 | 180 | 74.2 | 0.8 | 0.8 ± 0.041 |
| Sample 2 | 55.2 | 22.4 | 175 | 55.2 | 180 | 39.9 | 0.4 | ||
| Sample 3 | 55.2 | 22.1 | 175 | 55.2 | 180 | 18.3 | 0.2 | ||
| HJ16 | Sample 1 | 56.1 | 23.1 | 178 | 57.1 | 180 | 77.1 | 0.8 | 0.71 ± 0.019 |
| Sample 2 | 56.1 | 22.9 | 178 | 57.1 | 180 | 41.3 | 0.4 | ||
| Sample 3 | 56.2 | 23.1 | 178 | 57.1 | 180 | 18.7 | 0.2 | ||
a For the standard proteins, four concentrations (0.8–2.2 mg/ml) of gelsolin (80 KDa) in EGTA Tris buffer and three concentrations (0.6–1.8 mg/ml) of GAPDH in tetrameric state (147 kDa) in PBS were used to estimate I0 values at zero angles.
TABLE 2.
SAXS data analysis-based structural parameters deduced for the gp120-reactive IgG1 mAbs
The concentrations of the mAbs were estimated considering the mass of the individual mAb to be ∼150 kDa. NSD values are of the 10 models computed for the gp120-reactive mAbs.
| Unliganded mAbs (gp120-reactive non-neutralizing IgG1 mAbs) | Sample | Guinier analysis |
P(r) analysis |
I0/(c*m) | NSD ± variation | ||||
|---|---|---|---|---|---|---|---|---|---|
| Rg | RC | L | Rg | Dmax | I0 | ||||
| Å | Å | Å | mg/ml | ||||||
| A32 | Sample 1 | 61.7 | 21.2 | 200 | 61.9 | 205 | 63.7 | 0.7 | 0.67 ± 0.009 |
| Sample 2 | 61.7 | 21.1 | 200 | 61.9 | 205 | 32.2 | 0.3 | ||
| Sample 3 | 61.8 | 21.1 | 200 | 62.0 | 205 | 15.1 | 0.2 | ||
| F425_a1g8 | |||||||||
| Sample 1 | 63.9 | 23.2 | 205 | 69.1 | 205 | 75.3 | 0.8 | 0.66 ± 0.022 | |
| Sample 2 | 63.9 | 23.2 | 205 | 69.0 | 205 | 36.8 | 0.4 | ||
| Sample 3 | 63.9 | 23.2 | 205 | 69.1 | 205 | 17.5 | 0.2 | ||
| F425_b4a1 | |||||||||
| Sample 1 | 64.9 | 23.6 | 210 | 72.9 | 210 | 90.1 | 1.0 | 0.67 ± 0.029 | |
| Sample 2 | 64.8 | 23.6 | 210 | 72.9 | 210 | 44.5 | 0.5 | ||
| Sample 3 | 64.8 | 23.2 | 210 | 73.0 | 210 | 22.1 | 0.2 | ||
| F105 | Sample 1 | 63.7 | 23.2 | 205 | 72.9 | 207 | 90.1 | 1.0 | 0.64 ± 0.031 |
| Sample 2 | 63.7 | 23.2 | 205 | 72.9 | 207 | 44.5 | 0.5 | ||
| Sample 3 | 63.3 | 23.1 | 205 | 73.0 | 207 | 22.1 | 0.2 | ||
| 48D | Sample 1 | 62.8 | 23.1 | 202 | 72.9 | 200 | 90.1 | 1.0 | 0.64 ± 0.012 |
| Sample 2 | 62.8 | 23.2 | 202 | 72.9 | 200 | 44.5 | 0.5 | ||
| Sample 3 | 62.8 | 23.2 | 202 | 73.0 | 200 | 22.1 | 0.2 | ||
TABLE 3.
SAXS data analysis-based structural parameters deduced for the gp41-reactive IgG1 mAbs
NSD values are of the 10 models computed for the gp41-reactive mAbs.
| Unliganded mAbs | Sample | Guinier analysis |
P(r) analysis |
I0/(c*m) | NSD ± variation | ||||
|---|---|---|---|---|---|---|---|---|---|
| Rg | RC | L | Rg | Dmax | I0 | ||||
| Å | Å | Å | mg/ml | ||||||
| gp41-reactive neutralizing IgG1 mAbs | |||||||||
| 4E10 | Sample 1 | 55.8 | 23.6 | 175 | 53.9 | 175 | 77.3 | 0.8 | 0.67 ± 0.014 |
| Sample 2 | 55.8 | 23.6 | 175 | 53.9 | 175 | 38.3 | 0.4 | ||
| Sample 3 | 55.9 | 23.6 | 175 | 53.9 | 175 | 21.2 | 0.2 | ||
| 2F5 | Sample 1 | 57.4 | 24.4 | 180 | 57.4 | 185 | 91.9 | 0.9 | 0.70 ± 0.048 |
| Sample 2 | 57.3 | 24.4 | 180 | 57.4 | 185 | 45.5 | 0.5 | ||
| Sample 3 | 57.4 | 24.4 | 180 | 57.4 | 185 | 23.2 | 0.2 | ||
| Z13e1 | Sample 1 | 56.7 | 24.3 | 178 | 56.8 | 180 | 64.9 | 0.7 | 0.66 ± 0.020 |
| Sample 2 | 56.7 | 24.3 | 178 | 56.8 | 180 | 32.7 | 0.3 | ||
| Sample 3 | 56.7 | 24.3 | 178 | 56.9 | 180 | 16.8 | 0.2 | ||
| gp41 reactive non-neutralizing IgG1 mAbs | |||||||||
| 5F3 | Sample 1 | 59.2 | 20.5 | 192 | 59.2 | 200 | 75.7 | 0.8 | 0.75 ± 0.029 |
| Sample 2 | 59.1 | 20.5 | 192 | 59.2 | 200 | 35.7 | 0.4 | ||
| Sample 3 | 59.2 | 20.5 | 192 | 59.2 | 200 | 17.5 | 0.2 | ||
| 98-6 | Sample 1 | 63.4 | 24.7 | 202 | 63.7 | 205 | 49.0 | 0.5 | 0.67 ± 0.022 |
| Sample 2 | 63.4 | 24.7 | 202 | 63.6 | 205 | 23.8 | 0.3 | ||
| Sample 3 | 62.4 | 24.6 | 202 | 63.7 | 205 | 12.2 | 0.1 | ||
| F-240 | Sample 1 | 64.3 | 24.2 | 206 | 64.1 | 210 | 88.8 | 1.0 | 0.64 ± 0.028 |
| Sample 2 | 64.5 | 24.1 | 206 | 64.0 | 210 | 43.7 | 0.5 | ||
| Sample 3 | 64.5 | 24.1 | 206 | 64.0 | 210 | 20.8 | 0.2 | ||
Kratky analyses of the SAXS data sets were performed to gain insight into the extent of inherent disorder in the global shape of the unliganded mAbs (Fig. 2B). Peak profiles in the Kratky plot support a globular shape, and a hyperbolic profile indicates a system with higher inherent disorder (36, 37). Multiple peaks at almost the same position for 17b, b12, VRC01, VRC03, and HJ16 indicated that these mAbs have similar shapes, particularly in the sense of spatial disposition of their three domains (Q ∼0.03, 0.07, and 0.16 Å−1; marked with arrows). On the other hand, the Kratky plot for 2G12 showed one dominant peak at Q ∼0.04 Å−1 and two minor peaks at Q ∼0.13 and 0.19 Å−1, suggesting that the three domains are differentially positioned in this nmAb. In sharp contrast, the Kratky analysis for the non-neutralizing gp120-reactive mAbs studied (A32, F425_a1g8, F425_b4a1, F105, and 48D) were different from neutralizing mAbs and showed a main peak at Q ∼0.07 Å−1 and one shoulder at 0.03 Å−1 and one smaller peak at 0.15–0.17 Å−1. A similar profile of Kratky plots for gp41 binding nmAbs suggests that 2F5 and Z13e1 have their domains similarly positioned as 17b, b12, VRC01, VRC03, and HJ16, but the presence of an additional peak at Q ∼0.13 Å−1 indicated that they might have some feature similar to 2G12. Interestingly, 4E10 appeared to have a different shape profile because the two peaks at 0.03 and 0.07 Å−1 were not distinct as seen in other nmAbs. Further, Kratky plots for gp41-reactive non-nmAbs (5F3, 98-6, and F-240) comparable with those seen for gp120 reactive non-nmAbs supported a similarity in their global shapes regardless of their preferential ligand. Overall, Kratky analysis of unliganded mAbs revealed that there was a distinct difference in the relative spatial positioning of the three domains in the predominant solution shape of the non-neutralizing versus neutralizing Abs.
Additionally, to estimate the structural parameters of the global shape of the unliganded mAbs, indirect Fourier transformation of the measured I(Q) profiles were carried out using data over a Q range of (0.008–0.5 Å−1), which provided probability distribution of interatomic vectors of different dimensions contributing to the scattering shape of the mAbs in solution (Fig. 2C). In P(r) curve analysis, vectors arising from adjacent domains would lead to closely associated peaks, and domains separated by a distance would lead to gap between peaks due to the low probability of finding vectors representing intermediate range. The two-peaked P(r) solved for b12 was similar to that reported earlier by us (3), with Dmax and Rg of 180 and 53.4 Å, respectively. Interestingly, SAXS data for 17b, VRC01, VRC03, and HJ16 also solved P(r) curves with two peaks as seen for b12, and the deduced Dmax and Rg values for these four mAbs were also comparable with those calculated for b12 (Tables 1–3). However, the P(r) curve computed for 2G12 clearly differed from those of the other five gp120-reactive nmAbs. In comparison, smaller Dmax and Rg values of 2G12 (∼155 and 44 Å, respectively) indicated that the overall size of the 2G12 molecule is smaller than that the other five gp120-reactive nmAbs. Moreover, the P(r) for 2G12 adopted a peak and shoulder profile, further suggesting that the three domains of 2G12 were packed very different from those of 17b, b12, VRC01, VRC03, and HJ16. On the other hand, P(r) curves computed for gp120-reactive non-nmAbs A32, F425_a1g8, F425_b4a1, 48D, and F105 showed a three-peak profile with a clear gap between the peak at 40 Å and two close peaks at 100 and 140 Å, implying a very open shape or a gap between three domains. Interestingly, the gp41-reactive mAbs also indicated a clear difference in the computed P(r) curves for nmAbs versus lacking ones. The P(r) curves for the 2F5 and Z13e1 showed a clear two-peak profile, but 4E10 adopted a peak and shoulder profile, supporting a more compact global shape for the latter. In sharp contrast (and similar to non-neutralizing gp120-reactive mAbs), the P(r) curves for the non-neutralizing gp41-reactive mAbs also adopted a three-peak profile with a gap between one peak at 40 Å and two close peaks at ∼100 and 120–140 Å. The peak at 40 Å being present in both neutralizing and non-neutralizing gp120-gp41-reactive mAbs indicated that it may arise from common shape feature(s) in both sets of mAbs, possibly from the vectors present in the Fab or Fc domains alone. The disparities in the P(r) profiles of neutralizing and non-neutralizing mAbs in the higher dimensions occur due to differences in interatomic vectors arising from different positioning of domains. For both gp120- and gp41-binding mAbs, the presence of domains and the clear separation between the three peaks in the P(r) curves for non-nmAbs suggest a distal positioning of domains of non-nmAbs and a compact shape in the nmAbs, regardless of their ligand.
Visualizing Global Shape of Unliganded mAbs
Using the measured SAXS I(Q) profiles in the Q range of 0.008–0.5 Å−1, 10 independent models of each mAb were generated using dummy residues and both uniform density and chain ensemble modeling approaches. Finally, all 10 models for each mAb were compared with each other and clustered into similar shapes, and each group was averaged to obtain the predominant solution shape of individual unliganded mAb. In all cases, only one cluster was formed with all 8–10 models in each set; results were averaged and are presented in Fig. 3, A and B. It is pertinent to mention here that the results from uniform density and chain ensemble modeling were very similar. For chain-ensemble modeling, we had to input dummy residue numbers that were roughly equal to the amino acids present in mAbs. At same time, no such input was required for uniform density modeling, and thus we opted to analyze and present structure reconstruction results from a uniform density approach. Mean normalized spatial discrepancy values in the range of 0.65–0.8, with low S.D. suggested that the restored models agree with each other and constituted part of the same ensemble of shapes accessible to each mAb in solution (Tables 1–3).
FIGURE 3.

Global shapes restored for unliganded mAbs. A, dummy residue-based shapes restored for the gp120-reactive neutralizing and non-neutralizing mAbs. The mesh view model represents averaged common shape features from 10 independent solutions. Domains from the crystal structure of b12 have been arranged inside the envelope to compare the volumes of individual domains in the SAXS data-based models. Models below the dotted line are a top view of the same models. B, shapes restored for gp41-reactive neutralizing and non-neutralizing mAbs. Top views of the models are separated by the dotted line.
A quick comparison of the SAXS-based restored shapes showed that whereas non-nmAbs adopted an open shape, the nmAbs adopted a shape where the two Fab arms were not extended away from the central Fc. 17b adopted a shape where one Fab arm appeared extended away, and the other remained nestled close to the Fc, as reported by us for b12 (3). We have previously published (3) the SAXS data-based global shape of unliganded IgG1 b12 and thus have not presented the b12 model again. In contrast, the 2G12 Fab arms could not be distinguished from Fc, which correlated with the compact shape seen from Kratky and P(r) curve analysis, and previously published EM studies. Similar to 17b and b12 (3), other gp120-reactive nmAbs, VRC01, VRC03, and HJ16, also adopted a global shape, where only one Fab arm appeared extended. In the case of the gp41-binding nmAb 4E10, although the positioning of two Fab arms can be perceived from the model, they appeared closer to Fc or unextended, thus explaining the partial similarity in the pattern of its Kratky and P(r) curve to those seen for 2G12. Further, the model for 2F5 suggested asymmetric positioning of the two Fab arms, and the Fab-Fc linkers were not as open or extended as in other non-nmAbs. Interestingly, the model of Z13e1 indicated that one Fab was extended away, as seen in gp120-reactive nmAbs: 17b, b12, VRC01, VRC03, and HJ16. The open shapes of the non-nmAbs explained the gap seen between the peaks in the computed P(r) curves for these mAbs.
Alternatively, we also employed a Web-based genetic algorithm-based search protocol, SHAPEUP (38), to explore the three-dimensional shapes that best represented scattering shapes of unliganded mAbs encoded in the SAXS I(Q) profiles. In all cases, the top five models were in the same shape cluster with a correlation coefficient in range of 0.8–0.9. The averages of these models with the highest correlation coefficient are been shown in Fig. 4A. The mean correlation coefficient values were as follows: 17b, 0.8; 2G12, 0.9; VRC01, 0.9; VRC03, 0.8; HJ16, 0.8; b12, 0.9; A32, 0.8; F425_a1g8, 0.9; F425_b4a1, 0.8; F105, 0.8; 48D, 0.9; 4E10, 0.9; 2F5, 0.8; Z13e1, 0.8; 5F3, 0.8; 98-6, 0.8; F-240, 0.8. Importantly, these results provided similar shape profiles for each mAb as obtained by dummy residue-based modeling, thus excluding any artifact from modeling approaches employed. The primary finding from both restoration approaches was that there is a difference in the global shape of the mAbs capable of neutralizing HIV-1 and lacking neutralizing ability, and these differences are prevalent regardless of the epitopes recognized.
FIGURE 4.

SAXS intensity profile-based global shapes of unliganded mAbs using the SHAPEUP server. Shown are predominant shapes of the gp120-reactive neutralizing and non-neutralizing mAbs (A) and gp41-reactive neutralizing and non-neutralizing mAbs (B). C, coin shapes representing the relative positioning of the Fab and Fc domains of the mAbs in solutions from rigid body modeling. The representations for neutralizing and non-neutralizing mAbs highlight the differential spacing of Fab arms from central Fc and the perpendicular positioning of the Fc portion in neutralizing mAbs, as seen in the crystal structure of IgG1 b12 (PDB entry 1HZH). Similar representations for 2G12 and 4E10, whose solutions differed from other mAbs, are shown on the right.
Along with dummy residue modeling, we performed rigid body modeling by using intact structures of Fab and Fc portions from the crystal structure of IgG1 b12 (PDB code 1HZH). Briefly, two Fab and Fc domains were abstracted by removing the linkers between Fab and Fc domains from the crystal structure of IgG1 b12, and then the disjointed domains were repositioned in space relative to each other until the theoretical SAXS profile of the composite model was similar to the experimentally measured SAXS data. The only constraint employed was that the distance between the end of the two CH1 domains and the start of CH2 domains was less than 50 Å. The final χ2 values of the SASREF program-based rigid body modeling were as follows: 17b, 1.5; 2G12, 1.2; VRC01, 1.4; VRC03, 1.5; HJ16, 1.3; b12, 1.4; A32, 1.9; F425_a1g8, 1.6; F425_b4a1, 1.8; F105, 1.3; 48D, 1.5; 4E10, 1.3; 2F5, 1.5; Z13e1, 1.7; 5F3, 1.9; 98-6, 1.9; F-240, 1.6. Importantly, the primary observation was that for most nmAbs, regardless of their reactivity to gp120 or gp41, one Fab was positioned close to the Fc, whereas the other Fab arm was solved in position further away from Fc (Fig. 4C). The only exceptions were 2G12 and 4E10, where the two Fab arms in the final solution were packed close to each other on top of Fc and symmetrically closely stacked on two sides of the Fc, respectively. For these two mAbs, one reactive to gp120 and the other to gp41, models indicated no space between the Fc and Fab domains. On the other hand, similar to solutions from dummy residue- and genetic algorithm-based modeling, both Fab arms were distally located from the central Fc in the case of all of the non-nmAbs.
Another peculiar difference between the non-neutralizing and neutralizing Abs was that the Fc domain always solved at 90° to the axis of the Fab arms in the case of nmAbs, whereas the Fc and Fab domains were in the same plane (depicted by lines in the top views of schematic representations in Fig. 4C). Interestingly, this feature is also present in the crystal structure of the nmAb b12 (PDB code 1HZH). The sideways positioning of the Fc in nmAbs and the proximity of one Fab and Fc imply some kind of interaction between Fab and Fc in the global shape of the nmAbs. This is an important result because Fc and lower hinge plays a key role in recruitment of C1q to initiate Fc-mediated neutralization events. Among the mAbs studied here, the shape of 2G12 supported the presence of strong Fab-Fab interaction along with Fab-Fc, an observation that has been reported before (7, 9). In summary, all SAXS data-based modeling approaches provided similar results indicating that the global shapes of mAbs differ significantly; non-nmAbs adopt an open shape, and known nmAbs adopt a shape where at least one or both Fabs are unextended from the central Fc, which was found in a twisted orientation.
Shape of gp120-bound mAbs
Gel filtration profiles of the ∼1:2 molar mixtures of mAb-gp120 are presented in Fig. 5A. (These studies could be done for only gp120-reactive mAbs because gp41 peptide was too small to obtain differential results in these experiments.) The size exclusion chromatography clearly revealed that the non-nmAbs formed 1:2 complexes with a predominant peak close to an apparent mass of ∼400 kDa (expected ∼370–390 kDa), and there was a small peak corresponding to that expected for unliganded gp120 (Fig. 5A). At the same time, the four gp120-reactive nmAbs formed only 1:1 complex, and there was a significant amount of unliganded gp120. Based on retention times, the apparent masses of 1:1 complexes of 17b, b12, and VRC01 with gp120 were ∼270 kDa, and the mass of binary complex of 2G12-gp120 was ∼220 kDa, implying a relatively more compact shape of the latter. Moreover, different incubation times prior to injection onto the column did not alter the finding that only binary complex was formed in the case of gp120-reactive nmAbs. To further confirm the observations, we repeated the gel filtration experiments by incubating purified mAbs with a different version of gp120 (HIV-1BaL) and found similar results in case both non-neutralizing and neutralizing mAbs. The peaks corresponding to the mAb-gp120 complexes were collected and re-evaluated by gel filtration after different time intervals (during which they were stored at 4 °C). The observation of single peaks in the elution profile at the same retention volumes confirmed the stability of the mAb-gp120 complexes up to 72 h. Earlier, 2G12 had been shown to bind to a single gp140 molecule (1:1 binding) using its conjoined Fab arms (8). Additionally, we estimated the apparent mass of the gp120-mAb complexes using SEC-MALLS experiments (Fig. 5B). Data analysis yielded molecular mass of ∼400 kDa for complexes of non-nmAbs and a molecular mass of ∼280 kDa for complexes of gp120 with nmAbs, confirming limited binding ability of nmAbs.
FIGURE 5.

Analysis of the gp120-reactive mAbs in complex with gp120. A, gel filtration profiles of the 1:2 molar mixtures of gp120 reactive mAbs. B, SEC-MALLS analysis of proteins and their complexes in solution. Left, proteins eluted at 8 and 12 ml corresponding to 1:2 complex and unliganded mAb or gp120 with a molar mass of 3.94 × 105 and 1.63 × 105 g/mol, respectively. Right, proteins eluted at 10 and 12 ml, corresponding to 1:1 complex and unliganded mAb or gp120 with a molar mass of 2.88 × 105 and 1.68 × 105 g/mol, respectively. C, SAXS data acquired from purified fractions of 1:1 and 1:2 complexes of gp120-bound mAbs. D, left two panels, Kratky plots of the SAXS data measured from purified samples of gp120-mAb complexes; right two panels, P(r) curves computed from the SAXS data acquired from the binary and ternary complexes of mAbs and gp120. E, results from multiphase dummy atom modeling for the binary and ternary complexes of gp120 and mAbs. The blue shape represents the gp120 component in the complexes, and the mAbs are represented by a red shape. The gray mesh represents the hydration layer.
To gain visual insight into the predominant shapes of the mAb-gp120 complexes and explore whether the shapes of the gp120-reactive mAbs alter drastically upon binding the ligand, SAXS I(Q) profiles were acquired from the samples of some mAbs bound to gp120 (Fig. 5C). This was an important experiment because IgG1 b12 was seen to adopt an asymmetric shape in the unliganded state from crystallography and SAXS (2, 3), but earlier EM studies reported b12 to adopt an open shape bound to two gp120 molecules (8). Measured I(Q) profiles suggested a lack of aggregation in the samples of mAb-gp120 complexes (Fig. 5C). Guinier approximations for globular and rodlike scattering shapes were employed to obtain the Rg and RC values for the predominant solution shape of the mAb-gp120 complexes, and these were utilized to estimate the long dimension, L, of the complexes (Table 4). There was a clear difference in the L values estimated for the complexes of gp120(s) with neutralizing versus non-neutralizing mAbs, where the former adopted smaller dimensions. Kratky analysis of the SAXS data sets indicated that the binary complexes of nmAbs with gp120 adopt globular scattering shapes, whereas the presence of a two-peak profile for the ternary complex of non-nmAbs with gp120 suggested a multidomain globular shape (Fig. 5D). Additionally, P(r) curve analysis of the mAb-gp120 complexes supported the above interpretation (Fig. 5D). Multiple peaks in the case of non-nmAb-(gp120)2 complexes suggested that the shapes of these complexes have domains separated by space. Importantly, the Rg and Dmax values estimated from indirect Fourier transformation of the data agreed well with the estimations from Guinier analysis and also suggested the increased dimensions of the ternary complexes of non-nmAbs in comparison with the complexes formed by nmAbs. Among the neutralizing ones, the dimension of the 2G12-gp120 complex appeared smallest of the complexes studied here, with Dmax of only 250 Å, whereas the binary complex of VRC01-gp120 adopted the longest dimension of 290 Å. On the other hand, the A32-(g120)2 ternary complex adopted a Dmax value much smaller than the complexes formed with F425_a1g8 and F425_b4a1, smaller by 65–75 Å (Table 4). Additionally, the estimated I0 value from the P(r) analysis helped in ascertaining the concentration of the mAb-gp120 complexes in solution (Table 4).
TABLE 4.
SAXS data analysis based structural parameters deduced for the purified gp120-mAb complexes
NSD values are of the 10 models computed for the complexes.
| mAb-gp120 complexes | Guinier analysis |
P(r) analysis |
Concentration | NSD ± variation | |||||
|---|---|---|---|---|---|---|---|---|---|
| Rg | RC | L | Rg | Dmax | I0 | ||||
| Å | Å | Å | mg/ml | ||||||
| Neutralizing mAbs-gp120 (1:1) (∼270 kDa) | |||||||||
| 17b-gp120 | 69.3 | 18.1 | 232 | 69.4 | 260 | 60.9 | 0.4 | 0.52 ± 0.022 | |
| b12-gp120 | 69.9 | 17.3 | 235 | 70.1 | 265 | 93.7 | 0.6 | 0.57 ± 0.031 | |
| VRC01-gp120 | 80.0 | 18.3 | 270 | 80.1 | 290 | 50.1 | 0.3 | 0.63 ± 0.017 | |
| 2G12-gp120 | 65.9 | 17.2 | 221 | 66.1 | 250 | 86.1 | 0.5 | 0.45 ± 0.012 | |
| Non-neutralizing mAbs/gp120 (1:2) (∼390 kDa) | |||||||||
| A32-(gp120)2 | 103.1 | 53.4 | 308 | 103.6 | 315 | 53.6 | 0.2 | 0.63 ± 0.017 | |
| F425_a1g8-(gp120)2 | 120.2 | 52.1 | 378 | 120.8 | 390 | 83.1 | 0.3 | 0.59 ± 0.033 | |
| F425_b4a1-(gp120)2 | 121.1 | 51.7 | 382 | 121.9 | 400 | 95.2 | 0.4 | 0.62 ± 0.037 | |
The SAXS I(Q) profiles of the complexes were employed to model the shape of the mAb-gp120 complexes. By using SAXS I(Q) profiles of the complex, unliganded mAb, and unliganded gp120 and employing the MONSA program, we computed 10 models for the shape of the two components in the overall shape of each complex and averaged them to obtain the predominant shape of the complex (Fig. 5E). The complex for 2G12-gp120 showed that the gp120 binds the two conjoined Fab arms of 2G12, an observation that was in agreement with reports published earlier (7). In contrast, the other nmAbs formed a complex with gp120 using only one Fab arm. At the same time, the two-component models solved for the ternary complexes of non-nmAbs with two gp120 molecules clearly showed that each Fab arm could bind one gp120. There appeared to be slight differences in the way the gp120s bind to the different non-nmAb, but our shape reconstruction confirmed that except 2G12 other nmAbs bind only one gp120 by utilizing one of their Fab arms, whereas both Fab arms of the non-nmAbs are capable of binding one gp120 molecule each. In summary, analysis of complexes of mAbs with gp120 revealed that nmAbs can bind only one ligand via their extended arm (conjoined arms for 2G12), and their shape does not undergo detectable shape changes upon binding gp120(s).
Open Versus Closed Disposition of Fab Arms; Correlation with Papain Digestion
To further support our above findings that Fab arms are differentially disposed in space, we carried out papain digestion of the antibodies because each Fab-Fc linker of all of the IgG1 mAbs has papain proteolysis sites (40, 41). We optimized conditions for digestion of mAbs by papain immobilized on beads, and the proteolysis products after different time points (0, 30, 60, and 120 min) were analyzed by gel filtration. A typical analysis of the proteolysis profile (digestion profile of b12) is presented in Fig. 6A. Comparison of the elution profiles with the standard protein samples indicated that three main products emerge from papain digestion: undigested mAb (∼150 kDa) and fragments corresponding to 100 and 50 kDa. For every mAb, the area under these three main peaks was measured from three independent runs of gel filtration and plotted as a function of time (Fig. 6A). Papain has two cutting sites in the heavy chain His224-Thr225 and Glu233-Leu234, essentially separated by nine amino acids. For both non-neutralizing and neutralizing mAbs, Edman sequencing significantly revealed the N-terminal sequence of the Fc-containing portion (eluting close to ∼50 kDa) to be LLGPS, suggesting that in our proteolysis conditions, papain was cutting at the second site. Plots shown in Fig. 6C (particularly the extent of digestion after 30 and 60 min) revealed that under similar conditions, nmAbs are slightly more resistant to papain digestion compared with non-nmAbs, and the delay in proteolysis is present in both gp120- and gp41-reactive nmAbs. Further, 4E10 showed the slowest response to degradation by papain, an observation that correlated with its global shape of both Fab arms squeezed close to Fc and thus least accessible. Western blot experiments confirmed the presence of the Fc portion in the 100- and 50-kDa fragments of mAbs after 60 min of papain digestion (Fig. 7A). Results revealed that the 100-kDa fragment of nmAbs had the Fc portion, suggesting that it was composed of one Fab and Fc. On the other hand, a higher readout for the Fc portion in the 50-kDa fragment of the non-nmAbs suggested that papain was able to cut both Fab arms from Fc in non-nmAbs. The only surprise was 2G12, where the 100-kDa fragment lacked any Fc component, an observation that was reported earlier because its Fab arms elute as dimer of about ∼100 kDa (7). In summary, our complete versus partial papain digestion results of the non-neutralizing versus neutralizing mAbs correlated with the open versus distorted shapes for the mAbs, regardless of whether they bind gp120 or gp41.
FIGURE 6.

Summary of the gel filtration profiles of the papain-digested products of mAbs as a function of time. A, the left panel shows different peaks that eluted after papain digestion of neutralizing mAbs, IgG1 b12. Black, red, blue, and magenta profiles represent digestion products after 0, 30, 60, and 120 min of incubation of b12 with beads immobilized with papain. The cyan, magenta, and yellow boxes represent the fraction collection zones for b12 digestion runs. B, the right panel shows a histogram representation of the elution profiles shown in the left panel: the area under different peaks (corresponding to 150 kDa (cyan), 100 kDa (magenta), and 50 kDa (yellow) at four different time points. C, for all other mAbs, the digestion by papain is represented in histogram form with the same color code. Error bars were computed by comparing three independent runs of the digestions.
FIGURE 7.

Characterization of papain-digested fragments of mAbs. A, left, estimation of the Fc portion in the 100- and 50-kDa fragments of the gp120-reactive mAbs (except 2G12). Both blots and their estimation based on mean pixel intensity are presented. Middle, estimation of the Fc portion in the 100- and 50-kDa papain-digested fragments of gp41-reactive mAbs. Right, the 100-kDa fragment of 2G12 lacked any Fc component; rather, it eluted in the peak corresponding to 50 kDa in mass. (Comparisons have been shown with A32 and 98-6). B, left, ELISA results showing ability of the Fc-containing 100-kDa fragment of and full-length neutralizing mAbs to bind immobilized gp120 or gp41. Right, ELISA results showing the ability of immobilized full-length gp120-reactive mAbs and their 50- and 100-kDa fragments to bind gp120. After the addition of gp120, 2G12 was added, and signal corresponding to the Fc portion of 2G12 was measured. C, SAXS data-based predominant solution shape of the Fab-Fc portion from neutralizing mAbs. The shape of Fab-Fc from b12 has been compared with the shape of the closed Fab arm and Fc in the crystal structure of full-length IgG1 b12. Also, the shape of the Fab dimer purified after papain digestion of 2G12 has been compared with its crystal structure. D, results from multiphase modeling of Fab (from the 50-kDa fragment of gp120-reactive neutralizing mAbs and the 100-kDa fragment of Fab dimer from 2G12). Blue and red shapes represent bound gp120 and Fab arm, respectively. In the case of 2G12, the red shape resides in the Fab dimer. Error bars were computed by comparing three independent binding experiments.
Characterization of the Papain-digested Fragments of mAbs
We tested the ability of the gel filtration-purified 100 kDa Fab-Fc and 50-kDa Fab or Fc fragment from the mAbs to bind their respective ligands (Fig. 7B). Briefly, gp120 or MPER peptide was coated on ELISA plates, and the papain-digested fragments were allowed to bind these ligands. Later, bound Fc component was probed using anti-IgG1 Fc antibodies. Results brought forth that the 100-kDa fragments from all nmAbs showed only 5–10% binding to their ligands in comparison with the full-length mAbs (Fig. 7B, left), concluding that the Fab-Fc portions from nmAbs are not capable of binding their ligands. These results somewhat explained our SAXS-based findings that gp120-reactive nmAbs could bind only one gp120 molecule (Fig. 5). Additionally, for gp120 mAbs, we carried out another experiment where we coated the papain-digested 100- and 50-kDa fragments in separate wells in an ELISA plate and then added gp120, followed by the addition of 2G12, and its Fc portion was detected with anti-Fc antibodies (Fig. 7B, right). We found that full-length mAbs (17b, b12, and VRC01) and their 50-kDa fragments can bind to gp120. In the case of 2G12, as expected, the 100-kDa fragment composed of Fab dimer could bind gp120, and the Fab lacking the 50-kDa fragment of 2G12 (i.e. Fc) could not bind to gp120. These results raised a question, whether the papain digestion somehow altered the shape of the Fab-Fc, rendering it incapable of binding its ligand. To answer this, we purified the Fab-Fc fragment, concentrated the samples, and acquired SAXS data from them (Fig. 7C). The I(Q) profiles confirmed that the Fab-Fc fragments were monodisperse and lacked any aggregation (data not shown). Kratky analysis of Fab-Fc fragment except for 2G12 showed a two-peak profile, suggesting that the Fab-Fc fragment still retained some extent of rigidity. Further, the P(r) curves computed for the Fab-Fc fragment showed that most have a linear dimension close to 120–130 Å (Table 5). The Fab-Fc from b12 showed a clear two-domain shape, whereas the others except for 2G12 showed mainly a single domain shape. Interestingly, the 100-kDa fragment from 2G12 lacking the Fc portion solved a P(r) curve with two distinct peaks suggesting a two-domain shape (data not shown). Structure reconstruction showed that the Fab-Fc fragment from b12, 17b, VRC01, 4E10, 2F5, and Z13e1 adopted an L-shaped profile. Interestingly, the Fab-Fc from b12 compared well with the closed Fab arm and Fc of the crystal structure of the b12 antibody. Similarly, the 100-kDa fragment from 2G12, lacking Fc, adopted a shape that compared well with the dimeric crystal structure of the Fab solved earlier (42). The shape of the Fab-Fc fragment from other IgG1 mAbs suggested that this 100-kDa fragment resulted from limited proteolysis of the extended Fab arm of the mAbs by papain.
TABLE 5.
SAXS data analysis-based structural parameters deduced for the 100-kDa fragment of the mAbs
The concentrations of the digested mAbs were estimated considering the mass of the larger fragment as ∼100 kDa. NSD values are of the 10 models computed for the fragment.
| 100-kDa fragment | Guinier analysis |
P(r) analysis |
Concentration | NSD ± variation | |||||
|---|---|---|---|---|---|---|---|---|---|
| Rg | RC | L | Rg | Dmax | I0 | ||||
| Å | Å | Å | mg/ml | ||||||
| gp120-reactive neutralizing IgG1 mAbs | |||||||||
| 17b | 44.1 | 22.6 | 131 | 44.1 | 130 | 115.9 | 1.8 | 0.52 ± 0.02 | |
| b12 | 43.1 | 24.1 | 123 | 43.1 | 130 | 82.1 | 1.4 | 0.55 ± 0.03 | |
| 2G12 | 36.1 | 15.8 | 112 | 36.1 | 120 | 102.8 | 1.6 | 0.61 ± 0.02 | |
| VRC01 | 41.5 | 21.4 | 123 | 41.5 | 130 | 118.2 | 1.8 | 0.56 ± 0.03 | |
| gp41-reactive neutralizing IgG1 mAbs | |||||||||
| 4E10 | 41.2 | 21.2 | 122 | 71.9 | 130 | 42.2 | 0.7 | 0.42 ± 0.01 | |
| 2F5 | 40.6 | 20.8 | 120 | 69.1 | 130 | 49.6 | 0.8 | 0.54 ± 0.02 | |
| Z13e1 | 40.9 | 20.8 | 121 | 72.9 | 130 | 59.4 | 1.0 | 0.47 ± 0.03 | |
On the other hand, our ELISA showed that the Fab arm isolated from the fragments of gp120-reactive mAbs that eluted close to 50 kDa could easily bind gp120. (For 2G12, we used the Fab dimer that eluted close to 100 kDa). We mixed the Fab portion with gp120, purified from gel filtration, and acquired SAXS data from the gp120-Fab complex. I(Q) profiles confirmed no aggregation in samples and a monodisperse nature of scattering molecules in solution (data not shown). Kratky profiles of the SAXS profiles indicated that the Fab-gp120 complex from 2G12 adopted a tight globular shape, whereas the Fab-gp120 complex from other mAbs adopted a more spread out globular shape. Similar results were seen in the P(r) curves computed for the Fab-gp120 complexes, which showed a single lobe shape for the complex from 2G12, whereas complexes of Fabs from other mAbs (17b, b12, and VRC01) implied a two-domain shape (Table 6). Structure reconstruction was done by the MONSA program and averaged to interpret the shape profile of the gp120 and Fab components in the complex. The models suggested that the isolated Fab arms from the nmAbs could bind gp120 and that the conjoined Fab arms of 2G12 together bind a single gp120 molecule, very similar to what was seen earlier with full-length mAbs (Fig. 7D).
TABLE 6.
SAXS data analysis-based structural parameters deduced for the 1:1 complex of fragmented Fab arm (50-kDa fragments) and gp120
The concentrations of the Fab-gp120 complexes were estimated considering mass of the complex to be ∼170 kDa. NSD values are of the 10 models computed for the complex.
| Fab-gp120 complex | Guinier analysis |
P(r) analysis |
Concentration | NSD ± variation | ||||
|---|---|---|---|---|---|---|---|---|
| Rg | RC | L | Rg | Dmax | I0 | |||
| Å | Å | Å | mg/ml | |||||
| b12 | 57.8 | 20.6 | 187 | 57.9 | 185 | 87.6 | 0.8 | 0.77 ± 0.014 |
| 2G12 (220 kDa) | 54.7 | 20.1 | 176 | 54.4 | 195 | 104.1 | 0.7 | 0.75 ± 0.048 |
| 17b | 53.7 | 24.3 | 195 | 53.8 | 190 | 73.5 | 0.7 | 0.61 ± 0.020 |
| VRC01 | 61.1 | 24.4 | 195 | 60.9 | 190 | 88.1 | 0.8 | 0.72 ± 0.020 |
Can the Global Shape of nmAbs Be Affected by Buffer Composition, Mainly Ionic Strength and pH?
To explore whether the unusual global shapes of non-neutralizing and neutralizing mAbs can be influenced by ionic strength and pH of the buffers, we acquired SAXS intensity profiles from purified mAbs in buffers containing increasing amounts of NaCl (from 0.1 to 1 m at pH 7.4) and buffers having 0.137 m NaCl with varying pH (from pH 8 to 4). Importantly, we found that the broad range of salt concentrations that we explored did not induce any significant change in the scattering shape of the mAbs, both non-neutralizing and neutralizing mAbs. No change in the SAXS data analysis-based estimated Rg, Dmax, and P(r) curve profiles confirmed this observation (e.g. see b12 in Fig. 8A). Our results are in agreement with an earlier report, which explored the effect of salt type and concentration on two IgG1 mAbs (43). Their SAXS experiments showed that the shapes of the mAb remain unchanged with a broad range of chaotropic and kosmotropic salts. Similarly, our SAXS data analysis revealed that dialysis of purified mAbs against low pH buffer did not alter the shape profile or parameters of non-nmAbs, reactive to either gp120 or gp41. In sharp contrast, our SAXS experiments revealed that the shape parameters of the nmAbs increased upon decreasing buffer pH (Table 7). P(r) analysis showed that the two-peak profile of the interatomic vectors constituting the predominant solution shape of the b12 changed into a three-peak one at pH 5.5. Guinier and P(r) analysis revealed that the Rg and Dmax of b12 increased at pH 5.5 and 5. Similarly, for other nmAbs, exposure to low pH caused an increment in the shape parameters of the nmAbs (Fig. 8A and Table 7). Comparable I0 values estimated from the area under the P(r) curves confirmed that because the concentrations of nmAbs were kept similar, molecular masses of nmAbs were not increasing, which ruled out low pH-induced partial association.
FIGURE 8.

Analysis of the shape of unliganded mAbs under different buffer and salt conditions and its effect on ligand binding. A (left), SAXS data-based P(r) analysis for unliganded IgG1 b12 under different salt conditions. Middle, SAXS I(Q) profiles of b12 at different pH are plotted here (the inset shows variation of Rg as a function of pH). Right, the P(r) curves computed from SAXS data sets of b12 at different pH are shown here. B, global shapes of nmAbs observed at higher and lower pH (pH 8 and 4, respectively). C, results of the rigid body modeling of conformationally distinct populations that vary in shape and size specifically with respect to positioning of Fab arms and twisted Fc, presented here in schematic mode.
TABLE 7.
SAXS data analysis-based structural parameters deduced for the gp41-gp120-reactive IgG1 nmAbs at different pH conditions
| pH | Buffer | Rg (range) | Dmax (range) |
|---|---|---|---|
| Å | Å | ||
| 8 | HEPES | 55–58 | 175–180 |
| 7.4 | PBS | 55–58 | 175–180 |
| 6 | Tris-Cl | 56–59 | 175–180 |
| 5.5 | Citrate buffer | 57–60 | 175–180 |
| 5 | Citrate buffer | 60–62 | 195–200 |
| 4 | Sodium acetate | 61–63 | 195–200 |
Data analysis and structure restoration revealed that at pH 6, 7, and 8, nmAbs existed in the typical closed or semiclosed conformation characterized by Dmax values of ∼175 Å and shorter distances between Fab-Fab and Fab-Fc domains and, in most cases, asymmetric disposition of the Fab arms (Fig. 8C). However, at lower pH values of 4 and 5, the predominant solution conformation of nmAbs became open, characterized by Dmax values close to 200 Å, and models clearly showed both Fab arms to be stretched away from the central Fc. A recent SAXS data analysis-based publication showed that low pH led to open or increased shape parameters of the IgG1 mAbs (44). Importantly, in our case, the low pH-induced shape changes in nmAbs were found to be reversible because with an increase in pH, nmAbs with increased structural parameters and open shape in low pH again adopted smaller parameters, and the extended Fab arms closed as they did at pH 8. Earlier, we reported that low pH can induce reversible shape changes in two Ca2+ ion-binding proteins, gelsolin and calmodulin (36, 37). Thus, we checked any possible influence of divalent ions, particularly Ca2+ ions, on the shape of nmAb and non-nmAbs. SAXS experiments showed that there was no change in the shapes of the non-neutralizing or neutralizing Abs when the free Ca2+ ions were changed from 0.1 μm to 2 mm, indicating that the shape of nmAbs is sensitive to buffer pH. As done before, we attempted rigid body modeling of the shape of nmAbs at low pH using SAXS data as a reference. This modeling revealed that at low pH, the open shaped nmAbs had the central Fc domain in the same plane as the Fab arms (Fig. 8D). Modeling of the nmAbs indicated that low pH induces disruption of ionic interactions between Fab and Fc domains, leading to an open shape which is accompanied or enabled by a twisting of Fc by 90°. Very interestingly, our results show that increase in pH can reverse the effects.
The open shape of nmAbs at low pH raised a question, whether open nmAb can bind two ligands. Thus, we incubated gp120-reactive nmAbs in low pH buffers (pH 5 and 4), mixed with gp120 in 1:1 and 1:2 molar ratios, and loaded them on gel filtration to explore whether nmAbs that could only bind one gp120 at pH 7.4 could bind two gp120s after being incubated at pH 5. Surprisingly, we found that, except for 2G12, all of the gp120-reactive nmAbs eluted at positions close to their unliganded state (i.e. the nmAbs appeared to lose the ability to form even binary complex after dialysis against low pH buffer). To explore the ability of nmAbs to bind gp120 or gp41, we carried out ELISA experiments (Fig. 9, A and B). Results showed that nmAbs could bind gp120 until pH 7, which decreased considerably at pH 6 to 4, except 2G12. Similarly, using MPER peptide, we found that nmAbs reactive to gp41 also lost the ability to bind their ligand with lowering of buffer pH. Similarly, we found that excess NaCl in buffer containing nmAbs also reduced the efficacy of mAbs to bind their ligands, both gp120 and gp41 (Fig. 9, C and D). In the range we studied, nmAbs exposed to buffers having 600 mm or more NaCl could not bind their ligands. We could not find any literature support for similar findings earlier, except for a publication that showed that the rate of neutralization of Cypridina luciferase by antiluciferase γ-globulin increased with a decrease in NaCl concentration and decreased upon increasing salt concentration (45), but in contrast to our results, they reported that neutralization rates were enhanced upon lowering pH from 7.0 to 5.4. They also reported that neutralization rates were sensitive to variation in pH at low salt concentration but became insensitive to pH at higher salt concentration. The only limitation is that they did not explore whether neutralization was lost due to diminished binding of antibody-antigen or due to interference in events that follow the primary step.
FIGURE 9.

ELISA results showing the effect of varying pH (A and B) and ionic strength (A and B) on gp120 and gp41 binding. Error bars were computed by comparing three independent binding experiments.
Differences in the Global Shape of Non-neutralizing and Neutralizing mAbs; How May They Affect Neutralization Function?
Antibody-mediated neutralization can occur by activating the antibody's effector function by binding to C1q and FcR; by effective binding to surface protein gp120 and/or its trimer, which interferes with binding of gp120 to the human CD4 receptor; and by stripping of gp120 from the spikes on the viral surface, leading to a decapacitated virus. It is well known that the spatial disposition of Fc in the antibody shape is crucial for C1q and FcR binding to the Fc domain, which in turn regulates effector response (46). Recent docking studies have shown that the asymmetric shape of IgG1 b12 (PDB code 1HZH) and the C1q globular head have shape complementarity, whereas the structure of Mcg (PDB code 1MCO), a human IgG1 with a deleted hinge, is unable to interact with C1q because the latter's Fab arm obstructs the C1q binding site (47). Also, employing analytical ultracentrifugation and neutron scattering studies, it has been reported that asymmetric positioning of the Fab arms relative to the central Fc domain in rabbit IgG leaves one side of Fc differentially more exposed to form a complex with C1q and FcR with relatively higher efficacy (46). Overall, studies have indicated that structural symmetry or asymmetry in the shape of IgG somewhat regulates its binding with C1q (47, 48).
To estimate the degree of this regulation, we calculated the accessible surface area of the upper portion of Fc (where C1q and FcR bind) in the asymmetric crystal structure of IgG1 b12 (Protein Data Bank entry 1HZH) and a symmetric model generated using domains of IgG1 b12 but reoriented using SAXS data of non-nmAb IgG1 A32. We found that when the probe radius was smaller, the overall accessible surface areas of the symmetric and asymmetric mAbs were comparable, but the differences increased substantially as the probe radius increased. To test our observations experimentally, we compared the binding ability of purified Fc, Fab-Fc fragment from nmAbs, and full-length mAbs to C1q in the presence and absence of ligand (Fig. 10). Regardless of whether they were abstracted from non-neutralizing or neutralizing mAbs, the Fc portion alone showed the highest ability to bind C1q, and as expected, due to lack of the epitope required for binding, none of the 50-kDa Fab fragments either from non-neutralizing or neutralizing mAbs displayed binding to C1q (Fab binding data not shown). Interestingly, in correlation with previous reports, we observed that non-nmAbs appeared to bind C1q with lesser efficacy than nmAbs despite the open shape seen for the non-neutralizing ones with exposed Fc and Fab-Fc linker. We also checked the ability of 1:2 molar mixtures of mAbs and their ligands to bind coated C1q, which showed that ligand-bound nmAbs could bind more efficiently to C1q as compared with unliganded nmAbs, but no change was observed in the case of non-nmAbs. Surprisingly, the 100-kDa (Fab-Fc) fragment from all nmAbs showed reactivity to immobilized C1q comparable with full-length nmAbs with or without ligand. These findings were interesting because Fab-Fc fragments could not bind their ligand (Fig. 7B) but could bind C1q with comparable efficacy.
FIGURE 10.
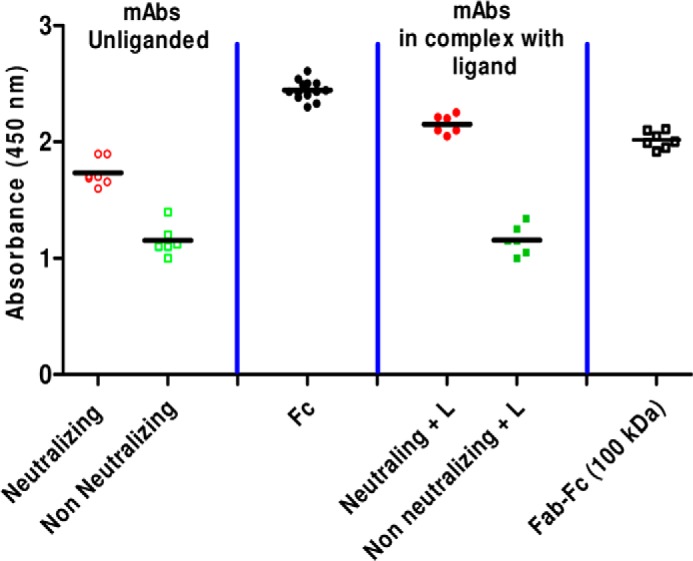
ELISA results showing the ability of full-length mAbs and their fragments unliganded or complexed to their ligands to bind coated C1q. Results show the ability of isolated Fc from neutralizing and non-neutralizing mAbs, full-length neutralizing and non-neutralizing mAbs, and digested Fab-Fc fragment (100 kDa) to bind C1q. Also, comparative results are presented for neutralizing and non-neutralizing mAbs first incubated with a 1:2 (mAb/ligand) molar ratio.
Our SAXS data-based shape modeling showed that the central Fc is in a twisted orientation (90° to the plane formed by the Fab arms), a feature that can be better visualized in the crystal structure of IgG1 b12. It is very likely that this twisted orientation of Fc provides better access to C1q and/or FcR (even in the cases of 4E10 and 2G12, which had their Fab arms fused to Fc) and improves the probability of effector function-mediated neutralization by nmAbs. Having observed that the Fc domain twists from an orthogonal to an in-plane orientation in nmAbs in low pH, we wondered whether nmAbs dialyzed at low pH would bind to C1q. ELISA results showed that nmAbs dialyzed with buffer having pH 5 completely lost the ability to bind C1q. We were expecting a reduced binding, somewhat comparable with that of non-neutral mAbs, but a complete loss probably indicates additional changes in the surface properties of nmAbs at low pH because they lost the ability to bind their ligands as well. Overall, this suggests that the twisted orientation of Fc seen in the global shapes of nmAbs improves C1q- and FcR-mediated viral neutralization.
It is also worth highlighting here that over decades, HIV is continuously reducing the number of trimeric spikes of gp120-gp41 on its surface, key molecules for host CD4+ cells (49–51). The antigen size, density, and fluidity have important implications in the evolution of antibodies. Thus, it may be speculated that while HIV-1 evolved a low spike density to specifically thwart bivalent or effective binding of mAbs, antibodies co-evolved toward polyreactivity (52). Essentially, evolved polyreactive mAbs can bind two different epitopes: one in the envelope components of HIV and another possibly in the membrane of HIV-1. Having observed altered disposition of Fab arms in nmAbs, we feel that the differential shape profile contributes toward enabling the two Fab arms to bind different ligands. Interestingly, promiscuity for ligand or cross-reactivity to ligands is a common serological serologic feature of certain infections in humans, including HIV-1, Epstein-Barr virus, and hepatitis C virus (53, 54). Moreover, Diebolder et al. (55) recently reported that in due course of evolution, 1:1 complex formation of mAb-ligand is beneficial for effector components (C1q) binding and enhances the complement activation multifold. These authors engineered chimeric antibody, where one of the Fab arms was from IgG1 b12 and the other Fab arm was specific for some other ligand. Their results showed that 1:1 binding (i.e. only one Fab arm binding to its ligand) was better for activating the C1q-mediated complement system. In correlation, our results clearly show that among the gp120-reactive nmAbs studied here, all could bind only one gp120. There is a probability that by binding to another surface component, the nmAbs spend a longer time attached to the gp120 spike, thus interfering with its ability to engage with human receptor CD4, if not simultaneously activating C1q-mediated neutralizing steps.
Further, the ability to recognize different epitopes would allow nmAbs to bind more effectively to the viral surface proteins. Earlier, it was shown that HIV-1 evades mAb-mediated neutralization by wiggling conformations of the epitopes recognized by the mAbs (56). It has been reported that nmAbs reactive to gp120 or gp41 epitopes can induce shedding of gp120 from the viral surface, leading to defunct viral particles (57). On similar lines, engineered tetravalent antibody CD4-IgG2, which can bind simultaneously to multiple gp120s, induces shedding of gp120s at substantially higher rates than most known gp120- or MPER-reactive mAbs (57, 58). Differential global shapes of Fab arms and their ability to bind their ligands possibly enable polyreactivity in nmAbs that overcome the conformational variability-based strategy of virus to evade mAb-mediated neutralization. We currently do not have any data on this but speculate that an extended stay of polyreactive mAbs bound to the gp120-gp41 assembly either destabilizes gp120-gp41 or induces subtle shape changes that uncap the gp120 from the spike.
Finally, it is worth noting that the unopen shapes for nmAbs might be the result of Fab-Fab or Fab-Fc interactions, or there may be some role of hinge region rigidity. Earlier, we correlated the asymmetric shape of IgG1 b12 with rigidity of the hinge region (3). A recent report showed an interesting observation that anti-HIV-1 mAb IgG1 F240 expressed in different cell lines yielded versions with substantially different neutralizing potency (59). The altered neutralization potency was correlated with differences in the glycosylation profiles that did not affect the binding of Fab to ligand, but it substantially altered the immunoreactivity. Additionally, reduced degrees of freedom in its hinges due to a high incidence of O-glycosylation and Pro residues, which rigidly position the Fab arms distally from the Fc, have been reported for human IgA1 (39). Partial rigidity paving the way for neutralizing potency could be seen in another recent report where, by introducing rigidity in the linker region, researchers could induce neutralizing potency in IgG3 mAbs (16). With the intention of studying SAXS-based change in global shape, we tried to alter the glycosylation pattern in a few gp120-reactive nmAbs by chemically removing N-linked glycans, but the experiments led to misfolded versions of the mAbs. Understanding how post-translation modification affects global shape of mAbs, hinge flexibility, ligand binding efficacy, and neutralizing potency will remain part of our future experiments. Very likely, we will also attempt to determine how neutralizing potency changes upon expressing mAbs in different cell lines. For now, we conclude that differences in global shape can be correlated with their neutralizing function, thus inviting a fresh look at the shape-function relationship of IgG1 antibodies.
Acknowledgments
We appreciate consistent support from the faculty and staff of CSIR-IMTECH. We thank the National Institutes of Health AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, for providing the reagents. Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DEAC02-98CH10886.
This work was supported by funds from CSIR-supported project OLP-0056 and the BioDiscovery and UNSEEN network projects. This is IMTECH communication 116/2013.
- nmAb
- neutralizing monoclonal antibody
- SEC-MALLS
- size exclusion chromatography-multiangle laser light scattering
- Fmoc
- N-(9-fluronyl)methoxycarbonyl
- SAXS
- small angle x-ray scattering
- PDB
- Protein Data Bank
- NSD
- normalized spatial discrepancy.
REFERENCES
- 1. Fouts T. R., Trkola A., Fung M. S., Moore J. P. (1998) Interactions of polyclonal and monoclonal anti-glycoprotein 120 antibodies with oligomeric glycoprotein 120-glycoprotein 41 complexes of a primary HIV type 1 isolate: relationship to neutralization. AIDS Res. Hum. Retroviruses 14, 591–597 [DOI] [PubMed] [Google Scholar]
- 2. Saphire E. O., Parren P. W., Pantophlet R., Zwick M. B., Morris G. M., Rudd P. M., Dwek R. A., Stanfield R. L., Burton D. R., Wilson I. A. (2001) Crystal structure of a neutralizing human IGG against HIV-1: a template for vaccine design. Science 293, 1155–1159 [DOI] [PubMed] [Google Scholar]
- 3. Ashish, Solanki A. K., Boone C. D., Krueger J. K. (2010) Global structure of HIV-1 neutralizing antibody IgG1 b12 is asymmetric. Biochem. Biophys. Res. Commun. 391, 947–951 [DOI] [PubMed] [Google Scholar]
- 4. Saphire E. O., Stanfield R. L., Crispin M. D., Parren P. W., Rudd P. M., Dwek R. A., Burton D. R., Wilson I. A. (2002) Contrasting IgG structures reveal extreme asymmetry and flexibility. J. Mol. Biol. 319, 9–18 [DOI] [PubMed] [Google Scholar]
- 5. Harris A. K., Bartesaghi A., Milne J. L., Subramaniam S. (2013) HIV-1 envelope glycoprotein trimers display open quaternary conformation when bound to the gp41 membrane-proximal external-region-directed broadly neutralizing antibody Z13e1. J. Virol. 87, 7191–7196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bartesaghi A., Merk A., Borgnia M. J., Milne J. L. S., Subramaniam S. (2013) Prefusion structure of trimeric HIV-1 envelope glycoprotein determined by cryo-electron microscopy. Nat. Struct. Mol. Biol. 20, 1352–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Calarese D. A., Scanlan C. N., Zwick M. B., Deechongkit S., Mimura Y., Kunert R., Zhu P., Wormald M. R., Stanfield R. L., Roux K. H., Kelly J. W., Rudd P. M., Dwek R. A., Katinger H., Burton D. R., Wilson I. A. (2003) Antibody domain exchange is an immunological solution to carbohydrate cluster recognition. Science 300, 2065–2071 [DOI] [PubMed] [Google Scholar]
- 8. Schülke N., Vesanen M. S., Sanders R. W., Zhu P., Lu M., Anselma D. J., Villa A. R., Parren P. W., Binley J. M., Roux K. H., Maddon P. J., Moore J. P., Olson W. C. (2002) Oligomeric and conformational properties of a proteolytically mature, disulfide-stabilized human immunodeficiency virus type 1 gp140 envelope glycoprotein. J. Virol. 76, 7760–7776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Roux K. H., Zhu P., Seavy M., Katinger H., Kunert R., Seamon V. (2004) Electron microscopic and immunochemical analysis of the broadly neutralizing HIV-1-specific, anti-carbohydrate antibody, 2G12. Mol. Immunol. 41, 1001–1011 [DOI] [PubMed] [Google Scholar]
- 10. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 11. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 12. Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Svergun D. I., Petoukhov M. V., Koch M. H. (2001) Determination of domain structure of proteins from x-ray solution scattering. Biophys. J. 80, 2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D. T., Konarev P. V., Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Cryst. 45, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Petoukhov M. V., Svergun D. I. (2005) Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 89, 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lu Y., Harding S. E., Michaelsen T. E., Longman E., Davis K. G., Ortega A., Grossmann J. G., Sandlie I., García de la Torre J. (2007) Solution conformation of wild-type and mutant IgG3 and IgG4 immunoglobulins using crystallohydrodynamics: possible implications for complement activation. Biophys. J. 93, 3733–3744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Humphrey W., Dalke A., Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graphics 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 18. Goddard T. D., Huang C. C., Ferrin T. E. (2007) Visualizing density maps with UCSF Chimera. J. Struct. Biol. 157, 281–287 [DOI] [PubMed] [Google Scholar]
- 19. Stults N. L., Asta L. M., Lee Y. C. (1989) Immobilization of proteins on oxidized crosslinked Sepharose preparations by reductive amination. Anal. Biochem. 180, 114–119 [DOI] [PubMed] [Google Scholar]
- 20. Veiga A. S., Pattenden L. K., Fletcher J. M., Castanho M. A., Aguilar M. I. (2009) Interactions of HIV-1 antibodies 2F5 and 4E10 with a gp41 epitope prebound to host and viral membrane model systems. ChemBioChem 10, 1032–1044 [DOI] [PubMed] [Google Scholar]
- 21. Albericio F. (ed) (2000) Solid-Phase Synthesis: A practical Guide, 1st Ed., CRC Press, Boca Raton, FL [Google Scholar]
- 22. Fields C. G., Lloyd D. H., Macdonald R. L., Otteson K. M., Noble R. L. (1991) HBTU activation for automated Fmoc solid-phase peptide synthesis. Pept. Res. 4, 95–101 [PubMed] [Google Scholar]
- 23. El-Faham A., Subirós Funosas R., Prohens R., Albericio F. (2009) COMU: a safer and more effective replacement for benzotriazole-based uronium coupling reagents. Chem. Eur. J. 15, 9404–9416 [DOI] [PubMed] [Google Scholar]
- 24. Huarte N., Lorizate M., Kunert R., Nieva J. L. (2008) Lipid modulation of membrane-bound epitope recognition and blocking by HIV-1 neutralizing antibodies. FEBS Lett. 582, 3798–3804 [DOI] [PubMed] [Google Scholar]
- 25. Lightle S., Aykent S., Lacher N., Mitaksov V., Wells K., Zobel J., Oliphant T. (2010) Mutations within a human IgG2 antibody form distinct and homogeneous disulfide isomers but do not affect Fc γ receptor or C1q binding. Protein Sci. 19, 753–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cavallo L., Kleinjung J., Fraternali F. (2003) POPS: a fast algorithm for solvent accessible surface areas at atomic and residue level. Nucleic Acids Res. 31, 3364–3366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Muster T., Steindl F., Purtscher M., Trkola A., Klima A., Himmler G., Rüker F., Katinger H. (1993) A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J. Virol. 67, 6642–6647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cardoso R. M., Zwick M. B., Stanfield R. L., Kunert R., Binley J. M., Katinger H., Burton D. R., Wilson I. A. (2005) Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity 22, 163–173 [DOI] [PubMed] [Google Scholar]
- 29. Trkola A., Purtscher M., Muster T., Ballaun C., Buchacher A., Sullivan N., Srinivasan K., Sodroski J., Moore J. P., Katinger H. (1996) Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J. Virol. 70, 1100–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burton D. R., Pyati J., Koduri R., Sharp S. J., Thornton G. B., Parren P. W., Sawyer L. S., Hendry R. M., Dunlop N., Nara P. L. (1994) Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science 266, 1024–1027 [DOI] [PubMed] [Google Scholar]
- 31. Zhou T., Xu L., Dey B., Hessell A. J., Van Ryk D., Xiang S. H., Yang X., Zhang M. Y., Zwick M. B., Arthos J., Burton D. R., Dimitrov D. S., Sodroski J., Wyatt R., Nabel G. J., Kwong P. D. (2007) Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature 445, 732–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Balla-Jhagjhoorsingh S. S., Corti D., Heyndrickx L., Willems E., Vereecken K., Davis D., Vanham G. (2013) The N276 glycosylation site is required for HIV-1 neutralization by the CD4 binding site specific HJ16 monoclonal antibody. PLoS One 10.1371/journal.pone.0068863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu X., Yang Z.-Y., Li Y., Hogerkorp C.-M., Schief W. R., Seaman M. S., Zhou T., Schmidt S. D., Wu L., Xu L., Longo N. S., McKee K., O'Dell S., Louder M. K., Wycuff D. L., Feng Y., Nason M., Doria-Rose N., Connors M., Kwong P. D., Roederer M., Wyatt R. T., Nabel G. J., Mascola J. R. (2010) Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329, 856–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang W., Godillot A. P., Wyatt R., Sodroski J., Chaiken I. (2001) Antibody 17b binding at the coreceptor site weakens the kinetics of the interaction of envelope glycoprotein gp120 with CD4. Biochemistry 40, 1662–1670 [DOI] [PubMed] [Google Scholar]
- 35. Jacques D. A., Trewhella J. (2010) Small-angle scattering for structural biology: expanding the frontier while avoiding the pitfalls. Protein Sci. 19, 642–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Garg R., Peddada N., Sagar A., Nihalani D., Ashish (2011) Visual insight into how low pH alone can induce actin-severing ability in gelsolin under calcium-free conditions. J. Biol. Chem. 286, 20387–20397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pandey K., Dhoke R. R., Rathore Y. S., Nath S. K., Verma N., Bawa S., Ashish (2014) Low pH overrides the need of calcium ions for the shape-function relationship of calmodulin: resolving prevailing debates. J. Phys. Chem. B 118, 5059–5074 [DOI] [PubMed] [Google Scholar]
- 38. Liu H. G., Hexemer A., Zwart P. H. (2012) The small angle scattering ToolBox (SASTBX): an open-source software for biomolecular small-angle scattering. J. Appl. Crystallogr. 45, 587–593 [Google Scholar]
- 39. Boehm M. K., Woof J. M., Kerr M. A., Perkins S. J. (1999) The Fab and Fc fragments of IgA1 exhibit a different arrangement from that in IgG: a study by x-ray and neutron solution scattering and homology modelling. J. Mol. Biol. 286, 1421–1447 [DOI] [PubMed] [Google Scholar]
- 40. Stelos P., Pressman D. (1962) Papain digestion of antigenantibody precipitates. J. Biol. Chem. 237, 3679–3685 [PubMed] [Google Scholar]
- 41. Lau H., Pace D., Yan B., McGrath T., Smallwood S., Patel K., Park J., Park S. S., Latypov R. F. (2010) Investigation of degradation processes in IgG1 monoclonal antibodies by limited proteolysis coupled with weak cation-exchange HPLC. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 878, 868–876 [DOI] [PubMed] [Google Scholar]
- 42. Calarese D. A., Lee H. K., Huang C. Y., Best M. D., Astronomo R. D., Stanfield R. L., Katinger H., Burton D. R., Wong C. H., Wilson I. A. (2005) Dissection of the carbohydrate specificity of the broadly neutralizing anti-HIV-1 antibody 2G12. Proc. Natl. Acad. Sci. U.S.A. 102, 13372–13377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lilyestrom W. G., Shire S. J., Scherer T. M. (2012) Influence of the cosolute environment on IgG solution structure analyzed by small-angle x-ray scattering. J. Phys. Chem. B 116, 9611–9618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tian X., Langkilde A. E., Thorolfsson M., Rasmussen H. B., Vestergaard B. (2014) Small-angle x-ray scattering screening complements conventional biophysical analysis: comparative structural and biophysical analysis of monoclonal antibodies IgG1, IgG2, and IgG4. J. Pharm. Sci. 103, 1701–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tsuji F. I., Davis D. L., Gindler E. M. (1962) Effect of sodium chloride and pH on the rate of neutralization of Cypridina luciferase by specific antibody. J. Immunol. 88, 83–92 [PubMed] [Google Scholar]
- 46. Rayner L. E., Kadkhodayi-Kholghi N., Heenan R. K., Gor J., Dalby P. A., Perkins S. J. (2013) The solution structure of rabbit IgG accounts for its interactions with the Fc receptor and complement C1q and its conformational stability. J. Mol. Biol. 425, 506–523 [DOI] [PubMed] [Google Scholar]
- 47. Guddat L. W., Herron J. N., Edmundson A. B. (1993) Three-dimensional structure of a human immunoglobulin with a hinge deletion. Proc. Natl. Acad. Sci. U.S.A. 90, 4271–4275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gaboriaud C., Juanhuix J., Gruez A., Lacroix M., Darnault C., Pignol D., Verger D., Fontecilla-Camps J. C., Arlaud G. J. (2003) The crystal structure of the globular head of complement protein C1q provides a basis for its versatile recognition properties. J. Biol. Chem. 278, 46974–46982 [DOI] [PubMed] [Google Scholar]
- 49. Zhu P., Liu J., Bess J., Jr., Chertova E., Lifson J. D., Grisé H., Ofek G. A., Taylor K. A., Roux K. H. (2006) Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature 441, 847–852 [DOI] [PubMed] [Google Scholar]
- 50. Liu J., Bartesaghi A., Borgnia M. J., Sapiro G., Subramaniam S. (2008) Molecular architecture of native HIV-1 gp120 trimers. Nature 455, 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Klein J. S., Gnanapragasam P. N., Galimidi R. P., Foglesong C. P., West A. P., Jr., Bjorkman P. J. (2009) Examination of the contributions of size and avidity to the neutralization mechanisms of the anti-HIV antibodies b12 and 4E10. Proc. Natl. Acad. Sci. U.S.A. 106, 7385–7390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Klein J. S., Bjorkman P. J. (2010) Few and far between: how HIV may be evading antibody avidity. PLoS Pathog. 6, e1000908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mouquet H., Scheid J. F., Zoller M. J., Krogsgaard M., Ott R. G., Shukair S., Artyomov M. N., Pietzsch J., Connors M., Pereyra F., Walker B. D., Ho D. D., Wilson P. C., Seaman M. S., Eisen H. N., Chakraborty A. K., Hope T. J., Ravetch J. V., Wardemann H., Nussenzweig M. C. (2010) Polyreactivity increases the apparent affinity of anti-HIV antibodies by heteroligation. Nature 467, 591–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Haynes B. F., Fleming J., St Clair E. W., Katinger H., Stiegler G., Kunert R., Robinson J., Scearce R. M., Plonk K., Staats H. F., Ortel T. L., Liao H. X., Alam S. M. (2005) Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science 308, 1906–1908 [DOI] [PubMed] [Google Scholar]
- 55. Diebolder C. A., Beurskens F. J., de Jong R. N., Koning R. I., Strumane K., Lindorfer M. A., Voorhorst M., Ugurlar D., Rosati S., Heck A. J. R., van de Winkel J. G. J., Wilson I. A., Koster A. J., Taylor R. P., Ollmann Saphire E. O., Burton D. R., Schuurman J., Gros P., Parren P. W. H. I. (2014) Complement is activated by IgG hexamers assembled at the cell surface. Science 343, 1260–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kwong P. D., Doyle M. L., Casper D. J., Cicala C., Leavitt S. A., Majeed S., Steenbeke T. D., Venturi M., Chaiken I., Fung M., Katinger H., Parren P. W., Robinson J., Van Ryk D., Wang L., Burton D. R., Freire E., Wyatt R., Sodroski J., Hendrickson W. A., Arthos J. (2002) HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 420, 678–682 [DOI] [PubMed] [Google Scholar]
- 57. Ruprecht C. R., Krarup A., Reynell L., Mann A. M., Brandenberg O. F., Berlinger L., Abela I. A., Regoes R. R., Günthard H. F., Rusert P., Trkola A. (2011) MPER-specific antibodies induce gp120 shedding and irreversibly neutralize HIV-1. J. Exp. Med. 208, 439–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rathore Y. S., Solanki A. K., Dhoke R. R., Ashish (2011) SAXS data analysis and modeling of tetravalent neutralizing antibody CD4-IgG2 −/+ HIV-1 gp120 revealed that first two gp120 bind to the same Fab arm. Biochem. Biophys. Res. Commun. 415, 680–685 [DOI] [PubMed] [Google Scholar]
- 59. Miranda L. R., Duval M., Doherty H., Seaman M. S., Posner M. R., Cavacini L. A. (2007) The neutralization properties of a HIV-specific antibody are markedly altered by glycosylation events outside the antigen-binding domain. J. Immunol. 178, 7132–7138 [DOI] [PubMed] [Google Scholar]