

Background: GPR126 plays critical roles in development, but its function in vessels is not well characterized.
Results: GPR126 regulates angiogenesis by modulating endothelial cell proliferation and migration via regulation of Vegfr2 expression.
Conclusion: GPR126 is important for physiological and pathological angiogenesis.
Significance: This finding provides a new functional mechanism for the regulation of angiogenesis.
Keywords: Angiogenesis, Endothelial Cell, G Protein-coupled Receptor (GPCR), Protein Kinase A (PKA), Vascular Endothelial Growth Factor (VEGF), VEGFR2
Abstract
Angiogenesis, the formation of new blood vessels from pre-existing ones, is essential for development, wound healing, and tumor progression. The VEGF pathway plays irreplaceable roles during angiogenesis, but how other signals cross-talk with and modulate VEGF cascades is not clearly elucidated. Here, we identified that Gpr126, an endothelial cell-enriched gene, plays an important role in angiogenesis by regulating endothelial cell proliferation, migration, and tube formation. Knockdown of Gpr126 in the mouse retina resulted in the inhibition of hypoxia-induced angiogenesis. Interference of Gpr126 expression in zebrafish embryos led to defects in intersegmental vessel formation. Finally, we identified that GPR126 regulated the expression of VEGFR2 by targeting STAT5 and GATA2 through the cAMP-PKA-cAMP-response element-binding protein signaling pathway during angiogenesis. Our findings illustrate that GPR126 modulates both physiological and pathological angiogenesis through VEGF signaling, providing a potential target for the treatment of angiogenesis-related diseases.
Introduction
Blood vessels are composed of the endothelium, which is a monolayer of cells that cover the vessel lumen, and a surrounding layer of mesenchymal cells such as vascular smooth muscle cells and pericytes (1). The development of the complicated vascular networks mainly requires two successive processes, vasculogenesis and angiogenesis (2). During vasculogenesis, mesoderm-derived hemangioblasts, which exhibit a dual hemangiopoietic potential, give rise to both hematopoietic cells and endothelial cells (EC)5 (3). The in situ differentiated ECs coalesce at the midline to form a lumenized vascular plexus. This primary network formed through vasculogenesis subsequently expands through angiogenesis, which is characterized by the growth of new blood vessels from pre-existing ones (1). During adulthood, most blood vessels remain quiescent for conducting blood flow to organs. However, during pathological conditions such as inflammation, tissue ischemia, and tumor progression, ECs are activated, and angiogenesis is initiated to form vessels to enhance blood supply (4). Control of blood vessel formation through both of these processes requires a finely tuned balance of molecular pathways (5).
Although vasculogenesis and angiogenesis are distinct processes, the basic molecular pathways that drive blood vessel formation are largely shared. One of the most important signaling cascades in blood vessel function is the vascular endothelial growth factor (VEGF) pathway. VEGF proteins stimulate cellular responses by binding to their tyrosine kinase receptors, known as VEGF receptors (VEGFR1–3) (6). Among the receptors, VEGFR2 mediates endothelial cell proliferation and survival and is the most important receptor implicated in all aspects of normal and pathological vascular endothelial cell biology (6). After binding with its ligands, VEGFR2 is activated through dimerization and trans-phosphorylation. The activated receptor then stimulates MAPK/ERK1/2, PI3K/AKT, FAK/Paxillin, and other signaling cascades that regulate endothelial cell proliferation, survival, adhesion, and migration (6). In addition to its activation, the expression of VEGFR2 is also tightly regulated. A number of transcriptional factors have been identified to regulate VEGFR2 transcription, including TFII-I (7, 8), Sp1/Sp3 (9), HIF-2α (10), and NANOG (11). Moreover, GATA2 mediates not only TGF-β inhibition of VEGFR2 expression but also antagonizes with TFII-1 to fine tune the tightly controlled extracellular matrix-induced VEGFR2 expression (7, 12).
G protein-coupled receptors (GPCRs) include the largest protein family of transmembrane receptors (13), sensing the extracellular stimuli and subsequently triggering intracellular signal transduction pathways and modulating a diverse array of physiological processes (14). A number of GPCR signaling pathways have been reported to play crucial roles in blood vessel development, such as lysophosphatidic acid/LPA4, sphingosine 1-phosphate/Edg-1, and lysophospholipid/GPR4 (15–17). During the process of angiogenesis, cross-talk between G protein and VEGFR2 pathways has been reported but not profoundly understood. It has been shown that Gαq/Gα11 proteins regulated VEGFR2 tyrosine phosphorylation through interaction with VEGFR2 (18). Moreover, thrombin has been shown to directly stimulate VEGFR1 and VEGFR2 expression (19), suggesting that the transcription of VEGF/VEGFR2 could be regulated by GPCRs.
The adhesion family of GPCRs is one of the five groups of the GPCR superfamily and contains a large extracellular region with common domains involved in protein-protein interactions and cell adhesion (13). Although few members of adhesion GPCRs have identified ligands or well defined biological functions, these GPCRs have attracted tremendous attention for the versatile functional domains contained in their long N terminus. GPR124, BAI1, and CD97 are distinct members that play pivotal roles in blood vessel development and pathological angiogenesis (20–25).
The physiological roles of the novel adhesion GPCR, GPR126, are not well characterized. Recently, Gpr126 was identified to be essential for Schwann cells to initiate myelination through induction of key transcriptional factors both in zebrafish and mice, suggesting its evolutionarily conserved functions (26, 27). Deletion of Gpr126 in mice leads to mid-gestation embryonic lethality due to internal hemorrhaging and failures in cardiovascular development (28). Although GPR126 is present in human umbilical vein endothelial cells and its expression is increased after the cells are challenged by lipopolysaccharide and thrombin (29), the precise roles of GPR126 in angiogenesis were not investigated. Here, we reported that GPR126 was enriched in both VEGFR2 positive cells, which were differentiated from embryonic stem cells, and endothelial cells from different mouse tissues. GPR126 deficiency significantly affected HMEC-1 cell proliferation, migration, tube formation, and three-dimensional angiogenic sprouting. Using Matrigel plug assays, we found that knockdown of Gpr126 prevented blood vessel formation to implanted plugs. In an oxygen-induced retinopathy mouse model, knockdown of Gpr126 in the retina dramatically impairs neovascular tuft formation. When the expression of Gpr126 interfered with specific morpholinos in zebrafish embryos, intersomitic vessel formation was severely impaired due to inhibition of endothelial cell proliferation and tip cell migration. Furthermore, we show both in vivo and in vitro evidence that GPR126 modulates VEGFR2 transcription through cAMP-induced GATA2 and STAT5 expression.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
HMEC-1 cells generously provided by Dr. F. J. Candal (Centers for Disease Control, Atlanta, GA) were maintained in MCDB-131 medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 10 ng/ml human epidermal growth factor (EGF) (Sigma), 1 μg/ml hydrocortisone, 5 mg/ml l-glutamine, and antibiotics (penicillin/streptomycin) at 37 °C with 5% CO2. For transfection, siRNA were mixed with Lipofectamine 2000 reagent (Invitrogen) according to the instruction manual. The siRNA sequences specifically targeting STAT5 mRNA were 5′-GCAGCAGACCAUCAUCCUG-3′ (STAT5 si1) and 3′-GACCCAGACCAAGUUUGCA-5′ (STAT5 si2). The sequences targeting GATA2 mRNA were 5′-GGCUCGUUCCUGUUCAGAATT-3′ (GATA2 si1) and 5′-GGAGGAGGAUUGUGCUGAUTT-3′ (GATA2 si2). All the siRNA duplexes were synthesized by the GenePharma Co. (Shanghai, China). The control siRNA for STAT5 and GATA2 is a scrambled sequence that will not lead to the specific degradation of any known cellular mRNA afforded by the GenePharma Co.
Cell Proliferation and BrdU Incorporation Assays
For the cell growth curve assay, cells were seeded at 1 × 104 cells/well and grown in 12-well plates continuously for 1–7 days in triplicate. The cells were harvested and counted every day. In bromodeoxyuridine (BrdU) incorporation assays, infected cells were incubated in the presence of 10 μm BrdU (Sigma) for 10 h after starvation overnight. Cells were then fixed with 4% paraformaldehyde and treated with 2 m HCl containing 1% Triton X-100. The cells were stained with monoclonal anti-BrdU (Sigma) antibody and then stained with FITC-conjugated goat anti-mouse antibodies and DAPI. The green immunofluorescence indicated BrdU staining. The total numbers of DAPI-positive cells, as well as BrdU-labeled cells, were counted in 5–10 different fields of each well.
Monolayer Wound-healing Experiments
HMEC-1 cells were seeded in 6-well plates precoated with 0.1% gelatin (Sigma) to full confluence and then incubated with 10 μg/ml mitomycin C at 37 °C and 5% CO2 for 2.5 h to inactivate cells. Then a linear wounding zone was created using a 200-μl pipette tip and washed with PBS. MCDB-131 medium supplemented with 0.5% FBS was added to the wells with VEGF (4 ng/ml). Images were taken after about 8–10 h of incubation at 37 °C, 5% CO2. The migrated cells were counted in the wounding zone determined by a predefined frame. The experiments were performed in triplicate and repeated at least three times.
Immunohistochemistry
Immunostainings were performed according to standard procedures. Staining was developed using 3,3′-diaminobenzidine/peroxidase substrate (brown precipitate). Slides were counterstained with hematoxylin. Tissues were obtained and fixed with 4% paraformaldehyde and embedded with paraffin. A blood vessel staining kit (CD31, Millipore) was used to stain blood vessels in 5-μm sections. Images of the stained blood vessels were taken using a Leica DM 4000B photomicroscope.
Lentivirus-based Overexpression and Knockdown
For virus-based overexpression of GPR126, STAT5, and GATA2, the ORFs of the three genes were subcloned into pLVX-IRES-ZsGreen1 vector, respectively. The virus-based knockdown was conducted using pll3.7 vector. The shRNA oligos targeting human GPR126 were CCTATCTTACATCCAAATCTA (Sh1) and GCTCATTCAGACAACTTCTAT (Sh2). The oligo targeting STAT5 was GTACTTCATCATCCAGTAC. The oligo targeting mouse Gpr126 was gacagctttctcctactca and was subcloned into modified pll3.7 in which the GFP was substituted by DsRed. Virus was harvested from the supernatant of 293T cells 48 h post-transfection before being used to infect target cells (2 × 105) with 5 × 106 TCID50. The cells were not used for proliferation assay and angiogenesis or Western blot experiments until the cells were cultured as described under “Cell Culture and Transfection” without virus for 24 h. High titer virus was prepared by ultracentrifugation at 1.5 × 105 g at 4 °C before being resolved in PBS.
In Vivo Matrigel Plug Assay
Matrigel plug assay was performed with modifications as described (30). Lentivirus expressing 1 × 109 pfu of pll3.7 or mouse Gpr126 shRNA1 (mSh1) was mixed in the Matrigel solution at 4 °C containing 500 ng/ml VEGF-A (Sigma) and 100 μg/ml heparin (Sigma). A total of 500 μl of Matrigel containing lentivirus was injected subcutaneously into the abdomen of male C57BL/6 mice. Six mice were employed for each group. The mice were killed 7 days after the injection. The Matrigel plugs with adjacent subcutaneous tissues were recovered by en bloc resection, and images were then taken using a stereomicroscope (Leica). Thereafter, each sample was embedded in paraffin before being sectioned at a thickness of 5 μm. Then the sections were stained with CD31 antibody. Images were taken with Leica microscope. The number of blood vessels was analyzed using Image-Pro Plus 6.0 software.
Oxygen-induced Retinopathy Model
All experiments conformed to regulations drafted by Association for Assessment and Accreditation of Laboratory Animal Care in Shanghai and were approved by East China Normal University Center for Animal Research (Project License No. 2611). Oxygen-induced retinopathy was performed in C57/BL6 mice according to the protocol (31). Briefly, 7-day-old (P7) mouse pups and their nursing mothers were exposed to 75% oxygen mixed with 25% N2-humidified medical grade oxygen in a sealed chamber containing bedding, food, and water for 5 days. On postnatal day 12 (P12), the mice were returned to room air after being injected with 5 μl of lentivirus (2 × 109 pfu) into the retina. The mice received intravitreal injections for lentiviral infection. Control virus was injected into the left eye, and the knockdown virus targeting mouse Gpr126 mRNA was injected into the right eye. On postnatal day 17, FITC-dextran (50 mg/ml in PBS, Sigma) was infused transcardially via the left ventricle and allowed to exit via the punctured right atrium. Eyes were fixed (4% formaldehyde; 24 h at 4 °C) for dissection before being scanned by confocal microscopy both at channel 488 and 533 nm. ImageJ software was used for quantification of avascular area.
Zebrafish and Morpholinos
Wild type (WT) AB, Tg(flk1:EGFP), Tg(fli1:EGFP)y1, and Tg(fli1:nEGFP)y7 zebrafish lines were maintained in Core Facility of Zebrafish Research, School of Basic Medicine, Chongqing Medical University. Embryos were injected at the one- to two-cell stage with 2.5 ng of a morpholino antisense oligonucleotide targeting zebrafish Gpr126 (5′-ACAGAATATGAATACCTGATACTCC-3′) (26). Control embryos were injected at the one- to two-cell stage with 2.5 ng of a translation-blocking antisense oligonucleotide as described (32). For confocal imaging, embryos were mounted in 1.0% low melting point agarose before being imaged by a confocal microscope of Zeiss LSM 510 (Oberkochen, Germany) or Olympus FV1000-MPE (Tokyo, Japan). In rescue experiments, the morpholino-resistant capped RNAs (mRNAs) were synthesized using T7 mMessage machine kit (Ambion) according to the manufacturer's instructions.
Western Blot Analysis
Western blots were performed with standard protocol with specific antibodies as follows: anti-GPR126 (Abcam, catalog no. ab75356); FAK (Cell Signaling, catalog no. 3285); phospho-FAK (Tyr-397) (Cell Signaling, catalog no. 3283); p44/42 MAPK (ERK1/2) (Cell Signaling, catalog no. 9102); phospho-ERK1/2 (Thr-202/Tyr-204) (Cell Signaling, catalog no. 4376); STAT5 (Cell Signaling, catalog no. 9363); STAT6 (Cell Signaling, catalog no. 9362); GATA2 (Epitomics, catalog no. 3348-1); VEGFR2 (Cell Signaling, catalog no. 2479); Tie2 (Cell Signaling, catalog no. 4224); CREB (Cell Signaling, catalog no. 9197); phospho-CREB (Ser-133) (Cell Signaling, catalog no. 9198), and β-actin (Sigma, catalog no. A5441). Specific peroxidase-conjugated secondary antibodies or IRDye® 800CW conjugated goat (polyclonal) anti-mouse (or rabbit) IgGs were used to detect protein expression by enhanced chemiluminescence kit (Pierce) or by Odyssey infrared imaging system (LI-COR Bioscience), respectively.
RT-PCR and qRT-PCR Analysis
Total RNAs were extracted from cells by using TRIzol reagent (Invitrogen). For the synthesis of the first strand of cDNA, 1 μg of total RNA was reverse-transcribed in 20 μl of reaction solution using PrimeScriptTM RT reagent kit (TaKaRa). The cDNA samples were then amplified by PCR using ExTaq DNA polymerase (TaKaRa) or by qRT-PCR assay performed with the MxPro-Mx3005P real time PCR system using SYBR Premix Ex TaqTM according to the manufacturer's instructions. All primers and PCR conditions used were shown in Table 1.
TABLE 1.
Primers for RT-PCR/qPCR assay
F is forward primer; R is reverse primer; M is Mus musculus; and D is Danio rerio.
| Tal1/scl-F | CTCGGCAGCGGGTTCTTTGG | M-VEGFR2-R | CAAAGCATTGCCCATTCGAT |
| Tal1/scl-R | GCCTGCTTCACCACCTTCTTG | M-Tie2-F | CAGGCTGATTGTTCGGAGATG |
| Foxc1-F | TAAGCCCATGAATCAGCCG | M-Tie2-R | TGCATTCCCCGGTATCTTCA |
| Foxc1-R | GCCGCACAGTCCCATCTCT | M-Nanog-F | AGGGTCTGCTACTGAGATGCTCTG |
| KLF2-F | CACCGACGACGACCTCAACA | M-Nanog-R | CAACCACTGGTTTTTCTGCCACCG |
| KLF2-R | CGCACAGATGGCACTGGAAT | M-Oct4-F | CTGTAGGGAGGGCTTCGGGCACTT |
| Etv2-F | AGTCGGACCGTGCCAGTTT | M-Oct4-R | CTGAGGGCCAGGCAGGAGCACGAG |
| Etv2-R | TCATGCCCGGCTTTCTCTT | M-GPR126-F | TTCCAGGAACAAATGCACCTTC |
| GATA2-F | GCACCTGTTGTGCAAATTGT | M-GPR126-R | GCACGCCATCACATAACTCACG |
| GATA2-R | GCCCCTTTCTTGCTCTTCTT | VEGFR1-F | ATTTGTGATTTTGGCCTTGC |
| Foxo1-F | AAGGATAAGGGTGACAGCAACAG | VEGFR1-R | CAGGCTCATGAACTTGAAAGC |
| Foxo1-R | TTGCTGTGTAGGGACAGATTATGAC | VEGFR2-F | GTGACCAACATGGAGTCGTG |
| Foxo3-F | AAGCCAGCTACCTTCTCTTCCA | VEGFR2-R | CCAGAGATTCCATGCCACT |
| Foxo3-R | GTGGCTAAGTGAGTCCGAAGTGA | VEGFR3-F | CATCCAGCTGTTGCCCAGG |
| Foxc2-F | GCCCAGCAGCAAACTTTCC | VEGFR3-R | GAGCCACTCGACGCTGATGAA |
| Foxc2-R | CCCGAGGGTCGAGTTCTCA | FGFR1-F | GGAGGATCGAGCTCACTCGTGG |
| STAT1-F | CCCAGAATGCCCTGATTAATG | FGFR1-R | CGGAGAAGTAGGTGGTGTCAC |
| STAT1-R | CTGCAGCTGATCCAAGCAAG | FGFR2-F | GTTAACACCACGGACAAAGAGATTGAG |
| STAT2-F | GTGGTTCAGGAAAGGGCAG | FGFR2-R | AACACGGTTAATGTCATAGG |
| STAT2-R | GGAGGGTGTCCGTTTTCAG | FGFR3-F | CGTGGGGCGAGCGGCAGAAGTC |
| STAT3-F | GGCCCCTCGTCATCAAGA | FGFR3-R | TCCGCTCGGGCCGTGTCCAGTAAG |
| STAT3-R | TTTGACCAGCAACCTGACTTTAGT | FGFR4-F | GTGCAGAAGCTCTCCCGCT |
| STAT4-F | CACCTGCCACATTGAGTCAACTA | FGFR4-R | TGAGCTTGACTTGCCGGAA |
| STAT4-R | TAAGACCACGACCAACGTACGA | EGFR-F | GCGTCTCTTGCCGGAATGT |
| STAT5-F | CGAGTGCAGTGGTGAGATCC | EGFR-R | CTTGGCTCACCCTCCAGAAG |
| STAT5-R | TCCTCTGTCACGGACTCTGC | GPR126-F | ACCTTTCTGTTGGGCATGACA |
| STAT6-F | CTGCCCTCTTCAAGAACCTG | GPR126-R | GAAATAGGTGGTAGAGCTGGA |
| STAT6-R | CATGAACGATGACCACCAAG | D-gpr126-F | CCTGACCGTGGAAACTGCACTAA |
| Hey1-F | GCTGGTACCCAGTGCTTTTGAG | D-gpr126-R | GATGATCCTGAAAGAGCCCCGTC |
| Hey1-R | TGCAGGATCTCGGCTTTTTCT | D-flk1-F | ACGACACTGGCATCTACTCATGC |
| Hey2-F | AGATGCTTCAGGCAACAGGG | D-flk1-R | GCCCTTGTCCTCCATTGTCACAT |
| Hey2-R | CAAGAGCGTGTGCGTCAAAG | D-myod1-F | CTCAAGAGATGCACGTCCACCAA |
| β-Actin-F | GTACGCCAACACAGTGCTG | D-myod1-R | GTCGGATTCGCCTTTTTCTGCTG |
| β-Actin-R | CGTCATACTCCTGCTTGCTG | D-β-actin-F | CGAGCAGGAGATGGGAACC |
| M-VEGFR2-F | GCCCTGCCTGTGGTCTCACTAC | D-β-actin-R | CAACGGAAACGCTCATTGC |
| M-CD31-F | TGCAATTTGAATCCGGACAG | ||
| M-CD31-R | TGCAATTTGAATCCGGACAG |
Tube Formation Assays
Matrigel was thawed at 4 °C, and each well of prechilled 24-well plates was coated with 200 μl of Matrigel and incubated at 37 °C for 45 min. Human dermal microvascular endothelial cells (5 × 104) were added in 500 μl of culture medium. After 4–6 h of incubation at 37 °C and 5% CO2, endothelial cell tubular structure formation was quantified by calculating the tube length of high power fields (200×) with an Olympus inverted microscope.
Spheroid-based Angiogenesis Assay
HMEC-1 cells were seeded in nonadherent round-bottom 96-well plates (Corning Glass) after being suspended in culture medium containing 0.2% (w/v) carboxymethylcellulose (Sigma). In these conditions, all the suspended cells grow to form a single spheroid per well of defined cell number (800 cells/spheroid). Spheroids were generated for 24 h before being embedded into collagen gels. The spheroid-containing gel was rapidly transferred into 24-well plates and incubated at 37 °C for 30 min to polymerize. Then 200 μl of culture medium was added on top of the gel. After 24 h, in vitro angiogenesis was evaluated by measuring the cumulative length of the sprouts that had grown out of each spheroid using digital imaging software (Image-Pro Plus 6.0), with nine spheroids analyzed per experimental group.
Live Cell Imaging
For live migration assay, HMEC-1 cells (1 × 104) were seeded on 3.5-cm gelatin-coated dishes and incubated for 16 h. Cell medium was then changed to a serum-free medium, and cells were incubated for an additional 8 h. For HMEC-1 migration kinetics, time-lapse images were taken every 10 min. After 10 h, the images were analyzed in sequence using Image-Pro Plus (version 6.0.0.260) software. The migration tracks of each cell in each group were plotted after normalizing the start point to x = 0 and y = 0. The coordinates of each cell from the photo sequences were determined by the “track objects” tool of Image-Pro Plus.
Chromatin Immunoprecipitation Assay
ChIP was performed as described (33). Briefly, HMEC-1 cells growing in 10-cm dishes were treated with formaldehyde, and the DNA-protein complex was extracted. Chromatin was incubated with or without antibody (anti-STAT5 antibody or anti-GATA2 antibody), respectively, for immunoprecipitation. After reversing the protein/DNA cross-links, DNA was recovered and amplified by specific primers (Table 2). The PCR products were resolved on a 2.0% agarose gel for photographing, and the sequences were confirmed by sequencing.
TABLE 2.
Primers for PCR in chromatin immunoprecipitation assays
| Predicted site on promoter | Forward primer | Reverse primer |
|---|---|---|
| STAT5 site (−6243 to −6235) on VEGFR2 promoter | CCTGCTCAAATGATGTGCTC | TATGTTCCAGTCCCAAACTC |
| STAT5 site (−3285to −3277) on VEGFR2 promoter | GCCTCCAGCTTTGTTCTTTT | ATTCAATACCATCCCCATCA |
| STAT5 site (1387–1395) on VEGFR2 promoter | GTGAGAAAGGGGAAAGGGTCA | TCCTGGTACTGTATGTGGGTG |
| GATA2 site on VEGFR2 | GTAAATGGGCTTGGGGAGCTG | GGCGGCTGCAGGGGCGTCT |
| CRE sites (-3381/ −3334) on STAT5 promoter | TGTACCACCTCCCTCCACCC | TAGCTGAGATCACGCCGTTG |
| CRE site (−3262) on STAT5 promoter | GGCAGACCTGATCGTCAAGG | TAGCTGAGATCACGCCGTTG |
| CRE site (−3053 ) on STAT5 promoter | ATATTGGCCAGGCTGATCTC | GCTCTATTGAATGGGACAGTG |
| CRE site (−1736) on GATA2 promoter | CTCTGTGCGGCGATTTTCTG | ACTGCGGTAGTGGAGGTGGC |
| CRE site (−776) on GATA2 promoter | GGACAGACAAACGGACGCCAGAC | AATCCGAGGAGGGAGGAAAGC |
| CRE site (−659) on GATA2 promoter | AAAGTTCATTTCCTGGCGAATC | GGGATTGACAGTCGGTAATTGG |
Luciferase Assay
The reagent and lysis buffer were obtained from Promega Corp. The human VEGFR2 promoter sequence was amplified and cloned into pGL3-enhancer plasmid with following primers. The primers for wild type STAT5 consensus containing construct (WT-Luc) were 5′-gggtctagaGCCTCCAGCTTTGTTCTTTT-3′ (forward) and 5′-cccggatccATTCAATACCATCCCCATCA-3′ (reverse). PCR products were digested with XbaI and BamHI restriction enzymes followed by ligation into the modified minCMV promoter containing the luciferase vector. The site-directed mutagenesis PCR primers used for mutant STAT5 consensus containing the construct were as follows: mutant-VEGFR2-Luc (Mut-Luc), 5′-TTAAAGTAGTTTTTTCCAACCCTGTCAAGAAAGTCATTGG-3′ (forward), and 5′-CAAGCTACCAATGACTTTCTTGACAGGGTTGGAAAAAACTAC-3′ (reverse);
Electrophoretic Mobility Shift Assay
The TnT® quick-coupled transcription/translation system was used for in vitro synthesis of STAT5 protein according to the instructions. Human VEGFR2 promoter-derived STAT5-binding site oligonucleotides were synthesized and 5′-end-labeled using T4 polynucleotide kinase and [γ-32P]ATP. EMSAs were performed following standard protocol, and protein DNA complexes were visualized by autoradiography.
Data Analysis
Data are expressed as means ± S.E. The means of two groups were compared using Student's t test (unpaired, two-tailed), with p < 0.05 considered to be statistically significant. Unless indicated in the figure legends, all the experiments were performed at least three times with similar results.
RESULTS
Gpr126 Is Highly Enriched in Endothelial Cells and Induced by Pro-angiogenic Factors in HMEC-1 Cells
Embryoid body (EB) formation and subsequent differentiation of embryonic stem cells (ESCs) supply an efficient in vitro system to study developmental events and related molecular mechanisms, such as the process of angiogenesis (34–36). Using an established EB model of mouse ES cell differentiation (37), we isolated Flk1/Vegfr2-positive cells at day 4 of EB formation, and we then screened for the expression of about 100 orphan GPCRs during ESC differentiation. We identified Gpr126 mRNA expression as being highly enriched in Flk1-positive cells at day 4 of EB formation (Fig. 1A). Furthermore, mRNA expression of Gpr126 was increased from the beginning of differentiation and peaked at day 4, similar to the expression profile of Vegfr2 (Fig. 1B). Other endothelial markers, including CD31 and Tie2, were also robustly induced, although Oct4 and Nanog, the markers of pluripotent ESCs, were rapidly down-regulated during differentiation (data not shown). These results suggested that Gpr126 is highly expressed in the early endothelial cell lineage during ESC differentiation.
FIGURE 1.

Gpr126 is highly enriched in endothelial cells and is induced by pro-angiogenic growth factors in HMEC-1. A, Gpr126 expression is highly enriched in Flk1 (Vegfr2)-positive (Flk1+) ES cells. Gpr126 mRNA was detected by qRT-PCR in sorted Flk1-positive (Flk1+) and Flk1-negative (Flk1−) ES cells at day 4 in an EB differentiation model. B, dynamic expression pattern of Gpr126 and Vegfr2 during ES cell differentiation. Gpr126 and Vegfr2 mRNA changes were monitored by qRT-PCR during differentiation of ES cells in an EB model. Expression was normalized to β-actin. C, regulation of GPR126 by different pro-angiogenic growth factors in HMEC-1 cells. The protein levels of GPR126 were detected with Western blot analysis in HMEC-1 cells treated with (+) or without (−) VEGF (50 ng/ml), FGF-2 (50 ng/ml), IGF-1 (50 ng/ml), and NGF (50 ng/ml) for 24 h. The numbers show the relative protein level of GPR126. D, protein levels of GPR126 were measured by Western blotting in HMEC-1 cells induced by VEGF (50 ng/ml) at the indicated time points. The numbers show the relative protein level of GPR126. E, expression of Gpr126 in different tissues using specific anti-Gpr126 antibody. Immunohistochemistry images of anti-Gpr126 (upper panel) and anti-CD31 (lower panel) antibodies in mouse limb buds (E15.5 embryos), lung, heart, and kidney are as indicated. Scale bar, 50 μm.
Considering the increased expression of GPR126 in human umbilical vein endothelial cells when challenged by LPS and thrombin (29), we speculated whether other factors could also stimulate GPR126 expression. The protein level of GPR126 was induced by VEGF, fibroblast growth factor-2 (FGF-2), and insulin-like growth factor 1 (IGF-1) but faintly by nerve growth factor (NGF) in human microvascular endothelial cells (HMEC-1) (Fig. 1C). Moreover, GPR126 was also induced by VEGF in a time-dependent manner in HMEC-1 cells (Fig. 1D). To further identify the expression of Gpr126 in tissues, immunohistochemical staining using a validated commercial antibody against Gpr126 was employed. Gpr126-positive cells were specifically detected in the endothelial cells of vessels of the embryonic limb bud, lung, heart, and kidney, and the distribution correlated with that of the endothelial cell marker CD31 (Fig. 1E).
Knockdown of GPR126 Impairs Endothelial Sprout Formation
The distinct expression pattern of GPR126 in the endothelium of tissues implies its roles in endothelial cell biology, such as cell sprouting, migration, proliferation, and tube formation. The initial step of angiogenesis is the sprouting of tip cells from existing vessels. To examine the roles of GPR126 in angiogenesis, we used lentivirus-mediated RNA interference (RNAi) to generate GPR126-deficient HMEC-1 cells by targeting two independent sequences of GPR126 mRNA (Fig. 2A). Then the cells were subjected to a three-dimensional spheroidal assay to mimic the sprouting step in vitro. Knockdown of GPR126 significantly impaired the outgrowth of capillary-like structures and attenuated the sprout number (Fig. 2B) and length (Fig. 2C), implicating that endogenous GPR126 is an important regulator of endothelial sprout formation during angiogenesis.
FIGURE 2.

Knockdown of Gpr126 impairs angiogenesis in vitro. A–C, knockdown of GPR126 impaired endothelial sprout formation. RNAi efficiency was determined by Western blotting in HMEC-1 cells. The numbers show the relative protein level of GPR126 compared with the negative control (NC) (A). Three-dimensional in vitro angiogenesis assays with collagen gel-embedded spheroids of vector control (NC) or GPR126 shRNAs (Sh1 and Sh2)-infected HMEC-1 cells. Representative micrographs and the quantification of the sprout number of each spheroid are shown (B). A statistical summary of cumulative sprout length of nine spheroids of each group are shown (C). D–F, regulation of endothelial cell proliferation by GPR126. D, vector control (NC) or GPR126 shRNA (Sh1 and Sh2)-infected HMEC-1 cells were seeded in 12-well plates and grown for 7 days. The cell number was counted every day. Knockdown of GPR126 affects BrdU incorporation and cell cycle progression in HMEC-1 cells. HMEC-1 cells were infected with vector control (NC) and GPR126 shRNAs (Sh1 and Sh2), respectively. BrdU was supplemented into culture medium for 10 h, and then the cells were fixed for immunofluorescence with anti-BrdU-specific antibody. The percentage of BrdU-positive cells was calculated (E). Control or GPR126 shRNA-infected HMEC-1 cells were subjected to flow cytometry for cell cycle analysis (F). Error bars represent S.E.; **, p < 0.01; ***, p < 0.001. G and H, knockdown of GPR126 inhibited HMEC-1 cell migration in wound healing assays. Dashed lines indicate the edge of the “wound” right after scratch (G). Relative cell numbers migrated to the “wound” were presented (H). I and J, GPR126 regulates the migration of endothelial cells. The migration tracks of 12 empty vector-infected (NC) or 12 GPR126 knockdown (Sh2) HMEC-1 cells were plotted after normalizing the start point to x = 0 and y = 0 (I). The coordinate of each cell from the photo sequences were determined by Image-Pro Plus (version 6.0.0.260). The column diagram showed a cumulative path length of 12 control and 12 GPR126-knockdown cells (J). K and L, capillary tube formation of endothelial cells infected with vector control (NC) or GPR126 shRNAs (Sh1 and Sh2) were seeded onto Matrigel. After 4–6 h, cells were fixed, and tubular structure was quantified by calculating the tube length of high power fields (×200). All error bars represent S.E. *, p < 0.05; **, p < 0.01 compared with control.
GPR126 Controls the Angiogenic Activity of Endothelial Cells
To elucidate the endothelial cell behavior controlled by GPR126, we further investigated the features of GPR126-deficient endothelial cells. First, we tested the role of GPR126 in endothelial cell proliferation by counting the number of cells and BrdU incorporation assays (Fig. 2, D and E). In addition, knockdown of GPR126 resulted in G1/S phase cell cycle arrest (Fig. 2F). These data suggest that GPR126 is actively involved in endothelial cell proliferation. Next, we evaluated the migration properties of endothelial cells with down-regulation of GPR126. Considering the role of GPR126 in cell proliferation, we pretreated the cells with mitomycin C to inhibit cell proliferation before wound healing and Boyden chamber cell migration assays. The number of migrated cells in GPR126-deficient cells decreased significantly compared with the control cells in both wound-healing migration assay (Fig. 2, G and H) and Boyden chamber cell migration assay (data not shown). Time-lapse fluorescence confocal microscopy analysis was also adopted to evaluate the real time effect of GPR126 knockdown on endothelial cell migration. Twelve cells of each group were monitored for a consecutive 10 h (Fig. 2I). The accumulated migration length of the cells of the GPR126 knockdown group was significantly less than that of the control group (Fig. 2J). Tube formation is an important parameter of endothelial cell function in angiogenesis. Knockdown of GPR126 inhibited the tube formation of endothelial cells with a decreased number of intercellular contacts and overall complexity of the network (Fig. 2K). The accumulated length of tubes was significantly less than that of the control (Fig. 2L). Taken together, these results demonstrated that GPR126 is a critical regulator that modulates angiogenic behavior of endothelial cells in vitro.
Inhibition of GPR126 Expression Impairs Physiological and Pathological Angiogenesis
To further confirm the physiological roles of Gpr126 in animal models, Matrigel mixed with VEGF, heparin, and shRNA lentivirus targeting mouse Gpr126 mRNA or control shRNA lentivirus was injected subcutaneously into the C57BL/6 strain mice following the well established protocols (30). The RNA interference efficiency was determined using NIH3T3 cells (Fig. 3A). In Matrigel plug assay, the color of the plugs containing lentiviral shRNA against Gpr126 was lighter than those plugs containing control lentivirus (Fig. 3B), suggesting that fewer red blood cells entered the plugs containing lentiviral shRNA against Gpr126. Through immunohistochemistry using anti-CD31 antibody, it is clearly shown that fewer blood vessels formed in mGpr126 shRNA virus-infected plugs (Fig. 3, B and C). The RNA interference efficiency of lentiviral shRNA against Gpr126 was determined by quantitative PCR assays using RNA extracted from Matrigel plugs treated with the indicated lentivirus (Fig. 3D).
FIGURE 3.

Knockdown of Gpr126 suppresses angiogenesis in Matrigel plugs and hypoxia-induced retinal neovascularization. A, RNAi efficiency was determined by Western blotting in NIH3T3 cells. B–D, effect of Gpr126 knockdown by mSh1 (mSh) on angiogenesis in Matrigel plugs. Gross photographs of typical plugs are shown, and Matrigel plugs were sectioned and stained with anti-CD31 antibody to indicate neovascularization (B). Quantification of vessel number per high power field (×200) is shown in C. The mRNA levels of Gpr126 in the cells extracted from indicated Matrigel plugs were detected by qRT-PCR (D). E, knockdown of Gpr126 in mouse retina affects hypoxia-induced retinal angiogenesis. Control or Gpr126 lentiviral shRNA was delivered through intravitreal injection into neonatal mice that were then subjected to OIR model. After perfusion with FITC-dextran (green), retinal flat mounts were prepared and analyzed by confocal microscopy. The red fluorescence indicates the lentiviral infection efficiency. Arrows show pathological neovascular tufts. Avascular areas are surrounded by white lines. F, quantification of relative ratio of avascular area versus total retinal area of each retina was determined as described under “Experimental Procedures.” ImageJ software was used for quantification of avascular area. Error bars represent S.E. *, p < 0.05 compared with control.
To test the angiogenic role of Gpr126 in pathological conditions, we took advantage of an oxygen-induced retinopathy mouse model that mimics ischemia-induced angiogenesis (31). As shown in Fig. 3E, the red fluorescence indicates the infection efficiency, and the green fluorescence indicates the blood vessel visualized by FITC-dextran. As expected, wild type retinas displayed few vessels in the central area (avascular area) accompanied by an overgrowth of perfused vessels in the periphery (Fig. 3E). In contrast, fewer neovascular complexes and smaller central avascular areas could be found in mGpr126-shRNA infected retinas (Fig. 3, E and F). The decreased susceptibility of retinas infected with lentiviral mGpr126-shRNA to oxygen-induced retinopathy suggests that Gpr126 regulates retinal angiogenesis in pathological conditions. Taken together, these findings suggest Gpr126 as a critical modulator that conveys an angiogenic signal to the formation and growth of the new vessels.
Silencing of Gpr126 Causes Angiogenesis Deficiency during Embryogenesis
Zebrafish is an excellent model organism to study blood vessel development due to its unique advantages, such as optical clarity and well established endothelium-specific reporter transgenic lines. The expression of Gpr126 in the endothelial cells of zebrafish was confirmed in Flk1-positive cells that were sorted from Tg(flk1:EGFP) zebrafish embryos by RT-PCR (Fig. 4A). The expression of Gpr126 was reduced in zebrafish embryos with morpholinos (MOs), the sequences of which were reported by Monk et al. (26). Control MOs or Gpr126-MOs were injected into one- or two-cell stage embryos of Tg(fli1:EGFP)y1 zebrafish, respectively. The knockdown efficiency was confirmed by Western blotting (Fig. 4B). None of the injected MOs induced any gross morphological defects as examined under bright field microscopy (Fig. 4C, left panel). During zebrafish blood vessel development, intersomitic vessels (ISVs) were grown from the dorsal aorta through angiogenesis. Compared with zebrafish treated with control MOs, the ISVs were severely impaired in Gpr126 morphants at 30 hpf (Fig. 4, C and D) and 36 hpf (Fig. 4D). The angiogenic defects caused by Gpr126 MO were significantly alleviated when human GPR126 mRNA was co-injected (Fig. 4C, bottom panel). These data suggest that GPR126 is evolutionarily conserved and required for proper vascular growth and development.
FIGURE 4.

Interference of Gpr126 expression impairs angiogenesis and vessel formation during zebrafish embryogenesis. A, Tg(flk1:EGFP) embryos at 24 hpf were dechorionated followed by removing yolk. Cells prepared from embryos by digestion using trypsin were filtered and sorted. RNA was extracted from in-isolated EGFP-positive cells, and RT-PCR was performed using gene-specific primers. Lane 1, sorted EGFP-positive cells; lane 2, the whole mount embryos; lane 3, no DNA template. B, RNAi efficiency of the morpholinos targeting zebrafish Gpr126 was detected by immunoblotting. Ctr MO, control morpholino; Gpr126 MO, morpholino against GPR126. C, lateral views of Tg(fli1:EGFP)y1 zebrafish embryos injected with indicated MOs or mRNA. Bright field images revealed no major changes in gross morphology (C, left panel). Confocal images of fli1:EGFP embryos of each treatment are shown (C, right panel). D, percentage of defective ISVs in each group at 30 and 36 hpf. 10 ISVs per embryo were quantified. Numbers above the column represent the number of defective ISVs/the number of total ISVs. E and F, confocal images of Tg(fli1:nEGFP)y7 embryos injected with MOs at 36 hpf. Endothelial cell number of ISVs was labeled. F, quantification of ISV endothelial cell number in Control MO (Ctr mo) and GPR126 MO-injected embryos at 36 (left panel) and 48 hpf (right panel). G, Gpr126 regulates the ISV tip cell polarization and migration. Images from high resolution two-photon laser scanning microscopy showed the difference of the ISV tip cell polarization and migration between the control MO and Gpr126 MO-injected embryos at 30, 36, or 48 hpf. The ISV tip cells of the Gpr126 morphants were unpolarized and arrested at the horizontal myoseptum (bottom arrows). Lateral views, anterior is to the left, and dorsal is up.
To investigate the cell proliferation process in detail, the endothelial cell numbers in segmental arteries were observed by a high resolution two-photon imaging microscope and quantified. In control Tg(fli1:nEGFP)y7 embryos at 36 hpf, most segmental arteries contained three to five cells, whereas Gpr126 morphants displayed dramatically reduced cell numbers (Fig. 4, E and F). Similar results were obtained at 48 hpf (Fig. 4F). Furthermore, knockdown of Gpr126 in zebrafish arrested the sprouting tip cells to spread horizontally along the myoseptum (Fig. 4G). The phenotypes of Gpr126 deficiency in zebrafish showed that the ECs sprouting from the dorsal aorta to the horizontal myoseptum were obviously affected, and the subsequent endothelial tip cell migration and proliferation were severely inhibited.
GPR126 Regulates VEGFR2 Expression in Vitro and in Vivo
Because the VEGF pathway is the pivotal signal for modulation of angiogenesis, we tested whether GPR126 participated in the regulation of the VEGF signaling pathway. As shown in Fig. 5A, VEGF-induced ERK and FAK activities measured at the phosphorylation level were distinctly down-regulated in GPR126-deficient HMEC-1 cells. Furthermore, when the expression of GPR126 was reduced, the protein level of VEGFR2 was severely down-regulated, although the expression of TIE2 did not change (Fig. 5B). To test whether GPR126 specifically regulates VEGFR2 transcription, semi-quantitative RT-PCR assays were performed. VEGFR2 mRNA was dramatically decreased in the GPR126 knockdown HMEC-1 cells, although the mRNA levels of VEGFR1, VEGFR3, EGF receptors, and FGF receptors did not exhibit notable changes (data not shown). Meanwhile, forced expression of GPR126 in HMEC-1 cells increased the VEGFR2 protein level (Fig. 5C). In Gpr126-deficient zebrafish embryos, the mRNA level of Flk1 (VEGFR2) was significantly down-regulated (Fig. 5D). These results indicated that GPR126 regulates the angiogenic process by regulating the expression of VEGFR2.
FIGURE 5.
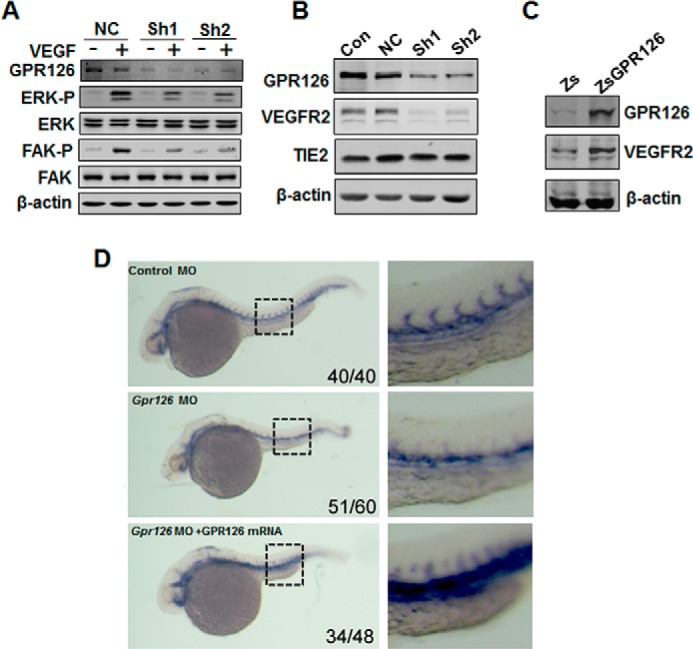
GPR126 regulates VEGFR2 expression in vitro and in vivo. A, regulation of ERK and FAK phosphorylation by GPR126. Immunoblot of lysates from HMEC-1 cells infected with control or GPR126 shRNAs (Sh1 and Sh2) in the presence (+) or absence of VEGF (−). VEGF-induced ERK and FAK activation in GPR126 shRNA-infected HMEC-1 cells (Sh1 and Sh2) was inhibited compared with the vector control (NC). B, down-regulation of VEGFR2 in GPR126-deficient cells. Western blot analysis showed that the protein level of VEGFR2, but not that of TIE2, decreased in GPR126 shRNAs (Sh1 and Sh2)-infected HMEC-1 cells compared with noninfected (Con) or vector control cells (NC). C, forced expression of GPR126 in HMEC-1 cells increased VEGFR2 expression. Zs, control vector; ZsGPR126, GPR126 overexpression construct. D, knockdown of zebrafish Gpr126 decreased Flk1 (Vegfr2) mRNA expression during embryogenesis. Whole mount in situ hybridization was performed with Flk1-specific probe. Flk1 mRNA expression was detected in zebrafish embryos with indicated treatment. Control MO (upper panel), Gpr126 MO (middle panel), or Gpr126 MO with human GPR126 mRNA coinjection (lower panel). Higher magnifications are shown (right panel). The number in the right bottom corner of each panel represents the number of representative phenotype as shown/the number of total embryos conducted with injection.
GPR126 Stimulates VEGFR2 Transcription via STAT5 and GATA2
To further assess the mechanism whereby GPR126 participates in transcriptional regulation of VEGFR2, we investigated the expression levels of numerous transcription factors that were either reported to regulate VEGFR2 expression or involved in angiogenesis. As shown in Fig. 6A, the expression levels of STAT5 and GATA2 were strikingly decreased in GPR126-deficient HMEC-1 cells, although no notable change has been observed for the expression of STAT3 and STAT6. The changes were confirmed by qRT-PCR assays at the mRNA levels (data not shown). To confirm the key role of GATA2 in mediating VEGFR2 expression, we examined VEGFR2 expression in cells transfected with GATA2 siRNAs. As shown in Fig. 6B, knockdown of GATA2 suppressed VEGFR2 expression in HMEC-1 cells. Meanwhile, ChIP assays showed that GATA2 bound directly to the promoter of VEGFR2 (Fig. 6C) as reported previously (7).
FIGURE 6.

GPR126 stimulates VEGFR2 transcription via STAT5 and GATA2. A, effects of GPR126 on the expression of transcription factors involved in angiogenesis. Protein levels were analyzed by immunoblot using specific antibodies in HMEC-1 cells (Con) and in cells infected with vector control (NC) or GPR126 shRNA (Sh1 and Sh2). B and C, regulation of VEGFR2 expression by GATA2. VEGFR2 protein level was analyzed by immunoblot in HMEC-1 cells (Con), cells transfected with control siRNA (Scramble), or GATA2 siRNA (GATA2 si1 and GATA2 si2) (B). Chromatin of HMEC-1 cells was immunoprecipitated with anti-GATA2 antibody or IgG control. The extracted DNA was used for PCR amplifications with VEGFR2 promoter-specific primers (C). D–F, regulation of VEGFR2 expression by STAT5. VEGFR2 protein level was analyzed by immunoblot in HMEC-1 cells (Con), cells transfected with control shRNA (NC), or STAT5 shRNA vector (STAT5Sh) (D), or control siRNA (Scramble), and STAT5 siRNA (ST5 si1 and ST5 si2) (E), respectively. F, forced expression of STAT5 increased VEGFR2 expression in HMEC-1 cells. Zs, control vector; ZsSTAT5, STAT5 overexpression vector. G, chromatin of HMEC-1 cells was immunoprecipitated with anti-STAT5 antibody or IgG control. The extracted DNA was used for PCR amplifications with VEGFR2 promoter-specific primers. STAT5 strongly bound to the predicted site (−3285 to −3277, TTCTGTGAA) in the VEGFR2 promoter. The assays were conducted at least three times. H, wild type (WT-Luc) or STAT5-binding site mutant (Mut-Luc) VEGFR2 promoter luciferase reporter was transfected with increasing doses of STAT5 expression plasmid, and the luciferase activity was determined. I, in vitro translated STAT5 protein was incubated with hot STAT5-binding element derived from VEGFR2 for EMSAs. Cold WT (Cold) or mutant STAT5-binding site (M-probe) probes were subjected for competition. J–L, GPR126 regulated STAT5 and GATA2 expression through cAMP-activated PKA-CREB pathway. Phospho-CREB (Ser-133) was activated in HMEC-1 cell with forskolin (20 μm) stimulation and inhibited with H-89 (10 μm) treatment (J). K, forskolin increased the STAT5 and GATA2 protein levels in a dose-dependent manner in HMEC-1 cells. L, ChIP assays of the CRE site in the STAT5 and GATA2 promoter. Top panel showed the predicted conserved CRE site or half-CRE site in the STAT5 promoter. Middle panel showed that of the GATA2 promoter. After immunoprecipitation of the cross-linked complexes, DNA was recovered by phenol/chloroform extraction. Then the DNA were amplified by PCR using indicated primers. The PCR bands in bottom panel showed the DNA fragment precipitated by anti-CREB antibody in the promoter region of STAT5 (bottom left) and GATA2 (bottom right), respectively.
Although it was not reported that STAT5 directly regulated VEGFR2 expression, activation of STAT5 in ES cells facilitated the generation of Flk1-positive cells within 5 days of coculture on OP9 (38), and STAT5 also mediated FGF-induced angiogenesis (39). To test whether STAT5 is a novel factor that binds to the VEGFR2 promoter and regulates its expression, we examined the relationship between STAT5 and VEGFR2 expression in endothelial cells. Knockdown of STAT5 by siRNA or shRNA down-regulated VEGFR2 protein levels (Fig. 6, D and E), although overexpression of STAT5 significantly increased VEGFR2 expression in HMEC-1 cells (Fig. 6F). We further analyzed the STAT5-binding sites in the VEGFR2 promoter region and confirmed that there was a binding site of STAT5 in the promoter of VEGFR2 using ChIP assays (Fig. 6G), luciferase assay (Fig. 6H), and EMSA (Fig. 6I).
Because cAMP was proposed to be the second messenger mediated by GPR126 in vivo and forskolin, which raises cAMP levels, restored the phenotype in GPR126-deficient zebrafish embryos (26), we assumed that GPR126 could regulate STAT5 and GATA2 expression through the cAMP-activated PKA-CREB pathway. Our data indicate that forskolin increased CREB activity and induced the expression of STAT5 and GATA2 in a dose-dependent manner in HMEC-1 cells (Fig. 6, J and K). Furthermore, to examine whether GPR126 regulates STAT5 and GATA2 expression through the cAMP-PKA-CREB signaling pathway, we predicated multiple conserved cis-elements for CREB binding in the proximal promoter regions of STAT5 and GATA2 by aligning the genomic sequences from rats, mice, and humans. We demonstrated strong binding of CREB in the promoters of the two transcription factors using ChIP assays (Fig. 6L).
STAT5 and GATA2 Restored the Angiogenic Activity of ECs Attenuated by Silencing of GPR126 in Vivo and in Vitro
To further verify that STAT5 and GATA2 are functional downstream targets of GPR126-regulated angiogenesis, we evaluated the effects of STAT5 and GATA2 in vitro using tube formation assays. Strikingly, overexpression of STAT5 and/or GATA2 in GPR126 knockdown HMEC-1 cells restored the number of cell-cell contacts and the overall complexity of the tubular network (Fig. 7, A and B). Furthermore, we performed rescue experiments in zebrafish. Co-injection of STAT5 mRNA or GATA2 mRNA with Gpr126 MO partially restored the growth of ISVs which were disrupted by Gpr126 MO alone (Fig. 7, C and D). When STAT5 and GATA2 mRNAs were introduced with Gpr126 MO at the same time, the impaired ISV morphology was more greatly rescued than individual injection of STAT5 mRNA or GATA2 mRNA, indicating that these two genes have synergistic effects during angiogenesis. This finding indicates that GPR126 modulates angiogenesis in zebrafish through the activation of the STAT5 and GATA2 axis. Taken together, our data indicate that GPR126 regulates angiogenesis by targeting VEGFR2 expression through the PKA-CREB-GATA2/STAT5 signaling pathway.
FIGURE 7.

STAT5 and GATA2 restored the angiogenic activity of ECs attenuated by GPR126 knockdown. A, forced expression of STAT5 and/or GATA2 in GPR126 knockdown HMEC-1 cells restored two-dimensional tube formation on Matrigels. pll3.7, control vector of shRNA; Zs, control vector for overexpression; GPR126-Sh, GPR126 shRNA; ST5, STAT5 overexpression; GA2, GATA2 overexpression. B, statistical summary of relative tube length of each group in A. C, restoration of angiogenic activity of endothelial cells in zebrafish embryos. Bright field images showed the morphology of 30 hpf Tg(fli1:EGFP)y1 zebrafish embryos in each injection treatment. Confocal fluorescence images showed ISVs sprouting of Tg(fli1:EGFP)y1 embryos of each treatment. D, statistical summary of percentage of embryos with ISV defect in each group in C. The numbers above the column represent the number of embryos with ISV defect/the number of embryos analyzed totally.
DISCUSSION
In this study, we have shown that GPR126 is an endothelial specific transmembrane receptor that modulates many aspects of endothelial cell biology, including cell proliferation, sprouting, migration, and tube formation in vitro. We further demonstrated that GPR126 is required for the outgrowth of ISVs in zebrafish embryos, the hypoxia-induced neovascularization of mouse retinal angiogenesis, and the formation of capillary-like structures from endogenous endothelial cells in Matrigel plugs. Furthermore, GPR126 modulates the expression of VEGFR2 and its downstream signaling by targeting GATA2 and STAT5 through the cAMP-PKA-CREB signaling pathway (Fig. 8). The identification of an endothelium-specific adhesion GPCR that positively modulates angiogenesis suggests an advance that has implications for many aspects of biology, including development, tissue injury response, and tumor progression.
FIGURE 8.

Diagram of putative mechanisms of GPR126 function in endothelial cells. GPR126 stimulates STAT5 and GATA2 transcriptional activities through cAMP-activated PKA-CREB pathway. The activated STAT5 and GATA2 independently bind to the VEGFR2 promoter and activate its expression. Up-regulation of VEGFR2 protein levels amplifies downstream angiogenic cascades, such as activation of FAK and ERK. Thus, GPR126 promotes VEGF signaling and angiogenesis by modulating VEGFR2 expression through STAT5 and GATA2 in endothelial cells.
ES cells are totipotent cells that are capable of differentiating into a variety of cell types, and ES cell-derived Flk1-positive cells were demonstrated to serve as vascular progenitors (40). Using an established EB model of mouse ES cells, we found that the expression of Gpr126 was dramatically elevated in Flk1-positive cells (Fig. 1A). We further demonstrated that Gpr126 is specifically expressed in endothelial cells from multiple tissues. This work shows for the first time the expression of Gpr126 in mammalian tissues through an immunohistochemical method. Furthermore, we also found that VEGF and other factors could induce the expression of Grp126 in HMEC-1 cells. These results are concordant with a report that demonstrated that Gpr126 is expressed in endothelial cells, and the expression could be induced by angiogenic factors (29). Severe cardiovascular defects that result in embryonic lethality were observed in the Gpr126 knock-out mouse (28). Waller-Evans et al. (28) did find internal hemorrhaging in mutant embryos, although no obvious vasculogenesis defect was identified. These findings suggest that Gpr126 could play a key role in another process during vessel development, angiogenesis.
The zebrafish embryo is an excellent model for investigating vascular development (41). In zebrafish, the dorsal aorta originates from angioblasts that migrate out from the lateral plate mesoderm and fuse to form blood vessels through vasculogenesis. The subsequent sprouting and extension of endothelial cells to form ISVs from the dorsal aorta is considered as angiogenesis (42). In Gpr126 knockdown zebrafish, no obvious defect was observed in the formation of the dorsal aorta, but the extension of ISVs was dramatically hindered, suggesting that Gpr126 plays a key role in the proliferation and migration of ECs during sprouting angiogenesis. To address the pathological roles of Gpr126 in angiogenesis, we employed a mouse oxygen-induced retinopathy model and Matrigel plug assays. Our data from these pathological models suggest that Gpr126 functions not only in vessel development but also in pathological angiogenesis such as retinopathy and tumor angiogenesis, implying its potential implications in pro- or anti-angiogenic therapies.
Increasing evidence has shown that GPCR signaling can cross-talk with VEGF pathways (18, 19, 43). GPR126 activates the cAMP-PKA signaling pathway, and PKA regulates the differentiation potential of vascular progenitors to be endothelially competent via induction of VEGFR2, suggesting that GPR126 might regulate angiogenesis through the VEGF pathway (26, 44). Our results demonstrated that GPR126 modulates VEGFR2 transcription through GATA2 and STAT5. Both STAT5 and GATA2 were involved in modulating the behavior of hematopoietic and angiogenic cells (7, 45, 46). GATA2 directly binds to the VEGFR2 promoter and regulates its transcription, and the regulation is sensitive to extracellular matrix elasticity as well as soluble VEGF (7, 12). STAT5 has been shown to modulate the endothelial cell and smooth muscle cell migration and proliferation, mediating FGF-induced angiogenesis (15, 39, 47, 48). VEGF activates STAT5 through JAK2 phosphorylation, but how STAT5 regulates VEGFR2 is not determined (49, 50). To our knowledge, the identification of a functional binding site of STAT5 in the VEGFR2 promoter and the direct regulation of VEGFR2 by STAT5 in this study are the first to demonstrate the direct relationship between STAT5 and VEGFR2.
GPR126 is a seven-transmembrane receptor that is evolutionarily conserved not only in the protein sequence but also in synteny of the surrounding genomic region in vertebrates (28). Besides the conserved protein sequences, recent studies have reported that GPR126 has conserved physiological functions during peripheral nerve development in fish and mammals (26, 27). In both Gpr126 mutant zebrafish and mice, peripheral myelination was dramatically affected due to Schwann cell differentiation arrest at the promyelinating stage through down-regulation of cAMP production and the expression of Pou3f1, Desert hedgehog, and Egr2 (26, 27). Another independent group reported mid-gestation lethality of Gpr126 mutant embryos, and the authors observed intraembryonic hemorrhage in about 50% of Gpr126-depleted embryos (28). It suggests that vascular malfunction could occur with Gpr126 depletion. In this study, we demonstrated that the physiological function of Gpr126 is conserved during angiogenesis in different species. As shown in Fig. 4C, the ISV defect in Gpr126 morphants in zebrafish could be rescued by human GPR126 mRNA. The phenotypes of Gpr126 deficiency can be rescued by forced expression of GATA2 and STAT5 in both fish and mammalian cells, further suggesting the conserved signaling pathways downstream of GPR126.
In summary, we have demonstrated that GPR126 plays a key role in angiogenesis by regulating the expression and activity of VEGFR2 through the PKA-CREB-GATA2/STAT5 signaling pathway. Our findings have important implications not only for vascular development but also for tumor biology. Because Gpr126 is an orphan G protein-coupled receptor, the elucidation of its endogenous ligands and antagonists will be a challenge in our future research.
Acknowledgments
We thank Dr. Anming Meng (Tsinghua University, China) for the generous gift of the Tg(flk1:EGFP), Tg(fli1:EGFP)y1, and Tg(fli1:nEGFP)y7 zebrafish lines.
This work was supported, in whole or in part, by National Institutes of Health Grant R15HL098931 from NHLBI. This work was also supported in part by State Key Development Programs of China grants 2012CB910400 and 2010CB945403), National Natural Science Foundation of China Grants 30800627, 30930055, and 31171318, and Science and Technology Commission of Shanghai Municipality Grant 14140900300.
- EC
- endothelial cell
- GPCR
- G protein-coupled receptor
- CREB
- cAMP-response element-binding protein
- EB
- embryoid body
- ESC
- embryonic stem cell
- hpf
- h post-fertilization
- oligo
- oligonucleotide
- MO
- morpholino
- ISV
- intersomitic vessel
- qRT
- quantitative RT.
REFERENCES
- 1. De Val S., Black B. L. (2009) Transcriptional control of endothelial cell development. Dev. Cell 16, 180–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schmidt A., Brixius K., Bloch W. (2007) Endothelial precursor cell migration during vasculogenesis. Circ. Res. 101, 125–136 [DOI] [PubMed] [Google Scholar]
- 3. Pardanaud L., Luton D., Prigent M., Bourcheix L. M., Catala M., Dieterlen-Lievre F. (1996) Two distinct endothelial lineages in ontogeny, one of them related to hemopoiesis. Development 122, 1363–1371 [DOI] [PubMed] [Google Scholar]
- 4. Pober J. S., Sessa W. C. (2007) Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 7, 803–815 [DOI] [PubMed] [Google Scholar]
- 5. Yancopoulos G. D., Davis S., Gale N. W., Rudge J. S., Wiegand S. J., Holash J. (2000) Vascular-specific growth factors and blood vessel formation. Nature 407, 242–248 [DOI] [PubMed] [Google Scholar]
- 6. Olsson A. K., Dimberg A., Kreuger J., Claesson-Welsh L. (2006) VEGF receptor signalling–in control of vascular function. Nat. Rev. Mol. Cell Biol. 7, 359–371 [DOI] [PubMed] [Google Scholar]
- 7. Mammoto A., Connor K. M., Mammoto T., Yung C. W., Huh D., Aderman C. M., Mostoslavsky G., Smith L. E., Ingber D. E. (2009) A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature 457, 1103–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jackson T. A., Taylor H. E., Sharma D., Desiderio S., Danoff S. K. (2005) Vascular endothelial growth factor receptor-2: counter-regulation by the transcription factors, TFII-I and TFII-IRD1. J. Biol. Chem. 280, 29856–29863 [DOI] [PubMed] [Google Scholar]
- 9. Meissner M., Stein M., Urbich C., Reisinger K., Suske G., Staels B., Kaufmann R., Gille J. (2004) PPARα activators inhibit vascular endothelial growth factor receptor-2 expression by repressing Sp1-dependent DNA binding and transactivation. Circ. Res. 94, 324–332 [DOI] [PubMed] [Google Scholar]
- 10. Elvert G., Kappel A., Heidenreich R., Englmeier U., Lanz S., Acker T., Rauter M., Plate K., Sieweke M., Breier G., Flamme I. (2003) Cooperative interaction of hypoxia-inducible factor-2α (HIF-2α) and Ets-1 in the transcriptional activation of vascular endothelial growth factor receptor-2 (Flk-1). J. Biol. Chem. 278, 7520–7530 [DOI] [PubMed] [Google Scholar]
- 11. Kohler E. E., Cowan C. E., Chatterjee I., Malik A. B., Wary K. K. (2011) NANOG induction of fetal liver kinase-1 (FLK1) transcription regulates endothelial cell proliferation and angiogenesis. Blood 117, 1761–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Minami T., Rosenberg R. D., Aird W. C. (2001) Transforming growth factor-β 1-mediated inhibition of the flk-1/KDR gene is mediated by a 5′-untranslated region palindromic GATA site. J. Biol. Chem. 276, 5395–5402 [DOI] [PubMed] [Google Scholar]
- 13. Yona S., Lin H. H., Siu W. O., Gordon S., Stacey M. (2008) Adhesion-GPCRs: emerging roles for novel receptors. Trends Biochem. Sci. 33, 491–500 [DOI] [PubMed] [Google Scholar]
- 14. Römpler H., Stäubert C., Thor D., Schulz A., Hofreiter M., Schöneberg T. (2007) G protein-coupled time travel: evolutionary aspects of GPCR research. Mol. Interv. 7, 17–25 [DOI] [PubMed] [Google Scholar]
- 15. Sumida H., Noguchi K., Kihara Y., Abe M., Yanagida K., Hamano F., Sato S., Tamaki K., Morishita Y., Kano M. R., Iwata C., Miyazono K., Sakimura K., Shimizu T., Ishii S. (2010) LPA4 regulates blood and lymphatic vessel formation during mouse embryogenesis. Blood 116, 5060–5070 [DOI] [PubMed] [Google Scholar]
- 16. Liu Y., Wada R., Yamashita T., Mi Y., Deng C. X., Hobson J. P., Rosenfeldt H. M., Nava V. E., Chae S. S., Lee M. J., Liu C. H., Hla T., Spiegel S., Proia R. L. (2000) Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J. Clin. Invest. 106, 951–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang L. V., Radu C. G., Roy M., Lee S., McLaughlin J., Teitell M. A., Iruela-Arispe M. L., Witte O. N. (2007) Vascular abnormalities in mice deficient for the G protein-coupled receptor GPR4 that functions as a pH sensor. Mol. Cell. Biol. 27, 1334–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zeng H., Zhao D., Yang S., Datta K., Mukhopadhyay D. (2003) Heterotrimeric Gαq/Gα11 proteins function upstream of vascular endothelial growth factor (VEGF) receptor-2 (KDR) phosphorylation in vascular permeability factor/VEGF signaling. J. Biol. Chem. 278, 20738–20745 [DOI] [PubMed] [Google Scholar]
- 19. Tsopanoglou N. E., Maragoudakis M. E. (1999) On the mechanism of thrombin-induced angiogenesis. Potentiation of vascular endothelial growth factor activity on endothelial cells by up-regulation of its receptors. J. Biol. Chem. 274, 23969–23976 [DOI] [PubMed] [Google Scholar]
- 20. Kuhnert F., Mancuso M. R., Shamloo A., Wang H. T., Choksi V., Florek M., Su H., Fruttiger M., Young W. L., Heilshorn S. C., Kuo C. J. (2010) Essential regulation of CNS angiogenesis by the orphan G protein-coupled receptor GPR124. Science 330, 985–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Anderson K. D., Pan L., Yang X. M., Hughes V. C., Walls J. R., Dominguez M. G., Simmons M. V., Burfeind P., Xue Y., Wei Y., Macdonald L. E., Thurston G., Daly C., Lin H. C., Economides A. N., Valenzuela D. M., Murphy A. J., Yancopoulos G. D., Gale N. W. (2011) Angiogenic sprouting into neural tissue requires Gpr124, an orphan G protein-coupled receptor. Proc. Natl. Acad. Sci. U.S.A. 108, 2807–2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cullen M., Elzarrad M. K., Seaman S., Zudaire E., Stevens J., Yang M. Y., Li X., Chaudhary A., Xu L., Hilton M. B., Logsdon D., Hsiao E., Stein E. V., Cuttitta F., Haines D. C., Nagashima K., Tessarollo L., St Croix B. (2011) GPR124, an orphan G protein-coupled receptor, is required for CNS-specific vascularization and establishment of the blood-brain barrier. Proc. Natl. Acad. Sci. U.S.A. 108, 5759–5764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang T., Ward Y., Tian L., Lake R., Guedez L., Stetler-Stevenson W. G., Kelly K. (2005) CD97, an adhesion receptor on inflammatory cells, stimulates angiogenesis through binding integrin counterreceptors on endothelial cells. Blood 105, 2836–2844 [DOI] [PubMed] [Google Scholar]
- 24. Nishimori H., Shiratsuchi T., Urano T., Kimura Y., Kiyono K., Tatsumi K., Yoshida S., Ono M., Kuwano M., Nakamura Y., Tokino T. (1997) A novel brain-specific p53-target gene, BAI1, containing thrombospondin type 1 repeats inhibits experimental angiogenesis. Oncogene 15, 2145–2150 [DOI] [PubMed] [Google Scholar]
- 25. Kaur B., Brat D. J., Devi N. S., Van Meir E. G. (2005) Vasculostatin, a proteolytic fragment of brain angiogenesis inhibitor 1, is an antiangiogenic and antitumorigenic factor. Oncogene 24, 3632–3642 [DOI] [PubMed] [Google Scholar]
- 26. Monk K. R., Naylor S. G., Glenn T. D., Mercurio S., Perlin J. R., Dominguez C., Moens C. B., Talbot W. S. (2009) A G protein-coupled receptor is essential for Schwann cells to initiate myelination. Science 325, 1402–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Monk K. R., Oshima K., Jörs S., Heller S., Talbot W. S. (2011) Gpr126 is essential for peripheral nerve development and myelination in mammals. Development 138, 2673–2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Waller-Evans H., Prömel S., Langenhan T., Dixon J., Zahn D., Colledge W. H., Doran J., Carlton M. B., Davies B., Aparicio S. A., Grosse J., Russ A. P. (2010) The orphan adhesion-GPCR GPR126 is required for embryonic development in the mouse. PLoS One 5, e14047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stehlik C., Kroismayr R., Dorfleutner A., Binder B. R., Lipp J. (2004) VIGR–a novel inducible adhesion family G-protein coupled receptor in endothelial cells. FEBS Lett. 569, 149–155 [DOI] [PubMed] [Google Scholar]
- 30. Tanaka A., Itoh F., Nishiyama K., Takezawa T., Kurihara H., Itoh S., Kato M. (2010) Inhibition of endothelial cell activation by bHLH protein E2–2 and its impairment of angiogenesis. Blood 115, 4138–4147 [DOI] [PubMed] [Google Scholar]
- 31. Connor K. M., Krah N. M., Dennison R. J., Aderman C. M., Chen J., Guerin K. I., Sapieha P., Stahl A., Willett K. L., Smith L. E. (2009) Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nat. Protoc. 4, 1565–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Y., Li Z., Xu P., Huang L., Tong J., Huang H., Meng A. (2011) Angiomotin-like2 is required for migration and proliferation of endothelial cells during angiogenesis. J. Biol. Chem. 286, 41095–41104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li D., Niu Z., Yu W., Qian Y., Wang Q., Li Q., Yi Z., Luo J., Wu X., Wang Y., Schwartz R. J., Liu M. (2009) SMYD1, the myogenic activator, is a direct target of serum response factor and myogenin. Nucleic Acids Res. 37, 7059–7071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hidaka M., Stanford W. L., Bernstein A. (1999) Conditional requirement for the Flk-1 receptor in the in vitro generation of early hematopoietic cells. Proc. Natl. Acad. Sci. U.S.A. 96, 7370–7375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vittet D., Prandini M. H., Berthier R., Schweitzer A., Martin-Sisteron H., Uzan G., Dejana E. (1996) Embryonic stem cells differentiate in vitro to endothelial cells through successive maturation steps. Blood 88, 3424–3431 [PubMed] [Google Scholar]
- 36. Fish J. E., Santoro M. M., Morton S. U., Yu S., Yeh R. F., Wythe J. D., Ivey K. N., Bruneau B. G., Stainier D. Y., Srivastava D. (2008) miR-126 regulates angiogenic signaling and vascular integrity. Dev. Cell 15, 272–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang Y., Yates F., Naveiras O., Ernst P., Daley G. Q. (2005) Embryonic stem cell-derived hematopoietic stem cells. Proc. Natl. Acad. Sci. U.S.A. 102, 19081–19086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schuringa J. J., Wu K., Morrone G., Moore M. A. (2004) Enforced activation of STAT5A facilitates the generation of embryonic stem-derived hematopoietic stem cells that contribute to hematopoiesis in vivo. Stem Cells 22, 1191–1204 [DOI] [PubMed] [Google Scholar]
- 39. Yang X., Qiao D., Meyer K., Friedl A. (2009) Signal transducers and activators of transcription mediate fibroblast growth factor-induced vascular endothelial morphogenesis. Cancer Res. 69, 1668–1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yamashita J., Itoh H., Hirashima M., Ogawa M., Nishikawa S., Yurugi T., Naito M., Nakao K. (2000) Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors. Nature 408, 92–96 [DOI] [PubMed] [Google Scholar]
- 41. Stainier D. Y., Fishman M. C. (1994) The zebrafish as a model system to study cardiovascular development. Trends Cardiovasc. Med. 4, 207–212 [DOI] [PubMed] [Google Scholar]
- 42. Childs S., Chen J. N., Garrity D. M., Fishman M. C. (2002) Patterning of angiogenesis in the zebrafish embryo. Development 129, 973–982 [DOI] [PubMed] [Google Scholar]
- 43. Leung T., Chen H., Stauffer A. M., Giger K. E., Sinha S., Horstick E. J., Humbert J. E., Hansen C. A., Robishaw J. D. (2006) Zebrafish G protein γ2 is required for VEGF signaling during angiogenesis. Blood 108, 160–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yamamizu K., Kawasaki K., Katayama S., Watabe T., Yamashita J. K. (2009) Enhancement of vascular progenitor potential by protein kinase A through dual induction of Flk-1 and neuropilin-1. Blood 114, 3707–3716 [DOI] [PubMed] [Google Scholar]
- 45. Pimanda J. E., Ottersbach K., Knezevic K., Kinston S., Chan W. Y., Wilson N. K., Landry J. R., Wood A. D., Kolb-Kokocinski A., Green A. R., Tannahill D., Lacaud G., Kouskoff V., Göttgens B. (2007) Gata2, Fli1, and Scl form a recursively wired gene-regulatory circuit during early hematopoietic development. Proc. Natl. Acad. Sci. U.S.A. 104, 17692–17697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zeoli A., Dentelli P., Rosso A., Togliatto G., Trombetta A., Damiano L., di Celle P. F., Pegoraro L., Altruda F., Brizzi M. F. (2008) Interleukin-3 promotes expansion of hemopoietic-derived CD45+ angiogenic cells and their arterial commitment via STAT5 activation. Blood 112, 350–361 [DOI] [PubMed] [Google Scholar]
- 47. Xu M., Nie L., Kim S. H., Sun X. H. (2003) STAT5-induced Id-1 transcription involves recruitment of HDAC1 and deacetylation of C/EBPβ. EMBO J. 22, 893–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dentelli P., Trombetta A., Togliatto G., Zeoli A., Rosso A., Uberti B., Orso F., Taverna D., Pegoraro L., Brizzi M. F. (2009) Formation of STAT5/PPARγ transcriptional complex modulates angiogenic cell bioavailability in diabetes. Arterioscler. Thromb. Vasc. Biol. 29, 114–120 [DOI] [PubMed] [Google Scholar]
- 49. Korpelainen E. I., Kärkkäinen M., Gunji Y., Vikkula M., Alitalo K. (1999) Endothelial receptor tyrosine kinases activate the STAT signaling pathway: mutant Tie-2 causing venous malformations signals a distinct STAT activation response. Oncogene 18, 1–8 [DOI] [PubMed] [Google Scholar]
- 50. Dudley A. C., Thomas D., Best J., Jenkins A. (2005) A VEGF/JAK2/STAT5 axis may partially mediate endothelial cell tolerance to hypoxia. Biochem. J. 390, 427–436 [DOI] [PMC free article] [PubMed] [Google Scholar]