

Background: Secreted antibacterial substances of fungi provide a rich source for new antibiotics.
Results: Copsin is a novel fungal antimicrobial peptide that binds in a unique manner to the cell wall precursor lipid II.
Conclusion: As part of the defense strategy of a mushroom, copsin kills bacteria by inhibiting the cell wall synthesis.
Significance: Copsin provides a novel highly stabilized scaffold for antibiotics.
Keywords: Antibiotic Resistance, Antibiotics, Antimicrobial Peptide (AMP), Fungi, Lipid II, Bacterial-Fungal Interaction (BFI), Fungal Secretome
Abstract
Fungi and bacteria compete with an arsenal of secreted molecules for their ecological niche. This repertoire represents a rich and inexhaustible source for antibiotics and fungicides. Antimicrobial peptides are an emerging class of fungal defense molecules that are promising candidates for pharmaceutical applications. Based on a co-cultivation system, we studied the interaction of the coprophilous basidiomycete Coprinopsis cinerea with different bacterial species and identified a novel defensin, copsin. The polypeptide was recombinantly produced in Pichia pastoris, and the three-dimensional structure was solved by NMR. The cysteine stabilized α/β-fold with a unique disulfide connectivity, and an N-terminal pyroglutamate rendered copsin extremely stable against high temperatures and protease digestion. Copsin was bactericidal against a diversity of Gram-positive bacteria, including human pathogens such as Enterococcus faecium and Listeria monocytogenes. Characterization of the antibacterial activity revealed that copsin bound specifically to the peptidoglycan precursor lipid II and therefore interfered with the cell wall biosynthesis. In particular, and unlike lantibiotics and other defensins, the third position of the lipid II pentapeptide is essential for effective copsin binding. The unique structural properties of copsin make it a possible scaffold for new antibiotics.
Introduction
Fungi and bacteria co-exist in a variety of environments, where fungi directly compete with heterotrophic bacteria. One of the best studied types of bacterial-fungal interactions is the antibiosis, where secreted substances play a key role in combating other microorganisms to defend a nutritional niche (1, 2). Secondary metabolites are the best characterized group of defense molecules, with the most prominent member being penicillin from the Penicillium chrysogenum mold (3). Various other metabolites discovered in studies of bacterial-fungal interactions have pharmaceutical applications, emphasizing the importance of gaining knowledge on molecular mechanisms of bacterial-fungal interactions.
Antimicrobial peptides (AMPs)2 are a class of defense molecules that participate in competing bacteria and other microbes as a part of an innate immune system. AMPs are a large and highly diverse group of low molecular mass proteins (<10 kDa), produced by both prokaryotes and eukaryotes. They are broadly classified according to their structure and amino acid sequence. Often, they are characterized by an amphipathic composition resulting in hydrophobic and cationic clusters (4). Plectasin was the first fungal defensin identified in the secretome of the ascomycete Pseudoplectania nigrella (5). It shows high similarity to plant and insect defensins with a core structural motif of a cysteine stabilized α/β-fold (CSαβ). Plectasin acts bactericidal by binding to the lipid II precursor and by inhibiting the peptidoglycan synthesis of predominantly Gram-positive bacteria (6). The cell wall biosynthetic pathways are a very effective target for antibacterial substances, as shown for a number of clinically applied antibiotics. The far biggest group of these drugs are the β-lactams, which inhibit the transpeptidation step of the peptidoglycan layer (7). In an era of increasing bacterial resistance against many commercially available antibiotics, AMPs are considered promising candidates for a new generation of antibiotics (8).
Here, we studied the interaction of the basidiomycete Coprinopsis cinerea with different bacterial species. To model the lifestyle of this coprophilous fungus, a system was developed, where the fungus was grown on medium-submerged glass beads. This method allowed for a co-cultivation with different bacterial species and made it possible to analyze the secretome of both fungus and bacteria.
The analysis of secreted proteins revealed that C. cinerea secreted AMPs that acted as a defense line against bacteria. One of these peptides, hereafter called copsin, was characterized further at a molecular and structural level, and its mode of action was determined both in vitro and in vivo.
EXPERIMENTAL PROCEDURES
Chemicals and Fungal Strains
The C. cinerea strain AmutBmut (A43mut B43mut pab1.2) was used for all experiments involving the fungus. All chemicals, if not otherwise mentioned, were bought at the highest available purity from Sigma-Aldrich.
C. cinerea in Competition with Bacteria on Glass Beads
Preparation of the glass bead plates was adapted from a previously published protocol (9). In brief, 40 g of sterile borosilicate glass beads (5 mm) were poured into a Petri dish, and 15 ml of C. cinerea minimal medium (CCMM) was added (composition per liter medium: 5 g of glucose, 2 g of asparagine, 50 mg of adenine sulfate, 1 g of KH2PO4, 2.3 g of Na2HPO4 (anhydrous), 0.3 g of Na2SO4, 0.5 g of C4H12N2O6, 40 μg of thiamine-HCl, 0.25 g of MgSO4·7H2O, and 5 mg of p-aminobenzoic acid).
C. cinerea was grown on 1.5% (w/v) agar plates containing YMG (0.4% (w/v) yeast extract (Oxoid, Basingstoke, UK), 1% (w/v) malt extract (Oxoid), 0.4% (w/v) glucose) for 4 days at 37 °C in the dark. Afterward, two mycelial plugs were cut from the margin of the mycelium and transferred to the center of a glass bead plate in a distance of 1 cm. After 60 h of letting the fungus grow on the beads (37 °C, dark), 4 ml of the medium underneath the fungus was replaced by 4 ml of a bacterial suspension (Bacillus subtilis 168, Pseudomonas aeruginosa 01, or Escherichia coli BL21) grown previously in CCMM to an A600 of 0.2. Both organisms were grown in competition for 48 h (37 °C, dark). For the fungal growth control, the medium was replaced by the same amount of CCMM (4 ml). As control for the bacterial growth, the three bacterial strains were grown in the beads system independently for 48 h. Growth of the bacteria was monitored by measuring the A600 of the medium.
Purification of AMPs from Fungal Secretome
C. cinerea was grown in a glass bead plate (19-cm diameter) with 200 g of glass beads and 80 ml of CCMM for 5 days (at 37 °C in the dark). Subsequently, the medium was extracted and concentrated to 4 ml by lyophilization. The proteins were then precipitated with 2.2 m ammonium sulfate at 4 °C for 30 min. After centrifugation at 20,000 × g for 15 min, the pellet was dissolved in PBS, pH 7.4, and the proteins were digested with 100 ng/μl proteinase K (Roche) at 60 °C for 2 h. The solution was loaded in a 10-kDa molecular mass cutoff dialysis cassette (Thermo Scientific, Waltham, MA) and dialyzed against PBS, pH 7.4, at 4 °C for 24 h. The remaining proteins were applied to a Resource S cation exchange column (GE Healthcare) and eluted with a 100–600 mm NaCl gradient in 50 mm sodium phosphate buffer, pH 7.4. 0.5-ml fractions were collected, and the effluent monitored by absorbance at 280 nm on an ÄKTA purifier 10 system (GE Healthcare). The antibacterial activity of each fraction was tested in a disk diffusion assay. Therefore, an overnight B. subtilis culture grown in LB-Lennox medium was diluted in 8 ml of prewarmed (45 °C) water agar (1%) to an A600 of 0.2 and poured on a LB agar plate. 40 μl of each fraction was loaded on a paper disk (Oxoid) placed on the solidified bacterial lawn. The diameter of the inhibition zone was determined after incubation at 37 °C for 24 h. The flow through and fractions showing activity were subjected to a reduction with 10 mm DTT at 37 °C for 45 min and alkylation with 40 mm iodoacetamide at 25 °C for 30 min in the dark. A proteolytic digest was performed with 0.25 μg/ml trypsin (Promega, Madison, WI) for 16 h at 37 °C, and the peptides were desalted on C18 ZipTip columns (Millipore, Billerica, MA). The LC-MS analysis was performed on a hybrid Velos LTQ Orbitrap mass spectrometer (Thermo Scientific) coupled to an Eksigent-nano-HPLC system (Eksigent Technologies, Framingham, MA). Separation of peptides was done on a self-made column (75 μm × 80 mm) packed with C18 AQ 3 μm resin (Bischoff GmbH, Essen, Germany). Peptides were eluted with a linear gradient from 2 to 31% acetonitrile in 53 min at a flow rate of 250 nl/min. MS and MS/MS spectra were acquired in the data-dependent mode with up to 20 collision induced dissociation spectra recorded in the ion trap using the most intense ions. All MS/MS spectra were searched against the p354_filteredMod_d C. cinerea database using the Mascot search algorithm v2.3 (Matrix Science, Boston, MA) with oxidation (methionine) as variable modification and carbamidomethyl (cysteine) as fixed modification. Further statistical validation was performed with Scaffold 4.0 (Proteome Software, Portland, OR) with a minimum protein probability of 90% and a minimum peptide probability of 50%. This program was also used for determining the total non-normalized spectral counts for a protein identified in the fractions active against B. subtilis and in the flow through.
cDNA Synthesis and Cloning of the Copsin Precursor
To extract RNA, 20 mg of lyophilized mycelium was lysed with 25 mg of 0.5-mm glass beads in three FastPrep steps, cooling the sample for 5 min on ice between each step. RNA was extracted with 1 ml of Qiazol (Qiagen) and 0.2 ml of chloroform. After a centrifugation at 12,000 × g at 4 °C for 15 min, RNA was recovered in the aqueous phase, washed on-column using the RNeasy Lipid Tissue Mini Kit (Qiagen), and eluted in RNase-free water. cDNA was synthesized from 2 μg of extracted RNA using the Transcriptor Universal cDNA Master kit (Roche) following the manufacturer's instructions.
The coding sequences of the copsin precursor protein were amplified from cDNA by PCR with the Phusion high fidelity DNA polymerase (Thermo Scientific) according to standard protocols (49): forward primer, 5′-CCGGAATTCATGAAACTTTCTACTTCTTTGCTCG-3′, and reverse primer, 5′-ACGCGTCGACTTAACAACGAGGGCAGGGG-3′.
The PCR product was cloned into the pPICZA expression plasmid (Invitrogen) containing a Zeocin resistance gene using the EcoRI and SalI (Thermo Scientific) restriction sites. The resulting plasmid was linearized by the SacI restriction enzyme and transformed into Pichia pastoris (strain NRRLY11430) by electroporation with 1.2 kV of charging voltage, 25 microfarad of capacitance, and 129 Ω of resistance (10). Positive clones were selected on YPD plates (1% (w/v) yeast extract, 2% (w/v) peptone, 2% (w/v) glucose, 2% (w/v) agar) containing 100 μg/ml Zeocin (LabForce, Muttenz, Switzerland).
Production and Purification of Recombinant Copsin
P. pastoris transformants were inoculated in BMGY medium (1% (w/v) yeast extract, 2% (w/v) peptone, 1.3% (w/v) YNB without amino acids (BD Biosciences), 100 mm potassium phosphate buffer, pH 6, 1% (v/v) glycerol) and cultured at 30 °C for 48 h. The cells were harvested by centrifugation at 3000 × g for 10 min; resuspended in P. pastoris minimal medium (1.3% (w/v) YNB without amino acids, 100 mm potassium phosphate buffer, pH 6, 0.4 μg/ml biotin, 0.5% (w/v) NH4Cl, 1% (v/v) MeOH); and cultured at 30 °C for 72 h. Methanol was added to 1% (v/v) in a time interval of 12 h, and NH4Cl was added to 0.5% (w/v) in a time interval of 24 h.
The culture broth was centrifuged at 3000 × g for 10 min, and the supernatant was concentrated in a 3.5-kDa Spectra/Por dialysis membrane (Spectrum Laboratories, Rancho Dominguez, CA) by a treatment with polyethylene glycol 6000 at 4 °C. The concentrated supernatant was dialyzed against 20 mm sodium phosphate, 50 mm NaCl buffer, pH 7 (buffer A), at 4 °C for 24 h. The protein solution was sterile-filtered and loaded on a self-made SP Sephadex cation exchange column equilibrated with buffer A. The column was washed with 180 mm NaCl, and bound proteins were eluted with 400 mm NaCl in 20 mm sodium phosphate buffer, pH 7. The eluent was subjected to a size exclusion chromatography for further polishing. The separation was performed on a Superdex75 column (HiLoad 16/60; GE Healthcare) equilibrated with 20 mm sodium phosphate, 50 mm NaCl buffer, pH 6. The effluent was monitored by absorbance at 210 nm. The fractions containing copsin were combined, and the molecular mass was determined by electrospray ionization-MS.
Production of 15N/13C-labeled Copsin in P. pastoris
The production of isotope-labeled copsin was adapted from a previously published protocol (11). In brief, P. pastoris transformants were inoculated in minimal medium supplemented with 0.5% (w/v) 15NH4Cl (98 atom % 15N) and 13C3-glycerol (99 atom % 13C), respectively, and cultivated in shake flasks at 30 °C to an A600 of ∼35. After centrifugation at 3000 × g for 10 min, the cell pellet was dissolved in P. pastoris minimal medium containing 0.5% (w/v) 15NH4Cl (98 atom % 15N) and [13C]methanol (99 atom % 13C). The culturing was performed at 30 °C for 3 days. [13C]methanol was added to 1% (v/v) in a time interval of 12 h, and 15NH4Cl was added to 0.5% (w/v) in a time interval of 24 h. Purification of 15N/13C copsin was performed as described for the unlabeled product.
NMR Preparation of NMR Samples
Uniformly 15N/13C isotope-labeled copsin in 20 mm sodium phosphate, 50 mm NaCl buffer was prepared as described above.
NMR Spectroscopy
Sequence specific resonance assignments were obtained using a set of two-dimensional [15N,1H] and [13C,1H] heteronuclear single quantum coherence, constant time [13C,1H] heteronuclear single quantum coherence, [1H,1H] total correlation spectroscopy (mixing time τm = 80 ms), exclusive correlation spectroscopy, [1H,1H] NOESY (τm = 40 ms), three-dimensional HN(CO)CA, and 15N-resolved [1H,1H] total correlation spectroscopy (τm = 40 ms) spectra recorded on Bruker Avance III 500-, 600-, 700-, and 900-MHz NMR spectrometers at a temperature of 293 K. The data were processed with TopSpin 3.0 (Bruker, Billerica, MA) and analyzed with CARA 1.8.4. The protonation state of histidine residues was determined by recording a [15N,1H] heteronuclear multiple-quantum correlation spectroscopy spectrum with a long (22 ms) INEPT transfer period (12). Proline residues were assigned to cis and trans conformations based on chemical shifts of 13Cβ and 13Cγ nuclei (13).
Structure Calculation
Structure calculation was performed using two-dimensional [1H,1H] NOESY measured in H2O and D2O solutions and three-dimensional 13C,15N-resolved [1H,1H] NOESY spectra (τm = 40 and 60 ms), recorded on Bruker Avance 700- and 900-MHz spectrometers. Eight hydrogen bonds, identified based on correlations in long range HNCO spectra (14), were included as restraints. The NOEs were manually picked; the resulting peak list, the amino acid sequence, and the NOESY spectra were used as input for a structure calculation with the software CYANA 3.0 (15), using the simulated annealing protocol. In each of the seven cycles, 800 structures were calculated. The 40 structures with the lowest target function were then selected and used to calculate the final structure. At the outset of the structure calculation, no assumptions on disulfide bond topology were used, and the cysteines were defined as negative charged without a Hγ atom. After the identification of the disulfide bonds (see next paragraph), the final structure calculation was performed, and the best 20 of 200 structures were subjected to constrained energy minimization using the software AMBER (16). The nonstandard N-terminal amino acid pyroglutamate was added to the library of AMBER. The determination of the secondary structure elements was based on the 20 final structures using the definitions of PyMOL. All 14 amide protons in the corresponding hydrogen bonds exchange slowly in D2O (Δt > 10 min), and eight were contained in the long range HNCO experiment (see above). TALOS+ and PROCHECK determined a slightly higher content of secondary structure but contained the ones from PyMOL.
NMR, Identification of Disulfide Bonds
In addition to the visual analysis of structures calculated without disulfide bonds, three established methods for the determination of disulfide bonds based on NMR data were used: (a) ambiguous intersulfur restraints (17), (b) probability of a certain disulfide bond ensemble (18), and (c) the averaged target function (19). (a) The cysteines were forced to form disulfide bonds by the CYANA algorithm using ambiguous distance restraints, for every cysteine, between one specific cysteine and all other cysteines (15). Clustering of at least two sulfur atoms together satisfies the restraint. Standard distances between the sulfur and carbon atoms Sγ-Sγ (upper/lower limit, 2.1/2.0 Å), Sγ-Cβ (3.1/3.0 Å), and Cβ-Sγ (3.1/3.0 Å) were used. The topology of the disulfide bond of the best 40 structures selected on the basis of their energy was analyzed. (b) An initial set of 800 conformers was calculated with no explicit constraint for disulfide bonds and the sulfur Hγ atoms removed. Subsequently the interatomic distances between all cysteine Cβ atoms were extracted and averaged over the 40 final lowest energy structures. Based on the initial structure ensemble, all conceivable disulfide patterns were formed (10–40; 18–48; 22–50; 35–54; 3–32; 25–57/10–40; 18–48; 22–35; 50–54; 3–32; 25–57/10–40; 18–48; 22–54; 35–50; 3–32; 25–57/10–48; 18–40; 22–50; 35–54; 3–32; 25–57/10–48; 18–40; 22–35; 50–54; 3–32; 25–57/10–48; 18–40; 22–54; 35–50; 3–32; 25–57/10–18; 48–40; 22–50; 35–54; 3–32; 25–57/10–18; 48–40; 22–35; 50–54; 3–32; 25–57/10–18; 48–40; 22–54; 35–50; 3–32; 25–57/10–40; 18–22; 48–50; 35–54; 3–32; 25–57/10–40; 18–50; 48–22; 35–54; 3–32; 25–57/10–40; 18–48; 22–50; 35–54; 3–25; 32–57/10–40; 18–48; 22–50; 35–54; 3–57; and 32–25), and a weighting factor representing the probability of a specific disulfide bond topology was then calculated for all these possibilities (Table 1). (c) The averaged target function of the 40 best CYANA structures was compared for conceivable fixed disulfide bond connectivities, and the number of violations was analyzed (Table 1).
TABLE 1.
Elucidation of disulfide bonds by target function and intercysteine distances analysis
Characterization of bundles of the 40 best conformers of copsin with different disulfide topologies. The disulfide pattern of the final structure (Fig. 3B) is represented in the top row of the first column for the two methods. Different other conceivable disulfide patterns are listed on following lines, and deviations in cysteine connectivities from the favored one are indicated in bold. Target function, amount of violations, and weights based on averaged Cβ–Cβ distances are shown for the different cysteine pairings. Best target function and weight factors were obtained for the cysteine pairings denoted in the first row (see footnote e).
| Disulfide bond pattern | Target functiona | Violationsa (distancesb/VdWc) | Weight factord |
|---|---|---|---|
| 10–40; 18–48; 22–50; 35–54; 3–32; 25–57e | 0.14 | 0/1 | 0.0648 |
| 10–40; 18–48; 22–35; 50–54; 3–32; 25–57 | 0.18 | 0/1 | 0.0283 |
| 10–40; 18–48; 22–54; 35–50; 3–32; 25–57 | 0.60 | 5/1 | 5.3654e-7 |
| 10–48; 18–40; 22–50; 35–54; 3–32; 25–57 | 0.16 | 0/1 | 1.3644e-4 |
| 10–48; 18–40; 22–35; 50–54; 3–32; 25–57 | 0.20 | 0/1 | 5.9653e-4 |
| 10–48; 18–40; 22–54; 35–50; 3–32; 25–57 | 0.95 | 6/2 | 1.1294e-9 |
| 10–18; 48–40; 22–50; 35–54; 3–32; 25–57 | 0.15 | 0/1 | 0.0044 |
| 10–18; 48–40; 22–35; 50–54; 3–32; 25–57 | 0.22 | 0/1 | 0.0019 |
| 10–18; 48–40; 22–54; 35–50; 3–32; 25–57 | 0.82 | 8/1 | 3.6487e-8 |
| 10–40; 18–22; 48–50; 35–54; 3–32; 25–57 | 0.54 | 3/1 | 1.7643e-5 |
| 10–40; 18–50; 48–22; 35–54; 3–32; 25–57 | 1.29 | 9/2 | 3.1976e-10 |
| 10–40; 18–48; 22–50; 35–54; 3–25; 32–57 | 0.15 | 0/1 | 0.0091 |
| 10–40; 18–48; 22–50; 35–54; 3–57; 32–25 | 0.30 | 5/1 | 7.5591e-5 |
a In target function and violation analysis, for every topology a structure calculation was performed using CYANA (15). The disulfides were given as fixed constraints determined by the standard three upper and lower distance limits (see text). Target function and violations are shown averaged over the 40 selected best energy structures.
b Distance violations.
c van der Waals (VdW) violations.
d Characterization of disulfide bond patterns by cysteine Cβ–Cβ distance analysis as previously described (18). Averaged distances were extracted from the structure ensemble generated without any disulfide constraints. Subsequently weights were assigned to every disulfide pattern. The weight is directly proportional to the likelihood of certain disulfide bond patterns to be realized in the final structure.
e Disulfide bonding connectivity of the final structure as described under “Results.” Variations of single disulfide bonds from this assignment are shown in bold in the first column.
Effect of Temperature, pH, and Proteases on Copsin Activity
The pH stability of copsin was determined after incubation for 1 h at room temperature in a pH gradient of buffered solutions, including 250 mm KCl/HCl buffer (pH 2), 100 mm sodium acetate buffer (pH 4 and 5), 250 mm sodium phosphate buffer (pH 6), and 250 mm HEPES buffer (pH 8). As a control, copsin form stock solution in 20 mm sodium phosphate, 50 mm NaCl buffer, pH 6, was loaded on one disk. Thermal stability was tested in PBS buffer, pH 7.4, at 4, 25, 50, 70, and 90 °C for 1 h of incubation.
The effect of proteases such as pepsin, trypsin, and proteinase K on the activity of copsin was investigated by incubation with respective proteases at 37 °C for 3 h in a reaction mixture containing a ratio of 1:10 (w/w) of protease to copsin. For pepsin, the reaction was performed in 250 mm KCl/HCl buffer, pH 2, and for trypsin, proteinase K, and the control without any protease in 100 mm Tris-HCl buffer, pH 8. Copsin was also incubated in 5 mm DTT at pH 8. To ensure that the proteases and buffers themselves do not contribute to any inhibition, each of the proteases alone in the corresponding buffer was used a control. After incubation, the samples were centrifuged at 12,000 × g, and the antibacterial activity of the supernatant was tested by a disk diffusion assay on B. subtilis.
Antimicrobial Activity
The minimal inhibitory concentrations (MICs) and minimal bactericidal concentrations (MBCs) were determined by the microdilution broth method (20). In brief, bacteria were grown to an A600 of 0.1–0.2 in Mueller-Hinton broth (MHB; BD Biosciences). The bacterial suspension was diluted to 105–106 cfu/ml and added to a 2-fold dilution series (0.1–64 μg/ml) of copsin in a 96-well microtiter plate (Enzyscreen, Haarlem, The Netherlands). The plates were incubated at 37 °C for 20–24 h. For all bacteria, the assays were performed in MHB pH 7.3. For Listeria spp. and Corynebacterium diphtheriae, MHB was supplemented with 3% laked horse blood (Oxoid). For B. subtilis and Staphylococcus carnosus, the assay was additionally performed in MHB buffered with 50 mm sodium phosphate, pH 6. The cfu/ml was determined by plating serial dilutions on LB agar plates. The MIC is defined as the lowest concentration of copsin, where no visible growth was observed. The MBC is defined as the concentration of polypeptide that killed 99.9% of bacteria after 20–24 h of incubation. All determinations were performed at least in duplicate.
Killing Kinetics
B. subtilis was grown in MHB to an A600 of 0.2 and diluted to an A600 of 0.1 in MHB supplemented with 50 mm sodium phosphate buffer adjusted to pH 6 or 7.3. Copsin was added to 4 μg/ml (4× MIC), and the cultures were incubated at 37 °C for 5 h. The cfu/ml were determined in intervals of 30 min by plating serial dilutions on LB agar plates. The A600 was measured at 0, 2, and 5 h.
Light Microscopy
For TAMRA labeling, copsin was dissolved in 1 m NaHCO3 buffer, pH 8.0, to 1.6 mg/ml. TAMRA (Invitrogen) was added to a final concentration of 1 mg/ml and incubated at room temperature for 1 h. The excess dye was removed by passing the solution through a prepacked Sephadex G-25 M PD-10 desalting column (GE Healthcare) in 20 mm sodium phosphate, 50 mm NaCl buffer, pH 6.0.
Bacteria were grown in LB medium to an A600 of 0.3 and incubated with TAMRA-copsin at 0.5× MIC (0.5 μg/ml) for B. subtilis and 2× MIC (16 μg/ml) for S. carnosus for 10 min. The cells were washed twice with PBS and then immobilized on a poly-l-lysine-coated coverslip. After washing away the unbound cells with sterile water, BODIPY-vancomycin (Invitrogen) was used at 1 μg/ml concentration for staining on the coverslip. The cells were observed under a 100× oil immersion objective (Zeiss, Oberkochen, Germany) on a spinning disk confocal microscope (Visitron, Puchheim, Germany) and imaged with an Evolve 512 EMCCD camera (Photometrics, Tucson, AZ) using standard filter sets for GFP (excitation/emission, 488/525 nm) and rhodamine (540/575 nm). Images were processed using Image J v1.46.
Binding of Copsin to Cell Wall Precursors
2 nmol of each purified C55P, lipid I, lipid II (21), or lipid III (22) were incubated in the presence of increasing molar copsin concentrations from 1:0.5–1:4 (lipid:copsin precursor) in a total volume of 30 μl. After incubation for 20 min at 25 °C, the mixture was extracted with 2:1 (v/v) n-butanol/pyridine acetate (pH 4.2), analyzed by TLC as described earlier (23, 24). Analysis was carried out by phorbol phosphomolybdic acid staining.
Synthesis of UDP-MurNAc Peptides by Staphylococcus aureus MurA-F Enzymes
UDP-MurNAc-pp was synthesized as described (6) with modifications. UDP-GlcNAc (100 nmol) was incubated with 15 μg of each of the recombinant histidine-tagged muropeptide synthetases MurA to MurF in 50 mm Bis-Tris propane, pH 8, 25 mm (NH4)2SO4, 5 mm MgCl2, 5 mm KCl, 0.5 mm DTT, 2 mm ATP, 2 mm phosphoenolpyruvate, 2 mm NADPH, 1 mm of each amino acid (l-Ala, d-Glu, l-Lys, and d-Ala), 10% Me2SO in a total volume of 100 μl for 120 min at 30 °C. 33 μl of the reaction mixture, corresponding to 25 nmol of UDP-MurNAc-PP, were used in a MraY synthesis assay without further purification (23). UDP-MurNAc-peptide variants with shortened stem peptide, i.e. UDP-MurNAc-dipeptide and -tripeptide, were synthesized in the presence of the corresponding subset of muropeptide synthetases and were used for the synthesis of lipid I (dipeptide) and lipid I (tripeptide), respectively. After synthesis, lipid intermediates were incubated at room temperature with increasing molar ratios of copsin for 20 min. TLC analysis was performed as described above.
Carboxyfluorescein (CF) Efflux from Lipid II Containing Liposomes
Large unilamellar vesicles were prepared by the extrusion technique, essentially as previously described (25). Vesicles were made of 1,2-dioleoyl-sn-glycero-3-phosphocholine supplemented with 0.5 mol % lipid II (referring to the total amount of phospholipid). CF-loaded vesicles were prepared with 50 mm CF and then diluted in 1.5 ml of buffer (50 mm MES-KOH, 100 mm K2SO4, pH 6.0) at a final concentration of 25 μm phospholipid on a phosphorous base. After addition of 1 μm peptide (nisin or copsin), the increase of fluorescence intensity was measured at 520 nm (excitation at 492 nm) on an RF-5301 spectrophotometer (Shimadzu, Kyoto, Japan) at room temperature. Leakage was documented relative to the total amount of marker release after solubilization of the vesicles by addition of 10 μl of 20% Triton X-100.
RESULTS
Interaction of C. cinerea and Bacteria
Dung of herbivores, the natural substrate of C. cinerea, is a rather complex environment. The fungus grows on a solid substrate in high humidity and is confronted with a complex diversity of bacteria and other microbes (26). To mimic this substrate and to study the interactions of C. cinerea with bacteria, an artificial system was applied in which the fungus was grown on glass beads submerged in liquid medium (9). Because the fungal mycelium was kept in place by the beads layer, the medium supporting fungal growth could be replaced and also allowed for the co-cultivation with either B. subtilis (strain 168), P. aeruginosa (strain PA01), or E. coli (strain BL21). In addition, the growth of both organisms in competition could be monitored easily (Fig. 1).
FIGURE 1.

Interactions between C. cinerea and bacteria. A, vegetative mycelium of C. cinerea was grown on submerged glass beads in minimal medium. After 60 h, 4 ml of medium was replaced by a suspension of B. subtilis, E. coli, or P. aeruginosa. 0 and 48 h after addition of the bacteria, the plates were photographed. Fungal growth is indicated by the white mycelium. B, bacterial growth was monitored by measuring A600 (OD600) over 48 h. As controls, bacteria and fungus were grown independently in the beads system. All data points were acquired in three biological replicates and are displayed with the standard deviation. C. cinerea exhibited a bactericidal effect in competition with B. subtilis and was strongly inhibited by P. aeruginosa.
The Gram-negative bacteria P. aeruginosa and E. coli had an inhibitory effect on the growth of C. cinerea (Fig. 1A). B. subtilis, the Gram-positive species tested, did not affect the expansion of the fungal mycelium, but C. cinerea had an inhibitory impact on B. subtilis (Fig. 1B). P. aeruginosa was not affected at the beginning of the co-cultivation, but the culture reached the stationary phase much earlier than the fungal-free control. The growth of E. coli did not display any dependence on C. cinerea. Our results showed that C. cinerea was indeed interacting in a species-specific way with bacteria. In particular, we noticed an antagonistic behavior in competition with B. subtilis and P. aeruginosa.
Identification and Recombinant Production of Secreted AMPs
The co-cultivation experiments suggested a secreted fungal substance with a negative effect on B. subtilis growth (Fig. 1B). To examine whether the fungus in axenic culture secretes an antibacterial substance, unchallenged C. cinerea was grown in the glass beads system in minimal medium. After 5 days, the medium was collected and concentrated, and an antibacterial activity was shown in a disk diffusion assay on B. subtilis. To identify putative AMPs involved in this inhibition, proteins were precipitated with ammonium sulfate and digested with proteinase K at 60 °C. The remaining proteins were separated on a cation exchange column and evaluated according to their activity against B. subtilis in a disk diffusion assay (Fig. 2, A and B). Fractions that displayed an inhibition zone against B. subtilis were treated with reducing agent followed by a tryptic digest and subjected to a MS measurement. Five C. cinerea-derived proteins were identified in the active fractions. These proteins were quantified by spectral counting, and the relative amount of each protein was correlated to the activity in the disk diffusion assay (Fig. 2C). The relative concentrations of the predicted protein CC1G_13813 were congruent to the activity profile.
FIGURE 2.
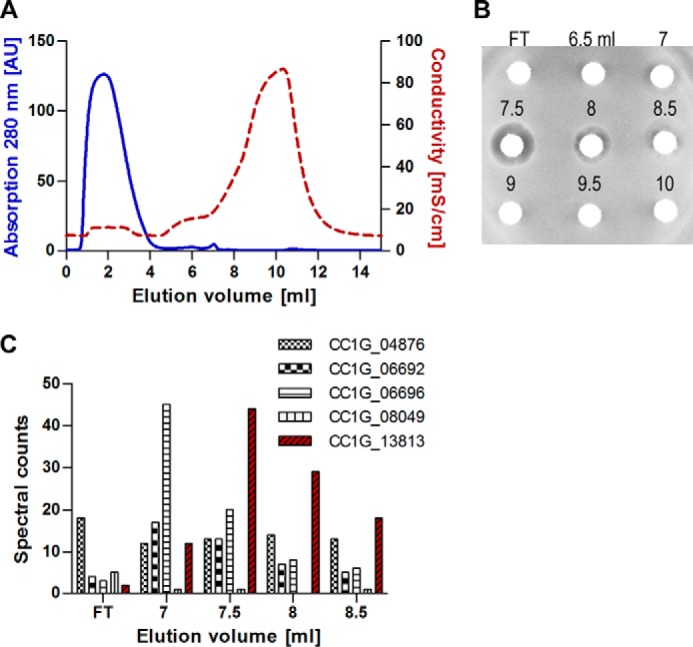
Identification of an AMP in the secretome of C. cinerea. A, proteins were extracted from the unchallenged C. cinerea secretome, digested with proteinase K, and the remaining proteins were fractionated on a cation exchange column. The effluent was monitored at 280 nm (solid line), and the conductivity was measured (dashed line). B, fractions collected (0.5 ml) during the run were spotted in a disk diffusion assay against B. subtilis. Activity was detected from 7 to 8.5 ml of elution volume. C, proteins in the active fractions and flow through (FT) were identified by electrospray ionization-MS/MS and quantified by spectral counting. The relative counts corresponding to the protein CC1G_13813 (copsin) were best correlating to the diameter of the inhibition zones displayed against B. subtilis.
The transcript of the CC1G_13813 locus was analyzed by reverse transcriptase-initiated PCR. The cDNA sequence revealed an open reading frame encoding a precursor protein with in total 184 amino acids. In silico analysis suggested a signal peptide (position 1–23), a pro-peptide (position 24–127), and a C-terminal domain of 57 amino acids corresponding to the mature AMP, which we termed copsin.
To obtain pure copsin, the cDNA encoding the native signal sequence was cloned in a pPICZA expression vector and transformed in P. pastoris. The secreted recombinant product was purified by cation exchange chromatography and yielded a mature polypeptide with a monoisotopic mass of 6059.5 Da, determined by electrospray ionization-MS (theoretical monoisotopic mass, 6059.5 Da).
Sequencing of the N terminus by electrospray ionization-MS/MS of recombinant copsin and of the native polypeptide from C. cinerea revealed the same peptide, both with an N-terminal glutamine converted to a pyroglutamate (data not shown). These findings showed that the recombinant prepro-protein was processed in P. pastoris in the same way as in the host fungus C. cinerea. Mature copsin contained 12 Cys residues, all involved in a disulfide bond as displayed by a mass shift of 12 Da after a reduction with DTT. This was further confirmed by characteristic 13C NMR chemical shifts, which differ from reduced Cys residues. [1H,15N] heteronuclear multiple bond correlation spectroscopy NMR spectra recorded at pH 6.8 and 7.4 exhibited a stable positive charge on the His26 side chain and a delta protonated, uncharged His21. Thus, a net positive charge of +7 was assigned to the polypeptide. A sequence comparison on the AMP database and by the Blastp algorithm exhibited maximum identities of 20–27% of copsin with defensins from invertebrates, fungi, and plants, such as plectasin from P. nigrella or eurocin from Eurotium amstelodami, both assigned to the fungal phylum of ascomycota (Fig. 3A) (5, 27, 28).
FIGURE 3.

Three-dimensional structure of copsin and similarity to other AMPs. A, a sequence alignment of copsin with AMPs exhibited the highest sequence identities with defensins from plants, fungi, and invertebrates (5, 28, 41, 45). Disulfide bonds shown in solid lines form the core structural motif CSαβ. Disulfide bonds in dashed lines were additionally detected in copsin. Regions of the α-helix and the two β-strands as well as the positions of the Cys residues are indicated according to the sequence of copsin. The alignment was performed with the ClustalW algorithm and visualized with the Jalview software (46, 47). B, cartoon representation of the structure of copsin determined by NMR spectroscopy. Left panel, ribbon diagram of the structure with the lowest energy after energy refinement with AMBER (16), highlighting secondary structure elements: α-helical region (red) and the β-sheet (green). The N and C termini are denoted by N and C, respectively, and selected residues are indicated with the residue type and sequence number. Middle panel, bundle of 20 conformers after energy refinement. Right panel, Cys residues are denoted by the sequence position and are colored in yellow for the conserved disulfide bonds and in orange for the three additional disulfide bonds of copsin.
Structural Features of Copsin
The three-dimensional structure was determined using NMR spectroscopy. An almost complete assignment of resonances was obtained (99.1% of backbone and 85.7% of side chain resonances assigned). The input data for the structure calculation and the characterization of the NMR structure are summarized in Table 2. The structure of copsin contains one α-helix (residues 15–23) followed by two β-strands (residues 36–40 and 46–50) and is stabilized by six disulfide bonds (Fig. 3B). The two β-strands form a small antiparallel sheet structure. These secondary structural elements exhibited a high sequence identity to known CSαβ defensins, whereas the loop regions and the termini were very unique in their length and composition. Standard identification of disulfide bonds following proteolytic digest could not be applied, because of a lack of single disulfide bond cleavage products. A comparison of a bundle of NMR structures calculated without disulfide constraints with the three-dimensional structures of other defensins strongly suggests three conserved disulfide bonds: these bonds connect the α-helix to the second β-strand (Cys18–Cys48 and Cys22–Cys50) and the N-terminal region to the first β-strand (Cys10–Cys40) (Fig. 3B). Three additional disulfides stabilize copsin, linking the termini to the loop between the α-helix and the β-sheet. Given that Cys22 is linked to Cys50, it follows that the two cysteines Cys35 and Cys54 are connected. For the remaining two disulfide bonds between the cysteines Cys3, Cys25, Cys32, and Cys57, no direct lead could be obtained from the structural data. To corroborate the four disulfide bonds obtained based on the structure and to find the most probable connectivities for the two remaining disulfides, the disulfide bond pattern was investigated using three established methods: target function analysis (19), ambiguous disulfide restraint (17), and cysteine-cysteine distance measurements (18). All three of these independent methods evaluated the same disulfide bonding pattern as the most likely one: Cys18–Cys48, Cys22–Cys50, Cys10–Cys40, Cys3–Cys32, Cys25–Cys57, and Cys35–Cys54 (Table 1). Thus, the four structural based disulfide bonds were confirmed, and for the two remaining ones the most likely combinations are Cys3–Cys32 and Cys25–Cys57.
TABLE 2.
Structural statistics of the 20 best NMR structures of copsin
Structure calculation by simulated annealing protocol of CYANA (15) after energy minimization using Amber (16) is shown. A, Structure calculation by simulated annealing protocol of CYANA (15) without disulfide constraints. B, Structure calculation by simulated annealing protocol of CYANA (15) with defined ensemble of disulfide bonds. C, Structure calculation parameters after energy minimization using Amber (16).
| Aa | Bb | Cc | |
|---|---|---|---|
| NMR restraints | |||
| Distance restraints | 809 | 823 | 823 |
| Intraresidual and sequential (|i − j| = 1) | 462 | 459 | 459 |
| Medium range (1 < |i − j| < 5) | 115 | 126 | 126 |
| Long range (|i − j|> = 5) | 232 | 230 | 230 |
| Hydrogen bondsd | 8 | 8 | 8 |
| Torsion angles | 0 | 0 | 0 |
| Disulfide bonds | 0 | 6 | 6 |
| Energy statisticse,f | |||
| Average distance constraint violations | 0 | 0 | |
| 0.1–0.2 Å | 8.8 ± 2.0 | ||
| 0.2–0.3 Å | 2.2 ± 1.2 | ||
| 0.3–0.4 Å | 0.2 ± 0.4 | ||
| >0.4 Å | 0.0 ± 0.0 | ||
| Maximal (Å) | 0.28 ± 0.03 | ||
| Average angle constraint violations | 1 | 1 | 0 |
| <5° | 0.0 ± 0.0 | ||
| >5° | 0.0 ± 0.0 | ||
| Maximal (°) | 0.0 ± 0.0 | ||
| Mean AMBER violation energy | |||
| Constraint (kcal mol−1) | 9.1 ±1.2 | ||
| Distance (kcal mol−1) | 9.1 ±1.2 | ||
| Torsion (kcal mol−1) | 0.0 ± 0.0 | ||
| Mean AMBER energy (kcal mol−1) | −2331.2 ± 5.2 | ||
| Mean deviation from ideal covalent geometry | |||
| Bond length (Å) | 0.0043 ± 0.0001 | ||
| Bond angle (°) | 1.728 ± 0.009 | ||
| Ramachandran plot statisticse,f,g | |||
| Residues in most favored regions (%) | 70.4 | 64.2 | 79.2 ± 1.9 |
| Residues in additionally allowed regions (%) | 29.6 | 35.8 | 20.7 ± 2.1 |
| Residues in generously allowed regions (%) | 0.1 | 0.0 | 0.1 ± 0.5 |
| Residues in disallowed regions (%) | 0.0 | 0.0 | 0.0 ± 0.0 |
| Root mean square deviation to mean structuree,f | |||
| Backbone atoms (Å) | 0.50 ± 0.09 | 0.43 ± 0.09 | 0.34 ± 0.09 |
| Heavy atoms (Å) | 0.98 ± 0.16 | 0.87 ± 0.11 | 0.78 ± 0.08 |
| Target function | 0.21 ±0.003 | 0.14 ±0.003 | |
a No constraint for disulfide linkage was used.
b Each disulfide bond was explicitly defined by upper and lower distance limit restraints between the sulfur and carbon atoms of the two linked cysteines: Cys3–Cys32, Cys10–Cys40, Cys18–Cys48, Cys22–Cys50, Cys25–Cys57, and Cys35–Cys54.
c 20 structures of column B with the lowest target function were energy minimized using Amber (16).
d H-bond constraints were identified from HNCO (14) experiments and slow exchanging amide protons in D2O.
e Statistics computed for the best 20 structures (see column C).
f Based on structured residue range, residues 3–57.
g Ramachandran plot, as defined by the program Procheck (48).
The compactness of the structure with its six disulfide bonds and the modifications at the termini are mainly responsible for the very high stability of copsin. The activity was completely retained after heat treatment or incubation at different pH values shown in a disk diffusion assay (Fig. 4, A and B). Copsin exhibited a strong resistance toward different proteases, such as proteinase K, trypsin, or pepsin (Fig. 4C). However, antibacterial activity was lost when treated with a reducing agent, supporting the pivotal role of disulfide bonds for the structural integrity of copsin.
FIGURE 4.
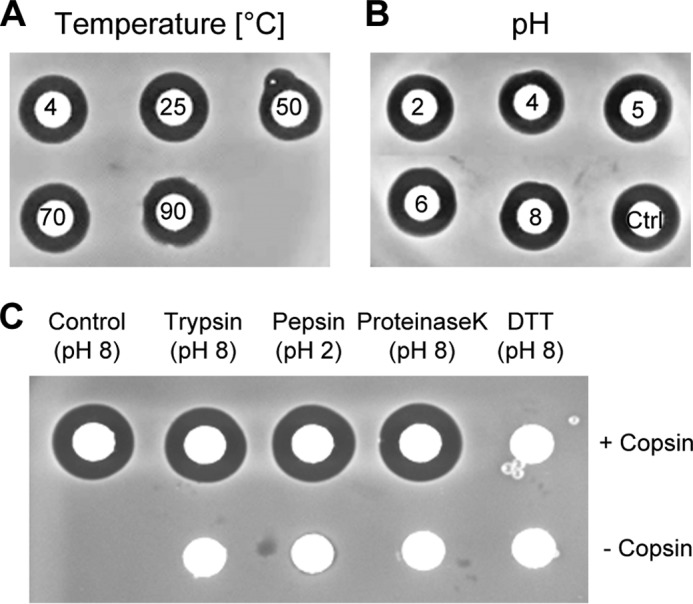
Temperature, pH, and protease stability of copsin. A and B, shown are disk diffusion assays on B. subtilis with copsin that was exposed to different temperatures (4, 25, 50, 70, 90 °C) and in a pH range of 2–8 for 1 h at 25 °C. C, to evaluate the protease resistance, copsin/protease mixtures in a ratio of 10:1 (w/w) were incubated at pH 8 for trypsin and proteinase K and at pH 2 for pepsin for 3 h. Copsin was also subjected to a treatment with 5 mm DTT. Additionally, the reaction mixtures were spotted without copsin. As control, untreated copsin was loaded on a paper disk. The activity of copsin was retained in the whole temperature and pH range tested, and it showed no sensitivity in a treatment with proteases. The only way to delete the activity of copsin was by disrupting the disulfide bonds.
Antibacterial Profile of Copsin
The antibacterial activity of recombinant copsin was tested in disk diffusion assays against a variety of Gram-positive and Gram-negative bacteria. For selected species, the MICs and MBCs were determined by the microdilution broth method in MHB at pH 7.3 (Table 3). Copsin exhibited MIC values in the low microgram per milliliter range for Gram-positive bacteria, such as B. subtilis, Listeria spp., and Enterococcus spp., including a vanA type vancomycin resistant Enterococcus faecium strain. Most potent activity was detected against Listeria monocytogenes with MIC values of 0.25–0.5 μg/ml. A change in pH to 6 in MHB did not affect the activity of copsin, determined against B. subtilis and S. carnosus. Gram-negative species such as E. coli were not affected in viability when exposed to copsin.
TABLE 3.
Antibacterial profile of copsin
MICs and MBCs were determined by the microdilution broth method in MHB, pH 7.3. Active, displayed an inhibition zone in a disk diffusion assay. ND, not determined.
| Bacterium | Source | Copsin |
|
|---|---|---|---|
| MIC | MBC | ||
| μg/ml | |||
| B. subtilis 168 | 1 | 1 | |
| Streptococcus | |||
| S. pneumoniae | DSM 20566 | Active | ND |
| S. pyogenes | DSM 20565 | Active | ND |
| Staphylococcus | |||
| S. carnosus | DSM 20501 | 8 | 8 |
| S. epidermidis | DSM 20044 | 64 | >64 |
| S. aureus 113 | DSM 4910 | >64 | >64 |
| S. aureus | ATCC 29213 | >40 | >40 |
| Listeria | |||
| L. monocytogenes | WSLC 1042 | 0.5 | 1 |
| L. monocytogenes | WSLC 1001 | 0.25 | 0.5 |
| L. ivanovii | WSLC 3009 | 0.25 | 0.5 |
| L. innocua | WSLC 2012 | 0.5 | 1 |
| Enterococcus | |||
| E. faecium | DSM 20477 | 4 | 8 |
| E. faecium VRE | DSM 13590 | 2 | 2 |
| E. faecalis | DSM 20478 | 8 | 16 |
| Micrococcus luteus | DSM 1790 | 0.6 | ND |
| Escherichia coli BL21 | >64 | >64 | |
| Corynebacterium diphtheriae | DSM 44123 | 4 | ND |
MICs and MBCs did not differ more than 2-fold in their value indicating a bactericidal effect of copsin on Gram-positive bacteria. To verify this finding, a kill curve assay using B. subtilis was performed in MHB at pH 6 and 7.3 (Fig. 5). Independent of the pH, a clear reduction of the viable count was observed after 30 min of incubation, supporting a bactericidal activity of copsin.
FIGURE 5.
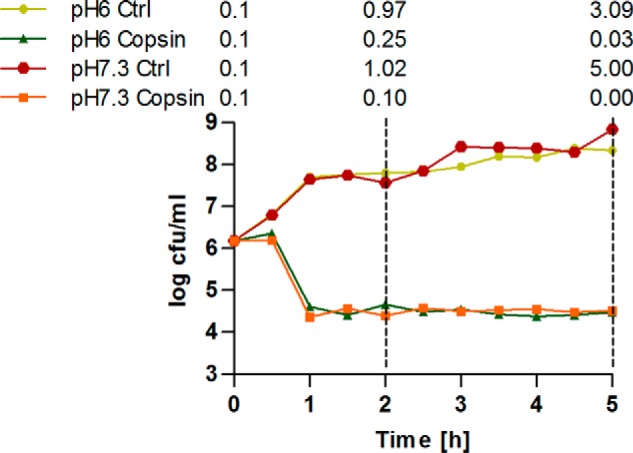
Killing kinetics of copsin. B. subtilis grown in MHB at pH 6 and 7.3 and incubated with and without (Ctrl) 4 μg/ml copsin (4× MIC). The A600 is shown above the spectrum, measured at 0, 2, and 5 h.
Molecular Target of Copsin in Bacteria
The antibacterial activity and specificity of copsin was comparable with cell wall biosynthesis interfering antibiotics such as plectasin or vancomycin (6). We therefore determined the cellular localization of copsin on B. subtilis and S. carnosus cells during exponential growth phase, using TAMRA-labeled copsin and BODIPY-FL-labeled vancomycin as a control. Fig. 6 shows a dual staining of BODIPY-vancomycin- and TAMRA-labeled copsin. Vancomycin is known to localize specifically to sites of active cell wall synthesis (29). Copsin exhibited binding at the cell surface, preferably in curved regions of the rod-shaped bacterium B. subtilis and similarly distributed on the spherical-shaped S. carnosus cells. A co-localization with vancomycin was found at the cell septa.
FIGURE 6.

Co-localization of copsin and vancomycin. B. subtilis and S. carnosus cells in the exponential growth phase were stained with TAMRA-copsin (red) and subsequently stained with BODIPY-vancomycin (green). Cells showed a co-localization of copsin with vancomycin at the cell septa. Scale bars, 2 μm.
To gain further insights on the molecular targets, we studied the binding affinity of copsin to essential precursors of bacterial cell wall synthesis in vitro. Lipid I was synthesized as N-acetylmuramic acid-pentapeptide (MurNAc-pentapeptide) linked to the bactoprenol-pyrophosphate lipid carrier (bactoprenol-pyrophosphate, C55-PP). The pentapeptide was composed of l-alanyl-γ-d-glutamyl-l-lysyl-d-alanyl-d-alanine, a composition found in Staphylococcus spp. For lipid II, GlcNAc was attached to the MurNAc moiety of lipid I (Fig. 7A). Binding studies included bactoprenol-phosphate (C55-P) and lipid III, a wall teichoic acid synthesis precursor consisting of GlcNAc linked to C55-PP (30). Copsin was incubated with the synthesized compounds in a defined molar ratio for 20 min, and the free substrate was analyzed by TLC (Fig. 7B). It was found that copsin bound in a 1:1 molar ratio to lipid I and II but had no affinity for C55-P and lipid III.
FIGURE 7.
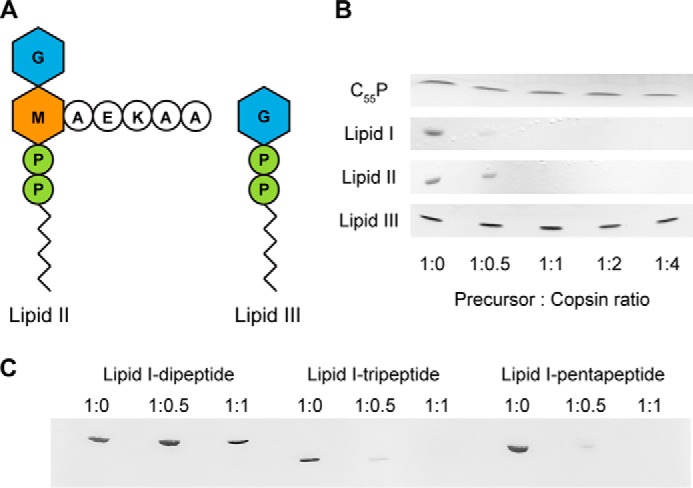
Binding of copsin to cell wall precursors. A, schematic of the peptidoglycan precursor lipid II and the wall teichoic acid precursor lipid III. M, MurNAc; G, GlcNAc; P, phosphate. B, purified cell wall precursors were incubated with increasing molar ratios of copsin. After extraction with n-butanol/pyridine acetate (pH 4.2), the samples were analyzed by TLC, which displayed a binding of copsin to lipids I and II. C, truncated versions of lipid I (lipid I-dipeptide and lipid I-tripeptide) as well as lipid I-pentapeptide were synthesized, using purified enzymes from S. aureus. Copsin was added in increasing molar ratios to the lipid I versions. After incubation for 20 min at room temperature, samples were extracted as described and analyzed by TLC. The result showed that copsin binds to the third position of the pentapeptide.
These findings suggested specific interaction of copsin with lipids I and II and indicated that MurNAc-pentapeptide might be pivotal for a stable complex. To examine whether the peptide chain was necessary for binding to copsin, truncated versions of the pentapeptide were synthesized, and the binding affinity to copsin was analyzed (Fig. 7C). Copsin showed a strongly reduced affinity for the lipid I-dipeptide (l-Ala–d-Glu) in comparison to the lipid I-tripeptide (l-Ala–d-Glu–l-Lys). This result demonstrated that the third position of the pentapeptide is essential for stable binding of copsin to the peptidoglycan precursor.
Based on the hypothesis that copsin bound to lipid II, we wanted to know whether copsin was able to permeabilize the bacterial membrane, similar to the mode of action of nisin, a pore-forming lantibiotic (25, 31). Two assays were performed: the first relied on CF efflux from lipid II containing liposomes (Fig. 8A), and the second relied on the potassium efflux of B. subtilis cells (data not shown). In both assays, no pore formation was detected. Importantly, preincubation of the lipid II containing liposomes with copsin inhibited the action of nisin, which was used as a positive control (Fig. 8B). These results showed that copsin interacted specifically with lipid II at the extracellular side of the bacterial membrane and prevented the binding of nisin but did not lead to pore formation.
FIGURE 8.
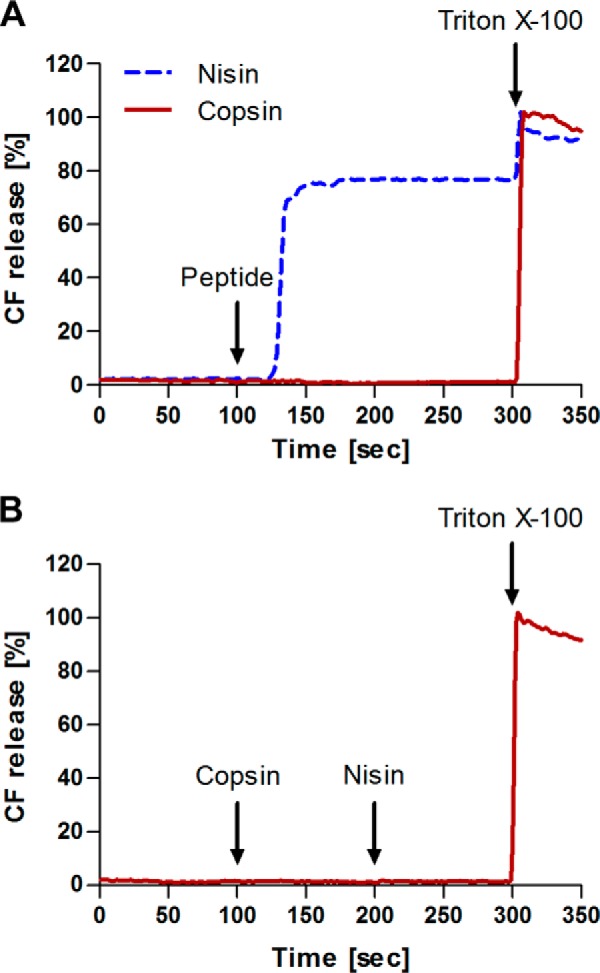
Carboxyfluorescein efflux from lipid II containing liposomes. Activity of copsin and nisin against unilamellar liposomes made of 1,2-dioleoyl-sn-glycero-3-phosphocholine supplemented with 0.1 mol % lipid II. Peptide-induced marker release from liposomes with entrapped CF was measured. The 100% leakage level was determined by addition of Triton X-100 after 300 s. A, 1 μm copsin (solid line) or nisin (dashed line) was added after 100 s. B, first, 1 μm copsin was added (100 s) following the addition of 1 μm nisin (200 s) to the same sample. Copsin did not permeabilize the membrane, but it blocked the action of nisin.
DISCUSSION
Co-cultivation studies of C. cinerea with bacteria led to the identification of the peptide-based antibiotic copsin, to our knowledge the first defensin identified and characterized in the fungal phylum of basidiomycota. Growing fungi on the surface of inert glass beads submerged in liquid medium provides a reproducible and easy to handle model system for studying the interaction with bacteria. In comparison to a confrontation assay on an agar plate or in a pure liquid culture, this setup has the big advantage of combining a solid surface and a humid environment, two pivotal factors for growth of bacteria and fungi in nature. Furthermore, it allows for a repeatable extraction of secreted fungal and bacterial substances to perform an appropriate analysis at the metabolomic and proteomic level. For our assays, B. subtilis, E. coli, and P. aeruginosa were selected as competitors, which are well characterized and are known to interact with fungi (1, 32, 33). Under the conditions tested, we observed an antagonistic behavior of the fungus with B. subtilis and P. aeruginosa and a more commensal association with E. coli. These differentiated interactions suggest an interdependent community of C. cinerea and bacteria on herbivorous dung to preserve their nutritional niche. Different studies demonstrated that AMPs act as regulators of microbial diversity, for example, α-defensins in mice or arminin peptides in Hydra species (34, 35). Based on the inhibition found against B. subtilis, we developed a novel workflow for the purification and identification of highly stable and active antibacterial peptides and proteins. This analytical method involved a quantitative MS measurement, where we finally selected copsin for further investigations.
P. pastoris is a highly efficient system for the heterologous production of secreted proteins at high yields in shake flasks and bioreactors (36). It ensured also a correct processing of copsin, because P. pastoris expresses a Kex2 protease that cleaves the pro-region at the lysine-arginine site, frequently identified in defensins (37). The three-dimensional structure of recombinant copsin was solved by NMR and revealed a CSαβ fold with a net charge of +7 at neutral pH. The CSαβ structural motif is a major characteristic of defensins of plants, invertebrates, and fungi and is differentiating them from vertebrate defensins with a common motif of a triple-stranded β-sheet structure (38). Copsin is stabilized by a unique connectivity of six cysteine bonds in contrast to most other CSαβ defensins, which are linked by three or four disulfide bonds. The structural compactness combined with an N-terminal pyroglutamate and a C-terminal cysteine involved in a disulfide bond render copsin extremely stable in a wide pH and temperature range and insensitive toward proteases.
Analysis of predicted fungal proteomes revealed different classes of CSαβ defensins primarily in the phylum of ascomycota (37, 39). Based on the structural similarity, an orthologous relation of copsin to these fungal defensins seems to be likely and is further supported by the presence of CSαβ-like bacteriocins as a possible common ancestor (38, 39). However, because the similarity is limited to the secondary structural elements, we cannot exclude a structural convergence toward a particular function such as a highly stabilized fold. More comprehensive studies are needed to conclusively define the relationship between different classes of fungal defensins.
Microscopy studies and binding assays with cell wall precursors revealed lipid II as the molecular target of copsin. Lipid II is a highly conserved molecule throughout the bacterial kingdom and is accessible at the extracellular side of the bacterial membrane. Binding to this essential building block is an effective way to interfere with a proper cell wall synthesis and consequently to kill a bacterium (40). In addition, it is unlikely that toxic effects on, for example, mammalian cells would occur because of the unique structural composition of this peptidoglycan precursor. Antibiotics found to interact with lipid II exert their actions over defined binding sites, such as vancomycin, which interacts with the d-Ala residues of the pentapeptide (29). In depth studies of plectasin exhibited that the N-terminal GFGC part is essential for a correct binding to pyrophosphate and d-Glu of lipid II (6). This motif is found in many other defensins such as MGD-1 or eurocin but is absent in copsin (28, 41). Binding studies of copsin with truncated versions of lipid I revealed that, instead of d-Ala and d-Glu, the third amino acid in the pentapeptide side chain is crucial for binding to copsin, independently of whether l-Lys or diaminopimelic acid is located at this position. This finding is consistent with the strong activity of copsin against a vancomycin-resistant E. faecium strain, where d-Lac is located at the C-terminal site of lipid II instead of d-Ala (42).
Copsin exhibited a distinct antibacterial profile differentiating it from other fungal defensins such as plectasin or micasin (5, 37). Besides Enterococcus spp., copsin exerted its most potent activity against L. monocytogenes, a food-borne pathogen causing severe forms of listeriosis in animals and humans (43, 44). The exceptional stability of copsin and the potent activity against bacteria are important features for further applications in clinics or food industry.
Acknowledgments
We thank the Functional Genomic Center Zurich for technical support, in particular S. Chesnov and R. Brunisholz. We are grateful to J. Hintze for support in recombinant production of copsin and R. Sieber for help in performing antibacterial assays.
This work was supported by the ETH Zurich.
The atomic coordinates and structure factors (code 2MN5) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The chemical shift assignments are available from the BioMagResBank under Number 19880.
- AMP
- antimicrobial peptide
- CSαβ
- cysteine stabilized α/β-fold
- CCMM
- C. cinerea minimal medium
- MIC
- minimal inhibitory concentration
- MBC
- minimal bactericidal concentration
- MHB
- Mueller-Hinton broth
- CF
- carboxyflourescein
- MurNAc
- N-acetylmuramic acid.
REFERENCES
- 1. Frey-Klett P., Burlinson P., Deveau A., Barret M., Tarkka M., Sarniguet A. (2011) Bacterial-fungal interactions: hyphens between agricultural, clinical, environmental, and food microbiologists. Microbiol. Mol. Biol. Rev. 75, 583–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Scherlach K., Graupner K., Hertweck C. (2013) Molecular bacteria-fungi interactions: effects on environment, food, and medicine. Annu. Rev. Microbiol. 67, 375–397 [DOI] [PubMed] [Google Scholar]
- 3. Fleming A. (1929) On the antibacterial action of cultures of a Penicillium, with special reference to their use in the isolation of B. influenzae. Br. J. Exp. Pathol. 10, 226–236 [Google Scholar]
- 4. Zasloff M. (2002) Antimicrobial peptides of multicellular organisms. Nature 415, 389–395 [DOI] [PubMed] [Google Scholar]
- 5. Mygind P. H., Fischer R. L., Schnorr K. M., Hansen M. T., Sönksen C. P., Ludvigsen S., Raventós D., Buskov S., Christensen B., De Maria L., Taboureau O., Yaver D., Elvig-Jørgensen S. G., Sørensen M. V., Christensen B. E., Kjaerulff S., Frimodt-Moller N., Lehrer R. I., Zasloff M., Kristensen H. H. (2005) Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus. Nature 437, 975–980 [DOI] [PubMed] [Google Scholar]
- 6. Schneider T., Kruse T., Wimmer R., Wiedemann I., Sass V., Pag U., Jansen A., Nielsen A. K., Mygind P. H., Raventós D. S., Neve S., Ravn B., Bonvin A. M., De Maria L., Andersen A. S., Gammelgaard L. K., Sahl H. G., Kristensen H. H. (2010) Plectasin, a fungal defensin, targets the bacterial cell wall precursor Lipid II. Science 328, 1168–1172 [DOI] [PubMed] [Google Scholar]
- 7. Schneider T., Sahl H. G. (2010) An oldie but a goodie: cell wall biosynthesis as antibiotic target pathway. Int. J. Med. Microbiol. 300, 161–169 [DOI] [PubMed] [Google Scholar]
- 8. Hancock R. E., Sahl H. G. (2006) Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24, 1551–1557 [DOI] [PubMed] [Google Scholar]
- 9. van Schöll L., Hoffland E., van Breemen N. (2006) Organic anion exudation by ectomycorrhizal fungi and Pinus sylvestris in response to nutrient deficiencies. New Phytol. 170, 153–163 [DOI] [PubMed] [Google Scholar]
- 10. Wu S., Letchworth G. J. (2004) High efficiency transformation by electroporation of Pichia pastoris pretreated with lithium acetate and dithiothreitol. BioTechniques 36, 152–154 [DOI] [PubMed] [Google Scholar]
- 11. Sugiki T., Ichikawa O., Miyazawa-Onami M., Shimada I., Takahashi H. (2012) Isotopic labeling of heterologous proteins in the yeast Pichia pastoris and Kluyveromyces lactis. Methods Mol. Biol. 831, 19–36 [DOI] [PubMed] [Google Scholar]
- 12. Blomberg F., Maurer W., Rüterjans H. (1977) Nuclear magnetic resonance investigation of 15N-labeled histidine in aqueous solution. J. Am. Chem. Soc. 99, 8149–8159 [DOI] [PubMed] [Google Scholar]
- 13. Güntert P., Mumenthaler C., Wüthrich K. (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273, 283–298 [DOI] [PubMed] [Google Scholar]
- 14. Dingley A. J., Nisius L., Cordier F., Grzesiek S. (2008) Direct detection of N-H[·]N hydrogen bonds in biomolecules by NMR spectroscopy. Nat. Protoc. 3, 242–248 [DOI] [PubMed] [Google Scholar]
- 15. Güntert P. (2004) Automated NMR structure calculation with CYANA. Methods Mol. Biol. 278, 353–378 [DOI] [PubMed] [Google Scholar]
- 16. Cornell W. D., Cieplak P., Bayly C. I., Gould I. R., Merz K. M., Ferguson D. M., Spellmeyer D. C., Fox T., Caldwell J. W., Kollman P. A. (1995) A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117, 5179–5197 [Google Scholar]
- 17. Nilges M. (1995) Calculation of protein structures with ambiguous distance restraints. Automated assignment of ambiguous NOE crosspeaks and disulphide connectivities. J. Mol. Biol. 245, 645–660 [DOI] [PubMed] [Google Scholar]
- 18. Klaus W., Broger C., Gerber P., Senn H. (1993) Determination of the disulphide bonding pattern in proteins by local and global analysis of nuclear magnetic resonance data. Application to flavoridin. J. Mol. Biol. 232, 897–906 [DOI] [PubMed] [Google Scholar]
- 19. Heitz A., Chiche L., Le-Nguyen D., Castro B. (1989) 1H 2D NMR and distance geometry study of the folding of Ecballium elaterium trypsin inhibitor, a member of the squash inhibitors family. Biochemistry 28, 2392–2398 [DOI] [PubMed] [Google Scholar]
- 20. Motyl M., Dorso K., Barrett J., Giacobbe R. (2006) Basic microbiological techniques used in antibacterial drug discovery. Curr. Protoc. Pharmacol. Chapter 13, Unit 13A.3 [DOI] [PubMed] [Google Scholar]
- 21. Brötz H., Bierbaum G., Reynolds P. E., Sahl H. G. (1997) The lantibiotic mersacidin inhibits peptidoglycan biosynthesis at the level of transglycosylation. Eur. J. Biochem. 246, 193–199 [DOI] [PubMed] [Google Scholar]
- 22. Müller A., Ulm H., Reder-Christ K., Sahl H. G., Schneider T. (2012) Interaction of type A lantibiotics with undecaprenol-bound cell envelope precursors. Microb. Drug Resist. 18, 261–270 [DOI] [PubMed] [Google Scholar]
- 23. Schneider T., Senn M. M., Berger-Bächi B., Tossi A., Sahl H. G., Wiedemann I. (2004) In vitro assembly of a complete, pentaglycine interpeptide bridge containing cell wall precursor (lipid II-Gly5) of Staphylococcus aureus. Mol. Microbiol. 53, 675–685 [DOI] [PubMed] [Google Scholar]
- 24. Rick P. D., Hubbard G. L., Kitaoka M., Nagaki H., Kinoshita T., Dowd S., Simplaceanu V., Ho C. (1998) Characterization of the lipid-carrier involved in the synthesis of enterobacterial common antigen (ECA) and identification of a novel phosphoglyceride in a mutant of Salmonella typhimurium defective in ECA synthesis. Glycobiology 8, 557–567 [DOI] [PubMed] [Google Scholar]
- 25. Wiedemann I., Breukink E., van Kraaij C., Kuipers O. P., Bierbaum G., de Kruijff B., Sahl H. G. (2001) Specific binding of nisin to the peptidoglycan precursor lipid II combines pore formation and inhibition of cell wall biosynthesis for potent antibiotic activity. J. Biol. Chem. 276, 1772–1779 [DOI] [PubMed] [Google Scholar]
- 26. Dix N. J., Webster J. (1995) Coprophilous fungi. In Fungal Ecology, pp. 203–224, Chapman and Hall, London [Google Scholar]
- 27. Wang G., Li X., Wang Z. (2009) APD2: the updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 37, D933–D937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oeemig J. S., Lynggaard C., Knudsen D. H., Hansen F. T., Nørgaard K. D., Schneider T., Vad B. S., Sandvang D. H., Nielsen L. A., Neve S., Kristensen H. H., Sahl H. G., Otzen D. E., Wimmer R. (2012) Eurocin, a new fungal defensin: structure, lipid binding, and its mode of action. J. Biol. Chem. 287, 42361–42372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sheldrick G. M., Jones P. G., Kennard O., Williams D. H., Smith G. A. (1978) Structure of vancomycin and its complex with acetyl-d-alanyl-d-alanine. Nature 271, 223–225 [DOI] [PubMed] [Google Scholar]
- 30. Xia G., Kohler T., Peschel A. (2010) The wall teichoic acid and lipoteichoic acid polymers of Staphylococcus aureus. Int. J. Med. Microbiol. 300, 148–154 [DOI] [PubMed] [Google Scholar]
- 31. Breukink E., Wiedemann I., van Kraaij C., Kuipers O. P., Sahl H. G., de Kruijff B. (1999) Use of the cell wall precursor lipid II by a pore-forming peptide antibiotic. Science 286, 2361–2364 [DOI] [PubMed] [Google Scholar]
- 32. Peleg A. Y., Hogan D. A., Mylonakis E. (2010) Medically important bacterial-fungal interactions. Nat. Rev. Microbiol. 8, 340–349 [DOI] [PubMed] [Google Scholar]
- 33. Nagórska K., Bikowski M., Obuchowski M. (2007) Multicellular behaviour and production of a wide variety of toxic substances support usage of Bacillus subtilis as a powerful biocontrol agent. Acta Biochim. Pol. 54, 495–508 [PubMed] [Google Scholar]
- 34. Salzman N. H., Hung K., Haribhai D., Chu H., Karlsson-Sjöberg J., Amir E., Teggatz P., Barman M., Hayward M., Eastwood D., Stoel M., Zhou Y., Sodergren E., Weinstock G. M., Bevins C. L., Williams C. B., Bos N. A. (2010) Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 11, 76–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Franzenburg S., Walter J., Künzel S., Wang J., Baines J. F., Bosch T. C., Fraune S. (2013) Distinct antimicrobial peptide expression determines host species-specific bacterial associations. Proc. Natl. Acad. Sci. U.S.A. 110, E3730–E3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cereghino J. L., Cregg J. M. (2000) Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol. Rev. 24, 45–66 [DOI] [PubMed] [Google Scholar]
- 37. Zhu S., Gao B., Harvey P. J., Craik D. J. (2012) Dermatophytic defensin with antiinfective potential. Proc. Natl. Acad. Sci. U.S.A. 109, 8495–8500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yeaman M. R., Yount N. Y. (2007) Unifying themes in host defence effector polypeptides. Nat. Rev. Microbiol. 5, 727–740 [DOI] [PubMed] [Google Scholar]
- 39. Zhu S. (2008) Discovery of six families of fungal defensin-like peptides provides insights into origin and evolution of the CSαβ defensins. Mol. Immunol. 45, 828–838 [DOI] [PubMed] [Google Scholar]
- 40. de Kruijff B., van Dam V., Breukink E. (2008) Lipid II: a central component in bacterial cell wall synthesis and a target for antibiotics. Prostaglandins Leukot. Essent. Fatty Acids 79, 117–121 [DOI] [PubMed] [Google Scholar]
- 41. Yang Y. S., Mitta G., Chavanieu A., Calas B., Sanchez J. F., Roch P., Aumelas A. (2000) Solution structure and activity of the synthetic four-disulfide bond Mediterranean mussel defensin (MGD-1). Biochemistry 39, 14436–14447 [DOI] [PubMed] [Google Scholar]
- 42. Arthur M., Courvalin P. (1993) Genetics and mechanisms of glycopeptide resistance in Enterococci. Antimicrob. Agents Chemother. 37, 1563–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Farber J. M., Peterkin P. I. (1991) Listeria monocytogenes, a food-borne pathogen. Microbiol. Rev. 55, 476–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Arias C. A., Murray B. E. (2012) The rise of the Enterococcus: beyond vancomycin resistance. Nat. Rev. Microbiol. 10, 266–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fant F., Vranken W., Broekaert W., Borremans F. (1998) Determination of the three-dimensional solution structure of Raphanus sativus antifungal protein 1 by 1H NMR. J. Mol. Biol. 279, 257–270 [DOI] [PubMed] [Google Scholar]
- 46. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 47. Waterhouse A. M., Procter J. B., Martin D. M., Clamp M., Barton G. J. (2009) Jalview Version 2a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Laskowski R. A., Rullmann J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 49. Sambrook J., Russell D. W. (2001) Molecular Cloning, 3rd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]