

Background: Listeria monocytogenes scavenges iron from heme and human hemoglobin using the Hbp1 and Hbp2 proteins.
Results: Crystal structures and heme transfer studies of Hbp2 reveal an unusual binding mechanism.
Conclusion: Hbp2 is a novel hemoglobin-binding hemophore that rapidly delivers hemin to Hbp1 and other Hbp2 proteins.
Significance: These studies provides insight into how L. monocytogenes captures heme iron.
Keywords: Bacterial Pathogenesis, Heme, Hemoglobin, Iron, X-ray Crystallography
Abstract
Iron is an essential nutrient that is required for the growth of the bacterial pathogen Listeria monocytogenes. In cell cultures, this microbe secretes hemin/hemoglobin-binding protein 2 (Hbp2; Lmo2185) protein, which has been proposed to function as a hemophore that scavenges heme from the environment. Based on its primary sequence, Hbp2 contains three NEAr transporter (NEAT) domains of unknown function. Here we show that each of these domains mediates high affinity binding to ferric heme (hemin) and that its N- and C-terminal domains interact with hemoglobin (Hb). The results of hemin transfer experiments are consistent with Hbp2 functioning as an Hb-binding hemophore that delivers hemin to other Hbp2 proteins that are attached to the cell wall. Surprisingly, our work reveals that the central NEAT domain in Hbp2 binds hemin even though its primary sequence lacks a highly conserved YXXXY motif that is used by all other previously characterized NEAT domains to coordinate iron in the hemin molecule. To elucidate the mechanism of hemin binding by Hbp2, we determined crystal structures of its central NEAT domain (Hbp2N2; residues 183–303) in its free and hemin-bound states. The structures reveal an unprecedented mechanism of hemin binding in which Hbp2N2 undergoes a major conformational rearrangement that facilitates metal coordination by a non-canonical tyrosine residue. These studies highlight previously unrecognized plasticity in the hemin binding mechanism of NEAT domains and provide insight into how L. monocytogenes captures heme iron.
Introduction
Listeria monocytogenes is a foodborne pathogen that causes listeriosis, an infection characterized by gastroenteritis, meningitis, encephalitis, and maternofetal infections in humans. It is highly virulent and after ingestion penetrates the gut, traverses the blood stream, and infects nervous tissues as an intracellular pathogen, resulting in 20–30% mortality (1). Like most bacteria, it requires iron for growth because this element functions as an essential cofactor in proteins that mediate microbial metabolism and physiology (1–3). In the wild and perhaps when inhabiting its host's intestinal track, L. monocytogenes imports iron-laden hydroxamate ferric siderophores that are produced by other microbes via an ABC3 transporter (FhuBCDG) (2–4). It also produces a citrate-inducible receptor for the uptake of ferric citrate, and it has been proposed to display a yet to be identified ferric iron reductase that may release soluble ferrous iron from catecholamines and heterologous siderophores (2). To establish an infection, L. monocytogenes must overcome innate nutritional immunity mechanisms that limit microbial access to iron by sequestering this precious element intracellularly and within extracellular proteins such as transferrin, lactoferrin, and hemopexin. Protoporphyrin IX + iron (heme) bound to human hemoglobin contains ∼70% of the human body's total iron. Two listerial uptake systems acquire heme, the ABC transporter HupDGC and the hemin/hemoglobin-binding proteins (Hbps) (4). An understanding of the molecular mechanism through which these systems function is of fundamental importance as it may lead to the development of novel antimicrobial agents that work by inhibiting heme acquisition (5).
L. monocytogenes uses both free hemin and hemoglobin (Hb) as iron sources. Two Fur-regulated listerial proteins are implicated in hemin uptake: hemin/Hb-binding protein 1 (Hbp1) and Hbp2 (originally named surface virulence-associated protein (SvpA)) (3, 4, 6). Hbp2 is expressed under iron-deficient conditions from the svpA-srtB operon, which also encodes a sortase (SrtB), components of an ABC transporter of unknown function (Lmo2182, Lmo2183, and Lmo2184), and three uncharacterized proteins (Lmo2178, Lmo2179, and Lmo2180) (see Fig. 1A). Based on their primary sequences, they are predicted to be covalently attached to the cell wall by the SrtB sortase because they contain appropriate C-terminal sorting signals (NPKSS and NAKTN, respectively) (7). This is consistent with mass spectrometry studies, which have shown that both proteins are surface-associated (8, 9). However, cell fractionation experiments have also shown that the majority of Hbp2 is secreted from the microbe and that only a small fraction of the protein is associated with the peptidoglycan matrix (3, 6, 9, 10). These and other studies suggest that Hbp2 acts as a hemophore that complexes extracellular hemin and mobilizes it for microbial uptake (4). Microbiological and [59Fe]hemin uptake assays clarified that Hbp2 is only required for hemin import at low external concentrations (<50 nm hemin) or when Hb is provided as an iron source (4). At higher hemin concentrations (>50 nm hemin), the HupDGC ABC transporter complex is sufficient for efficient hemin accumulation in the cytoplasm (3, 4). The role of Hbp1 in hemin acquisition remains unknown, but the gene encoding this protein is also located in the svpA-srtB operon where it is presumably co-expressed with Hbp2 in iron-depleted conditions. Based on primary sequence homology, the Hbp1 and Hbp2 proteins contain NEAr transporter (NEAT) domains, but the functions of these domains are not known (see Fig. 1B).
FIGURE 1.

Hbp1 and Hbp2 schematic and sequence alignment. A, a schematic representation of the chromosomal SrtB loci in S. aureus and L. monocytogenes encoding sortase SrtB (red) and the SrtB-anchored proteins IsdC and Hbp1 and Hbp2, respectively (light blue). In both organisms, the regulons also encode SrtA-anchored proteins IsdA, IsdB, Lmo2178, and Lmo2179 (dark blue) and ABC transporter IsdDEF and Lmo2182–2184, respectively (gray). A nearby chromosomal locus codes for the Hup ABC transporter (lmo2429–lmo2431), the primary hemin transporter in L. monocytogenes. B, a schematic representation of Hbp1 and Hbp2 with amino acid sequence numbering for each NEAT domain listed above. C, amino acid sequence alignment of NEAT domains from S. aureus, B. anthracis, and L. monocytogenes. Red highlighting indicates the conserved Tyr that coordinates the iron, yellow highlighting indicates the stabilizing Tyr that H-bonds to the coordinating Tyr, and orange indicates the novel Tyr observed for Hbp2N2.
Here we demonstrate that Hbp1 and Hbp2 bind to Hb and hemin. To gain insight into the mechanism of hemin binding, crystal structures of the central NEAT domain from Hbp2 (Hbp2N2; residues 183–303) and the Hbp2N2-hemin complex were determined at 2.7- and 1.8-Å resolution, respectively. The structures reveal a unique mode of metal ligation and an induced fit binding mechanism that have not been reported previously for a NEAT domain. The results of hemin transfer experiments are consistent with the notion that Hbp2 functions as an Hb-scavenging hemophore that delivers hemin to Hbp2 located within the cell wall. In vitro, this process occurs rapidly via the formation of an Hbp2-Hbp2 hemin transfer complex. The results of these studies provide new insight into the mechanism through which L. monocytogenes and other Gram-positive pathogens capture heme iron.
EXPERIMENTAL PROCEDURES
Cloning, Protein Expression, and Purification
DNA for subcloning was amplified from the L. monocytogenes EGD-e genome using the polymerase chain reaction (PCR) and cloned into the pHis-SUMO vector (11). Plasmids encoding proteins containing a removable N-terminal hexahistidine-small ubiquitin-like modifier (SUMO) tag were generated using standard cloning protocols. The following plasmids that express proteins with the SUMO tag (His6-SUMO tag) were constructed: pRM245 coding for amino acids 31–170 in Hbp2 (Hbp2N1), pRM246 coding for amino acids 183–303 in Hbp2 (Hbp2N2), pRM247 coding for amino acids 360–479 in Hbp2 (Hbp2N3), pRM248 coding for amino acids 31–479 in Hbp2, and pRM251 coding for amino acids 31–149 in Hbp1. Site-directed mutagenesis of His6-SUMO-Hbp2N2 was performed on plasmid pRM246 to generate plasmids pRM257 and pRM258 that express Hbp2N2(Y280A) and Hbp2N2(Y289A), respectively (QuikChange site-directed mutagenesis kit, Stratagene, La Jolla, CA). The nucleotide sequences of all plasmids were confirmed by DNA sequencing.
Proteins were expressed by transforming the aforementioned plasmids into Escherichia coli BL21(DE3) cells (New England BioLabs). Proteins were expressed overnight at 25 °C by adding 1 mm isopropyl β-d-thiogalactoside. Proteins were purified as described previously (12). Briefly, bacterial cells were harvested by centrifugation and ruptured by sonication, and the Hbps were purified using a cobalt (Co2+)-chelating column. The ULP1 protease was then used to cleave the N-terminal His6-SUMO tag. Hemin-saturated proteins were generated by adding hemin in 1.5-fold excess to purified protein solutions followed by removal of excess hemin using a Sephadex G-25 column (GE Healthcare) equilibrated with 20 mm Tris-HCl, pH 8.0. The hemin saturation level was checked by recording UV-visible spectra and calculating the ASoret/A280. To create the apo form of the proteins used in this study, hemin was removed from purified proteins by methyl ethyl ketone extraction (13).
Enzyme-linked Immunosorbent Assay (ELISA)
The ELISA was performed using 96-well plates (Corning, Sigma). Wells were coated with a volume of 100 μl of 10 μg/ml human hemoglobin (Sigma) dissolved in PBS, pH 7.4 and incubated on an orbital shaker overnight at 4 °C. Blocking of nonspecific sites was achieved by washing off coating solution with PBS, pH 7.4, 0.05% Tween 20 (Sigma) and applying 200 μl of a solution of PBS, pH 7.4, 1% bovine serum albumin (BSA), 0.2% nonfat dry milk, 0.05% Tween 20 for 1 h at room temperature. After blocking, 50 μl of each NEAT domain-containing protein solution was added to each well at a range of concentrations (0.5–250 pmol of total protein in PBS, pH 7.4, 0.05% Tween 20) and then incubated for 10 min on an orbital shaker at room temperature. After washing, 50 μl of monoclonal mouse IgG antibody against the hexahistidine tag (Abgent) was applied at a 1000-fold dilution in PBS, pH 7.4, 0.05% Tween 20 and incubated for 1 h on an orbital shaker at room temperature. After washing with PBS, pH 9, 0.05% Tween 20, 50 μl of monoclonal goat IgG antibody against mouse IgG conjugated to alkaline phosphatase (Sigma) was added at a 10,000-fold dilution in PBS, pH 9, 0.05% Tween 20 and incubated for 1 h on an orbital shaker at room temperature. After final washes in PBS, pH 9, 0.05% Tween 20, 50 μl of 1-Step p-nitrophenyl phosphate (Pierce) was added to develop the signal. The wells were incubated at room temperature for 1 h on an orbital shaker and then overnight at 4 °C on an orbital shaker. Optical densities were measured at 405 nm on an ELISA plate reader (Spectramax M5, Molecular Devices) at 30 min, 1 h, and overnight.
Hemin Binding, Transfer, and Dissociation Studies
Isothermal titration calorimetry (ITC) measurements were performed using a MicroCal iTC200 calorimeter (GE Healthcare). The apo forms of proteins to be studied were dialyzed into ITC buffer (50 mm NaH2PO4, pH 7.3, 5% DMSO). A fresh 4.1 mm hemin solution (Sigma) was immediately prepared before each titration by dissolving hemin into ITC buffer. The cell was filled with apoprotein, and the syringe was filled with hemin solution. Sixteen injections were performed using 2.5-μl aliquots (260 s between injections). The data were analyzed using MicroCal iTC200 analysis software (GE Healthcare). Hemin binding affinities for the following proteins were determined: Hbp1, Hbp2N2, Hbp2N3, Hbp2N2(Y280A), and Hbp2N2(Y289A).
A column-based approach was used to measure hemin transfer from Hbp2 to Hbp1. The apo form of His6-SUMO-Hbp1 and the holo form of Hbp2 were prepared according to aforementioned protocols. Both proteins were dissolved in transfer buffer (50 mm NaH2PO4, 300 mm NaCl, pH 7.0). They were allowed to incubate at room temperature for varying amounts of time after mixing; His6-SUMO-Hbp1 (15 μm) was mixed with holo-Hbp2 (15 μm). The proteins were then separated by applying them to a Co2+-chelating column that had been pre-equilibrated in transfer buffer. The column was then washed four times with 1.0 ml of transfer buffer followed by elution of the His6-SUMO-Hbp1 protein with 1 ml of the following buffer: 50 mm NaH2PO4, 300 mm NaCl, 250 mm imidazole, pH 7.0. Hemin content was quantified by determining the ASoret/A280 ratio for each of the fractions at the indicated time points, and this was then divided by the ratio representing heme-saturated protein. To monitor heme release into the solvent by different proteins, they were incubated with recombinant sperm whale myoglobin that contained H64Y and V68F mutations (H64Y/V68F Mb) (14). For these studies, proteins were exchanged into PBS buffer, pH 7.4. Heme-saturated donor proteins (Hbp1 or Hbp2) were added to H64Y/V68F Mb to a final concentration of 5 and 50 μm, respectively. Sucrose was also added to a final concentration of 450 μm to prevent protein aggregation as described previously (14). UV-visible spectra were then recorded between 260 and 700 nm for up to 24 h at 25 °C. The change in absorbance at 600 nm was plotted with time and used as a measure of hemin dissociation. All kinetics data were analyzed using the program GraphPad Prism.
Crystallization and Structure Determination
The apo form of Hbp2N2 (residues 183–303) was prepared as detailed above and dialyzed into a 10 mm HEPES, pH 8.0 buffer. The protein solution was concentrated to 150 mg/ml and crystallized at room temperature using the hanging drop vapor diffusion method in a reservoir solution of 100 mm sodium citrate tribasic dihydrate, pH 5.4, 200 mm potassium sodium tartrate tetrahydrate, 2.0 m ammonium sulfate. Large cubic crystals appeared within 3 days. Crystals were stored in liquid N2 after soaking in cryoprotectant solution that consisted of 2 m trimethylamine N-oxide dihydrate mixed 1:1 with the reservoir solution. The best diffracting crystals were obtained by soaking them in cryoprotectant solution for 2–5 min. Selenomethionine-substituted protein was produced by withholding native methionine and supplementing bacterial cells with selenomethionine upon induction with isopropyl β-d-thiogalactoside (Sigma). Selenomethionine-Hbp2N2 was purified as described above and crystallized similarly to the native protein. The crystals belonged to space group I23 with one protein molecule per asymmetric unit. All diffraction data were collected at the Advanced Photon Source (Argonne National Laboratory) beamline 24-ID-C using a Dectris Pilatus 6M pixel detector. Crystals were cryocooled to 100 K during the data collection. One hundred 1.0° oscillation frames were collected at a wavelength of 0.9795 Å. Data reduction and scaling were performed using XDS (15). The resolution of the data set was drawn at 2.7 Å (16). Experimental phases were obtained from the selenomethionine derivative using the multiwavelength anomalous dispersion method. Diffraction data sets were collected at two wavelengths on a selenomethionine derivative at the Advanced Photon Source (Argonne National Laboratory) beamline 24-ID-C. One data set was collected at the peak energy for selenium (0.9792 Å), and one was collected at a high energy remote (0.9686 Å). A data set at a low energy remote was collected in house using a Rigaku FR-E rotating copper anode (1.5418-Å wavelength) and an R-AXIS HTC imaging plate detector. The resulting electron density map was sufficiently clear to allow manual placement of a homologous structure, IsdX1, an anthrax hemophore (Protein Data Bank code 3SZ6) (17). The model was mutated to the Hbp2N2 sequence using the multiwavelength anomalous dispersion map as a guide. The model was then refined against the isomorphous native apo data set, which was at higher resolution than the selenomethionine derivative data. Methods for model building and refinement are discussed below.
The Hbp2N2-hemin complex used for crystallization trials was generated by adding free hemin to apo-Hbp2N2. Extensive effort was put forth to remove hemin aggregates prior to adding it to the protein. Briefly, in this procedure, 40 mg of hemin was dissolved in 4 ml of 0.1 m NaOH (4 °C) and then vortexed. A total of 4 ml of buffer A (1 m Tris-HCl, pH 8.0) was then added to the hemin solution, and it was then centrifuged at 18,000 × g for 10 min at 4 °C. The hemin solution was then diluted with 8 ml of buffer A and centrifuged for 10 min to pellet aggregated hemin. The hemin concentration of the cleared solution was 3 mm as determined by UV absorbance (assuming ϵ385 = 58.44 mm−1 cm−1). Hemin was then added slowly to the protein in 100-μl aliquots to a final concentration such that it was in 1.5-fold molar excess. The protein/hemin mixture was incubated at room temperature on a tube rotator for 1 h. The excess hemin was removed by separating the mixture using a Sephadex G-25 column. Fractions containing the Hbp2N2-hemin complex were then dialyzed overnight into buffer B (50 mm Tris-HCl, pH 7.4, 150 mm NaCl) solution. After concentration, the protein was then applied onto a Sephacryl S-100 XK 26/100 gel filtration column (GE Healthcare) equilibrated with buffer B, and pure fractions were pooled for crystallography trials. The Hbp2N2-hemin complex was concentrated to 53 mg/ml and crystallized at room temperature using the hanging drop vapor diffusion method in a reservoir solution of 100 mm MES, pH 6.0, 10 mm ZnCl2, 20% polyethylene glycol 6000. Crystals grew within 1 day and were cryoprotected by soaking the crystals briefly in reservoir solution that contained 35% glycerol. An x-ray diffraction data set was collected on beamline 24-ID-C as described above for apo-Hbp2N2. The crystals used to solve the structure were in the P21 space group and contained two protein-hemin complexes per unit cell. The structure of apo-Hbp2N2 was used as a search model for molecular replacement. The resolution limit of the data set was drawn at 1.8 Å. Models of apo-Hbp2N2 and the Hbp2N2-hemin complex were refined using REFMAC5 (18) and BUSTER-TNT (19) with TLS (translation, rotation, screw-rotation) parameterization of domain disorder (20). After each refinement step, the model was visually inspected in Coot using both 2Fo − Fc and Fo − Fc difference maps (21). Final structural models were validated using MolProbity (22), PROCHECK (23), ERRAT (24), and VERIFY3D (25).
RESULTS
Hbp1 and Hbp2 Bind Hemin with Nanomolar Affinity
Inspection of the primary sequences of Hbp1 and Hbp2 reveals that they contain NEAT domains (Fig. 1B). Because the functions of these domains are not known, we biochemically characterized their ligand binding properties. Initially, Hbp1 (residues Asp31–Ala149) and Hbp2 (residues Ser31–Ala479) polypeptides that contain the intact protein with the exception of the N- and C-terminal signal peptides and cell wall sorting signals, respectively (Fig. 1B), were purified. Upon overexpression and purification of these polypeptides from E. coli, it was immediately apparent that they interacted with hemin based on the presence of Soret peaks in their UV-visible spectra (Fig. 2A). To define the location of hemin binding in Hbp2, we purified polypeptides containing each of its three NEAT domains: Hbp2N1 (residues Ser31–Phe170), Hbp2N2 (residues Thr183–Lys303), and Hbp2N3 (residues Leu360–Ala479). As with the Hbp2 protein, upon purification, each domain had a reddish brown color, and UV-visible spectrum analysis revealed the presence of a Soret peak. Thus, all three NEAT domains within Hbp2 are capable of binding hemin.
FIGURE 2.

UV-visible spectra of Hbp1 and Hbp2. A, hemin added to a final concentration of 10 μm to 10 μm Hbp1 or Hbp2. B, representative ITC data for Hbp2N3. Fresh hemin was injected stepwise into a cell containing apoprotein at a constant temperature of 25 °C. The top panel shows the time course of the titration (black) and baseline (gray). The bottom panel shows the integrated isotherms. The program ORIGIN was used to fit the data using non-linear regression to a one-site binding model to derive thermodynamic parameters.
The hemin binding affinities of the isolated NEAT domains were determined using ITC. This analysis was performed using the apo form of each domain, which was produced using the methyl ethyl ketone organic extraction method (13). In these experiments, a syringe filled with hemin was incrementally injected into solutions containing the apo form of each domain, and the heat changes were monitored. Affinities were determined for all NEAT domains with the exception of the first domain from Hbp2 (Hbp2N1), which aggregated during hemin extraction. A representative hemin binding isotherm for the Hbp2N3 domain is shown in Fig. 2B, and similar high quality data were obtained for the other NEAT domains. As reported in Table 1, the Hbp1, Hbp2N2, and Hbp2N3 NEAT domains each bind one molecule of hemin with dissociation constants (KD) that range from 25 to 81 nm. Although the hemin affinity of the first NEAT domain from Hbp2 (Hbp2N1) could not be determined by ITC, it likely binds hemin with similar nanomolar affinity because when it is purified from E. coli it exhibits a similar level of hemin saturation as the other NEAT domains. These data along with hemin transfer experiments (discussed below; Fig. 3) indicate that all of the NEAT domains within the Hbp2 and Hbp1 proteins bind hemin with generally similar affinities.
TABLE 1.
Thermodynamic parameters and affinity data for hemin binding
| Protein | KD | ΔH | −TΔSa | ΔG | nb |
|---|---|---|---|---|---|
| nm | kcal/mol | kcal/mol | kcal/mol | ||
| Hbp1 | 25 ± 19 | −9.6 ± 0.1 | −1.3 | −11 ± 0.1 | 1 |
| Hbp2N2 | 81 ± 35 | −3.5 ± 0.1 | −6.3 | −9.8 ± 0.1 | 1 |
| Hbp2N3 | 48 ± 11 | −9.1 ± 0.1 | −0.9 | −10 ± 0.1 | 1 |
| Hbp2N2(Y289A) | 47,667 ± 14,445 | −1.8 ± 0.3 | −4.2 | −6.0 ± 0.3 | 1 |
| Hbp2N2(Y280A) | NDc | ND | ND | ND | ND |
a Temperature, 298 K.
b n refers to the molar ratio hemin:protein.
c ND refers to binding “not detected.”
FIGURE 3.

Demonstration of hemin transfer. A, a schematic of the hemin transfer experiment. A hemin donor (e.g. holo-Hbp2) is mixed with a histidine-tagged hemin acceptor (e.g. His6-SUMO-Hbp1). The proteins are then separated using a Co2+-chelating column, and the amount of hemin transferred to the acceptor is determined. A representative SDS-polyacrylamide gel shows complete separation of Hbp2 and His6-SUMO-Hbp1 after incubation for 60 min. W, wash; E, elution. B, hemin transfer from Hbp2 and Hbp1. The UV spectrum of the imidazole eluted His6-SUMO-Hbp1 protein before and after incubation with hemin-loaded Holo-Hbp2 (1:1 stoichiometry). Transfer from Hbp2 to His6-SUMO-Hbp1 is complete within 5 min of mixing the proteins. C, hemin transfer from holo-Hbp2N2 to His6-SUMO-Hbp1 (1:10 stoichiometry). The experiments were performed as shown in A. The amount of hemin captured by His6-SUMO-Hbp1 was determined by measuring the ASoret/A280 ratio of the eluted fractions, which was then divided by the ratio representing heme-saturated protein to determine the fraction of protein bound with hemin. The dashed line indicates the expected level of hemin saturation of the acceptor based on the dissociation constants measured by ITC. D, as in C but hemin transfer from holo-Hbp2N2 to His6-SUMO-Hbp2N3 was measured. E, as in C but hemin transfer from holo-Hbp1 to His6-SUMO-Hbp1 was measured. F, as in C but hemin transfer from holo-Hbp2N2 to His6-SUMO-Hbp2N1 was measured. There is no dashed line as ITC was not used to determine the hemin affinity of Hbp2N1. Error bars represent S.D. from three experiments.
Hemin Is Rapidly Transferred between Hbp1 and Hbp2
After scavenging hemin from the cell's surroundings, Hbp1 and Hbp2 may transfer it across the cell wall to the plasma membrane. To fulfill this task, they should ideally release hemin into the solvent very slowly but nonetheless rapidly transfer hemin between each other. The rate at which Hbp1 and Hbp2 spontaneously release hemin in the solvent was investigated using recombinant H64Y/V68F apomyoglobin (apo-Mb containing H64Y and V68F mutations; hereafter referred to as apo-Mb) (14). This protein is commonly used to measure hemin release from hemoproteins as its hemin-bound form has a unique UV-visible absorbance maxima at ∼600 nm. Mixing of hemin-saturated Hbp2 with a 10-fold excess of apo-Mb results in a measurable change in the UV-visible spectrum; the most significant changes occur at 600 nm, increasing as hemin is bound by apo-Mb (Fig. 4A). The time dependence of this change reveals that it occurs very slowly, compatible with a transfer mechanism in which hemin is first released from Hbp2 into the solvent followed by hemin binding by apo-Mb (the transfer process is biphasic with rate constants of 6.347 and 0.229 h−1) (Fig. 4B). Consistent with this conclusion, these values are similar to hemin dissociation rates from metHb (26). Similar experiments using hemin-saturated Hbp1 revealed little or no transfer to apo-Mb over the time course of the experiment, suggesting that it has slower hemin release kinetics than Hbp2. Taken together, these data indicate that the NEAT domains within Hbp1 and Hbp2 release hemin slowly into the solvent.
FIGURE 4.
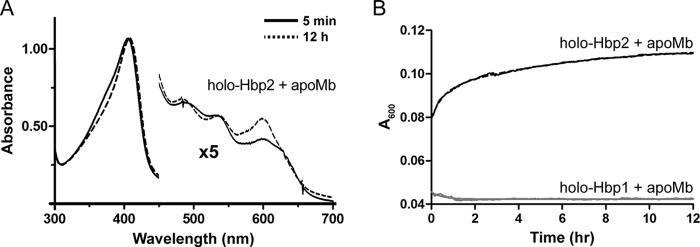
Hemin dissociation from Hbp1 and Hbp2. H64Y/V68F apomyoglobin was added at a final concentration of 50 μm to 5 μm Hbp1 or Hbp2. A, the absorbance spectra for reactions at 5 min and 12 h for Hbp2 are shown as solid and dotted lines, respectively. B, the panel shows the absorbance change at 600 nm, which is indicative of hemin scavenging by the H64Y/V68F myoglobin. For Hbp2, the black dotted line represents the raw data, and the solid line is the best fit using a double exponential function. The data for Hbp1 are shown in gray and could not be reliably fit because very little or no hemin is released from this protein.
Chromatography experiments indicate that hemin is transferred from holo-Hbp2 to apo-Hbp1. To investigate hemin transfer, holo-Hbp2 was mixed with an equal molar amount of an Hbp1 fusion protein that contained a hexahistidine-tagged small ubiquitin-like modifier protein at its N terminus (His6-SUMO-Hbp1), and the proteins were then separated using a Co2+-chelating column (Fig. 3A). Hemin transfer from holo-Hbp2 to His6-SUMO-Hbp1 was tracked by measuring the increase in UV absorbance of the separated His6-SUMO-Hbp1 (acceptor) protein after it was incubated with holo-Hbp2 (donor) for varying amounts of time because in a control experiment in which no holo-Hbp2 (donor) was provided minimal UV absorbance is seen at the Soret band of His6-SUMO-Hbp1 (Fig. 3B). This analysis reveals that hemin transfer is complete within 5 min of mixing the proteins, indicating that the rate of transfer is several orders of magnitude faster than the rate at which Hbp2 releases hemin into the solvent spontaneously (∼0.002 s−1) (Fig. 3B). This strongly suggests that Hbp1 and Hbp2 transfer hemin via an activated protein-protein complex.
Similar hemin transfer experiments were performed using holo-Hbp2N2 as the donor and either apo-Hbp1 (Fig. 3C) or apo-Hbp2N3 (Fig. 3D) as the acceptors. In all experiments, a donor:acceptor molar ratio of 1:10 was used, and the amount of hemin transferred to each acceptor was quantitatively determined my measuring the ASoret/A280 ratio of the imidazole-eluted fraction that contained the histidine-tagged acceptor protein. In both cases, hemin transfer occurs rapidly between the NEAT domains, and the amount of hemin captured by the acceptor protein is consistent with the heme affinity of each domain measured by ITC (indicated by a dashed line in each panel). In particular, relative to the Hbp2N3 domain, Hbp1 captures twice as much hemin from holo-Hbp2N2, which is compatible with its ∼2-fold higher affinity for hemin as measured by ITC (Table 1). The experiments were repeated using Hbp1 and His6-SUMO-Hbp1 proteins as the hemin donor and acceptor, respectively (Fig. 3E). The results reveal that homotypic hemin transfer between Hbp1 proteins also occurs rapidly and that the hemin partitions to the acceptor in a manner consistent with the ITC binding data (Fig. 3E). To gain insight into the hemin affinity of the Hbp2N1 domain whose hemin affinity was not quantified by ITC, the experiments were repeated using holo-Hbp2N2 as a donor and partially hemin-bound (15%) Hbp2N1 as the acceptor (Fig. 3F). As with the other domains, transfer to Hbp2N1 occurs rapidly from holo-Hbp2N2, and based on the amount of hemin transferred to Hbp2N1, it appears to have slightly higher affinity for hemin than Hbp2N2. Combined, these data indicate that the Hbp2 hemophore has the capacity to deliver hemin to the cell surface by transferring it to cell wall-associated Hbp1 and Hbp2 proteins.
Hbp1 and Hbp2 Are Hb Receptors
To investigate whether Hbp1 or Hbp2 can function as bacterial Hb receptors, microplate wells containing adhered Hb were incubated with varying amounts of Hbp1 or Hbp2 that was fused to a His6-SUMO tag. After washing, the amount of His6-SUMO-Hbp1 or His6-SUMO-Hbp2 retained in each well as a result of interacting with Hb was then determined using an immunoassay. The results of the ELISA indicate that both His6-SUMO-Hbp1 and His6-SUMO-Hbp2 are capable of binding Hb, whereas the negative control, the His6-SUMO tag, is unable to bind Hb (Fig. 5A). Binding is specific for Hb as little His6-SUMO-Hbp1 and His6-SUMO-Hbp2 was retained in the well in control experiments in which wells were coated only with BSA (data not shown). These data indicate that Hbp1 and Hbp2 bind to Hb with generally similar affinities because the wells are saturated when similar amounts of each protein are added (∼75 pmol). We estimate that Hbp1 and Hbp2 bind to Hb with KD values that are in the nanomolar range. This is because similar results are obtained in positive control experiments using the NEAT domain from the Staphylococcus aureus IsdH protein (IsdHN2; residues 326–466 of the IsdH protein) that has been shown to bind Hb with nanomolar affinity (27, 28). To determine which NEAT domain within Hbp2 mediates binding to Hb, we repeated the ELISA experiments using hexahistidine-tagged polypeptides containing its various NEAT domains (His6-SUMO-Hbp2N1, His6-SUMO-Hbp2N2, and His6-SUMO-Hbp2N3). This analysis revealed that the N-terminal (Hbp2N1) and C-terminal NEAT domains in Hbp2 bind Hb, whereas the central Hbp2N2 domain does not (Fig. 5B). Combined, the ELISA data indicate that both Hbp1 and Hbp2 can function as Hb receptors. In Hbp1, its single NEAT domain mediates Hb binding, whereas in Hbp2, the first and third modules within the tridomain protein are responsible for Hb binding.
FIGURE 5.
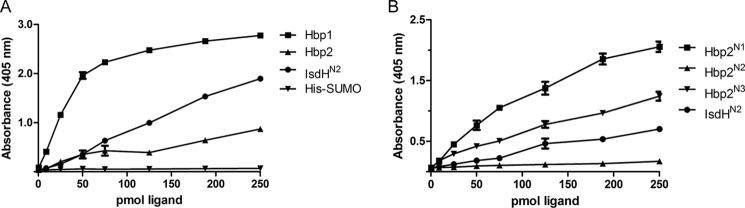
The Hbp1 and Hbp2 proteins interact with hemoglobin. A, the results of an ELISA experiment showing binding of Hbp1, IsdHN2, and to a lesser extent Hbp2 to Hb-coated wells. B, the results of an ELISA experiment showing binding of Hbp2N1, Hbp2N3, IsdHN2, and to a lesser extent Hbp2N2 to Hb-coated wells. Error bars represent S.D. from three experiments.
Crystal Structure of the Hbp2N2-Hemin Complex Reveals a Novel Coordination Mechanism
All studies reported to date have shown that NEAT domains use tyrosine residues within a conserved YXXXY motif to interact with hemin (Fig. 1C, colored red and yellow) (17, 29–35). In these structures, the hydroxyl group from the first tyrosine (red) residue in the motif functions as an axial ligand that coordinates the hemin metal, whereas the second tyrosine (yellow) stabilizes the positioning of the axial tyrosine through side chain-side chain hydrogen bonding. Upon examining the primary sequence of Hbp2, it is immediately apparent that its central NEAT domain (Hbp2N2) lacks the stabilizing tyrosine residue; it contains the sequence YXXXA instead of YXXXY (Fig. 1C). To determine whether this change significantly alters the mechanism of hemin binding, we used x-ray crystallography to determine the three-dimensional structures of apo-Hbp2N2 and the Hbp2N2-hemin complex at 2.7- and 1.8-Å resolution, respectively (Fig. 6, A and B). Initially, the structure of selenomethionine-labeled apo-Hbp2N2 was determined by the multiwavelength anomalous dispersion method (36). The structure of native apo-Hbp2N2 was obtained by refinement using the selenomethionine-labeled apo-Hbp2N2 as a starting model. A molecular replacement search was unnecessary because of the isomorphism between these two crystals. Finally, the structure of the Hbp2N2-hemin complex was determined by molecular replacement using the coordinates of native apo-Hbp2N2 as a search model (Fig. 6B). Table 2 contains statistics for these structures.
FIGURE 6.
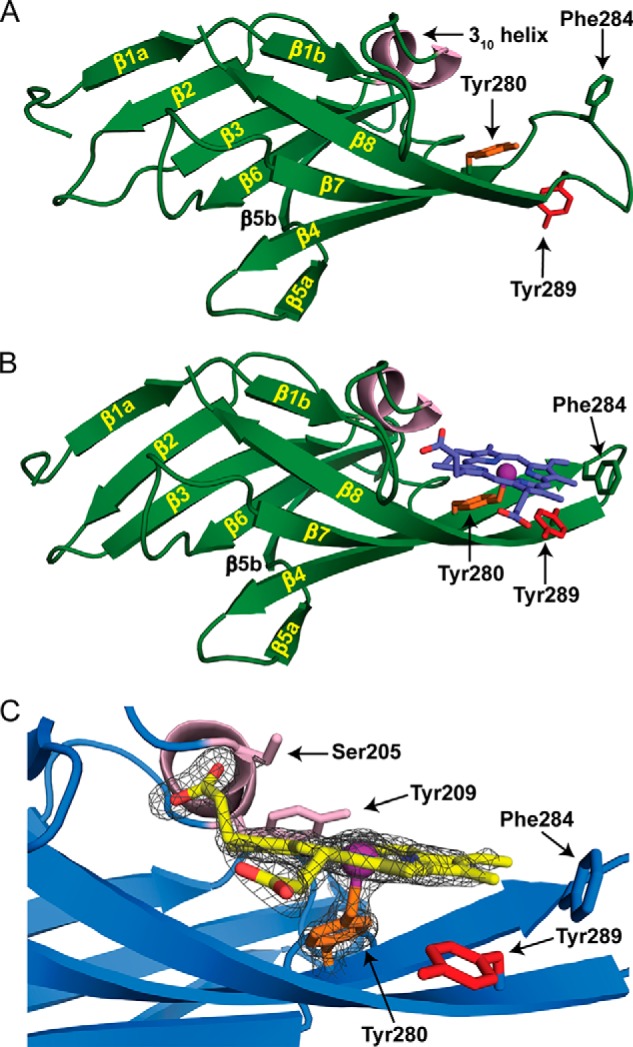
Crystal structures of apo- and holo-Hbp2N2. A and B, ribbon representation of apo-Hbp2N2 (A) and holo-Hbp2N2 (B). The β strands and helices are colored green and light pink, respectively. Residues are color-coded to match the sequence alignment in Fig. 1C. Secondary structural elements are labeled as described in the text. The hemin molecule is colored blue. C, the hemin binding pocket is shown with the 1.8-Å resolution holo-Hbp2N2 structure superimposed with a simulated annealing Fo − Fc omit map contoured to 5.0 σ shown as a dark gray mesh.
TABLE 2.
Data collection and refinement statistics
r.m.s., root mean square; CC, correlation coefficient.
| Apo-Hbp2N2 | Se-Hbp2N2 |
Holo-Hbp2N2 | |||
|---|---|---|---|---|---|
| Peak | High remote | Low remote | |||
| Data collection | |||||
| Space group | I23 | I23 | I23 | I23 | P21 |
| Cell dimensions | |||||
| a, b, c (Å) | 89.3, 89.3, 89.3 | 88.7, 88.7, 88.7 | 88.7, 88.7, 88.7 | 88.8, 88.8, 88.8 | 32.7, 67.7, 58.51 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 91.6, 90 |
| Resolution (Å) | 2.7 (2.85-2.70)a | 2.7 (2.76-2.70) | 2.75 (2.82-2.75) | 2.65 (2.74-2.65) | 1.8 (1.85-1.80) |
| Wavelength (Å) | 0.9795 | 0.9792 | 0.9686 | 1.5418 | 0.9792 |
| Rmerge | 0.052 (0.471) | 0.057 (0.723) | 0.055 (0.524) | 0.080 (0.709) | 0.046 (0.423) |
| Rr.i.m. | 0.053 (0.482) | 0.076 (0.876) | 0.068 (0.651) | 0.086 (0.791) | 0.055 (0.511) |
| Rp.i.m. | 0.012 (0.101) | 0.012 (0.141) | 0.011 (0.137) | 0.027 (0.251) | 0.030 (0.283) |
| I/σI | 35.3 (6.3) | 32.2 (3.3) | 33.6 (3.9) | 30.6 (3.7) | 14.1 (2.8) |
| CC1/2 (%) | 99.9 (98.4) | 99.9 (85.4) | 100.0 (91.7) | 100.0 (84.2) | 99.8 (84.7) |
| Completeness (%) | 100.0 (100.0) | 99.9 (99.4) | 100.0 (100.0) | 99.2 (100.0) | 96.7 (95.4) |
| Redundancy | 21.4 (22.8) | 20.2 (17.7) | 19.7 (11.1) | 10.1 (10.1) | 3.2 (3.1) |
| Refinement | |||||
| Resolution (Å) | 2.7 (3.02-2.70) | 1.8 (1.89-1.80) | |||
| No. reflections | 3,370 (929) | 22,884 (3,012) | |||
| Rwork | 0.254 (0.355) | 0.195 (0.227) | |||
| Rfree | 0.293 (0.414) | 0.225 (0.264) | |||
| CCwork | 0.909 (0.677) | 0.935 (0.807) | |||
| CCfree | 0.920 (0.429) | 0.882 (0.716) | |||
| CCa | 1.000 (0.996) | 1.00 (0.965) | |||
| No. atoms | |||||
| Protein | 947 | 1,865 | |||
| Sulfate | 1 | 0 | |||
| Hemin | 0 | 86 | |||
| Glycerol | 0 | 54 | |||
| Zinc | 0 | 4 | |||
| Water | 9 | 115 | |||
| B-factors (Å2) | |||||
| Protein | 125.4 | 41.0 | |||
| Sulfate | 216.5 | 0 | |||
| Hemin | 0 | 35.9 | |||
| Glycerol | 0 | 68.8 | |||
| Zinc | 0 | 60.4 | |||
| Water | 88.7 | 48.8 | |||
| Wilson B (Å2) | 109.4 | 33.3 | |||
| r.m.s deviations | |||||
| Bond lengths (Å) | 0.008 | 0.01 | |||
| Bond angles (°) | 1.0 | 1.1 | |||
| MolProbity | |||||
| Ramachandran outliers (%) | 0.0 | 0.0 | |||
| Ramachandran favored (%) | 94.12 | 97.86 | |||
| Bad backbone (%) | 0.15 | 0.14 | |||
| MolProbity score | 1.91 (99th percentile) | 1.01 (100th percentile) | |||
a Highest resolution shell is shown in parentheses.
The structure of the Hbp2N2-hemin complex reveals a mode of metal ligation not previously seen in the structures of other NEAT domain-hemin complexes (17, 29–34). Hbp2N2 in its free and hemin-bound states adopts a canonical NEAT domain fold that consists of eight β strands that form a β-sandwich (Fig. 6, A and B). One face of the structure contains the antiparallel strands β1a-β2-β3-β6-β5b, whereas the other face includes strands β1b-β8-β7-β4-β5a (35). Two molecules of Hbp2N2-hemin complexes are present in the asymmetric unit and have essentially identical conformations (the root mean square difference of their backbone atoms is 0.16 Å). In each complex, the hemin is wedged in between a short 310 helix (called a “lip”) and a β-hairpin formed by strands β7 and β8 (Fig. 6B). The structure precisely reveals the molecular basis of hemin binding as evidenced by a simulated annealing Fo − Fc omit map generated by deleting the coordinates of the hemin and axial Tyr ligand (Fig. 6C). In the structure, the hemin is pentacoordinate, and its planar face rests on the β-hairpin, positioning the hydroxyl oxygen from Tyr280 located in strand β7 to coordinate the iron from a distance of 2.2–2.4 Å. The majority of protein interactions with the hemin are non-polar. Val278 packs against the methyl of hemin pointing into the core of the protein. Tyr209 located in the 310 helix packs against the vinyl moiety, and the methyl group of Thr234 packs against the methyl of the hemin. Ala282 of the β7 strand also packs against the methyl group as well as the methine bridge and vinyl groups in the hemin. Phe284 at the end of the β7 strand forms non-polar contacts to the methyl and vinyl groups of hemin. This residue appears to have a capping function, holding the hemin in the pocket so its other end can be wedged underneath the 310 helix. Polar interactions also occur in the complex as the side chains of Ser204 and Ser205 hydrogen bond to the propionate group in hemin. Additionally, the backbone amide of Ser205 donates a hydrogen bond to the propionate of hemin.
The use of Tyr280 in strand β7 to coordinate the hemin is very surprising because in all other NEAT-hemin complexes studied to date metal ligation occurs via a highly conserved tyrosine residue located in strand β8 (Fig. 1C, colored red). Interestingly, Hbp2N2 also contains this conserved tyrosine (Tyr289), but instead of coordinating the metal its side chain π stacks against the hemin to project its hydroxyl group into the solvent (Fig. 7B). To probe the energetics of binding, ITC was used to measure hemin binding affinities of two single amino acid mutants of Hbp2N2 (Table 1). These include a Y280A mutant of Hbp2N2 that removes the hydroxyl group that coordinates the hemin metal in the crystal structure of the Hbp2N2-hemin complex and a Y289A mutant that alters the highly conserved tyrosine residue that in other NEAT domains functions to coordinate the metal. The Y280A mutant exhibited no detectable hemin binding as no significant heat changes were observed even after 1 mm hemin was added to a cell containing Hbp2N2(Y280A). The severity of this mutation is compatible with the crystal structure as the hydroxyl of Tyr280 coordinates the metal of the hemin, and ITC studies of other NEAT domains have shown similar large reductions in affinity when mutations are introduced that disrupt the axial bond to the hemin metal (37). The Y289A mutation also reduces hemin affinity significantly albeit to a lesser extent than the Y280A mutation. The loss in affinity caused by the Y289A mutant presumably occurs because it disrupts favorable hydrophobic interactions with the protoporphyrin molecule.
FIGURE 7.

Superimposition of apo- and holo-Hbp2N2. A, ribbon representation of superimposition of apo- (green) and holo-Hbp2N2 (blue). Residues shown as sticks are color-coded for holo-Hbp2N2 to match those in Fig. 6. B, superimposition of the hemin binding pocket. C, superimposition of holo-Hbp2N2 and S. aureus IsdC in blue and green, respectively. The hemes bound to holo-Hbp2N2 and IsdC are shown in stick format in pink and green, respectively. Iron atoms are shown as spheres. The residues corresponding to holo-Hbp2N2 and IsdC are shown in black and green, respectively. D, holo-Hbp2N2 shown as a surface representation. E, a model of the possible transfer complex formed by two molecules of Hbp2N2 generated by superimposing chains A and B via their heme molecules that was then energy-minimized with Rosetta and the Crystallography and NMR System (CNS) software suite.
A comparison of the structures of apo-Hbp2N2 and the Hbp2N2-hemin complex reveals that hemin binding causes major changes in the conformation of Hbp2N2. Fig. 7A shows an overlay of the backbone atoms of the protein in its free and hemin-bound states. The largest heme-dependent changes occur in the β-hairpin. These changes are primarily localized in the loop that connects the β strands, which is shortened from 10 to 4 amino acids as a result of hemin binding. Specifically, upon hemin binding, residues in the loop alter their structure such that strand β7 is lengthened to include residues Glu281–Phe284, and similarly, strand β8 is extended to include residues Ser287–Lys290. The loop may undergo a disordered to ordered transition upon binding hemin because in the apo form of the protein its backbone atoms exhibit slightly elevated temperature factors as compared with the remainder of the protein (∼124 versus 100 Å2), whereas in the Hbp2N2-hemin complex the backbone atoms have uniform B-factors. Ordering of the hairpin structure is presumably driven by hemin contacts to Tyr289 and Phe284, which are rotated toward the hemin in the complex to interact with hemin. Specifically, Tyr289 weakly π stacks against the pyrrole ring and makes van der Waals contacts with the methine bridge and methyl group of hemin, whereas the side chain of Phe284 packs against the methyl and vinyl groups in the hemin (Fig. 7B).
DISCUSSION
Nearly all bacteria require iron to grow, and during infections many pathogens obtain this metal from hemin, which contains ∼70% of the human body's total iron. Gram-positive pathogens acquire hemin iron using surface-displayed or secreted proteins that contain NEAT domains (38). Based on their primary sequences, two proteins in L. monocytogenes contain NEAT domains: Hbp1 (Lmo2186) and Hbp2 (Lmo2185) (4) (Fig. 1B). Hbp2 has been implicated in hemin acquisition as hbp2− strains are impaired in their ability to import hemin and use it as a source of iron (4). However, the biochemical function of these proteins in hemin acquisition is not well understood. One reason for this is because previous studies have shown that NEAT domains can bind different types of ligands, which enables them to perform distinct tasks in the acquisition process. For example, some NEAT domains interact with only host hemoproteins or only hemin, or they bind to hemin and additional ligands including plasma and extracellular matrix proteins (39). To understand the function of Hbp1 and Hbp2, we systematically characterized the ligand binding properties of their isolated NEAT domains. Hbp1 and Hbp2 contain one and three NEAT domains, respectively (Fig. 1B). Based on ITC measurements, the NEAT domains in Hbp1 and Hbp2 (Hbp2N2 and Hbp2N3) bind hemin with KD values ranging from 25 to 81 nm. These affinities are generally similar to those reported previously for the full-length Hbp2 protein (KD = 12 nm; Ref. 4) as well as estimates of the overall Km for the hemin transport system in intact bacterial cells (Km = 1 nm; Ref. 4). However, hemin has a propensity to aggregate, which may cause all of these approaches to underestimate the true KD of hemin binding (34, 40). This notion is compatible with hemin transfer experiments that showed that Hbp1 transfers little hemin to apo-Mb, a well characterized hemin-binding protein that is believed to bind hemin with subnanomolar affinity (41). Beyond hemin binding, the ELISA experiments indicate that the Hbp1 and Hbp2 proteins have dual functions as both proteins are also capable of binding Hb. In addition to hemin, all of the NEAT domains tested interact with Hb with the exception of the middle NEAT domain from Hbp2 (Hbp2N2) (Fig. 5). Finally, our hypothesis is consistent with previously reported results that indicate that ∼95% of the total Hbp2 protein produced by the cell is secreted into the surrounding milieu, whereas 5% of total Hbp2 associates with the cell wall (3, 10). Therefore, our in vitro binding data indicate that Hbp2 functions as an Hb-binding hemophore that scavenges hemin from the cell's surroundings and that it also functions as a surface receptor for Hb and heme.
The hemin transfer and affinity measurements reported herein provide insight into the function of Hbp2. The secreted Hbp2 hemophore scavenges external hemin with high affinity and rapidly transfers it to peptidoglycan-associated Hbp2 (Fig. 8). This notion is supported by in vitro experiments that show that the isolated NEAT domains of Hbp2 exchange hemin rapidly among one another but otherwise release hemin very slowly into the solvent (Figs. 3 and 4B). This proposed route of soluble Hbp2-mediated transfer of hemin to cell wall-associated Hbp2 is likely biologically relevant as a Δhbp2− strain was shown to be deficient in growth because it was unable to acquire hemin from purified Hbp2 protein (4). The role of Hbp1 in hemin uptake remains unclear. Its in vitro ligand properties are compatible with it playing an active role in hemin acquisition as it binds Hb, has higher affinity for hemin than any of the individual domains within Hbp2 (Table 1), and can rapidly receive hemin from Hbp2. Moreover, hbp1 resides upstream of hbp2 in the Fur-regulated srtB operon of EGD-e, so Hbp1 is presumably concomitantly expressed with Hbp2. Because mass spectrometry analyses have detected both Hbp1 and Hbp2 associated with the cell surface (8, 9), our data suggest that Hbp1 and Hbp2 form hetero- and homotypic protein-protein heme transfer complexes that provide a kinetically favorable conduit through which hemin can traverse the peptidoglycan to the cell membrane (Fig. 8). A similar pathway has been proposed for S. aureus (42) and is consistent with our finding that the isolated NEAT domains from Hbp1 and Hbp2 transfer hemin rapidly among one another in vitro (Fig. 3). However, as the hemin exchange reactions are a second order process, the proteins would need to be positioned proximally to one another within the cell wall to make this an effective mechanism of hemin transfer. After arrival at the membrane, hemin may be imported across the membrane by the previously characterized HupDGC complex and/or by proteins encoded by the lmo2182–2184 genes in the svpA-srtB locus that share primary sequence homology with ABC transporters (43). Either hemin could then directly enter metabolic biosynthetic pools for incorporation into proteins, or the iron may be released through energy-requiring oxidative degradation of the porphyrin ring. Conversely, when hemin concentrations reach toxic levels, it is likely exported from the cell (44). The ITC and hemin transfer data are consistent with the NEAT domains having the following order of affinities for hemin: Hbp1 > Hbp2N3 > Hbp2N1 > Hbp2N2 (ranked from highest to lowest affinity). However, these affinity differences are small so it is unclear whether they a play a significant role in directing hemin flow from hemoglobin to the cell surface.
FIGURE 8.
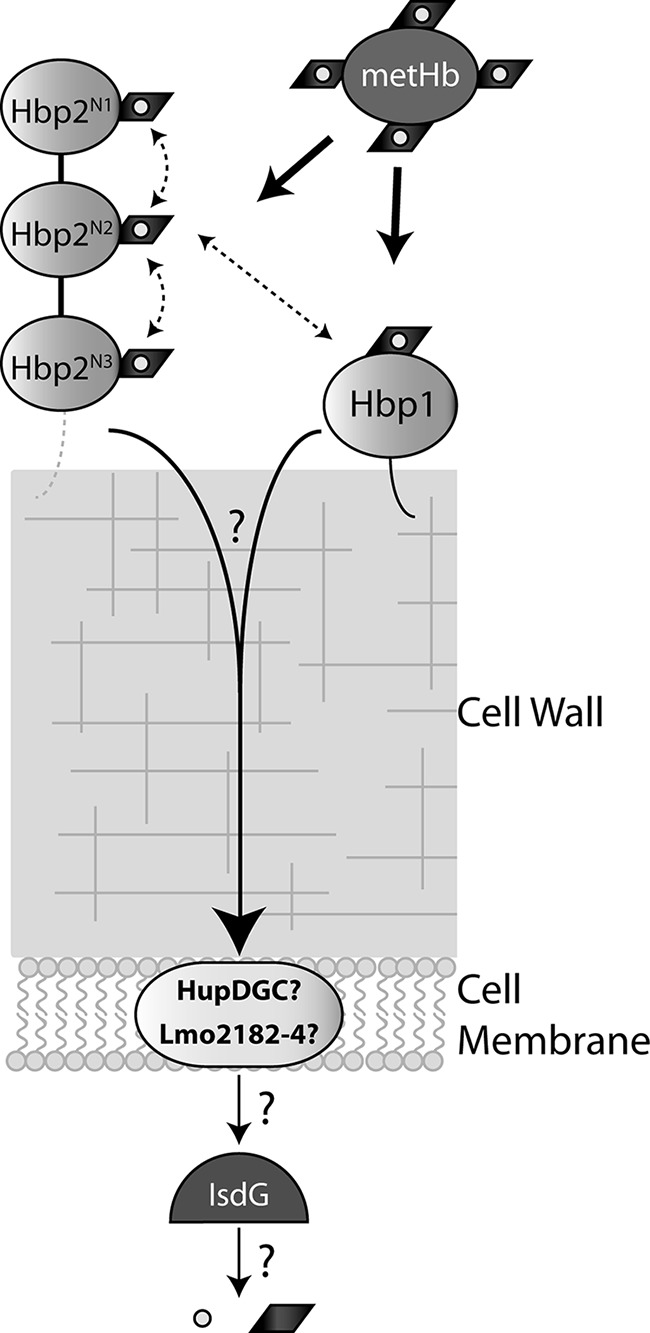
A model showing how heme is transported into L. monocytogenes. The majority of Hbp2 is secreted and binds Hb but may also bind Hb on the surface via Hbp2 or Hbp1. The dotted lines represent our observation that Hbp2 is also secreted. Heme can be transferred to cell wall-associated Hbp1 or Hbp2. Heme is then transferred to the ABC transporter HupDGC or Lmo2182–4 where it is used directly by the bacterium, or the porphyrin ring may be cleaved by an uncharacterized heme monooxygenase to release free iron for use by the bacterium.
The heme acquisition systems used by Bacillus anthracis, Streptococcus pyogenes, and S. aureus have been characterized previously, and similar to L. monocytogenes they utilize NEAT domain-containing proteins (5, 45). L. monocytogenes appears most closely related to B. anthracis as both microbes capture hemin using hemophores, whereas in S. pyogenes and S. aureus acquisition is mediated by surface-associated proteins (46). B. anthracis secretes two hemophores, IsdX1 and IsdX2, which contain one and five NEAT domains, respectively (47). Interestingly, similar to Hbp1 and Hbp2, many of the NEAT domains in these B. anthracis proteins have dual functions as they bind to both hemin and Hb. This is distinct from previously studied Hb-binding NEAT domains from the S. aureus IsdH and IsdB proteins that are incapable of binding hemin (35, 48, 49). In S. aureus, the IsdB and IsdH proteins use a conserved bidomain unit to extract hemin from Hb in which the NEAT domains function synergistically (12, 49, 50). Whether the multi-NEAT domain-containing Hbp2 protein uses a similar mechanism remains to be determined. In B. anthracis, two distinct cell wall-associated proteins are poised to receive hemin from its IsdX1 and IsdX2 hemophores (the IsdC and BslK proteins) (51–53). Therefore, it seems possible that proteins in addition to Hbp1 and Hbp2 will perform this function in L. monocytogenes, and in this regard the proteins encoded by the lmo2178 and lmo2179 genes that are located downstream of hbp1 and hbp2 are of interest for further studies.
Our results reveal that the Hbp2 hemophore coordinates the metal in hemin through a novel mechanism. In all reported structures of NEAT domains bound to hemin, residues within a conserved YXXXY motif mediate binding. These structures include the hemin complexes of the S. aureus IsdA (30), IsdC (32), IsdHN3 (34), and IsdBN2 (29) proteins as well as the B. anthracis IsdX1 (17) and IsdX2N5 (31) proteins. In these structures, both tyrosines in the motif are located on the β8 strand of the protein. The first tyrosine directly coordinates the metal in hemin, whereas the second tyrosine stabilizes the positioning of the coordinating tyrosine through side chain-side chain hydrogen bonding. Both of these interactions are important as tyrosine mutation significantly reduces affinity (37). Interestingly, our hemin binding measurements indicate that the central NEAT domain within Hbp2 (Hbp2N2) interacts with hemin with high affinity even though it lacks the second tyrosine in the YXXXY motif (in Hbp2N2 the motif is replaced with YXXXA). As part of our goal to understand the mechanism through which Hbp2 scavenges hemin, we determined the three-dimensional structure of Hbp2N2 in its free and hemin-bound states. The structure of the complex reveals a novel coordination mechanism in which Tyr280 located in strand β7 coordinates the iron instead of a canonical tyrosine residue used by all other previously characterized NEAT domains that is housed on the β8 strand (Fig. 1C). An overlay of the structures of the Hbp2N2-hemin and IsdC-hemin complexes reveals that the non-canonical coordinating tyrosine in Hbp2N2 (Tyr280) approaches the metal from a unique direction (Fig. 7C). In the Hbp2N2-hemin complex, Tyr280 located in strand β7 packs against pyrrole ring C, whereas in conventional hemin-binding NEAT domains typified by IsdC the coordinating tyrosine in strand β8 is positioned adjacent to pyrrole ring B that is located more distal to the 310 helix. As compared with the prototypical S. aureus IsdC-hemin complex, the hemin molecule bound to Hbp2N2 is tilted by ∼25°, which appears to increase its accessibility to solvent (43% of its surface area is exposed in Hbp2N2 as compared with 35% exposed in IsdC). Interestingly, despite originating from different β strands as compared with other NEAT domains, the coordinating tyrosine residue in Hbp2N2 is located a similar distance from the metal (the tyrosyl oxygen-Fe3+ distance is 2.2–2.4 Å), and the tyrosyl side chain packs against the porphyrin at a similar angle (∼60°). Like Hbp2N2, the primary sequences of several other NEAT domains lack the second tyrosine in the YXXXY motif and therefore may coordinate the metal in an unusual manner (e.g. B. anthracis Hal protein, the Clostridium perfringens CPE0221 protein, the Clostridium botulinum CLH3066 protein, and the first NEAT domain of the Shr protein from S. pyogenes). However, the mechanism through which they bind hemin remains to be determined. Interestingly, a distinct mode of metal ligation has recently been documented for the S. aureus IsdB protein, which unlike other NEAT domains contains a hexacoordinate heme iron that interacts with tyrosine and methionine ligands (29). The latter binding mechanism is generally similar to that used by the structurally unrelated HmuY protein from the Gram-negative bacterium Porphyromonas gingivalis as this β-sheet-containing protein coordinates the central iron atom in the bound hemin molecule using two histidine residues (54). Combined, these results highlight previously unrecognized plasticity in the hemin binding mechanism of NEAT domains.
Hbp2N2 undergoes a major conformation rearrangement upon binding hemin not seen previously in other NEAT domains. Structures of three NEAT domains have been determined in both their hemin-bound and free states (B. anthracis IsdX1 (17), S. aureus IsdA (30), and IsdHN3 from S. aureus (34)). In these structures, the proteins contain a preformed binding pocket for hemin, and only modest structural changes in the binding pocket occur upon ligand binding. IsdHN3 exhibits the largest change in its binding pocket when hemin binds as it closes slightly; the β7/β8 hairpin moves as a rigid unit ∼4 Å toward the 310 helix (34). NMR relaxation studies are also compatible with a similar rigid body closure motion occurring when the S. aureus IsdC protein binds hemin (33). In contrast, in Hbp2N2, residues forming the tip of the β7/β8 hairpin undergo a major conformational rearrangement upon binding hemin (Fig. 7, A and B). This key difference is caused by non-conservative changes in the primary sequence of Hbp2N2 that preclude the formation of many non-covalent interactions that stabilize the end of the β7/β8 hairpin in the structures of NEAT domains that contain a preformed binding pocket. In particular, in the structures of IsdA, IsdX1, and IsdHN3 determined in the absence of hemin, several residues at the tip of the β7/β8 hairpin form a network of stabilizing interactions. The side chains of the tyrosine residues in the YXXXY motif hydrogen bond with one another either directly or via a water molecule. The side chain of the coordinating tyrosine residue also forms non-polar interactions with residues that contain large hydrophobic side chains that are located in the β7 strand and the turn that connects the β7 and β8 strands. Collectively, these interactions may stabilize the β7/β8 hairpin portion of the pocket, holding it in a binding-competent configuration. In contrast, in Hbp2N2 non-conservative amino acids occur at these positions, precluding these stabilizing interactions. As a result, residues in the turn of the β7/β8 hairpin adopt a distinct, more flexible conformation in the absence of hemin. Notably, the β7/β8 hairpin forms a packing interface in the crystal of the complex, but these contacts are unlikely to cause the large observed differences in the hairpin conformation as the contacts are not present in the crystal of the apo form of the protein.
Interestingly, the asymmetric unit of the crystal of the Hbp2N2-hemin complex contains two copies of the complex in which the hemin binding pockets are juxtaposed (Fig. 7D). Hemin stacking interactions bridge the two proteins that are related by a 2-fold symmetry axis that is coplanar with their porphyrin rings. At the interface, hemin pockets approach one another such that the turn that is in the β7/β8 hairpin of one protein is adjacent to the 310 helix on the opposing protein. This general orientation may resemble the activated protein-protein complex through which Hbp2N2 rapidly transfers hemin to Hbp1, Hbp2N1, or Hbp2N3. Presumably, in the transfer complex, the single hemin molecule bound to Hbp2N2 is transferred when the β7/β8 hairpin in the accepting NEAT domain simultaneously contacts the face of the porphyrin ring (Fig. 7E). The specific mechanism of transfer remains to be elucidated, but the central metal has been proposed to be simultaneously coordinated by tyrosine residues from the donor and acceptor proteins (55, 56). Structurally similar complexes may mediate hemin transfer among other NEAT domains as our NMR studies of the IsdA-IsdC NEAT domain complex suggest that it adopts a related pseudosymmetric “hand clasp” structure that forms transiently (57). Moreover, kinetic measurements of other NEAT domains indicate that they transfer hemin rapidly among one another, and in many instances NEAT-hemin complexes have been shown to form similar crystal packing interactions (17, 31, 32, 34). Future work will be directed toward elucidating the molecular basis of hemin transfer, identifying other proteins in L. monocytogenes that facilitate hemin capture, and will seek to understand their relationship to disease. These experiments will shed light onto how pathogens acquire iron and may lead to the development of novel antimicrobial agents that work by inhibiting heme uptake.
Acknowledgments
We thank members of the Clubb laboratory for useful comments. We also thank Dr. John Olson for the generous gift of H64Y/V68F apomyoglobin.
This work was supported, in whole or in part, by National Institutes of Health R01 Grants AI52217 (to R. T. C.) and GM53836 (to P. E. K. and S. M. N.) and Grant F31GM101931 (to M. S.). This work was also supported by National Science Foundation Grant MCB0952299 (to P. E. K. and S. M. N.).
The atomic coordinates and structure factors (codes 4MYP and 4NLA) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- ABC
- ATP-binding cassette
- Hbp
- hemin/hemoglobin-binding protein
- Isd
- iron-regulated surface determinant
- NEAT
- NEAr transporter
- heme
- protoporphyrin IX + iron
- SUMO
- small ubiquitin-like modifier
- SvpA
- surface virulence-associated protein
- ITC
- isothermal titration calorimetry
- Mb
- myoglobin.
REFERENCES
- 1. Camejo A., Carvalho F., Reis O., Leitão E., Sousa S., Cabanes D. (2011) The arsenal of virulence factors deployed by Listeria monocytogenes to promote its cell infection cycle. Virulence 2, 379–394 [DOI] [PubMed] [Google Scholar]
- 2. Klebba P. E., Charbit A., Xiao Q., Jiang X., Newton S. M. (2012) Mechanisms of iron and haem transport by Listeria monocytogenes. Mol. Membr. Biol. 29, 69–86 [DOI] [PubMed] [Google Scholar]
- 3. Jin B., Newton S. M., Shao Y., Jiang X., Charbit A., Klebba P. E. (2006) Iron acquisition systems for ferric hydroxamates, haemin and haemoglobin in Listeria monocytogenes. Mol. Microbiol. 59, 1185–1198 [DOI] [PubMed] [Google Scholar]
- 4. Xiao Q., Jiang X., Moore K. J., Shao Y., Pi H., Dubail I., Charbit A., Newton S. M., Klebba P. E. (2011) Sortase independent and dependent systems for acquisition of haem and haemoglobin in Listeria monocytogenes. Mol. Microbiol. 80, 1581–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cassat J. E., Skaar E. P. (2013) Iron in infection and immunity. Cell Host Microbe 13, 509–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Borezée E., Pellegrini E., Beretti J. L., Berche P. (2001) SvpA, a novel surface virulence-associated protein required for intracellular survival of Listeria monocytogenes. Microbiology 147, 2913–2923 [DOI] [PubMed] [Google Scholar]
- 7. Mariscotti J. F., García-del Portillo F., Pucciarelli M. G. (2009) The Listeria monocytogenes sortase-B recognizes varied amino acids at position 2 of the sorting motif. J. Biol. Chem. 284, 6140–6146 [DOI] [PubMed] [Google Scholar]
- 8. Calvo E., Pucciarelli M. G., Bierne H., Cossart P., Albar J. P., García-Del Portillo F. (2005) Analysis of the Listeria cell wall proteome by two-dimensional nanoliquid chromatography coupled to mass spectrometry. Proteomics 5, 433–443 [DOI] [PubMed] [Google Scholar]
- 9. Mujahid S., Pechan T., Wang C. (2007) Improved solubilization of surface proteins from Listeria monocytogenes for 2-DE. Electrophoresis 28, 3998–4007 [DOI] [PubMed] [Google Scholar]
- 10. Bierne H., Garandeau C., Pucciarelli M. G., Sabet C., Newton S., Garcia-del Portillo F., Cossart P., Charbit A. (2004) Sortase B, a new class of sortase in Listeria monocytogenes. J. Bacteriol. 186, 1972–1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Senturia R., Faller M., Yin S., Loo J. A., Cascio D., Sawaya M. R., Hwang D., Clubb R. T., Guo F. (2010) Structure of the dimerization domain of DiGeorge critical region 8. Protein Sci. 19, 1354–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Spirig T., Malmirchegini G. R., Zhang J., Robson S. A., Sjodt M., Liu M., Krishna Kumar K., Dickson C. F., Gell D. A., Lei B., Loo J. A., Clubb R. T. (2013) Staphylococcus aureus uses a novel multidomain receptor to break apart human hemoglobin and steal its heme. J. Biol. Chem. 288, 1065–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ascoli F., Fanelli M. R., Antonini E. (1981) Preparation and properties of apohemoglobin and reconstituted hemoglobins. Methods Enzymol. 76, 72–87 [DOI] [PubMed] [Google Scholar]
- 14. Hargrove M. S., Singleton E. W., Quillin M. L., Ortiz L. A., Phillips G. N., Jr., Olson J. S., Mathews A. J. (1994) His64(E7) → Tyr apomyoglobin as a reagent for measuring rates of hemin dissociation. J. Biol. Chem. 269, 4207–4214 [DOI] [PubMed] [Google Scholar]
- 15. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Karplus P. A., Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ekworomadu M. T., Poor C. B., Owens C. P., Balderas M. A., Fabian M., Olson J. S., Murphy F., Bakkalbasi E., Honsa E. S., He C., Goulding C. W., Maresso A. W. (2012) Differential function of lip residues in the mechanism and biology of an anthrax hemophore. PLoS Pathog. 8, e1002559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 19. Blanc E., Roversi P., Vonrhein C., Flensburg C., Lea S. M., Bricogne G. (2004) Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr. D Biol. Crystallogr. 60, 2210–2221 [DOI] [PubMed] [Google Scholar]
- 20. Winn M. D., Murshudov G. N., Papiz M. Z. (2003) Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374, 300–321 [DOI] [PubMed] [Google Scholar]
- 21. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 24. Colovos C., Yeates T. O. (1993) Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 2, 1511–1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lüthy R., Bowie J. U., Eisenberg D. (1992) Assessment of protein models with three-dimensional profiles. Nature 356, 83–85 [DOI] [PubMed] [Google Scholar]
- 26. Hargrove M. S., Whitaker T., Olson J. S., Vali R. J., Mathews A. J. (1997) Quaternary structure regulates hemin dissociation from human hemoglobin. J. Biol. Chem. 272, 17385–17389 [DOI] [PubMed] [Google Scholar]
- 27. Dryla A., Gelbmann D., von Gabain A., Nagy E. (2003) Identification of a novel iron regulated staphylococcal surface protein with haptoglobin-haemoglobin binding activity. Mol. Microbiol. 49, 37–53 [DOI] [PubMed] [Google Scholar]
- 28. Dryla A., Hoffmann B., Gelbmann D., Giefing C., Hanner M., Meinke A., Anderson A. S., Koppensteiner W., Konrat R., von Gabain A., Nagy E. (2007) High-affinity binding of the staphylococcal HarA protein to haptoglobin and hemoglobin involves a domain with an antiparallel eight-stranded β-barrel fold. J. Bacteriol. 189, 254–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gaudin C. F., Grigg J. C., Arrieta A. L., Murphy M. E. (2011) Unique heme-iron coordination by the hemoglobin receptor IsdB of Staphylococcus aureus. Biochemistry 50, 5443–5452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Grigg J. C., Vermeiren C. L., Heinrichs D. E., Murphy M. E. (2007) Haem recognition by a Staphylococcus aureus NEAT domain. Mol. Microbiol. 63, 139–149 [DOI] [PubMed] [Google Scholar]
- 31. Honsa E. S., Owens C. P., Goulding C. W., Maresso A. W. (2013) The near-iron transporter (NEAT) domains of the anthrax hemophore IsdX2 require a critical glutamine to extract heme from methemoglobin. J. Biol. Chem. 288, 8479–8490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sharp K. H., Schneider S., Cockayne A., Paoli M. (2007) Crystal structure of the heme-IsdC complex, the central conduit of the Isd iron/heme uptake system in Staphylococcus aureus. J. Biol. Chem. 282, 10625–10631 [DOI] [PubMed] [Google Scholar]
- 33. Villareal V. A., Pilpa R. M., Robson S. A., Fadeev E. A., Clubb R. T. (2008) The IsdC protein from Staphylococcus aureus uses a flexible binding pocket to capture heme. J. Biol. Chem. 283, 31591–31600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Watanabe M., Tanaka Y., Suenaga A., Kuroda M., Yao M., Watanabe N., Arisaka F., Ohta T., Tanaka I., Tsumoto K. (2008) Structural basis for multimeric heme complexation through a specific protein-heme interaction: the case of the third neat domain of IsdH from Staphylococcus aureus. J. Biol. Chem. 283, 28649–28659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pilpa R. M., Fadeev E. A., Villareal V. A., Wong M. L., Phillips M., Clubb R. T. (2006) Solution structure of the NEAT (NEAr transporter) domain from IsdH/HarA: the human hemoglobin receptor in Staphylococcus aureus. J. Mol. Biol. 360, 435–447 [DOI] [PubMed] [Google Scholar]
- 36. Hendrickson W. A. (1991) Determination of macromolecular structures from anomalous diffraction of synchrotron radiation. Science 254, 51–58 [DOI] [PubMed] [Google Scholar]
- 37. Vu N. T., Moriwaki Y., Caaveiro J. M., Terada T., Tsutsumi H., Hamachi I., Shimizu K., Tsumoto K. (2013) Selective binding of antimicrobial porphyrins to the heme-receptor IsdH-NEAT3 of Staphylococcus aureus. Protein Sci. 22, 942–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wandersman C., Delepelaire P. (2012) Haemophore functions revisited. Mol. Microbiol. 85, 618–631 [DOI] [PubMed] [Google Scholar]
- 39. Braun V., Hantke K. (2011) Recent insights into iron import by bacteria. Curr. Opin. Chem. Biol. 15, 328–334 [DOI] [PubMed] [Google Scholar]
- 40. Moriwaki Y., Terada T., Caaveiro J. M., Takaoka Y., Hamachi I., Tsumoto K., Shimizu K. (2013) Heme binding mechanism of structurally similar iron-regulated surface determinant near transporter domains of Staphylococcus aureus exhibiting different affinities for heme. Biochemistry 52, 8866–8877 [DOI] [PubMed] [Google Scholar]
- 41. Hargrove M. S., Barrick D., Olson J. S. (1996) The association rate constant for heme binding to globin is independent of protein structure. Biochemistry 35, 11293–11299 [DOI] [PubMed] [Google Scholar]
- 42. Abe R., Caaveiro J. M., Kozuka-Hata H., Oyama M., Tsumoto K. (2012) Mapping ultra-weak protein-protein interactions between heme transporters of Staphylococcus aureus. J. Biol. Chem. 287, 16477–16487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grigg J. C., Vermeiren C. L., Heinrichs D. E., Murphy M. E. (2007) Heme coordination by Staphylococcus aureus IsdE. J. Biol. Chem. 282, 28815–28822 [DOI] [PubMed] [Google Scholar]
- 44. McLaughlin H. P., Xiao Q., Rea R. B., Pi H., Casey P. G., Darby T., Charbit A., Sleator R. D., Joyce S. A., Cowart R. E., Hill C., Klebba P. E., Gahan C. G. (2012) A putative P-type ATPase required for virulence and resistance to haem toxicity in Listeria monocytogenes. PLoS One 7, e30928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hood M. I., Skaar E. P. (2012) Nutritional immunity: transition metals at the pathogen-host interface. Nat. Rev. Microbiol. 10, 525–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nobles C. L., Maresso A. W. (2011) The theft of host heme by Gram-positive pathogenic bacteria. Metallomics 3, 788–796 [DOI] [PubMed] [Google Scholar]
- 47. Maresso A. W., Garufi G., Schneewind O. (2008) Bacillus anthracis secretes proteins that mediate heme acquisition from hemoglobin. PLoS Pathog. 4, e1000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Krishna Kumar K., Jacques D. A., Pishchany G., Caradoc-Davies T., Spirig T., Malmirchegini G. R., Langley D. B., Dickson C. F., Mackay J. P., Clubb R. T., Skaar E. P., Guss J. M., Gell D. A. (2011) Structural basis for hemoglobin capture by Staphylococcus aureus cell-surface protein, IsdH. J. Biol. Chem. 286, 38439–38447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pilpa R. M., Robson S. A., Villareal V. A., Wong M. L., Phillips M., Clubb R. T. (2009) Functionally distinct NEAT (NEAr transporter) domains within the Staphylococcus aureus IsdH/HarA protein extract heme from methemoglobin. J. Biol. Chem. 284, 1166–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dickson C. F., Kumar K. K., Jacques D. A., Malmirchegini G. R., Spirig T., Mackay J. P., Clubb R. T., Guss J. M., Gell D. A. (2014) Structure of the hemoglobin-IsdH complex reveals the molecular basis of iron capture by Staphylococcus aureus. J. Biol. Chem. 289, 6728–6738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tarlovsky Y., Fabian M., Solomaha E., Honsa E., Olson J. S., Maresso A. W. (2010) A Bacillus anthracis S-layer homology protein that binds heme and mediates heme delivery to IsdC. J. Bacteriol. 192, 3503–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Honsa E. S., Fabian M., Cardenas A. M., Olson J. S., Maresso A. W. (2011) The five near-iron transporter (NEAT) domain anthrax hemophore, IsdX2, scavenges heme from hemoglobin and transfers heme to the surface protein IsdC. J. Biol. Chem. 286, 33652–33660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fabian M., Solomaha E., Olson J. S., Maresso A. W. (2009) Heme transfer to the bacterial cell envelope occurs via a secreted hemophore in the Gram-positive pathogen Bacillus anthracis. J. Biol. Chem. 284, 32138–32146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wójtowicz H., Guevara T., Tallant C., Olczak M., Sroka A., Potempa J., Solà M., Olczak T., Gomis-Rüth F. X. (2009) Unique structure and stability of HmuY, a novel heme-binding protein of Porphyromonas gingivalis. PLoS Pathog. 5, e1000419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Grigg J. C., Mao C. X., Murphy M. E. (2011) Iron-coordinating tyrosine is a key determinant of NEAT domain heme transfer. J. Mol. Biol. 413, 684–698 [DOI] [PubMed] [Google Scholar]
- 56. Ran Y., Malmirchegini G. R., Clubb R. T., Lei B. (2013) Axial ligand replacement mechanism in heme transfer from streptococcal heme-binding protein Shp to HtsA of the HtsABC transporter. Biochemistry 52, 6537–6547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Villareal V. A., Spirig T., Robson S. A., Liu M., Lei B., Clubb R. T. (2011) Transient weak protein-protein complexes transfer heme across the cell wall of Staphylococcus aureus. J. Am. Chem. Soc. 133, 14176–14179 [DOI] [PMC free article] [PubMed] [Google Scholar]